

Abstract
Evidence suggests that the basal forebrain (BF) cholinergic system degenerates early in the course of Alzheimer’s disease (AD), likely due to the vulnerability of BF cholinergic neurons to tau pathology. However, it remains unclear whether the presence of tauopathy is the only requirement for initiating the BF degeneration in asymptomatic subjects at risk for AD (AR-AD), and how BF structural deficits evolve from normal aging to preclinical and prodromal AD. Here, we provide human in vivo magnetic resonance imaging evidence supporting that abnormal cerebrospinal fluid levels of phosphorylated tau (T+) are selectively associated with bilateral volume loss of the nucleus basalis of Meynert (nbM, Ch4) in AR-AD individuals. Spreading of atrophy to medial septum and vertical limb of diagonal band Broca (Ch1–Ch2) occurred in both preclinical and prodromal AD. With the exception of A+, all groups revealed significant correlations between volume reduction of BF cholinergic compartments and atrophy of their innervated regions. Overall, these results support the central role played by tauopathy in instigating the nbM degeneration in AR-AD individuals and the necessary coexistence of both AD proteinopathies for spreading damage to larger BF territories, thus affecting the core of the BF cholinergic projection system.
Keywords: basal forebrain; biomarkers, cholinergic system; magnetic resonance imaging; nucleus basalis Meynert; preclinical Alzheimer’s disease
Introduction
Research based on cytoarchitectonics, cytochemistry, and connectivity patterns has revealed that basal forebrain (BF) cholinergic neurons provide the major source of acetylcholine to the cerebral cortex, hippocampus, and amygdala. Large cholinergic neurons of the nucleus basalis Meynert (nbM), termed Ch4, send projections to the entire neocortical mantle and the amygdala, while hippocampal cholinergic inputs arise from the medial septum and vertical limb of the diagonal band Broca, corresponding to Ch1 and Ch2, respectively (Mesulam et al. 1983; Mesulam and Geula 1988). Given the widespread distribution of acetylcholine (ACh) receptors in the brain, the BF cholinergic system (BFCS) serves as a major cholinergic hub for brain regions implicated in cortical activation, attention, sensory processing, motivation, and memory processes, playing a central role in modulating neural dynamics underlying arousal regulation and cognitive operations (Baxter and Chiba 1999; Zaborszky et al. 2018).
Accumulated evidence has supported significant dysfunctions of central cholinergic neurons in Alzheimer’s disease (AD) (Davies and Maloney 1976; Perry et al. 1977; White et al. 1977). Pivotal experiments showed a deficiency of choline acetyl transferase (ChAT) in the neocortex, amygdala, and hippocampus of AD patients, revealing that cortical regions containing the greatest density of neurofibrillary tangles (NFTs) exhibited the maximum reduction of ChAT (Davies and Maloney 1976). Since then, numerous studies have reported substantial BF cholinergic deficits in mild cognitive impairment (MCI; Mufson et al. 2002; Peng et al. 2004; Ginsberg et al. 2011, 2016) and AD patients (Whitehouse et al. 1981; McGeer et al. 1984; Arendt et al. 1985; Vogels et al. 1990), which have been further associated with the severity of the cognitive impairment (Bierer et al. 1995; Baskin et al. 1999; Pappas et al. 2000). Human tissue-based research has also provided copious evidence of selective vulnerability of nbM neurons to NFT pathology (Mesulam et al. 2004). Furthermore, recent findings suggest that the link between tau pathology and cholinergic deficits may be initiated earlier than expected, when toxic tau oligomeric species associated with pretangle pathology accumulate in vulnerable nbM neurons (Tiernan et al. 2018).
With the advent of AD biomarkers, many studies have identified AD pathology with in vivo techniques years before the development of symptoms (e.g., Jack et al. 2014; Gordon et al. 2016), leading to different classifications of preclinical AD states (Jack et al. 2010; Sperling et al. 2011; Dubois et al. 2016). According to the revised criteria from Dubois et al. (2016), asymptomatic individuals at risk for AD (AR-AD) are characterized by the presence of one of the AD biomarkers (either Amyloid (A+) or Tau (T+)), whereas preclinical (asymptomatic) and prodromal AD (MCI) states require the presence of both AD proteinopathies (A+T+). Previous studies have revealed that BF atrophy correlates with amyloid β burden (Kerbler et al. 2014) and precedes AD pathology in the entorhinal cortex (Schmitz et al. 2018) and symptom onset in cognitively normal subjects (Butler et al. 2018). Moreover, recent experiments have shown that individuals with subjective cognitive decline, a risk population for preclinical AD, exhibit smaller volume of Ch1–Ch2 and Ch4 subdivisions of the BF (Scheef et al. 2019). However, human in vivo experiments explicitly assessing the vulnerability of the BFCS to isolated AD pathology in AR-AD subjects or to both proteinopathies in preclinical and prodromal AD are still lacking. It is also unknown when the coupling between structural deficits of BF cholinergic compartments and the atrophy of their innervated regions (i.e., amygdala, hippocampus, and neocortex) starts developing in the course of AD, considering that structural covariance between atrophy of BF cholinergic compartments and patterns of cortico-amygdalar degeneration is already present in prodromal AD (Cantero et al. 2017; Schmitz et al. 2018). All together, this information may be of relevance for the prevention of cognitive deficits by designing tailored interventions aimed at delaying BF cholinergic degeneration and preserving the cholinergic transmission to the cortical mantle and limbic system.
To address these questions, we have compared in vivo magnetic resonance imaging (MRI) measures of gray matter (GM) volume of different BF cholinergic compartments (Ch1–Ch2 and Ch4) between cognitively normal individuals with negative AD biomarkers in cerebrospinal fluid (CSF; A-T-, control), AR-AD (either A+ or T+), preclinical (A + T+, asymptomatic), and prodromal AD (A + T+, MCI). Based on evidence supporting the role of tau oligomers and NFTs in degeneration of nbM cholinergic neurons (e.g., Mesulam et al. 2004; Tiernan et al. 2018), our prediction is that BF structural deficits will occur first in asymptomatic T+ rather than in A+ subjects and that the extent of cholinergic damage will be driven by the synergistic toxic effect of both proteinopathies (A+T+). We further expect that selective atrophy of nbM in T+ and A+T+ individuals parallels GM deficits of their innervated regions, likely revealing cholinergic denervation of these structures.
Materials and Methods
Subjects
According to the revised criteria from Dubois et al. (2016), cognitively normal subjects with either amyloid (A+) or tau pathology (T+) were considered AR–AD individuals Participants with both proteinopathies (A+T+) were classified as having preclinical or prodromal AD, depending on the absence or presence of subtle cognitive decline, respectively. Based on these criteria, the study sample consisted of 120 subjects: A−T− (n = 33), A+ (n = 33), T+ (n = 29), preclinical AD (A+T+, n = 8), and prodromal AD (A+T+, n = 17). They were recruited at the Hospital Santa Creu i Sant Pau (Barcelona, Spain) and the University Hospital Marques de Valdecilla (Santander, Spain), as part of a cross-sectional multicenter study called the “SIGNAL project.” Participants underwent a neurological exam and extensive neuropsychological assessment. Education, defined in years of education, was assessed by self-report or close family members. Subjects gave their informed consent prior to their inclusion in the study, which was previously approved by the local ethics committee at each center according to the Declaration of Helsinki.
CSF Measures
CSF was obtained through lumbar puncture following international consensus recommendations (Alcolea et al. 2015). Briefly, CSF was collected in the morning between 09:00 AM and 12:00 PM in polypropylene tubes and immediately centrifuged for 10 min (1900–2000g). The first 1 cc was discarded to avoid hematic contamination. CSF samples were aliquoted (0.5 mL) into polypropylene tubes and frozen at −80°C. All CSF samples were shipped in dry ice to the Hospital Santa Creu i Sant Pau, where they were analyzed. Commercially available enzyme-linked immunosorbent assay kits were used to determine the levels of Aβ1–42 (Innotest β-amyloid1–42; Fujirebio Europe) and phosphorylated tau (p-tau; Innotest Phospho-Tau181P; Fujirebio Europe), following the manufacturer’s recommendations. The presence of amyloid (A+) and p-tau (T+) pathology was defined as CSF Aβ1–42 ≤ 550 pg/mL and CSF p-tau ≥61 pg/mL, respectively (Alcolea et al. 2015).
MRI Acquisition and Cortical Thickness Estimation
Participants were scanned in two different centers (Hospital del Mar and University Hospital Marqués de Valdecilla) using the same protocol and identical MRI scanners (Philips Achieva 3T, 8-channel receive head coil). High-resolution structural cerebral scans were obtained with a 3D T1-weighted magnetization-prepared rapid gradient echo sequence acquired in the sagittal plane (TR/TE = 8.2/3.8 ms, flip angle = 8°, voxel resolution = 1 mm isotropic, no gap between slices). Neuroimaging analyses were all performed in the Laboratory of Functional Neuroscience at the Pablo de Olavide University (Seville, Spain).
