

Keywords: SARS-CoV-2, Alignment-free, Correlation measure, DNA sequence
Abstract
Coronaviruses (especially SARS-CoV-2) are characterized by rapid mutation and wide spread. As these characteristics easily lead to global pandemics, studying the evolutionary relationship between viruses is essential for clinical diagnosis. DNA sequencing has played an important role in evolutionary analysis. Recent alignment-free methods can overcome the problems of traditional alignment-based methods, which consume both time and space. This paper proposes a novel alignment-free method called the correlation coefficient feature vector (CCFV), which defines a correlation measure of the L-step delay of a nucleotide location from its location in the original DNA sequence. The numerical feature is a 16 × L-dimensional numerical vector describing the distribution characteristics of the nucleotide positions in a DNA sequence. The proposed L-step delay correlation measure is interestingly related to some types of L + 1 spaced mers. Unlike traditional gene comparison, our method avoids the computational complexity of multiple sequence alignment, and hence improves the speed of sequence comparison. Our method is applied to evolutionary analysis of the common human viruses including SARS-CoV-2, Dengue virus, Hepatitis B virus, and human rhinovirus and achieves the same or even better results than alignment-based methods. Especially for SARS-CoV-2, our method also confirms that bats are potential intermediate hosts of SARS-CoV-2.
1. Introduction
The worldwide outbreak of the SARS-CoV-2 virus has necessitated a deeper understanding of the transmission path, evolutionary process, and other dynamics of viruses. Since the first appearance of novel pneumonia (COVID-19) in Wuhan, Hubei province, China, there has been a lot of discussion on the origin of the causative virus, SARS-CoV-2 (Sironi et al., 2020). SARS-CoV-2 is the seventh coronavirus known to infect humans: SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease. Hepatitis B virus (HBV) is another infectious virus that can establish a persistent and chronic infection in humans through immune energy. In 2016, worldwide estimates suggest that 257 million people are chronically infected with the Hepatitis B virus (HBV). About 15% to 25% of them may die from cirrhosis or liver cancer (Nelson et al., 2016). HBV is a partially double-stranded DNA virus and a member of the hepadnaviridae family. Dengue viruses can also spread to people via the bite of an infected Aedes species mosquito. About 40% of the world's population living in areas with a risk of dengue (Tsang et al., 2019). Human rhinoviruses is one of the most common viruses in humans and is the predominant cause of the common cold. Sequence analysis is a popular and effective technique for studying these viruses, and genetic sequence comparison has become a crucial procedure in many modern biological techniques.
Sequence comparison is traditionally performed by alignment-based methods. These methods often attempt to maximize an alignment score calculated as the sum of substitution scores minus gap penalties. The popular algorithms include Smith-Waterman, Needleman-Wunch, Muscle, etc (Edgar, 2004, Chookajorn, 2020). However, alignment-based methods are burdened by large memory and time consumption, and may be affected by high mutation and recombination rates (Vinga, 2014). These problems can be overcome by alignment-free methods, which are gaining attraction in biological fields. Alignment-free methods quantify sequence similarity/dissimilarity that does not use or produce alignment at any step of algorithm application. Since alignment-free methods do not rely on dynamic programming, they are computationally less expensive and therefore suitable for whole genome comparisons.
An alignment-free method called Subsequence Natural Vector (SNV) was proposed to subtyping HIV-1 genomes and achieved very high sensitivity and specificity (He et al., 2020). The alignment-free method Natural Vector has achieved very high accuracy in classification of viruses including Ebola, Zika and HBV, etc. (Li et al., 2017, Deng et al., 2011). The Chaos Game Representation has succeeded in species classification (Joel, 1990, Randhawa et al., 2020b). A machine learning method combined with digital signal processing was used for rapid classification of novel pathogens especially SARS-CoV-2 (Randhawa et al., 2020a). Among the alignment-free approaches, k−mer methods based on the frequencies of subsequences of a defined length k were widely used in sequence analysis (Kurtz et al., 2008). A new method to compute k-mer frequencies and its application to annotate large repetitive plant genomes (Zielezinski et al., 2019, Bernard et al., 2019). Such as, feature frequency profiles (FFP) is one popular k-mer based method that considers the frequencies, positions, and other properties of a k bp-length substring in a genome (Wu et al., 2009), and recently a new method which is based on k-mer is proposed, this method can be effectively used for sequence comparison (Sarkar et al., 2021).
