

Abstract
Background:
We previously showed that fibrinogen is a major determinant of the growth of a murine model of colorectal cancer (CRC).
Objective:
Our aim was to define the mechanisms coupling fibrin(ogen) to CRC growth.
Results:
CRC tumors transplanted into the dorsal subcutis of Fib− mice were less proliferative and demonstrated increased senescence relative to those grown in Fib+ mice. RNA-Seq analyses of Fib+ and Fib− tumors revealed 213 differentially regulated genes. One gene highly upregulated in tumors from Fib− mice was stratifin, encoding 14-3-3σ, a master regulator of proliferation/senescence. In a separate cohort, we observed significantly increased protein levels of 14-3-3σ and its upstream and downstream targets (i.e., p53 and p21) in tumors from Fib− mice. In vitro analyses demonstrated increased tumor cell proliferation in a fibrin printed 3D environment compared to controls, suggesting that fibrin(ogen) in the tumor microenvironment promotes tumor growth in this context via a tumor cell intrinsic mechanism. In vivo analyses showed diminished activation of focal adhesion kinase (FAK), a key negative regulator of p53, in Fib− tumors. Furthermore, NMR-based metabolomics demonstrated significantly reduced metabolic activity in tumors from Fib− relative to Fib+ mice. Together these findings suggest that fibrin(ogen)-mediated engagement of colon cancer cells activates FAK, which inhibits p53 and its downstream targets including 14-3-3σ and p21, thereby promoting cellular proliferation and preventing senescence.
Conclusions:
These studies suggest that fibrin(ogen) is an important component of the colon cancer microenvironment and may be exploited as a potential therapeutic target.
Keywords: Fibrinogen, p53, 14-3-3σ, Focal adhesion kinase(FAK), colorectal adenocarcinoma, cell senescence
INTRODUCTION
Multiple lines of evidence demonstrate a link between hemostatic system components and the colorectal cancer (CRC) pathogenesis [1, 2]. Elevation of plasma fibrinogen levels and circulating D-dimer correlate with a poor prognosis in colorectal cancer [3]. Patients homozygous for the prothrombotic mutation factor V Leiden display an increased incidence of colon cancer [4]. Murine models have shown that thrombin and fibrin(ogen) promote colonic tumorigenesis in colitis associated cancer through a mechanism involving fibrin(ogen)-mediated engagement of the leukocyte integrin αMβ2 [5]. Thrombin and fibrin(ogen) have also been shown to promote the growth of fully established colonic adenocarcinoma in mice,[6] but the mechanisms coupling fibrin(ogen) to tumor growth in this context have not been rigorously explored.
Focal adhesion kinase (FAK) is a cytoplasmic tyrosine kinase located in focal adhesion complexes that regulate integrin-mediated cell spreading, migration, and signaling events. Previous studies showed that FAK activation plays a significant role in cancer progression [7, 8]. Previous studies have also shown that fibrinogen can bind numerous integrin receptors including αIIbβ3, αVβ3, αXβ2, αMβ2, α5β1, and αVβ1[9–11]. In the current study we demonstrate that fibrin(ogen) matrices in the stroma of a murine colon cancer activate focal adhesion kinase (FAK). FAK activation drives tumor growth by promoting the ubiquitination of the tumor suppressor p53 [12, 13] leading to diminished expression of key downstream targets of p53, including 14-3-3 [14]. 14-3-3σ is one of 7 members of the 14-3-3 protein family that act as adapter proteins, binding to diverse signaling proteins to exert their biological functions [15, 16]. 14-3-3σ is the only isoform that has been described as having potent tumor suppressing activity [17]. It is well-established that 14-3-3σ interacts with p53 and has an adapter role in the regulation of p53 networks [14].14-3-3σ also is a master regulator of cellular metabolism and negatively regulates the cell cycle [18]. Consistent with this, we also observed upregulation of 14-3-3σ and stabilization of p53 in the absence of fibrinogen, as well as diminished tumor cell proliferation and increased senescence.