MRI data were processed using the analysis pipeline of Freesurfer v6.0 (https://surfer.nmr.mgh.harvard.edu/) that involves intensity normalization, registration to Talairach, skull stripping, white matter (WM) segmentation, tessellation of the WM boundary, and automatic correction of topological defects (Fischl and Dale 2000). Pial surface misplacements and erroneous WM segmentation were manually corrected on a slice-by-slice basis to enhance the reliability of cortical thickness measurements. Individual thickness maps were further transformed into the same spherical coordinate system of cortical surfaces and then resampled to the average spherical surface by aligning each individual cortical folding pattern to the average folding pattern of the entire population. This procedure has shown to enhance the spatial localization of cortical features among participants, thereby minimizing metric distortions (Fischl et al. 1999). Finally, cortical thickness maps were smoothed using nonlinear spherical wavelet-based denoising schemes, which have shown superior specificity and sensitivity at detecting local and global changes compared to Gaussian filter kernels (Bernal-Rusiel et al. 2008). Cortical thickness was defined as the average of the shortest distance between the pial surface and the GM–WM boundary at each vertex across the cortical mantle (Fischl and Dale 2000).
Volume Estimation of BF Cholinergic Compartments, Amygdala, and Hippocampus
Voxel-based MRI data were preprocessed and analyzed using SPM12 (Wellcome Trust Center for Neuroimaging; www.fil.ion.ucl.ac.uk/spm). Briefly, T1-weighted images were manually reoriented to the anterior commissure and further segmented into GM, WM, CSF, and skull/scalp compartments following the unified segmentation of SPM12. Next, the diffeomorphic anatomical registration through an exponentiated lie algebra (DARTEL) algorithm was applied to segmented brain images to obtain an enhanced inter-subject registration with improved realignment of smaller inner structures (Ashburner 2007). Individual flow fields obtained from the DARTEL registration to the reference template were used to warp the GM compartment, therefore preserving the total amount of GM volume before warping. All preprocessed GM maps were visually checked for overall segmentation and registration accuracy. GM maps were spatially normalized to the Montreal Neurological Institute (MNI) space, and normalized modulated GM images were smoothed with a Gaussian kernel of 4 mm.
The volume of BF cholinergic nuclei (i.e., Ch1–Ch2 and Ch4) was obtained with cytoarchitectonic probabilistic maps of BF magnocellular compartments (Zaborszky et al. 2008). Briefly, BF areas containing magnocellular cell groups within the septum, horizontal and vertical limbs of the diagonal band, and the sublenticular area were delineated in histological sections of 10 human autopsy brains by using a modified Gallyas silver method (Zaborszky et al. 2008). Ch1–Ch2 compartments correspond to the medial septal nucleus and the diagonal band of Broca, respectively. Given that boundaries between Ch1 and Ch2 were arbitrary, these two regions were merged into one for statistical purposes. The Ch4 compartment largely corresponds to the NbM, extending as far rostrally as cell aggregates underneath the nucleus accumbens. Large aggregates of darkly stained cell group within the ventrolateral edge of the bed nucleus of the stria terminalis at the border of the internal capsule/anterior commissure were also classified as Ch4. The Ch4 compartment further includes magnocellular cell groups within a well-defined area that begins caudal to the supraoptic nucleus at the level where the optic tract adjoins the internal capsule/cerebral peduncle and extends laterally up to the most caudal level of the medial mammillary nucleus and the central nucleus of the amygdala (Zaborszky et al. 2008). BF cholinergic compartments were registered to the single-subject template of the MNI atlas, and maximum probability maps of each BF compartment were obtained for statistical purposes (Eickhoff et al. 2005). We further used cytoarchitectonically verified maps of the human amygdala and hippocampus for statistical analysis (Amunts et al. 2005).
Statistical Analysis
The normality assumption of demographic, biochemical, and cognitive variables was first assessed with the Kolmogorov–Smirnov test using the Lilliefors correction. Group differences were evaluated with independent samples t-tests (continuous variables) and the chi-square test (categorical variables) using SPSS v22 (SPSS Inc.).
For each hemisphere, vertex-wise group differences in cortical thickness were first assessed with one-way analysis of covariance (ANCOVA) adjusted by age and sex. If the one-way ANCOVA showed a significant group effect, post hoc pairwise comparisons were then performed. Results were corrected for multiple comparisons using a previously validated hierarchical statistical model (Bernal-Rusiel et al. 2010). This statistical approach first controls the family-wise error (FWE) rate at the level of clusters (P < 0.05) by applying random field theory over smoothed statistical maps, and next controls the false discovery rate at the level of vertex (P < 0.05) over unsmoothed statistical maps. Cluster extent threshold was fixed at 90 vertices. We further assessed group differences in GM volume of each BF cholinergic compartment (i.e., Ch1–Ch2 and Ch4), hippocampus, and amygdala, separately, using the voxel-based morphometry (VBM) approach implemented in SPM12 (one-way ANCOVA adjusted by age, sex, and total intracranial volume [ICV], corrected for FWE, P < 0.05; cluster extend threshold >5 voxels). If the omnibus ANCOVA showed an overall difference across the groups, post hoc pairwise comparisons were then performed.
We next performed vertex-wise linear regression analyses to determine correlations between group differences in GM volume of each BF cholinergic compartment (i.e., Ch1–Ch2 and Ch4) and cortical thinning in each group separately, adjusting by age and sex. The results were corrected for multiple comparisons (cluster extend threshold = 90 vertices) using the hierarchical statistical model mentioned above (Bernal-Rusiel et al. 2010). Voxel-wise linear regression analyses were performed to assess correlations between group differences in GM volume of each BF cholinergic compartment (i.e., Ch1–Ch2 and Ch4) and group differences in GM volume of amygdala and hippocampus, separately. These analyses were also adjusted by age, sex, and ICV and corrected for FWE (P < 0.05; cluster extend threshold > 5 voxels).
We further assessed voxel-wise group differences in WM volume with the VBM approach implemented in SPM12 (one-way ANCOVA adjusted by age, sex, and ICV, corrected for FWE, P < 0.05; cluster extend threshold > 5 voxels). Post hoc pairwise comparisons were performed if the omnibus null hypothesis was rejected. Significant group differences in WM volume were displayed on specific WM tracts using the Johns Hopkins University (JHU) WM tractography atlas (Hua et al. 2008). Voxel-wise linear regression analyses allowed us to evaluate whether patterns of WM atrophy were related to volume loss of BF cholinergic compartments in each group. These analyses were also adjusted by age, sex, and ICV and corrected for FWE (P < 0.05; cluster extend threshold > 5 voxels).
For all the analyses, we have reported the extent of change (cluster size), the P-corrected value, the statistic for the peak vertex/voxel, and the effect size obtained with the partial eta-squared (Keppel 1991).
Confirmatory Analysis Based on the ADNI Dataset
In order to verify results obtained with our sample, we applied the same analytical approach to the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies, and nonprofit organizations, as a $60 million, 5-year public–private partnership. The primary goal of ADNI has been to test whether serial MRI, positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. The principal investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California—San Francisco. ADNI is the result of efforts of many coinvestigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the United States of America and Canada. More information can be found in the acknowledgements section (see also http://adni-info.org/).
We selected individuals with normal cognitive function and MCI patients who had baseline CSF and 3T MRI data in the ADNI database. Aβ1–42 and total tau (t-tau) levels were measured using the multiplex xMAP Luminex platform (Luminex) with Innogenetics (INNO-BIA AlzBio3) immunoassay kit-based reagents. In the analyses performed with the ADNI cohort, t-tau was used because it has shown a higher specificity than p-tau in the ADNI dataset (92.3% vs. 73.1%) (Shaw et al. 2009). Using published cutoffs (192 pg/mL for Aβ1–42 and 93 pg/mL for t-tau) (Shaw et al. 2009), we selected 256 subjects: A−T− (n = 78), A+ (n = 45), T+ (n = 10), preclinical AD (A+T+, n = 14), and prodromal AD (A+T+, n = 109).
Results
Demographic Characteristics, CSF Biomarkers, and Cognitive Function
Table 1 contains demographic, cognitive, and CSF data for each group. All groups were statistically comparable in sex distribution and education years. However, the one-way ANOVA showed group differences in age (F(4,115) = 8.99, P = 10−5), T+, preclinical, and prodromal AD being significantly older than controls (P < 0.001). The null hypothesis of differences across groups was rejected for all cognitive variables (Mini-Mental State Examination: F(4,115) = 20.53, P = 10−13; Boston Naming Test with education as covariate: F(4,114) = 13.79, P = 10−9; Rey figure: F(4,115) = 5.66, P = 0.0004; and VOSP: F(4,115) = 4.56, P = 0.002). Preclinical and prodromal AD patients showed lower scores in the Boston Naming Test than controls (P < 0.001), while for the remaining tests, T+, preclinical, and prodromal AD patients showed a significant impairment when compared to controls (P < 0.05). However, the cognitive function of individuals with A+ was comparable to that of controls. The ANOVAs also revealed significant differences among groups for the three CSF biomarkers (Aβ1–42: F(4,115) = 48.35, P = 10−24; p-tau: F(4,115) = 42.14, p = 10−21; t-tau: F(4,115) = 41.25, P = 10−21). As expected, Aβ1–42 levels were significantly lower in the three groups with amyloidosis as compared with controls (P = 10−6) and T+ (P = 10−11), while p-tau and t-tau were higher in the three groups with tauopathy as compared with controls (p-tau: P = 10−6; t-tau: P = 10−5) and A+ (p-tau: P = 10−8; t-tau: P = 10−6).