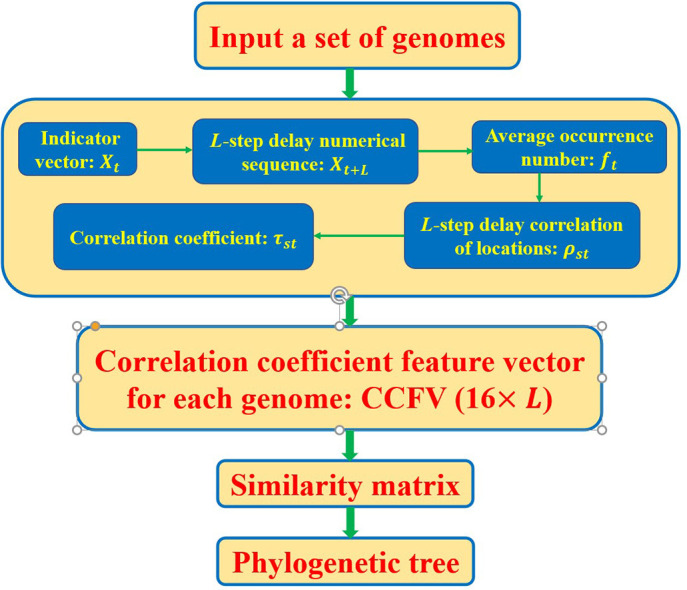
This paper proposes a numerical feature vector that describes the nucleotide distribution of viral gene sequences. A DNA sequence is first mapped to four binary vectors, whose elements are assigned 0 or 1 depending on the locations of the four nucleotides along the sequence. Second, these four vectors are processed by our proposed correlation coefficient feature vector (CCFV) method, which calculates the correlation coefficient and auto-correlation coefficient of an L-step delay from the original sequence. Third, from 1 to L-steps, these correlation coefficients are compiled into a 42 × L-dimensional vector. This method represents a sequence by a low-dimensional numerical vector, which greatly improves the construction speed of phylogenetic tree. The similarity between two viruses is then determined by the Euclidean distance between their 42 × L-dimensional vectors. Finally, a detailed evolutionary tree is drawn by the unweighted pair group method with arithmetic mean (UPGMA). Our method is shown to obtain the correct evolutionary relationship. According to biological features of bat and the very close evolutionary relationship between bat-nCoV and SARS-CoV-2 as shown in the phylogenetic tree constructed by our method, we demonstrate that bats seem to be the natural reservoir of SARS-CoV-2 causing the recent COVID-19 outbreak.
2. Methods
2.1. Location correlation coefficient
Let S = s 1 s 2 ⋯ N be a DNA sequence with s i ∈ {A, C, G, T}. To obtain the important information of a nucleotide t, t ∈ {A, C, G, T} in the sequence, we first convert the sequence into a indicator vector as follows: Let X t = (x t(1), x t(2), · , x t(N)), t ∈ {A, C, G, T}, where
| (1) |
We define a L-step delay numerical sequence X t+L of X t as:
| (2) |
where x t(N + 1) = ⋯ = x t(l + N) = 0, especially, X t = X t+0. Then we define the average occurrence number of t ∈ {A, C, G, T} as:
| (3) |
The L-step delay correlation of locations in nucleotide, ρ tt(L), is defined as:
| (4) |
Example: Given a sequence=“ACTGTGAGT”, we have N=9, and suppose that L = 2 and t is A and T, respectively. Then:
The detailed calculation process is shown in as shown in Fig. 1 .
