

Abstract
In late 2019, a novel coronavirus began spreading in Wuhan, China, causing a potentially lethal respiratory viral infection. By early 2020, the novel coronavirus, called SARS-CoV-2, had spread globally, causing the COVID-19 pandemic. The infection and mutation rates of SARS-CoV-2 make it amenable to tracking introduction, spread and evolution by viral genome sequencing. Efforts to develop effective public health policies, therapeutics, or vaccines to treat or prevent COVID-19 are also expected to benefit from tracking mutations of the SARS-CoV-2 virus. Here we describe a set of comprehensive working protocols, from viral RNA extraction to analysis using established visualization tools, for high throughput sequencing of SARS-CoV-2 viral genomes using a MinION instrument. This set of protocols should serve as a reliable “how-to” reference for generating quality SARS-CoV-2 genome sequences with ARTIC primer sets and long-read nanopore sequencing technology. In addition, many of the preparation, quality control, and analysis steps will be generally applicable to other sequencing platforms.
Keywords: SARS-CoV-2, sequencing, nanopore, MinION, COVID-19, genome
INTRODUCTION
COVID-19, an ongoing global pandemic, has taken the lives of countless people worldwide (https://www.worldometers.info/coronavirus/). Effective pandemic response and infection containment have remained challenging [1]. The ability of viruses to evolve quickly further underscores the need for constant surveillance and monitoring, especially at the genetic level for tracking emergence of novel variants of concern [2]. Viral genome sequencing can help address these challenges and shed light on the identity and evolution of genetic variants [3]. Additionally, viral genome sequencing can delineate conserved and mutable regions which would provide valuable insights in the development of effective treatments and vaccines [4-6].
Several different approaches exist to sequence the genome of SARS-CoV-2, the coronavirus that causes COVID-19, such as metagenomic sequencing, targeted enrichment sequencing and PCR-amplification based methods [6]. Metagenomic and target enrichment sequencing are prohibitively expensive to generate large-scale complete genomes and are associated with incomplete coverage and depth [7]. A solution to address these limitations is a PCR-based amplification which enriches and amplifies the target of interest at the same time [8]. By utilizing a tiling amplicon scheme with multiplex PCR, it is possible to generate enough coverage and depth to produce complete genome sequences of SARS-CoV-2 in a cost-effective manner [9]. This approach can even reconstruct complete genomes of samples that have partially degraded viral RNA genomes or low viral load.
Several groups have demonstrated the ability to sequence SARS-CoV-2 using nanopore sequencing with high accuracy at a consensus-level rate and high sensitivity for detecting single nucleotide variants (SNVs) [10-13]. Our lab adopted and modified protocols initially developed by the ARTIC Network [9] to sequence SARS-CoV-2 viral genomes using the MinION nanopore sequencing platform from Oxford Nanopore Technologies (ONT). We present here an entire validated workflow in a 96-well plate format, including a how-to guide for every step from viral RNA extraction to data visualization. Beyond compiling many protocols into a detailed how-to guide, we improved the interfacing and efficiency of individual steps. These include many small, or in some cases quite significant, changes to RNA extraction, cDNA synthesis, qPCR, ARTIC multiplex PCR, sequencing library preparation, and data analysis. These protocols have been validated and benchmarked through the successful sequencing of over 4000 complete SARS-CoV-2 genomes to date from the U.S. state of Illinois, which are available on the GISAID (Global Initiative on Sharing All Influenza Data) database and publicly viewable on Nextstrain (https://nextstrain.org/groups/illinois-gagnon-public/ncov/gagnon).
RESULTS AND DISCUSSION
The entire workflow to generate SARS-CoV-2 genomes (Fig. 1) is performed in a 96-well plate format. All detailed working protocols are provided in the supplemental material and are referenced throughout. The workflow begins with RNA extraction from viral transport media (VTM) containing patient-derived nasopharyngeal (NP) swabs or potentially other clinical sample sources like saliva. Extracted RNA is converted to complementary DNA (cDNA) and viral load approximated by quantitative PCR (qPCR). Following qPCR each sample under a certain cycle threshold (Ct) undergoes two multiplex PCR reactions using ARTIC V3 primer pools that specifically amplify the SARS-CoV-2 viral genome to generate overlapping amplicon products. PCR amplicons can then be optionally visualized by gel electrophoresis. The two separate ARTIC PCR product pools for each sample are pooled and the amplicons purified through a clean-up step with paramagnetic Solid Phase Reversible Immobilization (SPRI) beads. The purified PCR amplicons can be optionally quantified by Qubit fluorescence before library preparation, which entails an amplicon end-preparation, 96-well sample barcoding, barcoded amplicon pooling and clean-up, and adapter ligation and clean-up. Sample library is loaded onto a MinION nanopore sequencer (ONT) and sequenced. The bioinformatic analysis to generate whole genome consensus sequences involves basecalling of raw reads, demultiplexing of barcodes, filtering by length, alignment to the SARS-CoV-2 genome, variant calling and consensus sequence generation. Finally, lineage and clade of the consensus genomes can be assigned using online or locally implemented tools such as Pangolin and Nextclade (www.pangolin.cog-uk.io) [14]. Phylogenetic analyses can be performed using Nextstrain [15] and results visualized on nextstrain.org. An overview of workflow steps is provided and detailed working protocols for each step are included in Supplemental Materials.
Figure 1.