Table 1.
Demographic, cognitive, and CSF data of the study population
| A-T- controls | A+ amyloidosis | T+ SNAPs | A+T+ preclinical AD | A+T+ prodromal AD | |
|---|---|---|---|---|---|
| Age | 58.4 ± 6.7 | 61.5 ± 8.2 | 67.2 ± 8.5** | 69.9 ± 5.6** | 67.4 ± 5.3** |
| Sex (m/f) | 10/23 | 9/24 | 15/14 | 6/2 | 5/12 |
| Education, years | 13.4 ± 4.5 | 13 ± 4.7 | 12.8 ± 5.3 | 12.4 ± 5 | 12.1 ± 4.7 |
| ApoE4 (yes/no) | 6/27 | 17/16 | 9/20 | 6/2 | 14/3 |
| MMSE | 29.3 ± 0.8 | 29 ± 0.9 | 28.3 ± 1.3 | 29.1 ± 0.8 | 26.3 ± 2.0*** |
| Boston Naming Test | 55.1 ± 3.4 | 54.9 ± 3.6 | 53.8 ± 4 | 49.1 ± 4.4** | 46.3 ± 5.9*** |
| Rey figure | 33.0 ± 2.6 | 33.9 ± 1.3 | 30.5 ± 5.9* | 29 ± 5.6* | 29 ± 6.8* |
| VOSP | 9.6 ± 0.7 | 9.0 ± 0.8 | 8.9 ± 1.4* | 8.2 ± 1.5* | 8.2 ± 2.0** |
| Aβ1–42 (pg/mL) Range |
777 ± 145 554–1133 |
475 ± 65** 322–550 |
915 ± 263** 582–1531 |
437 ± 64*** 349–508 |
425 ± 71*** 276–515 |
| t-tau (pg/mL) Range |
200 ± 77 84–427 |
182 ± 110 64–534 |
441 ± 167*** 262–840 |
528 ± 271*** 379–1170 |
726 ± 286*** 352–1314 |
| p-tau (pg/mL) Range |
41 ± 11 24–60 |
32 ± 10 18–55 |
75 ± 17*** 61–133 |
88 ± 46*** 63–201 |
107 ± 42*** 64–230 |
Note: Results are expressed as mean ± standard deviation. m/f, male/female; SNAPs, suspected non-Alzheimer’s disease pathophysiology; MMSE, Mini-Mental State Examination; VOSP, Visual Object and Space Perception Battery. Asterisks indicate post hoc comparisons with respect to controls *P < 0.05; **P < 10−3; ***P < 10−6.
Group Differences in Volume of BF Cholinergic Compartments, Hippocampus, Amygdala, and Cortical Thickness
The one-way ANCOVA revealed that the volume of BF cholinergic compartment was significantly different among the five groups (Ch1–Ch2: F(4,113) = 4.79, P = 10−5; Ch4: F(4,113) = 5.31, P = 0.001). Results of these analyses are shown in Table 2 and illustrated in Figure 1. T+, preclinical, and prodromal AD individuals exhibited significant lower volume of Ch4 (i.e., nbM) than controls. The extent of BF atrophy reached Ch1–Ch2 territories (i.e., medial septum and the diagonal band of Broca) in preclinical and prodromal AD. GM volume of BF cholinergic compartments was comparable in controls and A+ subjects.
Table 2.
Extent of atrophy of BF cholinergic compartments, amygdala, and hippocampus in T+, preclinical AD (A+T+), and prodromal AD (A+T+) compared with controls (A−T−)
| Brain region | CS (mm3) | P | x | y | z | T |
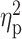
|
|---|---|---|---|---|---|---|---|
| Control > T+ | |||||||
| Basal forebrain | |||||||
| Right Ch4 | 91 | 10−3 | 20 | -4 | −10 | 4.7 | 0.72 |
| Left Ch4 | 33 | 10−2 | −19 | −5 | −12 | 3.8 | 0.76 |
| Amygdala | |||||||
| Right amygdala | 136 | 10−3 | 15 | −2 | −18 | 5.1 | 0.23 |
| Right amygdala | 39 | 10−2 | 22 | 5 | −21 | 5.4 | 0.15 |
| Left amygdala | 17 | 10−2 | −19 | −5 | −13 | 3.9 | 0.18 |
| Control > preclinical AD | |||||||
| Basal forebrain | |||||||
| Right Ch1–Ch2 | 24 | 10−2 | 3 | 2 | −9 | 3.8 | 0.16 |
| Left Ch4 | 161 | 10−4 | −23 | −4 | −12 | 7.3 | 0.66 |
| Right Ch4 | 181 | 10−4 | 24 | −4 | −12 | 7.1 | 0.65 |
| Amygdala | |||||||
| Left amygdala | 1457 | 10−6 | −23 | −4 | −13 | 7.9 | 0.28 |
| Right amygdala | 1870 | 10−7 | 25 | −6 | −12 | 7.8 | 0.26 |
| Hippocampus | |||||||
| Left hippocampus | 238 | 10−4 | −21 | −12 | −18 | 6.5 | 0.36 |
| Right hippocampus | 231 | 10−4 | 15 | −9 | −16 | 6.6 | 0.35 |
| Right hippocampus | 52 | 10−3 | 21 | −35 | −1 | 5.4 | 0.35 |
| Control > prodromal AD | |||||||
| Basal forebrain | |||||||
| Right Ch1–Ch2 | 201 | 10−3 | 0 | 6 | −8 | 5.5 | 0.47 |
| Right Ch1–Ch2 | 22 | 10−2 | 8 | 3 | −14 | 4.4 | 0.31 |
| Left Ch4 | 273 | 10−5 | −22 | −6 | −11 | 10.9 | 0.78 |
| Right Ch4 | 181 | 10−4 | 20 | −7 | −12 | 8.3 | 0.72 |
| Amygdala | |||||||
| Left amygdala | 2616 | 10−8 | −22 | −6 | −12 | 12.1 | 0.41 |
| Right amygdala | 2115 | 10−7 | 25 | −6 | −15 | 9.2 | 0.26 |
| Hippocampus | |||||||
| Left hippocampus | 2156 | 10−13 | 19 | −9 | −16 | 8.4 | 0.36 |
| Right hippocampus | 2638 | 10−14 | −22 | −10 | −20 | 8.3 | 0.35 |
Note: CS, cluster size (>5 voxels), it refers to the extent of atrophy; P,
cluster-level FWE correction (P < 0.05), peak MNI
coordinates (x-y-z); T, peak
t-value; 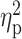 , peak partial eta-squared
showing the effect size.
, peak partial eta-squared
showing the effect size.
Figure 1 .
Group differences in GM volume of BF cholinergic compartments. Results were corrected for multiple comparisons (FWE, P < 0.05). L, left; R, right. Note that AD pathology, as revealed by abnormal CSF levels of Aβ1–42 and p-tau, determines the extent of BF atrophy.
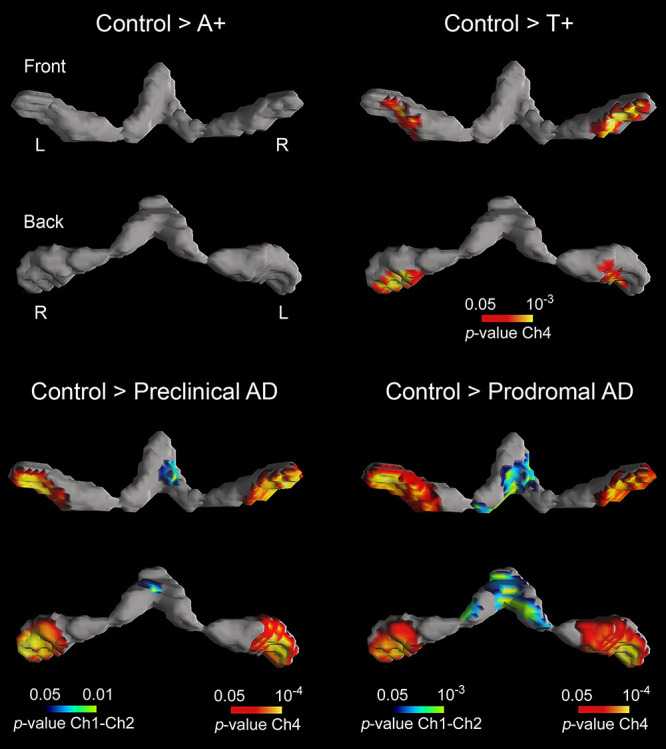
We next assessed group differences in structural integrity of brain regions that are synaptically connected to BF nuclei, such as the amygdala, hippocampus, and neocortex Table 2). The one-way ANCOVA yielded a significant group effect for the amygdala (F(4,113) = 5.78, P = 0.001) and the hippocampus (F(4,113) = 6.94, P = 10−11). While A+T+ subjects exhibited significant volume loss of amygdala and hippocampus compared to controls, atrophy only affected the amygdala in T+ subjects (see Fig. 2).