Fig. 1.

The process of calculating the Xt, Xt+2 and ft when t is A and T, respectively.
Similarly, we define the correlation ρ st(L) of nucleotides s and t in a sequence as follows:
| (5) |
To normalize the above correlation, we finally define the correlation coefficient τ st(L) between nucleotides s and t as:
| (6) |
The auto-correlation coefficient τ tt(l) of nucleotide t itself is defined as the normalized auto-correlation:
| (7) |
Finally, we get a 16 × L feature vector of nucleotides called the correlation coefficient feature vector (CCFV):
Therefore, if there’re n genomes, a n × (16 × L) matrix can be construct. Meanwhile, Euclidean distance is used to get the similarity matrix. Example: Continue with the example above, for sequence=“ACTGTGAGT”, we have N=9, and suppose that L = 2 and t is A and T, respectively. Then:
3. Results
The SARS-CoV-2 datasets were derived from GISAID (https://www.gisaid.org/) and NCBI (https://www.ncbi.nlm.nih.gov/). The Dengue virus, Hepatitis B virus (HBV) and Human rhinovirus virus (HRV) datasets were from NCBI. Our CCFV method was applied to the evolutionary analysis for these viruses. L was determined as 5 in all datasets. Computations were performed on a personal computer with an Intel Core i5-10210U CPU @ 1.60 GHz and 16 GB RAM.
3.1. SARS-CoV-2
China's first COVID-19 outbreak in Wuhan, Hubei Province, was reported in late 2019. The virus can cause severe pneumonia and has become a major global health threat (Chang et al., 2020, Benvenuto et al., 2020). The COVID-19 virus has been officially termed as SARS-CoV-2. The clinical symptoms of COVID-19 infection include (but are not limited to) dyspnea, fever, pneumonia, and renal failure. The SARS-CoV-2 virus rapidly mutates, has strong transmission ability, and produces similar symptoms to influenza. For these reasons, COVID-19 is difficult to control and diagnose. For example, COVID-19 recurred in Beijing's Xinfadi Market in July of 2020. As of August 29 2021, more than 216 million cases have been diagnosed and 4.4 million deaths have been reported worldwide, and the data continue to rise according to Johns Hopkins Coronavirus Resource Center (CRC) (https://coronavirus.jhu.edu/map.html).
3.1.1. Coronaviruses
According to epidemiological investigation and gene sequence comparisons, SARS-CoV-2 is 87.5% similar to Bat-SL-CoVZC45 and 87.6% similar to Bat-SL-CoVZXC21. Both variants are coronaviruses found in bats. Previous studies have shown that SARS-CoV-2 infection is transmitted by an intermediate host (bats or the civet Paguma larvata) before mutating and spreading among humans (Zhou et al., 2020, Zhu et al., 2020). In the present study, 80 whole genomes of coronaviruses were sourced from the GISAID and NCBI databases. The phylogenetic tree built from our CCFV using the UPGMA method is given in Fig. 2 . Among the 73 BetaCoV and 7 AlphaCoV, 5 SARS-CoV-2 from Wuhan and the bat coronavirus BatCoV RaTG13 belong to sister branches (red branches in Fig. 2), indicating maximum similarity among these 6 genomes. (Zhou et al., 2020) found that SARS-CoV-2 in early patient samples shared 96.2% sequence identity with bat-coronavirus (bat-nCoV) RaTG13 at the whole-genome level. In their phylogenetic tree obtained by sequence homology comparison, seven SARS-CoV-2 viruses from seven patients clustered together with bat-coronavirus (bat-nCoV) RaTG1 (Zhou et al., 2020). According to biological features of bat and the high identity sequence between bat-nCoV and SARS-CoV-2 demonstrated by a lot of additional research, bats are considered as the natural reservoir of SARS-CoV-2 (Wu et al., 2020). In our phylogenetic tree, the 5 SARS-CoV-2 viruses from Wuhan cluster with RaTG13 as well. Thus we conclude that bats seems to be the natural reservoir of SARS-CoV-2 by our method.