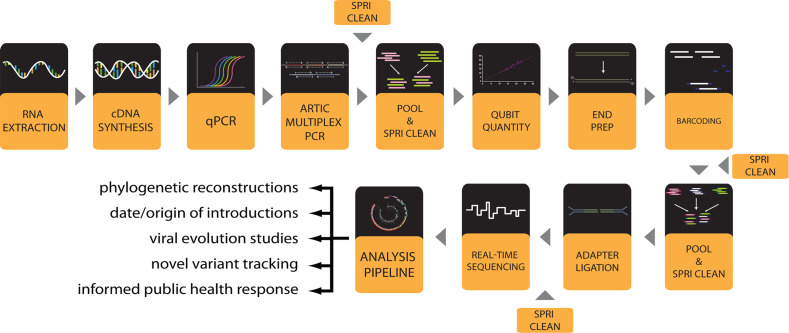
Workflow for SARS-CoV-2 viral genome sequencing with a MinION instrument.
Extraction of viral RNA genomes
The viral RNA genome is typically extracted from various VTM solutions of clinical COVID-19 samples. Samples should be stored at –80°C, thawed on ice, and kept cold to maintain viral genome integrity. To increase safety and compliance, our laboratory only receives samples from clinics or public health laboratories that have first been treated with a lysis buffer or inactivation solution [16] that is compatible with our RNA extraction protocol (Working protocol S1). The RNA extraction is carried out in a BSL-2 class biosafety cabinet. A magnetic bead-based extraction is performed using a 96-well magnetic plate following the manufacturer’s recommended protocol with some minor modifications. These include the use of home-made (HM) lysis (inactivation) solution (Working protocol S1) in place of, or mixed 1:1 with, the manufacturer’s lysis solution. Additionally, elution is performed in a buffer containing EDTA and an RNase inhibitor to help preserve the quality and integrity of RNA. Once RNA extraction is complete, RNA samples are kept on ice and converted to cDNA as soon as possible. The extracted RNA is then stored in heat-sealed plates at –80°C for long-term storage.
During the RNA extraction it is always recommended to include a negative control containing phosphate buffered saline (PBS) or clean VTM in place of a patient sample in at least one well of each 96-well plate. Negative control samples should proceed through the entire workflow and be sequenced to determine the extent of any contamination. It should be assumed that contaminating amplicons in the negative control might be present in all samples. Additionally, a positive commercially available SARS-CoV-2 RNA sample with a known genetic sequence can be used to validate the workflow at early stages but does not need to be included in every sequencing run. These controls can help identify general cross-contamination issues and ensure efficient extraction. When setting up the workflow for the first time, extracted RNA can be quantified using a spectrophotometer and resolved on denaturing agarose gels or analyzed using a bioanalyzer to evaluate RNA extraction efficiency and integrity. We do not provide protocols for these optional steps.
Reverse transcription of viral RNA to cDNA
Following extraction, RNA is converted to cDNA (Working protocol S2) as soon as possible, preferably without a freeze-thaw cycle, to prevent potential RNA degradation. Initially, we used ABI Hi-Capacity cDNA Synthesis Kit but have since switched to Lunascript RT SuperMix kit (New England Biolabs) because of the short reaction time and single master mix containing dye, which accelerated and simplified the workflow and reduced contamination potential. However, for the ABI Hi-Capacity cDNA synthesis reactions, we found 10 μl of RNA template yields comparable results to 5 μl of template when displayed on gel electrophoresis following ARTIC PCR (Fig. S1). When using Lunascript RT SuperMix for cDNA synthesis, we found that a constant volume (16 μl) of template in a 20 μl cDNA reaction was reproducible and reliable (Working protocol S2). After synthesis, cDNA can be stored temporarily at 4°C while awaiting qPCR and multiplex ARTIC PCR. For long-term stability, store cDNA at –20°C or –80°C after heat-sealing the 96-well plate.
qPCR to approximate viral load
To quantify approximate viral load in each sample, qPCR is performed using cDNA as template. To reduce costs and maintain consistency, we typically use only one primer-probe set, commercially available from Integrated DNA Technologies (IDT). This primer-probe set, N2, detects the nucleocapsid (N) gene and is identical to one of the primer-probe sets recommended early in the pandemic for qPCR-based patient diagnosis of probable COVID-19 infection [11]. We typically use the PrimeTime Gene Expression Master Mix (IDT) and manufacturer’s recommended protocol for qPCR reactions (Working protocol S3). To help standardize quantitation across plates, the same baseline threshold value is set for each run to consistently call Ct values (Fig. 2A). In our experience, the Ct values determined for any given sample are higher than those usually reported (when such data is available) by the facilities that provided patient samples. We calculated the mean Ct value of samples from the originating lab to be 20.71 while mean Ct value using our qPCR assay was 28.36 (Fig. S2). This is possibly due to a number of factors, including variations in kits used, freeze-thaw cycles, and sample handling. Our recommendation is to avoid freeze-thaw of purified RNA before cDNA synthesis to help reduce further loss of viral genome integrity.
Figure 2.