Figure 2 .
Group differences in GM volume of the amygdala (first and second rows) and hippocampus (third and fourth rows). Both limbic structures receive cholinergic projections from Ch4 and Ch1–Ch2, respectively. Results were corrected for multiple comparisons (FWE, P < 0.05). L, left; R, right. Note that only T+ (second column) and A+T+ subjects (preclinical and prodromal AD, third and fourth columns, respectively) showed structural deficits in these regions.
The ANCOVA also showed a significant group effect for the neocortex (left: F(4,113) = 4.18, P = 10−4; right: F(4,113) = 4.96, P = 0.001). Pairwise group differences in cortical thickness are included in Table 3 and Fig. 3. A+ subjects revealed divergent patterns of cortical thickness in different regions. Compared to controls, A+ exhibited bilateral thinning of posterior cingulate and right anterior cingulate cortex together with thickening of parieto-occipital regions. Patterns of cortical atrophy spread to frontoparietal networks and the cingulate cortex in T+ individuals. The spatial extent of cortical thinning differed between preclinical and prodromal AD patients. While preclinical AD subjects showed cortical deficits mostly affecting the right temporal lobe, prodromal AD patients revealed bilateral thinning of temporal, left inferior parietal, and medial orbitofrontal regions together with atrophy of the right anterior cingulate cortex. Results were slightly different when cortical GM volume was included as dependent variable instead of cortical thickness (see Supplementary Material and Supplementary Fig. 1).
Table 3.
Extent of cortical thickness changes in asymptomatic subjects AR-AD (either A+ or T+), preclinical AD (A+T+), and prodromal AD (A+T+) compared with controls (A−T−)
| Cortical region | CS (mm2) | Mean ± SD thickness | % Atrophy | P |
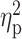
|
|
|---|---|---|---|---|---|---|
| Control > A+ | Control | A+ | ||||
| Left posterior cingulate | 757 | 2.45 ± 0.21 | 2.26 ± 0.24 | 8 | 10−4 | 0.14 |
| Left entorhinal | 566 | 3.06 ± 0.25 | 2.82 ± 0.26 | 8 | 10−5 | 0.16 |
| Right posterior cingulate | 883 | 2.16 ± 0.21 | 1.95 ± 0.25 | 10 | 10−6 | 0.17 |
| Right anterior cingulate | 496 | 2.74 ± 0.24 | 2.65 ± 0.26 | 3 | 10−4 | 0.04 |
| Right middle temporal | 483 | 3.12 ± 0.19 | 2.85 ± 0.3 | 9 | 10−4 | 0.21 |
| A+ > Control | A+ | Control | ||||
| Left cuneus | 290 | 1.77 ± 0.17 | 1.52 ± 0.15 | 14 | 10−7 | 0.38 |
| Left superior parietal | 902 | 2.05 ± 0.16 | 1.9 ± 0.16 | 7 | 10−4 | 0.2 |
| Right lateral occipital | 1489 | 2.02 ± 0.15 | 1.87 ± 0.14 | 8 | 10−5 | 0.25 |
| Right lingual gyrus | 1267 | 1.79 ± 0.14 | 1.65 ± 0.12 | 8 | 10−4 | 0.27 |
| Control > T+ | Control | T+ | ||||
| Left precuneus | 912 | 2.36 ± 0.16 | 2.08 ± 0.17 | 12 | 10−6 | 0.41 |
| Left supramarginal gyrus | 290 | 2.68 ± 0.18 | 2.42 ± 0.14 | 10 | 10−4 | 0.33 |
| Left posterior cingulate | 629 | 2.56 ± 0.19 | 2.34 ± 0.15 | 9 | 10−4 | 0.26 |
| Left pars triangularis | 339 | 2.52 ± 0.19 | 2.27 ± 0.2 | 10 | 10−4 | 0.23 |
| Right precentral | 598 | 2.35 ± 0.15 | 2.12 ± 0.16 | 10 | 10−4 | 0.33 |
| Right middle frontal | 497 | 2.31 ± 0.16 | 2.12 ± 0.14 | 8 | 10−5 | 0.29 |
| Right posterior cingulate | 387 | 2.22 ± 0.18 | 1.99 ± 0.22 | 10 | 10−5 | 0.19 |
| Right supramarginal gyrus | 179 | 2.75 ± 0.19 | 2.44 ± 0.24 | 11 | 10−4 | 0.24 |
| Right precuneus | 358 | 2.51 ± 0.2 | 2.25 ± 0.2 | 10 | 10−6 | 0.3 |
| Control > Preclinical AD | Control | Preclinical AD | ||||
| Right superior temporal | 592 | 3.27 ± 0.27 | 2.72 ± 0.3 | 17 | 10−6 | 0.39 |
| Right superior temporal | 393 | 2.87 ± 0.2 | 2.52 ± 0.25 | 12 | 10−4 | 0.32 |
| Right middle temporal | 322 | 2.77 ± 0.2 | 2.36 ± 0.15 | 15 | 10−5 | 0.44 |
| Right posterior cingulate | 276 | 2.19 ± 0.27 | 1.91 ± 0.34 | 13 | 10–4 | 0.14 |
| Control > Prodromal AD | Control | Prodromal AD | ||||
| Left middle temporal | 1428 | 2.74 ± 0.18 | 2.44 ± 0.23 | 11 | 10−7 | 0.26 |
| Left inferior parietal | 414 | 2.47 ± 0.19 | 2.16 ± 0.25 | 13 | 10−4 | 0.31 |
| Left medial orbitofrontal | 183 | 2.87 ± 0.36 | 2.49 ± 0.35 | 13 | 10−4 | 0.15 |
| Right middle temporal | 1057 | 2.77 ± 0.15 | 2.46 ± 0.18 | 11 | 10−5 | 0.27 |
| Right anterior cingulate | 430 | 2.82 ± 0.26 | 2.53 ± 0.21 | 10 | 10−4 | 0.17 |
Note: CS, cluster size, it refers to the extent of cortical thickness change; SD, standard
deviation; P, corrected P-value
(P < 0.05) in the peak vertex;
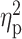 , peak partial eta-squared
showing the effect size.
, peak partial eta-squared
showing the effect size.
Figure 3 .
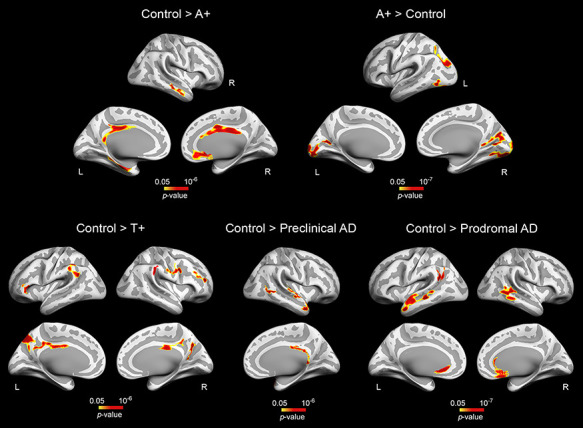
Group differences in cortical thickness. Results were corrected for multiple comparisons using a previously validated hierarchical statistical model (Bernal-Rusiel et al. 2010). L, left; R, right. Note that A+ participants exhibited opposite patterns of cortical thickness (top panel, left and right), whereas T+ and A+T+ (preclinical and prodromal AD) individuals showed thinning patterns spreading over different cortical territories (bottom panel).
The volume of cerebral WM was also significantly different across groups (F(4,113) = 9.25, P = 10−36). With the exception of A+, the remaining groups (i.e., T+, preclinical, and prodromal AD) exhibited patterns of WM atrophy compared with controls (Table 4 and Fig. 4). T+ individuals showed structural WM deficits in left cingulum hippocampus (P = 10−6) and right anterior thalamic radiation (P = 10−10). In the preclinical AD group, WM atrophy was evident in the inferior longitudinal fasciculus bilaterally (left, P = 10−8; right, P = 10−6), while in the prodromal group, WM deficits were extended to the right anterior thalamic radiation (P = 10−40), right inferior longitudinal fasciculus (P = 10−24), and left fronto-occipital fasciculus (P = 10−36/10−21). These patterns of WM atrophy were not associated with volume loss of BF cholinergic compartments for none of the groups studied.
Table 4.