Fig. 2.

UPGMA phylogenetic tree constructed from the nucleotide sequences of 80 complete coronavirus genomes (L = 5). MHV: murine hepatitis virus; PEDV: porcine epidemic diarrhea virus; TGEV: porcine transmissible gastroenteritis virus, SARS-CoV-2 and bat CoV RaTG13 are shown in bold and in red. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.1.2. SARS-CoV-2 variant
With the emergence of SARS-CoV-2 mutants, people began to pay attention to its variants. On the one hand, for the development of vaccine, on the other hand, it can provide beneficial help for clinic. GISAID divided SARS-CoV-2 into nine clades: S, O, L, V, G, GH, GR, GV, and GRY. The S and L clades were around at the beginning of the pandemic. But then later there were other variants.
We obtain 37 SARS-CoV-2 genomes from GISAID which include nine different clades. Using Muscle method, O,S,GR,SH are mixed with other clades, see Fig. 3 . The variant of SARS-CoV-2 usually contain only a few mutations, the model-based algorithm can not identify difference of variants in many cases. Our technique consider location correlation coefficient of the different nucleotide and achieve the better result shown in Fig. 4 .
Fig. 3.

UPGMA phylogenetic tree of 37 SARS-CoV-2 based on the Muscle method.
Fig. 4.

UPGMA phylogenetic tree of 37 SARS-CoV-2 based on the correlation coefficient feature vector (CCFV) method with L = 5.
3.2. Dengue virus
DENV is transmitted to humans through the bite of an infected mosquito of the Aedes genus. Dengue fever infects humans in more than 100 countries worldwide (https://www.cdc.gov/dengue/about/index.html) (Sirisena and Noordeen, 2014). Approximately three billion people are at risk of contracting DENV, and 400 million people are infected with the virus each year. Huma infection is caused by four serotypes called DENV 1, 2, 3, and 4 (Tsang et al., 2019). Therefore, research on this virus is urgently demanded. To predict the serotypes of an unknown virus, the evolutionary analysis of this virus should be performed. The phylogenetic trees constructed from the genomes of 330 Dengue viruses by NV and our CCFV method are presented in Fig. 5, Fig. 6 , respectively. The viruses are correctly placed into four types, but the type 3 doesn’t come together by the NV method. Our method obviously outperformed the NV method in prediction accuracy.
Fig. 5.
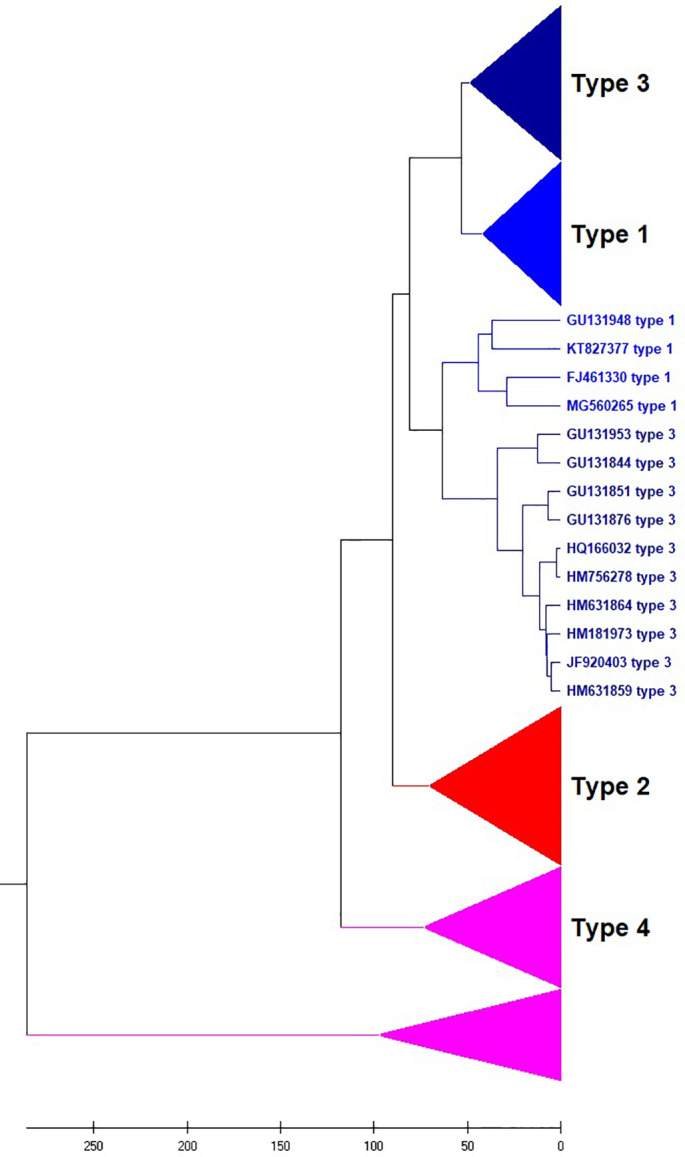
UPGMA phylogenetic tree of 330 Dengue viruses based on the NV method.