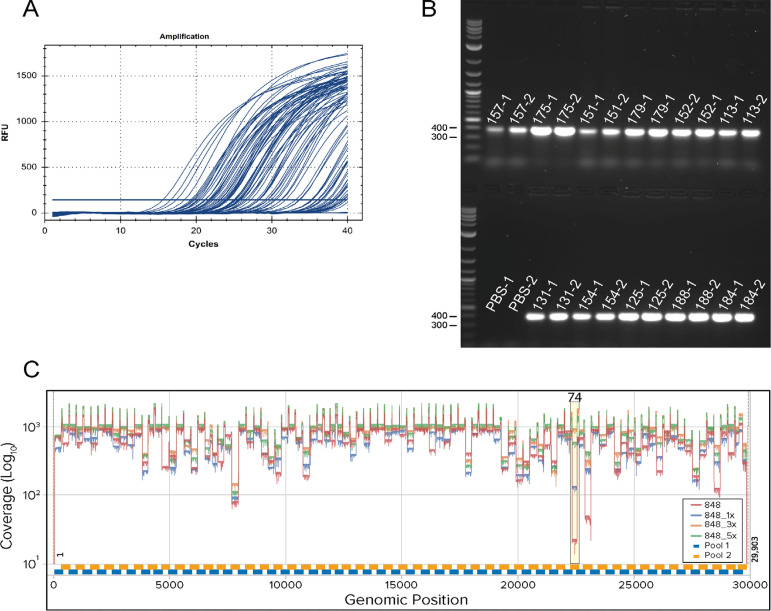
Representative qPCR and ARTIC PCR and genome sequencing coverage.A. Amplification curves from SARS-CoV-2 qPCR assay and IDT 2019-nCoV CDC approved N2 primers and probes. Baseline adjustment was manually set to 200. B. Representative agarose gel electrophoresis of ~400 bp PCR products amplified using ARTIC nCoV V3 Primers (IDT). Reaction pool 1 (labels with suffixes -1) and pool 2 (labels with suffixes -2) were run side by side with 5 μl of sample on the gel. PBS (no template) extraction controls for pool 1 and 2 show no contamination. C. Coverage plot with amplicon 74 dropout using ARTIC V3 Primers (bottom panel). The plot shows no spike-in (red), 1× (15 nM) (blue), 3× (45 nM) (orange) and 5x (75 nM) (green) of 74_Right and 74_Left for sample 848 (Ct = 20.68) with coverage for amplicon 74 of 18×, 126×, 929× and 541×, respectively. Experiments were conducted at the same time using identical reagents and protocols. Amplicon 74 region is highlighted in yellow.
Multiplex ARTIC PCR to amplify viral genome sequences
Once qPCR is performed, viral genome cDNA is amplified to increase the copy number of viral genomes. We use two pools of primers designed by the ARTIC Network [8,9], which are commercially available from IDT as the ARTIC nCoV-2019 V3 primer set. These require two pools of PCR reactions for every sample, with pool 1 containing 55 primer sets and pool 2 containing 54 primer sets. Two primer pools are used to create overlapping amplicons that reduce interference during PCR and prevent short overlapping products from being preferentially produced (Fig. 2B). Through this tiling amplicon scheme, complete coverage of the genome is achieved. We use the Q5 High-Fidelity DNA Polymerase from New England Biolabs (NEB) for PCR reactions (Working protocol S4).
We have optimized the PCR cycling parameters to generate higher and more consistent coverage from higher Ct samples than can typically be achieved with previously described ARTIC PCR protocols in our experience (data not shown) [8,9]. The new cycling uses the principle of touchdown PCR to continually reduce the annealing temperature slightly during the first 20 cycles from 65°C to 63°C to improve specificity. Despite modified cycling, one amplicon continually dropped out: amplicon 74 in primer pool 2. Others have previously noted the dropout of amplicon 74 and found that the abundance of amplicon 74 decreased as the annealing/extension temperature was decreased [17]. To address this issue, we spiked-in 1-, 3-, or 5-fold more of the primer set for amplicon 74 (which was purchased separately from IDT) and used a randomly selected sample (848) for testing. Coverage of amplicon 74 for no spike-in and spike-in 1-, 3- and 5-fold molar excess was determined to be 18×, 126×, 929× and 541×, respectively (Fig. 2C). After read-length filtering the number of reads for no spike-in, 1-, 3- and 5-fold molar excess that mapped to SARS-CoV-2 was found to be 65430, 61430, 79957 and 73248, respectively. None of these conditions appeared to reduce coverage of other amplicons from separate samples tested. In fact, coverage of amplicon 76, also often low, seemed to improve as well. Thus, we propose using 3-fold molar excess (45 nM) of amplicon 74 primers over the amount typically present. The details of our ARTIC PCR protocol are found in Working protocol S4. Recently, ARTIC Network has addressed amplicon dropouts due to mutation acquired by variants. Amplicon 74 appears to systematically dropout in the Beta variant due to a common deletion (241/243del), causing 74_Left to be affected. ARTIC Network has addressed this issue by releasing an updated ARTIC V4 primer set to address variant related dropouts (https://community.artic.network/t/sars-cov-2-version-4-scheme-release/312).
In principle, quantitation of viral genomes by qPCR should enable precise addition of specific amounts of cDNA for enrichment and amplification in multiplexed ARTIC PCR. However, in practice we have found that this does not necessarily improve quality or yield and increases hands-on time. We have determined that a single defined amount of cDNA volume per ARTIC PCR reaction, up to ¼ of the total reaction volume (5 μl), will provide greater than 20× coverage across 93.12% of the genome for samples up to a Ct value of 35 (generated in our lab) in our hands (Fig. S2B). This was calculated on samples we sequenced where the Ct value was provided from the originating lab (n = 1130). Thus, we use qPCR to triage samples that are unlikely to yield full genomes. Samples with a Ct value over 35 using our protocols are usually not processed further. ARTIC PCR reactions can be stored at –20°C or –80°C but are usually kept at 4°C while awaiting clean-up and quantification as described below.