RESULTS
Fibrin(ogen) in the tumor stroma promotes colon cancer growth and limits tumor cell senescence
We have previously shown that fibrin(ogen) promotes the growth of colonic adenocarcinoma cells (MC38) in immunocompetent mice [6]. In order to explore the mechanisms coupling fibrinogen to tumor growth in this context, we injected MC38 cells into the dorsal subcutis of fibrinogen deficient (Fib−) and control mice (Fib+). Consistent with previous results, tumors grew significantly more slowly in Fib− mice relative to controls. (Fig. 1A). To investigate the effect of fibrinogen on cell cycle regulation, we stained tumor tissues for Ki67 (a cell proliferation marker). There were significantly fewer Ki67+ tumor cell nuclei in Fib− tumors compared to Fib+ tumors, indicating that fibrin(ogen) in the tumor stroma promotes tumor cell proliferation (Figs. 1B & C). In order to determine the impact of fibrinogen on tumor cell survival, we quantified the expression of key apoptosis markers, including cleaved caspase-3 (CCA3), B-cell lymphoma-2 (BCL-2) and Bcl-2-associated X protein (BAX) in Fib+ and Fib− tumors. No significant differences were found in the expression of these apoptotic markers, suggesting that apoptosis does not significantly contribute to fibrinogen-dependent differences in MC38 tumor growth (Fig. 1C–F). In order to determine if fibrinogen is inhibiting tumor cell senescence, we stained tumor tissue for β-galactosidase (SA-β-Gal), an established marker of cellular senescence. SA-β-gal+ staining cells were only found in Fib− tumors (Fig. 1G), suggesting that fibrin(ogen) promotes tumor growth in this context by preventing tumor cell senescence. Consistent with our SA-β-gal staining data, we observed significantly higher protein expression of p21, a key inducer of senescence, in Fib− tumor tissues compared to controls (Fig. 1H &I; supplementary Figure 2A).
Figure 1. Loss of fibrin(ogen) causes cell cycle arrest and cell senescence.
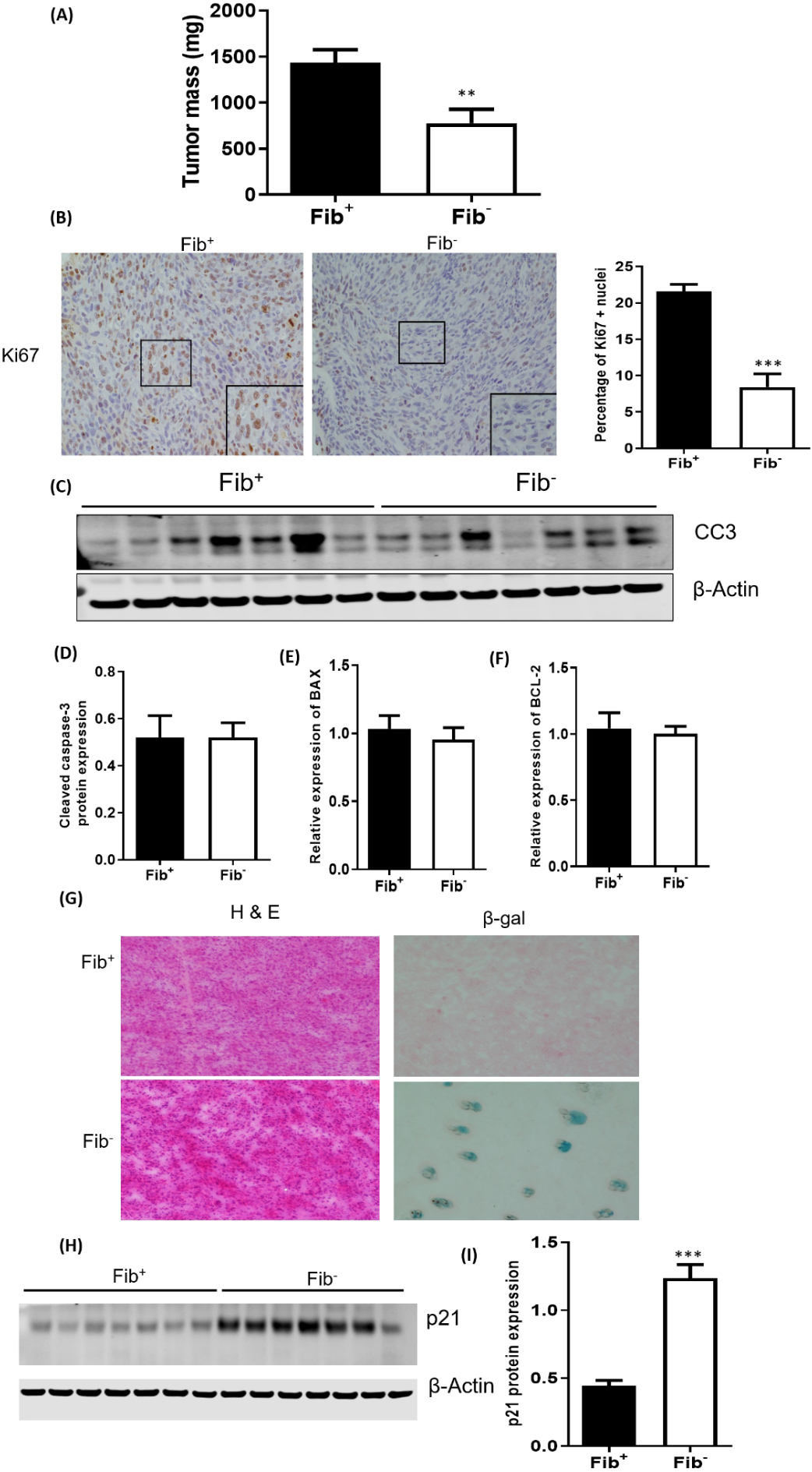
(A) MC38 tumor growth is significantly impeded in Fib− mice relative to Fib+ mice. Shown is tumor mass 21 days after inoculation. (B) Shown are representative sections of MC38 tumor tissue harvested from Fib+ and Fib− mice stained (brown) for Ki67 (C) Quantification of Ki67 positive nuclei in Fib+ and Fib− tumors revealed significantly more Ki67 staining in Fib+ tumors. Western blot (D) and densitometric analyses (E) showing similar cleaved caspase-3 levels in Fib+ and Fib− tumors. BAX (F) and BCL-2 (G) expression were also similar in Fib+ and Fib− tumors. (H) Beta-galactosidase staining in Fib+ and Fib− tumors. Western blot (I) and densitometric analyses (J) of p21 protein expression in Fib+ and Fib− tumors. Bars and error bars represent means and SEMs (n=7 per cohort, *p < 0.001, Mann Whitney U test).