Fig. 6.
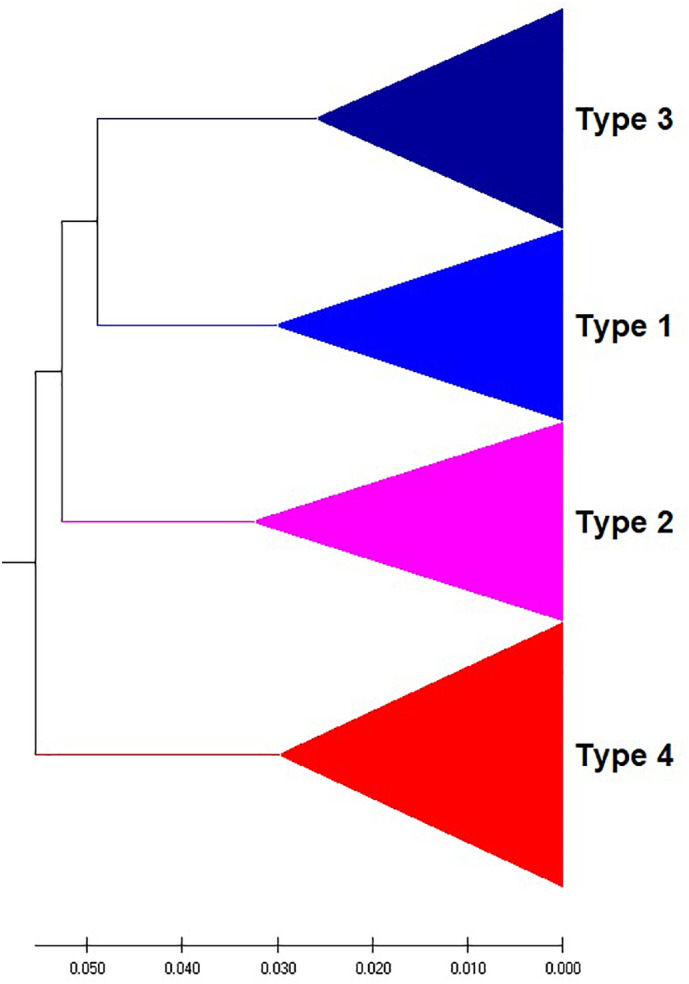
UPGMA phylogenetic tree of 330 Dengue viruses based on the CCFV method with L = 5.