Pooling and purification of ARTIC PCR products
Before library preparation, pool 1 and pool 2 of ARTIC PCR reactions are combined for every sample and the ~400 base-pair (bp) amplicons are purified from contaminating PCR reactants using SPRI AMPure XP beads (Beckman-Coulter). When first performing this workflow, it is recommended that 1/5 of the total volume of each pool’s PCR reaction be separately resolved by agarose gel electrophoresis to visualize a specific band for each pool at the correct size of ~400 bp. (Fig. 2B). The band intensity can be compared to Ct values from qPCR to approximate the efficiency of ARTIC PCR and adjust cutoff values for further processing. The remaining pool 1 and pool 2 for every sample are combined and then purified with paramagnetic SPRI beads in a 96-well format following the manufacturer’s recommended protocol (Working protocol S5). This protocol requires a 96-well magnetic plate. After this step, DNA can be frozen at –20°C or –80°C (heat sealed 96-well plates) but is most often temporarily stored at 4°C until Qubit quantification and end preparation reactions can be performed.
Fluorescence-based qubit quantification of pooled and purified ARTIC PCR products
The purified PCR pools are quantified using a Qubit 2.0 Fluorometer and Qubit dsDNA High Sensitivity (HS) Assay Kit following the manufacturer’s recommended protocol (Working protocol S6). This method is specific to double-stranded DNA and is suitable for detection of low levels of DNA. Quantification allows the same amount of the cleaned and pooled PCR product for each sample to be used in the next step. This ensures that similar number of reads from each barcode are loaded onto the flow cell and individual barcoded samples are not overrepresented during the sequencing run. When first performing this workflow, we recommend quantifying all samples with Qubit to quantify the amount of PCR product available and determine the level of recovery with respect to Ct values from qPCR. However, to reduce time and cost, we routinely only quantitate a small representative group of 10–20 samples to ensure expected results are being obtained. We have found that our multiplex ARTIC PCR protocol results in average concentrations ranging from 91 to 112 ng per samples when the Ct is 30 or below (Fig. S3). We usually estimate the volume of pooled and cleaned PCR product to use for end-prep reactions, which requires 60 ng, based on these quantification results.
Preparation of DNA ends for barcoding
Following the approximation of pooled PCR product concentration by Qubit, 60 ng of pooled and SPRI purified PCR amplicons from SPRI clean-up are added to the end preparation reactions designed to create compatible ends of the DNA amplicons for sample barcoding. These are referred to as end-prep reactions. The DNA is first end-repaired followed by dA-tailing and inactivation of end-repair enzymes. We use the Ultra II End-Prep kit from NEB following the manufacturer’s recommended protocol with minor modifications (Working protocol S7). After this step, DNA can be frozen at –20°C but is most often kept at 4°C for all downstream library preparation steps.
DNA library sample barcoding and adapter ligation
To sequence a full 96-well plate of samples, each sample must be uniquely barcoded and later demultiplexed by allocating reads to samples with the matching barcode. This reduces the overall cost per sample and allows sequencing of up to 96 samples in a single run. To achieve this, we use the Native Barcoding Expansion 96 kit (EXP-NBD196) from ONT. We use the NEB Blunt/TA Ligase Mix to ligate barcodes to the end-prepped DNA (Working protocol S8). After individual barcoding reactions are performed in a 96-well plate, all samples are pooled and cleaned with SPRI beads similar as described above to remove non-ligated barcodes through size selection. This clean-up step is essential to prevent free barcodes from ligating to the incorrect sample during adapter ligation in the next step. This SPRI clean-up requires a microcentrifuge tube magnetic rack. Barcoded samples are stored temporarily at 4°C while awaiting adapter ligation.
After barcoding and clean-up, ONT adapters are then ligated to each amplicon end for the pooled libraries in a single reaction. This reaction sequentially uses Adapter Mix II (AMII) from ONT, NEBNext Quick Ligation Reaction Buffer, and Quick T4 DNA Ligase (Working protocol S9). The final reaction is then cleaned up using SPRI beads and eluted. Short Fragment Buffer (SFB) is used for the last two clean-up steps rather than ethanol to reduce free barcode carry over and prevent degradation of the adapter-motor protein complex. After elution, the final library is stored at 4°C until Qubit quantification and priming and loading of a nanopore sequencing instrument, such as the MinION from ONT.
Loading and running the MinION
For optimal performance on a MinION instrument, 20 ng of adapter-ligated library should be loaded onto the flow cell. Overloading the flow cell results in lower throughput. Therefore, a final Qubit quantification is performed similar to that described above. The DNA sequencing library is then prepared using ONT reagents following recommended protocols (Working protocol S10). The flow cell is then primed using the Flow Cell Priming Kit (ONT) (Fig. 3A). Priming and loading is described in detail in Working protocol S10. To start the sequencing run, ONT’s MinKNOW software is used which can be downloaded and installed from ONT’s website (https://community.nanoporetech.com/downloads).
Figure 3.