Group differences in cerebral WM volume
| WM tract | CS (mm3) | P | x | y | z | T |
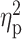
|
|---|---|---|---|---|---|---|---|
| Control > T+ | |||||||
| Right anterior thalamic radiation | 1270 | 10−10 | 17 | −36 | 0 | 6.9 | 0.25 |
| Left cingulum hippocampus | 477 | 10−6 | 11 | −41 | −2 | 5.8 | 0.34 |
| Control > preclinical AD | |||||||
| Left inferior longitudinal | 675 | 10−8 | −42 | −15 | −18 | 7.2 | 0.35 |
| Right inferior longitudinal | 355 | 10−6 | 38 | −2 | −30 | 7.4 | 0.44 |
| Control > prodromal AD | |||||||
| Right anterior thalamic radiation | 13 566 | 10−40 | 24 | 29 | 20 | 7.1 | 0.21 |
| Right inferior longitudinal | 6085 | 10−24 | 39 | −50 | −9 | 7.2 | 0.41 |
| Left inferior fronto-occipital (anterior) | 11 416 | 10−36 | −29 | 20 | 15 | 7.4 | 0.2 |
| Left inferior fronto-occipital (posterior) | 5173 | 10−21 | −36 | −26 | −12 | 9.1 | 0.4 |
Note: WM, white matter; CS, cluster size, it refers to the extent of WM atrophy; P,
cluster-level FWE correction (P < 0.05), peak MNI
coordinates (x-y-z); T, peak t-value; 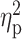 , peak partial eta-squared
showing the effect size. Location of affected WM regions was obtained with the JHU
white-matter tractography atlas (Hua et al.
2008).
, peak partial eta-squared
showing the effect size. Location of affected WM regions was obtained with the JHU
white-matter tractography atlas (Hua et al.
2008).
Figure 4 .
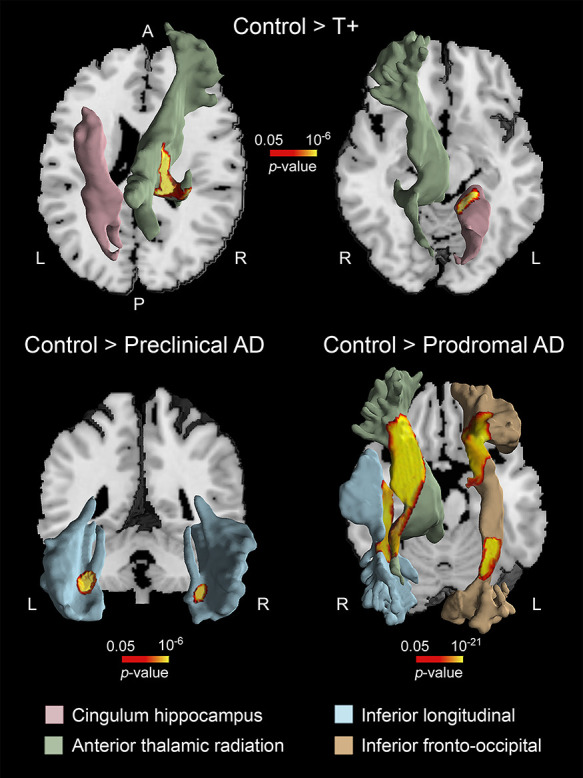
Patterns of WM atrophy in asymptomatic subjects AR-AD. Note that T+, preclinical, and prodromal AD subjects showed different regional patterns of WM deficits. The location of WM atrophy was obtained with the JHU WM tractography atlas (Hua et al. 2008). A, anterior; P, posterior; L, left; R, right.
Effect of AD Pathology on the Relationship between BF Cholinergic Compartments and Their Innervated Regions
Because cholinergic denervation mainly affects cerebral regions receiving BF cholinergic projections, we next asked whether volume loss of BF cholinergic compartments correlated with atrophy of amygdala, hippocampus, and neocortex in individuals at different risk for AD. Results showed significant associations between volume loss of Ch4 and atrophy of amygdala in T+ (left/right amygdala: P = 10−2/10−3), preclinical (left/right amygdala: P = 10−5/10−5), and prodromal AD (left/right amygdala: P = 10−6/10−6), whereas positive correlations between Ch1–Ch2 and hippocampus only affected prodromal AD patients (left/right hippocampus: P = 10−3/10−3) (Fig. 5).
Figure 5 .
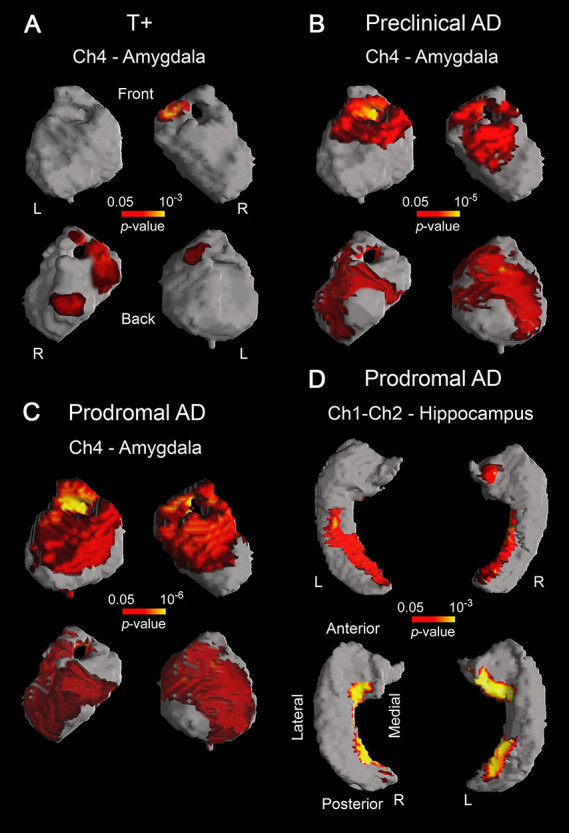
Significant correlations between GM volume loss of different BF cholinergic compartments and atrophy of their innervated regions in T+ (A), preclinical AD (B), and prodromal AD (C, D). Results were corrected for multiple comparisons (FWE, P < 0.05). L, left; R, right. Note that tauopathy (T+) is a requirement for the emergence of the structural coupling between Ch4 and amygdala (A), the spatial extent of this coupling being larger in preclinical/prodromal AD than in T+ (B, C). The structural coupling between Ch1–Ch2 and hippocampus only occurred in prodromal AD (D).
Confirmatory Results Based on the ADNI Dataset
Results obtained with the ADNI sample showed similarities and differences with those obtained in our sample (see Supplementary Figs 2–5). Disparities are likely caused by the small size of the T+ group in the ADNI database (N = 10 vs. N = 29 in our sample) and/or for the use of t-tau (in the ADNI dataset) instead of p-tau (in our sample) for determining tau pathology in CSF.
One-way ANCOVAs confirmed that structural integrity of brain structures assessed differed significantly across groups (BF: F(4,249) = 4.95, P = 0.001; amygdala: F(4,249) = 5.42, P = 0.001; hippocampus: F(4,249) = 6.4, P = 10−7; cortical thickness: left—F(4,249) = 4.86, P = 0.006; right—F(4,249) = 4.74, P = 0.004). Group differences in GM volume of BF compartments, amygdala, and hippocampus in the ADNI cohort were comparable to those observed in our sample, although T+ subjects of the ADNI database did not show atrophies of Ch4 and amygdala (Supplementary Figs 2 and 3). Individuals with preclinical and prodromal AD exhibited volume loss of Ch1–Ch2 (preclinical, left: P = 0.02, right: P = 0.03; prodromal, left: P = 0.001), Ch4 (preclinical, left: P = 0.02; prodromal, left: P = 10−4, right: P = 10−4), amygdala (preclinical, right: P = 0.04; prodromal, left: P = 10−6, right: P = 10−6), and hippocampus (preclinical, right: P = 0.03; prodromal, left: P = 10−16, right: P = 10−16). Similar to our sample, A+ subjects of the ADNI cohort did not exhibit volume changes in BF cholinergic compartments, hippocampus, or amygdala.
Changes of cortical thickness affected all ADNI groups with AD pathology, although the spatial distribution of these changes was not identical in both datasets (Supplementary Fig. 4). A+ subjects exhibited patterns of cortical thinning (left cingulate cortex: P = 10−6; right insula: P = 10−6) and thickening (left inferior/superior parietal: P = 10−4/10−3; right rostral cingulate: P = 10−3), while T+ and preclinical AD subjects showed atrophy of the left isthmus cingulate (P = 10−4) and the right superior temporal sulcus (P = 10−3), respectively. Patterns of cortical thinning spread over bilateral temporal (left: P = 10−12; right: P = 10−12), parietal (left: P = 10−8; right: P = 10−7), and frontal regions (left: P = 10−8; right: P = 10−7).
The relationship between volume loss of BF compartments and atrophy of hippocampus/amygdala in prodromal AD was comparable in the two cohorts, but it differed in T+ and preclinical AD groups (Supplementary Fig. 5). Thus, prodromal AD patients of the ADNI dataset showed significant correlations between Ch1–Ch2 and hippocampus (left/right hippocampus: P = 10−11/10−11) and between Ch4 and amygdala (left/right amygdala: P = 10−5/10−5). Interestingly, correlations between Ch4 atrophy and cortical thinning in prodromal AD only emerged in the ADNI cohort (left: P = 10−5; right: P = 10−4), supporting previous findings obtained with comparable sample sizes (Cantero et al. 2017).