3.3. Hepatitis B virus
The HBV causes a persistent chronic infection in immunocompromised humans (Lazarus et al., 2018). The HBV includes ten genotypes assigned A-J (Yuen et al., 2018, Juliette et al., 2013). Therefore, the rapid and accurate identification of HBV genotypes is a significant goal in clinical diagnosis. Here, we performed an evolutionary analysis of 152 complete HBV genomes extracted from the Hepatitis B virus Database (HBVdb). The phylogenetic tree by our CCFV method are shown in Fig. 7 . CCFV clearly assigned each genotype to a correct branch. As comparison, the UPGMA trees built by the currently popular alignment method Muscle are shown in Fig. 8 . Both methods give the same result, however, CCFV is much faster than Muscle, see Table 1 . This result demonstrates that the CCFV is superior than Muscle in terms of runtime.
Fig. 7.

UPGMA phylogenetic tree of 152 HBVs based on the CCFV method with L = 5.
Fig. 8.

UPGMA phylogenetic tree of 152 HBVs based on Muscle method.
Table 1.
Running time for Muscle, and CCFV method. “s”: seconds; “min”: minute.
| Method | Muscle | CCFV |
|---|---|---|
| HBV (152) | 11 min | 0.93 s |
| Dengue (330) | Larger than 30 min | 4.92 s |
| HRV (116) | Larger than 30 min | 1.2 s |
3.4. Human rhinovirus
Human rhinovirus (HRV) was first isolated in the 1950s from nasopharyngeal secretions of patients with the common cold. HRV belongs to the Picornaviridae family and genus Enterovirus. Past studies have shown that HRV consists of three genetically distinct groups: HRV-A, HRV-B, and HRV-C. In a previous study (Palmenberg et al., 2009), 116 complete genomes including 113 HRV and 3 outgroup HEV-C genomes were studied. The result for the 116 genomes based on CCFV is shown in Fig. 9 . The result based our technique is the same as the result in the previous study. However, our running time is only 1.20 seconds and the running time in the previous paper is very high because of usage of multiple sequence alignment for building the phylogenetic tree.
Fig. 9.

UPGMA phylogenetic tree of 116 HRVs based on the CCFV method with L = 5.
4. Discussion and conclusion
The proposed CCFV method maps a DNA sequence to four binary vectors, then determines the correlation coefficient and auto-correlation coefficient of an k step delay from the original sequence. The correlation coefficients after delaying the sequence by 1 to L steps are concatenated into a (16 × L)-dimensional vector. Describing each DNA sequence in this vector form, we measured the similarity between two sequences by calculating the Euclidean distance between the vectors representing those sequences. Using the UPGMA method, we finally constructed the phylogenetic trees of the genomes contained in two SARS-CoV-2 datasets, a DENV dataset, and a HBV dataset. Our method clustered together five SARS-CoV-2 variants from Wuhan. Moreover, this group formed two adjacent branches with the bat coronavirus suggesting that bats are an natural reservoir of SARS-CoV-2. The CCEV successfully established the evolutionary relationships for viruses SARS-CoV-2, DENV, HBV and HRV. In addition, we derive some interesting formulas to approximate the proposed correlation/auto-correlation coefficient. These formulas indicate the L step delay correlation coefficient is directly proportional to the frequency of some types of spaced L + 1-mers. We compare the approximate correlation coefficient with the true correlation coefficient in all datasets. The approximate error is very small and may be ignorable.
To illustrate the effectiveness of our approach for larger genomes, we collect 59 bacterial genomes with length ranging from 0.8 to 5 million bp. Then we build a phylogenetic tree by the CCFV technique. As shown in Fig. 10 , these bacterial genomes are separated into 14 families which are clearly separated from each other.
Fig. 10.

UPGMA phylogenetic tree of 59 bacteria genome sequences based on the CCFV method with L = 5.
For genome sequence S, NV method consider the number, the mean position and the mean position of nucleotide α ∈ A, C, G, T in S. For each nucleotide, our method considers not only the correlation of appearance of nucleotides, but also the correlation due to L-steps delay. Thus the NV method only contains the distribution of single nucleotide, but ignores the correlation of nucleotides. Therefore, we can extract more information of nucleotides in a genome.