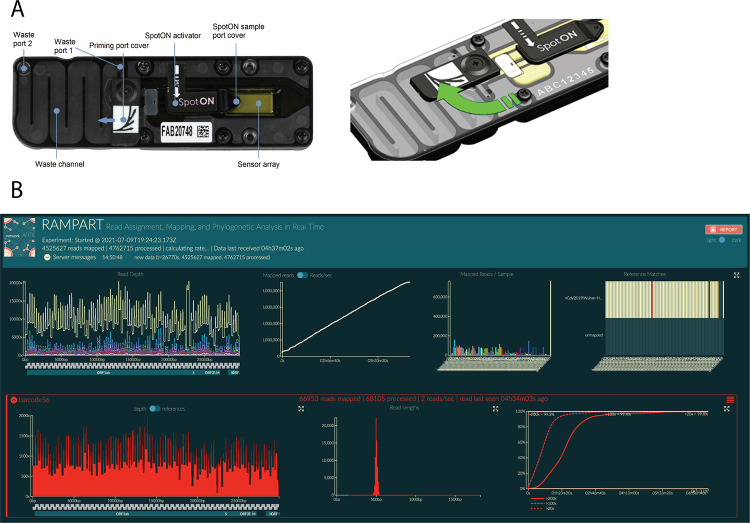
Loading and running the MinION for SARS-CoV-2 genome sequencing.A. Image of the R9.4.1 Flow Cell displaying the different components of the device (left panel). An illustration demonstrating the priming port opening to primer port cover turning clockwise to expose the opening of the priming port (right panel). B. Example of RAMPART display. Top from left to right: (1) Top left shows coverage across the genomes for all samples. (2) Number of mapped reads through time. (3) Number of reads mapped for each barcode sample. (4) Heatmap showing reads mapped to nCoV2019| Wuhan-Hu-1 (accession MN908947). Bottom from left to right: (5) The coverage across the genome for an individual sample. (6) Read length distribution for an individual barcoded sample showing the expected peak of ~400 bp. (7) Percent > 20×, > 100×, and > 200× as a function of time.
Basecalling is the first step to analyze nanopore sequencing electrical signals. This can be performed “live” in real-time while sequencing or “offline” after the sequencing run has completed. The advantage of running basecalling during sequencing is that it provides real-time results that can be used to monitor sequencing quality and progress. If sufficient data has been obtained, the run can be stopped and processed further. The disadvantage of live basecalling is that it utilizes large amounts of computational power and read/write resources. This can cause the sequencing run to slow and on rare occasion, cause crashes resulting in data loss. Therefore, it is recommended to acquire the necessary hardware to support live basecalling during sequencing.
To perform live basecalling during the sequencing run, basecalling should be selected in MinKNOW software with the high accuracy basecalling (HAC) model selected. Graphics processing units (GPUs) can be used to basecall in near real-time whereas central processing units (CPUs), such as a standard laptop for MinION, will not maintain real-time basecalling because of hardware and performance limitations. Real-time basecalling using the GPU in MinKNOW is possible by installing a stand-alone GPU version of Guppy and configuring it with MinKNOW. Details on how to install and configure Guppy GPU in MinKNOW are described in Working protocol S11. When selecting demultiplexing in MinKNOW, “two barcodes required” should be selected for use with native barcoding. Once the sequencing run has been started, Rampart can be used to monitor and gather insight on the sequencing run in real-time by pointing it to the fastq_pass directory where individual barcode directories will be found (Fig. 3B). Once sufficient coverage has been obtained, the sequencing run can be stopped.
To perform offline basecalling, no basecalling should be selected in MinKNOW. The raw data will be saved to the selected output folder as fast5 files. Fast5 files can be converted to Fastq files by basecalling offline using Guppy in the command terminal or subterminal after the sequencing run has been stopped. The basecalled fastq files can then be demultiplexed and visualized in Rampart after the sequencing run. The results can still be visualized in Rampart if basecalling is run after the sequencing run by pointing Rampart to the basecalled fastq files. Detailed protocol of how to perform real time or offline basecalling and further analysis are described in Working Protocol S11. The flow cell should be washed and stored appropriately (Working protocol S12).
Sequencing, real-time visualization, and data analysis
Recommendations for desktop requirements and software generally follow the recommendations from ONT. These include a minimal SSD storage of 1 TB, 16 GB of RAM and Intel i7 or Xeon with 4+ cores (https://nanoporetech.com/community/lab-it-requirements). sSince we have written our data analysis instructions using Linux OS, we recommend using Linux OS to analyze data when following the analysis we have provided. We also recommend using a graphics card from NVIDIA GPU to increase basecalling speed if high accuracy basecalling via Guppy is used.
As previously noted, we recommend implementing Rampart (Fig. 3B) to visualize coverage for each barcoded sample and gather qualitative insight during sequencing runs (https://artic.network/rampart). Rampart can be run concurrently with MinKNOW and be used to determine whether sufficient coverage and depth has been achieved. Since live coverage visualization using Rampart requires basecalling in near real-time, we recommend that MinKNOW is configured to run with the stand-alone GPU version of Guppy. Furthermore, this allows high accuracy basecalling and demultiplexing to occur simultaneously during the sequencing run, allowing for faster data turnaround times.