Fibrinogen structure/function and colon cancer growth.
To gain mechanistic insight into what aspects of fibrin(ogen) structure/function regulate CRC tumor growth, we used 4 complementary mouse strains expressing function-selective variants of fibrinogen: Fibγ390–396A mice, which lack the binding motif for the leukocyte integrin receptor αMβ2 [9], FibγΔ5 mice which lack the γ chain binding motif for the platelet integrin receptor αIIbβ3 [19], and FibAEK mice which lack the capacity to form fibrin polymer due to a germline knock-in mutation eliminating the Aα chain thrombin cleavage site [20]. The imposition of Fibγ390−396A had no impact on MC38 growth relative to FibWT mice, suggesting that fibrin(ogen)-mediated interactions with αMβ2 are not required to support the growth of these cells (Fig. 2A). No significant differences were observed in tumor growth between FibγΔ5 and FibWT mice, indicating that platelet-mediated binding to this established γ chain αIIbβ3 binding motif is dispensable for colonic tumor growth (Fig. 2B). Similarly, we found no difference in MC38 tumor growth between FibAEK and FibWT mice, suggesting that fibrinogen promotes tumor growth in this context even in the absence of stable fibrin polymer formation (Fig. 2C). To further interrogate the role of fibrin polymerization in colon cancer growth, we analyzed tumor growth in mice lacking FXIIIA [21]. Paralleling results in FibAEK mice, MC38 tumors grew similarly in FXIIIA−/− and WT mice (Fig. 2D), indicating that FXIII-transglutaminase activity is not essential for the growth of this colon cancer cell line.
Figure 2. Structure/function analyses of fibrinogen-driven MC38 tumor growth.

MC38 tumor growth was unaffected by the imposition of mutations in the native fibrinogen γ chain that (A) eliminate a binding motif for αMβ2 (Fibγ390−396A mice) or (B) αIIbβ3 (FibγΔ5 mice). (C) Tumor growth was also similar between WT and FibAEK mice, which express a mutant fibrinogen “locked” in the soluble state. (D) Elimination of FXIII had no impact on the growth of MC38 tumors. The data shown represent the mean and SEM. Number of mice included in these studies varies from 6 to 10 in each group (Fibγ390−396A mice, n= 9; FibγΔ5 mice, n=10; FibAEK mice, n=7; FXIII, n=6).
Fibrin(ogen) promotes colon cancer growth by limiting 14-3-3σ expression, resulting in downregulation of p53.