Our correlation coefficient measure has some limitations as well. First, the defined delay step number L is a variable parameter, which must be determined in multiple trial-and-error tests. Second, some viruses in the same region do not form a branch in the evolutionary tree by our method, possibly because they have mutated from viruses in other regions, or the dimension of the numerical vector is insufficiently high to capture the information loss. These problems should be investigated in further study.
CRediT author statement
Lily He and Peter K. Li conceived the initial planning, implemented the code and analysed the results. All authors wrote and reviewed the manuscript.
Declaration of Competing Interest
The authors declare that they have no conflicts of interest related to this manuscript.
Acknowledgements
This study is supported by Promotion plan for young teachers' scientific research ability of Beijing University of Civil Engineering and Architecture (X21026).
Appendix
A.1. Location correlation coefficient approximate formula
That is:
| (8) |
According to definition of the auto-correlation ρ tt(L) of nucleotide t, we obtain
When L = 1, , and the following relationship holds:
In this case, the value of represents the number of “tt” (2-mer) present. For general L, the value of represents the number of “t * ⋯ * t” L +1-mer present, denoted as n t*⋯*t. Here each t * ⋯ * t-mer has identical head and tail nucleotide t separated by L-1 undetermined nucleotides. In some studies, this kind of k-mer is called spaced k-mer.
Let n t(L) be the number of t occurrences in the first L locations of a DNA sequence. Then
| (9) |
Note that is less than . Since L is very small and the genome length N is very large, for example in this paper L = 5 and N is more than 10000, the last term is ignorable. Therefore the approximate formula for ρ tt(L) is as follows:
| (10) |
The ρ st(L) can be approximated as:
| (11) |
The l-step delay correlation coefficient τ st(L) can be approximated as:
| (12) |
The auto-correlation coefficient τ tt(L) of nucleotide t can be approximated as:
| (13) |
According to above approximate formulas, we can see that the proposed correlation coefficient is directly proportional to the ratio , which is the frequency of spaced L + 1-mer s * ⋯ * t in a sequence.
References
- Benvenuto D., Giovanetti M., Salemi M., Prosperi M., Flora C.D., Alcantara L.C.J., Angeletti S., Ciccozzi M. The global spread of 2019-ncov: a molecular evolutionary analysis. Pathog. Global Health. 2020;114:64–67. doi: 10.1080/20477724.2020.1725339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard G., Chan C.X., Chan Y.B., Chua X.Y., Cong Y., Hogan J.M., Maetschke S.R., Ragan M.A. Alignment-free inference of hierarchical and reticulate phylogenomic relationships. Brief. Bioinform. 2019;20:426–435. doi: 10.1093/bib/bbx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T.J., Yang D.M., Wang M.L., Liang K.H., Wang C.T. Genomic analysis and comparative multiple sequence of sars-cov2. J. Chin. Med. Assoc. 2020;83:1. doi: 10.1097/JCMA.0000000000000335. [DOI] [PubMed] [Google Scholar]
- Chookajorn T. Evolving COVID-19 conundrum and its impact. Proc. Natl. Acad. Sci. U.S.A. 2020;117:12520–12521. doi: 10.1073/pnas.2007076117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M., Yu C., Liang Q., He R.L., Yau S.S.-T. A novel method of characterizing genetic sequences: genome space with biological distance and applications. PLoS One. 2011;6:e17293. doi: 10.1371/journal.pone.0017293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. Muscle: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Dong R., He R.L., Yau S.T. A novel alignment-free method for HIV-1 subtype classification. Infect. Genet. Evol. 2020:77. doi: 10.1016/j.meegid.2019.104080. [DOI] [PubMed] [Google Scholar]