A general SARS-CoV-2 targeted amplification bioinformatics pipeline will include basecalling, demultiplexing, trimming, alignment, variant identification and consensus building. In order to reduce false variant calls, a fully validated pipeline should be used (Working protocol S11). We have implemented the ARTIC bioinformatics pipeline because of its wide use and ONT support (https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html). Once fast5 files are generated it is highly recommended that high accuracy basecalling is used in order to generate more accurate results of single nucleotide polymorphisms (SNPs). High accuracy basecalling results in better single molecule accuracy but is considerably slower. The basecalled data is then demultiplexed using Guppy with strict parameters to ensure that barcodes are at both ends of the read. This increases the number of reads binned into the “unclassified” reads but reduces barcode mismatches due to the presence of in silico chimera reads. To remove additional chimeras, length filtering is performed using ARTIC guppyplex and reads between 400–700 bp are retained and input into the ARTIC bioinformatics pipeline. There are two workflows developed by ARTIC network to generate consensus sequences and identify variants. Nanopolish uses fast5 signal data while the other, Medaka, does not. In our experience both workflows tend to give consistent results with high variant and single nucleotide variant (SNV) detection accuracy [10]. We have preferentially used Medaka because it only requires fastq files as the input, giving it a small speed improvement. Medaka can also identify heterozygous variants which point to multiple infections, contamination and/or cross contamination. By default, the pipeline minimum coverage is set to 20× to call sites. To detect variant sites more accurately, we aim for a minimum coverage of 100× across 90% of the genome by monitoring the run in real-time using Rampart. Otherwise, a masking model applies ambiguous bases (Ns) to sites with lower than the minimum required coverage. After the pipeline has run, a consensus genome sequence is generated which can be concatenated with other consensus genome sequences and further analyzed.
If variants are called from high Ct samples or partial genomes, the sequence should be carefully evaluated and visualized in a graphical viewer. Starting with very few genome copies may lead to artefactual error and false variant calling. Some enzymes may induce base errors that show up in the sequencing data and result in false positive mutations. To reduce such artifacts, high fidelity enzymes during cDNA synthesis and PCR are recommended [18]. If contamination is observed in the negative control, mutation sites should only be called when sequencing depth greatly exceeds the number of SARS-CoV-2 reads observed in the negative control. To assess amplicon dropouts, we recommend using CoV-GLUE [19] which identifies mismatches in sequencing primers/probe sets.
Phylogenetic analysis, variant calling, and database deposition of SARS-CoV-2 genomes
To process sequenced samples, they must first be properly prepared and formatted (Working protocol S13). A convenient way to observe sequences and determine the quality is to use the NextClade website (https://clades.nextstrain.org/). Dragging and dropping your sequence FASTA file onto the site will initiate an evaluation process that returns the number of mutations compared to the source SARS-CoV-2 strain, as well as the number of ambiguous nucleotides (Ns) and what “clade” each sequence corresponds to, which are the phylogenetic categories the Nextstrain pipeline separates sequences into. This application also allows searching of the aligned sequences for specific nucleotide or amino-acid mutations (the “filter” button), which is useful for quickly identifying unusual sequences for more careful analysis.
If planning to submit sequences to a global database, such as NCBI (National Center for Biotechnology Information) or GISAID, certain requirements must be met. GISAID requires metadata for each sequence, including collection date and location, the passage history of the sample, and the sequencing technology used. To submit samples in a batch format, more than one at a time, they must be provided with a FASTA file containing all assembled sequences, along with a comma-separated values (.csv) file containing all the metadata. Upon registering and requesting permission for batch upload (https://www.epicov.org/epi3/frontend), a template metadata file is provided explaining the correct formatting. The upload process will do a quality-control pass, alerting users to missing metadata, missing FASTA sequences, or other missing information. Additionally, GISAID is currently checking to ensure that any frameshift mutations present in submitted sequences are correct, and not simply the result of sequencing errors. Submitted sequences with frameshifts must be accompanied with a confirmation email or they will be rejected.
NCBI has similar requirements (https://submit.ncbi.nlm.nih.gov/sarscov2/). A date and location of collection must be provided. Submitted samples will be processed to trim ambiguous ends, and low-quality sequences (those with too many ambiguous nucleotides, or unacceptably short or long sequences) will be removed.
Finally, if the sequences are going to be placed into a phylogenetic context using the Nextstrain pipeline (https://nextstrain.org/sars-cov-2/), they must also meet requirements there as well. Nextstrain rejects any sample that lacks metadata (for example time and location of collection, length of sequence) or is too ambiguous. If more than 10% of the sequence is comprised of ambiguous nucleotides, the software will automatically discard it. Nextstrain also estimates the mutation rate of SARS-CoV-2. If samples contain an unlikely number of mutations based on their collection date, these samples will also be excluded from the analysis.
The Nextstrain software can be installed by following their SARS-CoV-2 analysis tutorial (https://nextstrain.github.io/ncov/). Briefly, after the software is installed, all sequence FASTA files must be added in bulk to a sequences.fasta file in the Nextstrain data directory, and the accompanying metadata for those sequences must be added to the metadata.tsv file in the same location. Then a profile must be built that directs the software to focus on those sequences. This can be done using the provided example profiles and is tutorialized in the same link above (Working protocol S13). Once this is completed, running the analysis is as straightforward as running a single command. It generates a file which contains a phylogenetic tree of the sequences, a map of collection locations, and a graph of the diversity of mutations in the sampled sequences. Results are in the form of a .json file, created in the auspice subdirectory, which can be viewed by dragging-and-dropping the file onto the Auspice website (https://auspice.us/). More permanent hosting for these analyses can be setup via the Nextstrain website (https://docs.nextstrain.org/en/latest/guides/share/nextstrain-groups.html). An example of our laboratory’s Nextstrain build with genome sequences from Illinois is shown in Figure 4.