To understand the mechanisms underlying diminished tumor growth in fibrinogen-deficient mice, we adopted an unbiased approach and harvested tumors from 4 Fib+ and 4 Fib− mice 14 days after injection of tumor cells into the dorsal subcutis. This time point was chosen because this is when MC38 tumor growth first begins to diverge in Fib+ mice relative to Fib− mice [6]. Consistent with these previous results, tumor size was similar in Fib+ and Fib− mice 14 days after inoculation (Fig. 3A). We performed RNA-seq analyses on these tumors and found differential expression of 213 genes between genotypes (Fig. 3B). Among the 213 genes, 127 genes were downregulated and 86 genes were upregulated in tumors harvested from Fib− mice relative to Fib+ mice (supplementary Table 2). Pathway enrichment analysis revealed that the differentially expressed genes in the tumors from Fib− mice were related to cellular proliferation, survival, migration and metabolism (Figs. 3C&D, supplementary Figure 1A). One of the genes highly upregulated in tumors harvested from Fib− mice relative to Fib+ tumors was SFN, which encodes 14-3-3σ. We validated the expression of 14-3-3σ at the protein level (Figs. 3E&F). We also validated the expression of other key genes, including UNC5B, MMP9, PTN, HSPB1, and GSTA4 by qPCR found to be differentially regulated in RNAseq analyses (supplementary Figure 1B–F).
Figure 3. Fibrinogen promotes tumor growth by altering expression of key genes involved in cell cycle regulation and metabolism.

(A) MC38 cells were injected into the dorsal subcutis of Fib+ and Fib− mice and harvested 14 days after inoculation for RNAseq analyses. Note that at this early time point there were no fibrinogen-dependent differences in tumor size. (B) Volcano plot illustrating differentially expressed genes. Red dots are genes upregulated in tumors from Fib− mice relative to Fib+, and green dots are downregulated genes compared to Fib+. 14-3-3σ is one of the most significantly upregulated genes in Fib− tumors. Also shown are gene ontology (GO) analyses of biological functions for genes downregulated (C) and upregulated (D) in Fib− tumors relative to Fib+ tumors. Western blot (E) and relative densitometry (F) confirming increased expression of 14-3-3σ in Fib− tumors relative to Fib. Error bars represent SEMs (*p < 0.05, Mann Whitney U test).
14-3-3σ is of particular interest as it is a potent cell cycle regulator and it protects p53 from mouse double minute 2 homolog (MDM2) mediated ubiquitination and degradation [14]. We hypothesized that in the absence of fibrinogen, the upregulation of 14-3-3σ and subsequent stabilization of p53 limits tumor growth. To test this hypothesis, we immunoblotted for 14-3-3σ and p53 in a separate validation cohort. We observed significantly increased protein expression of 14-3-3σ and p53 in tumors harvested from Fib− mice relative to controls (Figs. 