Discussion
Pioneering research in the 1970s identified a marked loss of BF cholinergic cells and altered ACh synthesis in the nbM of AD patients (Davies and Maloney 1976; Perry et al. 1977; White et al. 1977). The central role of ACh in cognitive function (Drachman and Leavitt 1974) and neuronal plasticity (Kilgard and Merzenich 1998) led to the hypothesis that AD-related cognitive decline was likely due to progressive BF presynaptic cholinergic deficits affecting central cholinergic transmission (Bartus et al. 1982). Despite the growing body of literature supporting this hypothesis, little research has examined whether volume loss of nbM precedes symptom onset (Kerbler et al. 2014; Schmitz et al. 2018; Butler et al. 2018), and none of these studies have specifically evaluated whether BF structural deficits initiate with either amyloidosis or tauopathy (or both) in AR-AD individuals. Here we showed, for the first time in humans using in vivo MRI, that volume loss of nbM occurs in asymptomatic subjects with tauopathy (T+) but not in individuals with isolated amyloidosis (A+). In preclinical and prodromal AD (A+T+), our results revealed that BF atrophy extends to the medial septum and vertical limb of the diagonal band Broca (Ch1–Ch2). Cerebral regions receiving BF cholinergic projections showed well-defined patterns of structural deficits with AD CSF biomarkers, but only T+ and A+T+ (both preclinical and prodromal AD) individuals exhibited significant associations between volume loss of BF cholinergic compartments and atrophy of cholinergically innervated limbic regions. These results were partially confirmed using the ADNI cohort. Overall, these findings suggest that tauopathy is required for the initiation of structural deficits and cholinergic denervation of the nbM in AR-AD subjects, and the extent of this damage to larger BF territories and other innervated structures seems to be dependent on the presence of both AD proteinopathies in later stages of the AD continuum.
Long-standing evidence shows that amyloid deposition is a key event in AD pathogenesis (Hardy and Higgins 1992) and that cholinergic dysfunctions play a central role in AD-related cognitive decline (Bartus et al. 1982). Research supporting the link between cholinergic deficits and Aβ deposits mainly comes from animal models (e.g., Inestrosa et al. 1996; Pettit et al. 2001; Boncristiano et al. 2002). In one of these studies, cortical amyloidosis paralleled decreased cholinergic fiber density in neocortex rather than reduced number of magnocellular neurons in the nbM (Boncristiano et al. 2002), in agreement with neuropathological studies in cognitively normal elderly subjects (Beach et al. 2000; for opposite results, see Cullen et al. 1997). However, we have failed to confirm any meaningful correlation between volume loss of Ch4 and atrophy of its innervated cerebral regions in A+ individuals. Several explanations may account for the lack of BF vulnerability to abnormal CSF levels of Aβ1–42 in the present study. One possibility is that higher aggregation (Hoshi et al. 1997), longer exposure (Tran et al. 2001), and/or larger cortical territories occupied by Aβ plaques (Cummings and Cotman 1995) lead to significantly higher neurotoxicity and inflammation favoring cholinergic neuronal loss in nbM and cortical cholinergic denervation. It may also happen that isolated amyloidosis does not generate an imbalance in the nerve growth factor (NGF) metabolic pathway sufficient to initiate an impairment in the BFCS (Cuello et al. 2019). The latter hypothesis finds experimental support in human postmortem MCI/AD studies showing that NGF signaling is resilient to brain amyloidosis but is altered with tau pathology (Perez et al. 2015; for opposite results, see Capsoni et al. 2000).
Despite the fact that BF cholinergic deficits are closely related to the tauopathy (Sassin et al. 2000; Mesulam et al. 2004), human in vivo evidence supporting this association is noticeably lacking. By using quantitative MRI techniques, we have shown atrophy of the nbM in asymptomatic T+ that may be due to the presence of tau oligomers associated with pretangle pathology within cholinergic nbM neurons (Tiernan et al. 2018). Previous studies have shown that cytoskeletal alterations are already present in the nbM from preclinical Braak I–II stages (Sassin et al. 2000). At this early stage of tauopathy, cholinergic axons of affected nbN neurons may become dystrophic affecting the structural integrity of their innervated regions (Mesulam 2013). This assumption is indirectly supported by findings of the present study. Thus, our T+ participants showed bilateral volume loss of nbM, amygdala, and neocortex in tandem with significant positive correlation between atrophy of nbM and amygdala. It is worth mentioning that both the Ch1–Ch2 compartments and the hippocampus were structurally preserved in the T+ group, presumably reflecting the integrity of this cholinergic circuitry during early tau pathology. However, MCI patients presented significant structural deficits in medial septum and vertical limb of the diagonal band Broca (i.e., Ch1–Ch2) together with hippocampal atrophy (see Figs 1 and 2), supporting recent results obtained with an independent MCI sample (Cantero et al. 2017). Accordingly, our results provide novel human in vivo evidence suggesting that structural deficits of BF cholinergic compartments arise before cognitive symptoms become evident. This damage affects the nbM in asymptomatic T+ subjects, while the extent of this damage depends on the synergistic toxic interaction between amyloid and tau pathology in preclinical and prodromal AD.
While WM pathology has been profusely described in AD patients (Brun and Englund 1986), only a few studies have specifically investigated structural changes of WM in asymptomatic AR-AD subjects. Here, we have shown that T+, preclinical, and prodromal AD subjects presented patterns of WM volume loss that were unrelated to atrophy of BF cholinergic compartments. Different lines of evidence support these findings. Firstly, postmortem studies have revealed that nondemented subjects with histopathological AD lesions have WM shrinkage comparable to that observed in AD patients (de la Monte 1989). Secondly, in vivo MRI experiments have shown that asymptomatic individuals with higher burden of AD pathology have specific patterns of WM atrophy (Cantero et al. 2018), deficits of WM microstructure (Gold et al. 2014; Hoy et al. 2017), and disrupted WM networks (Fischer et al. 2015). Therefore, this evidence suggests that WM degeneration precedes the onset of AD symptoms and may contribute to account for heterogeneity of AD.
These results may have implications for the identification of asymptomatic individuals AR-AD who might benefit most of cholinergic therapy. Previous research has revealed that a reduced number of neurons in the nbM parallels cortical GM atrophy in histopathologically confirmed AD patients (Cullen et al. 1997), suggesting a link between cholinergic denervation and atrophy of vulnerable cortical regions. In this context, we found no significant associations between volume loss of Ch4 and cortical thinning in p-tau+ individuals, indicating that cortical cholinergic innervation may be still preserved in isolated tauopathy.
Long-established research suggests a role for NGF metabolic dysfunction in AD progression (e.g., Fahnestock et al. 2001; Pedraza et al. 2005), mainly based on the strong dependency of cholinergic BF neurons on endogenous NGF levels (Cuello et al. 2007). More precisely, it has been shown that the imbalance between mature and precursor NGF may exacerbate AD-like retrograde degeneration of BF cholinergic neurons (Allard et al. 2018). Moreover, pharmacological evidence has strongly supported the link between NGF dysfunctions and structural deficits of the nbM. Accordingly, early experiments revealed positive correlations in NGF levels between the nbM and regions innervated by this cholinergic compartment (Korsching et al. 1985). nbM neurons have also shown to be highly dependent on the retrograde transport of NGF for the maintenance of their terminal synapses in the cerebral cortex (Cuello et al. 2007). Based on this evidence, it seems reasonable to search for therapeutic strategies aimed at restoration of NGF signaling to delay degeneration of the nbM and consequently preserve the cholinergic transmission in AR-AD subjects (Counts and Mufson 2005). While delivering mature NGF directly to the central nervous system seems to be the most straightforward approach, this strategy inevitably requires from invasive techniques and the biological action of NGF may be highly unspecific (Iulita and Cuello 2014). Further evidence has shown that targeting the inhibition of mature NGF degradation leads to the formation of new cholinergic boutons, providing an alternative strategy to slowing degeneration of the nbM (Allard et al. 2012). Although these results are promising, all the experiments conducted to date have been performed in rats and therefore require validation in humans. Much translational research needs to be done prior to transfer NGF-related therapeutic approaches to clinical trials.