Figure 4.
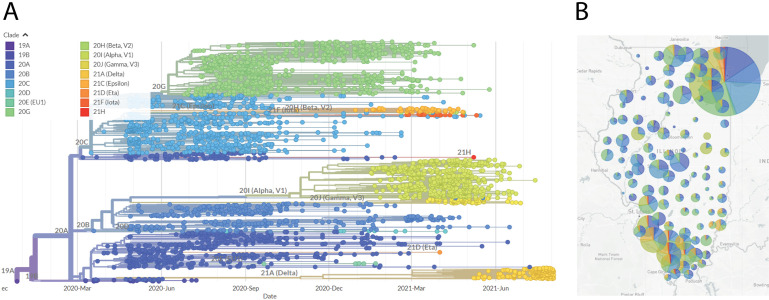
Nextstrain phylogenetic visualization and map view of SARS-CoV-2 genome sequences.A. Phylogenetic tree generated by the Nextstrain pipeline. Sequences are derived from samples taken in Illinois, from April 2020 through August 2021, and sequenced by our laboratory. Clade colors are indicated. B. Map of Illinois showing sample locations, by county. The size of the circle indicates the relative number of sequences derived from that county. The pie chart indicates the proportional distribution of Nextstrain clades at that location. Clade designation colors correlate with those designated in panel (A).
CONCLUSION
Here we presented a comprehensive and validated set of working protocols for sequencing high-quality SARS-CoV-2 genomes in high throughput from patient samples. These protocols implement the popular ARTIC PCR and MinION nanopore sequencing with 96 samples at a time. We have optimized or improved several steps for efficiency, amplicon coverage and higher recovery. This includes improvement to amplicon 74 coverage, constant volumes recommendations of template for cDNA and ARTIC PCR reactions, and ARTIC PCR cycling conditions. Many of the considerations we have covered in these protocols will translate to other sequencing platforms. This workflow and the accompanying supplemental protocols provide a reliable starting point and a reference for those seeking to generate SARS-CoV-2 genome sequences using nanopore sequencing technology, especially from the cost-effective MinION instrument.
Supplementary information
Figure S1. Effects of template volume on cDNA synthesis and ARTIC PCR for both primer pools.
Figure S2. Ct value differences between labs and genome recovery.
Figure S3. Correlation between Ct value and PCR product concentration.
Working Protocol S1. Viral RNA extraction.
Working Protocol S2. cDNA synthesis.
Working Protocol S3. qPCR.
Working Protocol S4. Multiplex ARTIC PCR.
Working Protocol S5. SPRI Clean-up of ARTIC PCR.
Working Protocol S6. Qubit quantification.
Working Protocol S7. Library end prep.
Working Protocol S8. Sample barcoding.
Working Protocol S9. Adapter ligation.
Working Protocol S10. MinION loading and running.
Working Protocol S11. Sequencing data analysis.
Working Protocol S12. Flows cell wash and store.
Working Protocol S13. Visualizing data in Nextclade and Nextstrain.
Supplementary information of this article can be found online at https://jbmethods.org/jbm/article/view/360.
Acknowledgments
We thank the Illinois Department of Public Health for access to patient samples. This work was funded by discretionary funds from the Southern Illinois University (SIU) School of Medicine Dean’s Office and the Office of the Vice Chancellor for Research at SIU Carbondale. This work was also funded by the Walder Foundation through a collaborative agreement with the Open Commons Consortium as part of the Chicago CAN initiative.
Footnotes
Author contributions
A.A.P. conceived of the project, developed and validated all protocols with assistance of other authors, supervised the project, analyzed data, interpreted results, prepared figures, and wrote the manuscript. M.S.B. guided protocol validation, interpreted sequencing data, built phylogenetic trees, uploaded to sequencing data to GISAID, prepared figures, and wrote the manuscript. A.A.W. assisted in most protocol development and validation, especially sequencing preparations like barcoding and clean-up, analyzed data, and interpreted results. R.J.S. handled patient samples, performed RNA extractions, cDNA syntheses, and performed multiple steps in the sequencing preparation workflow. S.E.B. helped develop and validate ARTIC multiplex PCR, as well as performed ARTIC PCR for the workflow. M.P. helped develop, validate, and perform cDNA syntheses for the workflow. K.N.O. helped develop and optimize qPCR quantification of viral load and helped validate RNA extraction protocols. P.E.M. helped validate and perform qPCR for the workflow. A.O.Y. helped validate and perform sample Qubit quantifications adapter ligations for the workflow. C.L.B. helped validate and perform MinION loading, washing and maintenance for sequencing runs. R.C. helped validate and perform barcoding and clean-up for sequencing library preparations. S.W.B. assisted A.A.P. in validation of 74 dropout corrections. M.M.H. assisted ARTIC PCR optimization and troubleshooting as well as Qubit quantification validation. K.T.G. conceived of the project, directed, and contributed experimentally to the project as well as prepared figures and wrote the manuscript.