- Joel J.H. Chaos game representation of gene structure. Nucleic Acids Res. 1990:2163–2170. doi: 10.1093/nar/18.8.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliette H., Fanny J., Gilbert D., Alan K., Fabien Z., Christophe C. HBVdb: a knowledge database for Hepatitis B virus. Nucleic Acids Res. 2013;41:D566–D570. doi: 10.1093/nar/gks1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S., Narechania A., Stein J.C., Ware D. A new method to compute k-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics. 2008;9:517. doi: 10.1186/1471-2164-9-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarus J.V., Timothy B., Brchot C., Anna K., Veronica M., Michael N., Capucine P., Ulrike P., Homie R., Thomas L.A.a. The Hepatitis B epidemic and the urgent need for cure preparedness. Nat. Rev. Gastroenterol. Hepatol. 2018;15:517–518. doi: 10.1038/s41575-018-0041-6. [DOI] [PubMed] [Google Scholar]
- Li Y., He L., He R.L., Yau S.S.T. Zika and flaviviruses phylogeny based on the alignment-free natural vector method. DNA Cell Biol. 2017;36:1–8. doi: 10.1089/dna.2016.3532. [DOI] [PubMed] [Google Scholar]
- Nelson N.P., Easterbrook P.J., McMahon B.J. Epidemiology of Hepatitis B virus infection and impact of vaccination on disease. Clinics Liver Dis. 2016;20:607–628. doi: 10.1016/j.cld.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmenberg A.C., Spiro D., Kuzmickas R., Wang S., Djikeng A. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science. 2009;324:55–59. doi: 10.1126/science.1165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa G., Soltysiak M., Roz H., Souza C., Hill K., Kari L. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLoS One. 2020;15:e0232391. doi: 10.1371/journal.pone.0232391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randhawa G.S., Hill K.A., Kari L. MLDSP-GUI: an alignment-free standalone tool with an interactive graphical user interface for DNA sequence comparison and analysis. Bioinformatics. 2020;36:2258–2259. doi: 10.1093/bioinformatics/btz918. [DOI] [PubMed] [Google Scholar]
- Sarkar B.K., Sharma A.R., Bhattacharya M., Sharma G., Lee S.S., Chakraborty C. Determination of k-mer density in a DNA sequence and subsequent cluster formation algorithm based on the application of electronic filter. Sci. Rep. 2021;11:13701. doi: 10.1038/s41598-021-93154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirisena P.D.N.N., Noordeen F. Evolution of dengue in Sri Lanka changes in the virus, vector, and climate. Int. J. Infect. Dis. 2014;19:6–12. doi: 10.1016/j.ijid.2013.10.012. [DOI] [PubMed] [Google Scholar]
- Sironi M., Hasnain S.E., Phan T., Luciani F., Gonzlez-Candelas F. SARS-CoV-2 and COVID-19: a genetic, epidemiological, and evolutionary perspective. Infect. Genet. Evol. 2020;84:104384. doi: 10.1016/j.meegid.2020.104384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang T.K., Ghebremariam S.L., Gresh L., Gordon A., Halloran M.E., Katzelnick L.C., Rojas D.P., Kuan G., Balmaseda A., Sugimoto J.a. Effects of infection history on dengue virus infection and pathogenicity. Nat. Commun. 2019;10:1246. doi: 10.1038/s41467-019-09193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinga S. Editorial: alignment-free methods in computational biology. Brief. Bioinform. 2014;15:341–342. doi: 10.1093/bib/bbu005. [DOI] [PubMed] [Google Scholar]
- Wu F., Su Z., Bin Y., Zhang Y.Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.A., Jun S.R., Sims G.E., Kim S.H. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. Proc. Natl. Acad. Sci. U.S.A. 2009;106:12826–12831. doi: 10.1073/pnas.0905115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen M.F., Chen D.S., Dusheiko G.M., Hla J., Dty L., Locarnini S.A., Peters M.G., Lai C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers. 2018;4:18035. doi: 10.1038/nrdp.2018.35. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Wang W., Liu Z., Liang C., Tan W. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020;11:3910. doi: 10.1038/s41467-020-17796-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielezinski A., Girgis H.Z., Bernard G., Leimeister C.A., Karlowski W.M. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019;20:144. doi: 10.1186/s13059-019-1755-7. [DOI] [PMC free article] [PubMed] [Google Scholar]