The current study has limitations that deserve mention. First, our findings were obtained with a relatively small sample, and the level of AD pathology was only established on the basis of CSF biomarkers. Although our results were partially confirmed with the ADNI cohort, they should be validated in further studies combining CSF biomarkers with amyloid and tau PET imaging. Moreover, it is worth noting that our A+ group was younger than T+ and A+T+ individuals, likely lessening the neurotoxic effect of isolated amyloidosis on the BFCS. Although all statistical analyses performed in the present study were adjusted by age, it may be the case that residual effects of age act synergistically with tauopathy boosting BF damage. Finally, our results rely on indirect volume MRI measurements of BF nuclei derived from cytoarchitectonic probabilistic maps of BF magnocellular compartments obtained in 10 subjects (Zaborszky et al. 2008). Consequently, BF volumetric changes cannot distinguish between glial and neuronal loss or between cell shrinkage and cell death. Due to the small size of BF cholinergic compartments, findings of the present study may also reflect tissue changes other than cholinergic cell degeneration likely due to inaccurate GM segmentation and/or spatial normalization effects. However, the high specificity of the BF atrophy pattern, the spatial extent of BF damage with AD pathology, and the relationship between specific BF cholinergic compartments and their innervated regions (i.e., Ch1–Ch2 with hippocampus, and Ch4 with amygdala) strongly argue against this possibility. Given that BF cholinergic deficits are not unique to AD, long-term follow-up studies combining tailored cholinergic and noncholinergic therapies with in vivo MRI measurements of BF cholinergic compartments are required to determine whether pharmacological interventions are indicated in AR-AD subjects, and whether BF imaging biomarkers are predictive of future cognitive changes in late life. Finally, we cannot rule out potential effects of lifetime alcohol consumption on cerebral changes reported in the present study (e.g., Gu et al. 2014; Downer et al. 2015) and specifically on volume reduction of the nbM (Floyd et al. 1997). Future experiments should include alcohol consumption as covariate in the statistical model to disentangle its effects on structural changes of the cholinergic circuitry in asymptomatic subjects AR-AD.
In conclusion, our results showed that abnormal CSF levels of phosphorylated tau (T+) parallel bilateral volume loss of the nbM in AR-AD individuals. The relationship between volume loss of nbM and atrophy of amygdala also emerged in T+ subjects, spreading over larger cholinergic territories in later stages of the AD continuum (A+T+). These results argue for a significant role of tauopathy in initiating BF degeneration and cholinergic denervation in asymptomatic subjects AR-AD. Whether T+ subjects respond best to cholinergic therapy, reducing the risk of, or completely preventing the clinical onset of AD, needs to be clarified in future studies.
Supplementary Material
Funding
Spanish Ministry of Economy and Competitiveness (SAF2017-85310-R to J.L.C., PSI2017-85311-P to M.A.); the Regional Ministry of Innovation, Science and Enterprise, Junta de Andalucia (P12-CTS-2327 to J.L.C.); the International Center on Aging CENIE-POCTEP (0348_CIE_6_E to M.A.); CIBERNED (SIGNAL collaborative study); Institute of Health Carlos III (grants PI11/02526, PI14/01126, and PI17/01019 to J.F.; PI13/01532 and PI16/01825 to R.B.; PI17/01895 to A.L.; PI08/0139, PI12/02288, and PI16/01652 to P.S.-J.); Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa”; Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED) (Program 1, AD to A.L., J.L.C., P.S.-J.); Pla Estratègic de Recerca i Innovació en Salut (grant SLT006/17/125 to D.A. and SLT006/17/00119 to J.F.); a Fundació La Marató de TV3 grant (20141210 to J.F. and 247/C/2016 to R.B.); Fundació Bancaria Obra Social La Caixa (DABNI project to R.B.); Fundación BBVA (grant to A.L.); Instituto de Investigación Marqués de Valdecilla and Joint Programme Neurodegenerative Disease (DEMTEST PI11/03028 to P.S.J.).
Notes
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledge Conflict of Interest: None declared.
References
- Alcolea D, Martinez-Lage P, Sanchez-Juan P, Olazaran J, Antunez C, Izagirre A, Ecay-Torres M, Estanga A, Clerigué M, Guisasola MC, et al. . 2015. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurology. 85:626–633. [DOI] [PubMed] [Google Scholar]
- Allard S, Leon WC, Pakavathkumar P, Bruno MA, Ribeiro-da-Silva A, Cuello AC. 2012. Impact of the NGF maturation and degradation pathway on the cortical cholinergic system phenotype. J Neurosci. 32:2002–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard S, Jacobs ML, Do Carmo S, Cuello AC. 2018. Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiol Aging. 67:10–20. [DOI] [PubMed] [Google Scholar]
- Amunts K, Kedo O, Kindler M, Pieperhoff P, Mohlberg H, Shah NJ, Habel U, Schneider F, Zilles K. 2005. Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: intersubject variability and probability maps. Anat Embryol. 210:343–352. [DOI] [PubMed] [Google Scholar]
- Arendt T, Bigl V, Tennstedt A, Arendt A. 1985. Neuronal loss in different parts of the nucleus basalis is related to neuritic plaque formation in cortical target areas in Alzheimer’s disease. Neuroscience. 14:1–14. [DOI] [PubMed] [Google Scholar]
- Ashburner J. 2007. A fast diffeomorphic image registration algorithm. Neuroimage. 38:95–113. [DOI] [PubMed] [Google Scholar]
- Bartus RT, Dean RL III, Beer B, Lippa AS. 1982. The cholinergic hypothesis of geriatric memory dysfunction. Science. 217:408–414. [DOI] [PubMed] [Google Scholar]
- Baskin DS, Browning JL, Pirozzolo FJ, Korporaal S, Baskin JA, Appel SA. 1999. Brain choline acetyltransferase and mental function in Alzheimer disease. Arch Neurol. 56:1121–1123. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Chiba AA. 1999. Cognitive functions of the basal forebrain. Curr Opin Neurobiol. 9:178–183. [DOI] [PubMed] [Google Scholar]
- Beach TG, Kuo YM, Spiegel K, Emmerling MR, Sue LI, Kokjohn K, Roher AE. 2000. The cholinergic deficit coincides with Abeta deposition at the earliest histopathologic stages of Alzheimer disease. J Neuropathol Exp Neurol. 59:308–313. [DOI] [PubMed] [Google Scholar]
- Bernal-Rusiel JL, Atienza M, Cantero JL. 2008. Detection of focal changes in human cortical thickness: spherical wavelets versus Gaussian smoothing. Neuroimage. 41:1278–1292. [DOI] [PubMed] [Google Scholar]
- Bernal-Rusiel JL, Atienza M, Cantero JL. 2010. Determining the optimal level of smoothing in cortical thickness analysis: a hierarchical approach based on sequential statistical thresholding. Neuroimage. 52:158–171. [DOI] [PubMed] [Google Scholar]
- Bierer LM, Haroutunian V, Gabriel S, Knott PJ, Carlin LS, Purohit DP, Perl DP, Schmeidler J, Kanof P, Davis KL. 1995. Neurochemical correlates of dementia severity in Alzheimer’s disease: relative importance of the cholinergic deficits. J Neurochem. 64:749–760. [DOI] [PubMed] [Google Scholar]
- Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, Stalder M, Phinney AL, Abramowski D, Sturchler-Pierrat C, Enz A, et al. . 2002. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci. 22:3234–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun A, Englund E. 1986. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol. 19:253–262. [DOI] [PubMed] [Google Scholar]
- Butler T, Harvey P, Deshpande A, Tanzi E, Li Y, Tsui W, Talos D, Devinsky O, Kuchna I, Nowicki K, et al. . 2018. Basal forebrain septal nuclei are enlarged in healthy subjects prior to the development of Alzheimer's disease. Neurobiol Aging. 65:201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero JL, Zaborszky L, Atienza M. 2017. Volume loss of the nucleus basalis of Meynert is associated with atrophy of innervated regions in mild cognitive impairment. Cereb Cortex. 27:3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantero JL, Atienza M, Sanchez-Juan P, Rodriguez-Rodriguez E, Vazquez-Higuera JL, Pozueta A, Gonzalez-Suarez A, Vilaplana E, Pegueroles J, Montal V, et al. . 2018. Cerebral changes and disrupted gray matter cortical networks in asymptomatic older adults at risk for Alzheimer's disease. Neurobiol Aging. 64:58–67. [DOI] [PubMed] [Google Scholar]
- Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. 2000. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 97:6826–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, Mufson EJ. 2005. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol. 64:263–272. [DOI] [PubMed] [Google Scholar]
- Cuello AC, Bruno MA, Bell KF. 2007. NGF-cholinergic dependency in brain aging MCI and Alzheimer's disease. Curr Alzheimer Res. 4:351–358. [DOI] [PubMed] [Google Scholar]
- Cuello AC, Pentz R, Hall H. 2019. The brain NGF metabolic pathway in health and in Alzheimer’s pathology. Front Neurosci. 13:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen KM, Halliday GM, Double KL, Brooks WS, Creasey H, Broe GA. 1997. Cell loss in the nucleus basalis is related to regional cortical atrophy in Alzheimer's disease. Neuroscience. 78:641–652. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Cotman CW. 1995. Image analysis of beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 346:1524–1528. [DOI] [PubMed] [Google Scholar]
- de la Monte SM. 1989. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer's disease. Ann Neurol. 25:450–459. [DOI] [PubMed] [Google Scholar]
- Drachman DA, Leavitt J. 1974. Human memory and the cholinergic system. A relationship to aging? Arch Neurol. 30:113–121. [DOI] [PubMed] [Google Scholar]
- Davies P, Maloney AJF. 1976. Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet. 2:1403. [DOI] [PubMed] [Google Scholar]