References
- 1.Daszak P, Keusch GT, Phelan AL, Johnson CK, Osterholm MT. Infectious Disease Threats: A Rebound To Resilience. Health Aff (Millwood). 2021February;40(2):204–11. https://doi.org/10.1377/hlthaff.2020.01544 10.1377/hlthaff.2020.01544PMID: [DOI] [PubMed] [Google Scholar]
- 2.Grubaugh ND, Hodcroft EB, Fauver JR, Phelan AL, Cevik M. Public health actions to control new SARS-CoV-2 variants. Cell. 2021March;184(5):1127–32.https://doi.org/10.1016/j.cell.2021.01.044 10.1016/j.cell.2021.01.044PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardy J, Loman NJ, Rambaut A. Real-time digital pathogen surveillance - the time is now. Genome Biol. 2015July;16(1):155. https://doi.org/10.1186/s13059-015-0726-x 10.1186/s13059-015-0726-xPMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khudyakov Y. Molecular surveillance of hepatitis C. Antivir Ther. 2012;17 (7 Pt B):1465–70. https://doi.org/10.3851/IMP2476 10.3851/IMP2476 PMID: [DOI] [PubMed] [Google Scholar]
- 5.McGinnis J, Laplante J, Shudt M, George KS. Next generation sequencing for whole genome analysis and surveillance of influenza A viruses. J Clin Virol. 2016June;79:44-50. https://doi.org/10.1016/j.jcv.2016.03.005 10.1016/j.jcv.2016.03.005PMID: [DOI] [PubMed] [Google Scholar]
- 6.Houldcroft CJ, Beale MA, Breuer J. Clinical and biological insights from viral genome sequencing. Nat Rev Microbiol. 2017March;15(3):183–92. https://doi.org/10.1038/nrmicro.2016.182 10.1038/nrmicro.2016.182PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomson E, Ip CL, Badhan A, Christiansen MT, Adamson W, Ansari MA, et al. Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes. J Clin Microbiol. 2016October;54(10):2470–84. https://doi.org/10.1128/JCM.00330-16 10.1128/JCM.00330-16PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc. 2017June;12(6):1261–76. https://doi.org/10.1038/nprot.2017.066 10.1038/nprot.2017.066PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyson JR, et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv : the preprint server for biology, (September4, 2020). [Google Scholar]
- 10.Bull RA, Adikari TN, Ferguson JM, Hammond JM, Stevanovski I, Beukers AG, et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat Commun. 2020December;11(1):6272. https://doi.org/10.1038/s41467-020-20075-6 10.1038/s41467-020-20075-6PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker DJ, Aydin A, Le-Viet T, Kay GL, Rudder S, de Oliveira Martins L, et al. CoronaHiT: high-throughput sequencing of SARS-CoV-2 genomes. Genome Med. 2021February;13(1):21. https://doi.org/10.1186/s13073-021-00839-5 10.1186/s13073-021-00839-5PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gohl DM, Garbe J, Grady P, Daniel J, Watson RH, Auch B, et al. A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genomics. 2020December;21(1):863. https://doi.org/10.1186/s12864-020-07283-6 10.1186/s12864-020-07283-6PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Wang H, Mao L, Yu H, Yu X, Sun Z, et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci Rep. 2020October;10(1):17492. https://doi.org/10.1038/s41598-020-74656-y 10.1038/s41598-020-74656-yPMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020November;5(11):1403–7. https://doi.org/10.1038/s41564-020-0770-5 10.1038/s41564-020-0770-5PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018December;34(23):4121–3. https://doi.org/10.1093/bioinformatics/bty407 10.1093/bioinformatics/bty407PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pastorino B, Touret F, Gilles M, Luciani L, de Lamballerie X, Charrel RN. Evaluation of Chemical Protocols for Inactivating SARS-CoV-2 Infectious Samples. Viruses. 2020June;12(6):624. https://doi.org/10.3390/v12060624 10.3390/v12060624PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itokawa K, Sekizuka T, Hashino M, Tanaka R, Kuroda M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS One. 2020September;15(9):e0239403. https://doi.org/10.1371/journal.pone.0239403 10.1371/journal.pone.0239403PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potapov V, Ong JL. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS One. 2017January;12(1):e0169774. https://doi.org/10.1371/journal.pone.0169774 10.1371/journal.pone.0169774PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singer J, Gifford R, Cotten M, Robertson D. CoV-GLUE: A Web Application for Tracking SARS-CoV-2 Genomic Variation. Preprints. 18June2020. https://doi.org/10.20944/preprints202006.0225.v1s 10.20944/preprints202006.0225.v1s [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of template volume on cDNA synthesis and ARTIC PCR for both primer pools.
Figure S2. Ct value differences between labs and genome recovery.
Figure S3. Correlation between Ct value and PCR product concentration.
Working Protocol S1. Viral RNA extraction.
Working Protocol S2. cDNA synthesis.
Working Protocol S3. qPCR.
Working Protocol S4. Multiplex ARTIC PCR.
Working Protocol S5. SPRI Clean-up of ARTIC PCR.
Working Protocol S6. Qubit quantification.
Working Protocol S7. Library end prep.
Working Protocol S8. Sample barcoding.
Working Protocol S9. Adapter ligation.
Working Protocol S10. MinION loading and running.
Working Protocol S11. Sequencing data analysis.
Working Protocol S12. Flows cell wash and store.
Working Protocol S13. Visualizing data in Nextclade and Nextstrain.