- Downer B, Jiang Y, Zanjani F, Fardo D. 2015. Effects of alcohol consumption on cognition and regional brain volumes among older adults. Am J Alzheimers Dis Other Demen. 30:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, Bakardjian H, Benali H, Bertram L, Blennow K, et al. . 2016. Proceedings of the Meeting of the International Working Group (IWG) and the American Alzheimer's Association on “The Preclinical State of AD”. Alzheimers Dement. 12:292-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K. 2005. A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage. 25:1325–1335. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. 2001. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 18:210–220. [DOI] [PubMed] [Google Scholar]
- Fischer FU, Wolf D, Scheurich A, Fellgiebel A, Alzheimer's Disease Neuroimaging Initiative . 2015. Altered whole-brain white matter networks in preclinical Alzheimer's disease. Neuroimage Clin. 8:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Tootell RB, Dale AM. 1999. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp. 8:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM. 2000. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA. 97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd EA, Young-Seigler AC, Ford BD, Reasor JD, Moore EL, Townsel JG, Rucker HK. 1997. Chronic ethanol ingestion produces cholinergic hypofunction in rat brain. Alcohol. 14:93–98. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, Mufson EJ. 2016. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Neurochem. 96:1401–1408. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, Che S. 2011. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Chem Neuroanat. 42:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold BT, Zhu Z, Brown CA, Andersen AH, LaDu MJ, Tai L, Jicha GA, Kryscio RJ, Estus S, Nelson PT, et al. . 2014. White matter integrity is associated with cerebrospinal fluid markers of Alzheimer's disease in normal adults. Neurobiol Aging. 35:2263–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BA, Blazey T, Su Y, Fagan AM, Holtzman DM, Morris JC, Benzinger TL. 2016. Longitudinal β-amyloid deposition and hippocampal volume in preclinical Alzheimer disease and suspected non-Alzheimer disease pathophysiology. JAMA Neurol. 73:1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Scarmeas N, Short EE, Luchsinger JA, DeCarli C, Stern Y, Manly JJ, Schupf N, Mayeux R, Brickman AM. 2014. Alcohol intake and brain structure in a multiethnic elderly cohort. Clin Nutr. 33:662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. 1992. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 256:184–185. [DOI] [PubMed] [Google Scholar]
- Hoshi M, Takashima A, Murayama M, Yasutake K, Yoshida N, Ishiguro K, Hoshino T, Imahori K. 1997. Non-toxic amyloid beta-peptide 1−42 suppresses acetylcholine synthesis. Possible role in cholinergic dysfunction in Alzheimer’s disease. J Biol Chem. 272:2038–2041. [DOI] [PubMed] [Google Scholar]
- Hoy AR, Ly M, Carlsson CM, Okonkwo OC, Zetterberg H, Blennow K, Sager MA, Asthana S, Johnson SC, Alexander AL, et al. . 2017. Microstructural white matter alterations in preclinical Alzheimer's disease detected using free water elimination diffusion tensor imaging. PLoS One. 12(3):e0173982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua K, Zhang J, Wakana S, Jiang H, Li X, Reich DS, Calabresi PA, Pekar JJ, Zijl PC, Mori S. 2008. Tract probability maps in stereotaxic spaces: analyses of white matter anatomy and tract-specific quantification. Neuroimage. 39:336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Alvarez A, Pérez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. 1996. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme. Neuron. 16:881–891. [DOI] [PubMed] [Google Scholar]
- Iulita MF, Cuello AC. 2014. Nerve growth factor metabolic dysfunction in Alzheimer's disease and Down syndrome. Trends Pharmacol Sci. 35:338–348. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. 2010. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, Lowe VJ, Senjem ML, Gunter JL, Preboske GM, et al. . 2014. Age-specific population frequencies of cerebral β-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 13:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel G. 1991. Design and analysis: a researcher’s handbook. Englewood Cliffs (NJ): Prentice Hall. [Google Scholar]
- Kerbler GM, Fripp J, Rowe CC, Villemagne VL, Salvado O, Rose S, Coulson EJ, Initiative A's DN. 2014. Basal forebrain atrophy correlates with amyloid β burden in Alzheimer's disease. Neuroimage Clin. 7:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. 1998. Cortical map reorganization enabled by nucleus basalis activity. Science. 279:1714–1718. [DOI] [PubMed] [Google Scholar]
- Korsching S, Auburger G, Heumann R, Scott J, Thoenen H. 1985. Levels of nerve growth factor and its mRNA in the central nervous system of the rat correlate with cholinergic innervation. EMBO J. 4:1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG, Suzuki J, Dolman CE, Nagai T. 1984. Aging, Alzheimer’s disease, and the cholinergic system of the basal forebrain. Neurology. 34:741–745. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, Wainer BH. 1983. Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical coa, diagonal band nuclei, connections of the septal areleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J Comp Neurol. 214:170–197. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Geula C. 1988. Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol. 275:216–240. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Shaw P, Mash D, Weintraub S. 2004. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 55:815–828. [DOI] [PubMed] [Google Scholar]
- Mesulam MM. 2013. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. J Comp Neurol. 521:4124–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI, et al. . 2002. Loss of basal forebrain P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol. 443:136–153. [DOI] [PubMed] [Google Scholar]
- Pappas BA, Bayley PJ, Bui BK, Hansen LA, Thal L. 2000. Choline acetyltransferase activity and cognitive domain scores of Alzheimer’s patients. Neurobiol Aging. 21:11–17. [DOI] [PubMed] [Google Scholar]
- Pedraza CE, Podlesniy P, Vidal N, Arévalo JC, Lee R, Hempstead B, Ferrer I, Iglesias M, Espinet C. 2005. Pro-NGF isolated from the human brain affected by Alzheimer's disease induces neuronal apoptosis mediated by p75NTR. Am J Pathol. 166(2):533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Wuu J, Mufson EJ, Fahnestock M. 2004. Increased proNGF levels in subjects with mild cognitive impairment and mild Alzheimer disease. J Neuropathol Exp Neurol. 63:641–649. [DOI] [PubMed] [Google Scholar]
- Perez SE, He B, Nadeem M, Wuu J, Scheff SW, Abrahamson EE, Ikonomovic MD, Mufson EJ. 2015. Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer's disease. Biol Psychiatry. 77:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry EK, Perry RH, Blessed G, Tomlinson BE. 1977. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet. 1:189. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. 2001. Beta-amyloid (1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 21:RC120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassin I, Schultz C, Thal DR, Rüb U, Arai K, Braak E, Braak H. 2000. Evolution of Alzheimer's disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 100:259–269. [DOI] [PubMed] [Google Scholar]
- Scheef L, Grothe MJ, Koppara A, Daamen M, Boecker H, Biersack H, Schild HH, Wagner M, Teipel S, Jessen F. 2019. Subregional volume reduction of the cholinergic forebrain in subjective cognitive decline (SCD). Neuroimage Clin. 21:101612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz TW, Mur M, Aghourian M, Bedard MA, Spreng RN, Initiative A’s DN . 2018. Longitudinal Alzheimer's degeneration reflects the spatial topography of cholinergic basal forebrain projections. Cell Rep. 24:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, et al. . 2009. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR Jr, Kaye J, Montine TJ, et al. . 2011. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiernan CT, Mufson EJ, Kanaan NM, Counts SE. 2018. Tau oligomer pathology in nucleus basalis neurons during the progression of Alzheimer disease. J Neuropathol Exp Neurol. 77:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran MH, Yamada K, Olariu A, Mizuno M, Ren XH, Nabeshima T. 2001. Amyloid beta-peptide induces nitric oxide production in rat hippocampus: association with cholinergic dysfunction and amelioration by inducible nitric oxide synthase inhibitors. FASEB J. 15:1407–1409. [DOI] [PubMed] [Google Scholar]
- Vogels OJ, Broere CA, Laak HJ, Donkelaar HJ, Nieuwenhuys R, Schulte BP. 1990. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging. 11:3–13. [DOI] [PubMed] [Google Scholar]
- White P, Hiley CR, Goodhardt MJ, Carrasco LH, Keet JP, Williams IE, Bowen DM. 1977. Neocortical cholinergic neurons in elderly people. Lancet. 1:668–671. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. 1981. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 10:122–126. [DOI] [PubMed] [Google Scholar]
- Zaborszky L, Hoemke L, Mohlberg H, Schleicher A, Amunts K, Zilles K. 2008. Stereotaxic probabilistic maps of the magnocellular cell groups in human basal forebrain. Neuroimage. 42:1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Gombkoto P, Varsanyi P, Gielow MR, Poe G, Role LW, Ananth M, Rajebhosale P, Talmage DA, Hasselmo ME, et al. . 2018. Specific basal forebrain-cortical cholinergic circuits coordinate cognitive operations. J Neurosci. 38:9446–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.