4A–C), consistent with the view that p53 is protected from ubiquitination by 14-3-3σ in the absence of fibrinogen. The p53 tumor suppressor protein plays a major role in the cellular response to DNA damage and other genomic aberrations, ultimately leading to cell cycle arrest. Acetylation of p53 at Lys379 is a key mechanism by which p53 accumulates in the cell to control the response to DNA damage under stress [22]. Acetylation of p53 enhances the binding of p53 to DNA, promoting p53 mediated transcription of downstream targets. Analyses of p53 acetylation at Lys379 showed increased acetylated p53 in tumors from Fib− mice relative to controls. (Fig. 4D).
Figure 4. Fibrinogen downregulates 14-3-3σ and p53 in MC38 colon cancer tumors.
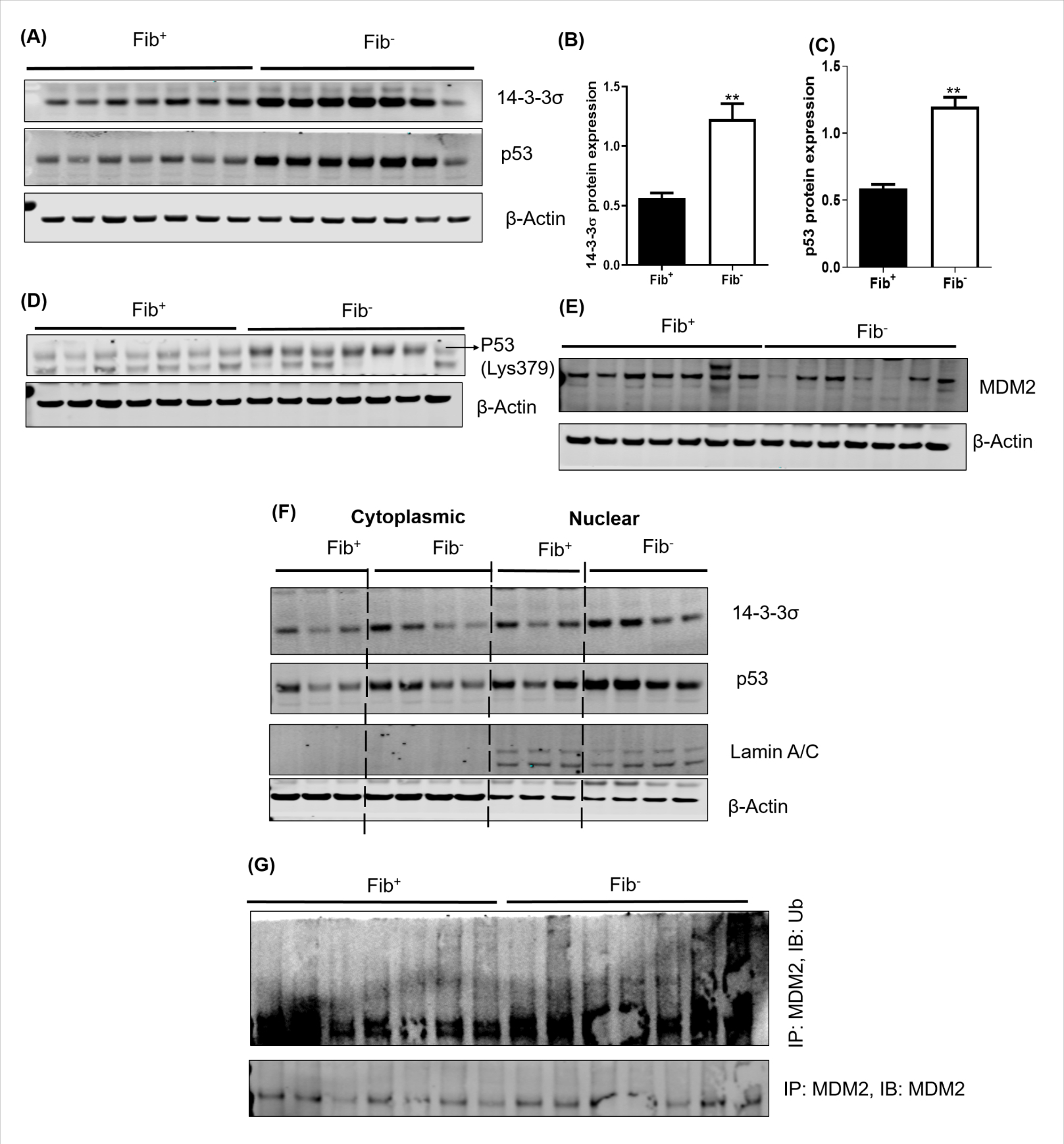
Shown are Western blots (A) and quantitative densitometry analyses of 14-3-3σ (B) and p53 (C) in Fib+ and Fib− tumors. (D) We observed decreased expression of acetylated P53 (Lys379) in Fib+ relative to Fib− tumors (E) Western blots showing the expression pattern of MDM2 in Fib+ and Fib− tumors (F) Nuclear-cytoplasmic fractions were isolated from freshly harvested tumor tissues and western blot experiments performed for 14-3-3σ and p53 in nuclear and cytoplasmic fractions of Fib+ and Fib− tumors. β-actin was used as a cytoplasmic control. Lamin A/C was used to determine the purity of nuclear preparations. (G) Fibrinogen deficiency increases MDM2 ubiquitination. Tumor lysates from Fib+ and Fib− were immunoprecipitated with an anti-MDM2 antibody and immunoblotted with an anti-ubiquitin antibody. Error bars represent SEM (B&C) (**p < 0.01, Mann Whitney U test).
Next we sought to understand whether the relative increase in 14-3-3σ seen in tumors from fibrinogen-deficient mice is directly coupled to diminished MDM2-mediated autoubiquitination of p53. We found significantly less expression of MDM2 in Fib− tumors relative to Fib+ tumors based on Western blot (Fig. 4E). We prepared nuclear and cytosolic fractions from Fib+ and Fib− tumors and performed p53 immunoblotting. Consistent with reduced MDM2, p53 was elevated in the nuclear fraction of Fib− tumors relative to that of Fib+ tumors (Fig. 4F). Furthermore, we executed coimmunoprecipitation of MDM2 followed by immunoblotting of ubiquitin (Ub) and found more ubiquitination of MDM2 in Fib− tumor samples as compared to controls (Fig. 4G). Together these findings suggest that the absence of fibrin(ogen) in the tumor microenvironment leads to upregulation of 14-3-3σ expression, which in turn protects p53 from MDM2-mediated ubiquitination by triggering the autoubiquitination of MDM2.
Fibrinogen activates focal adhesion kinase (FAK) in the colonic adenocarcinoma stroma
Fibrin(ogen) can bind multiple integrin and non-integrin receptors, making it capable of interacting with tumor cells directly, as well as multiple tumor stromal cells. To determine if fibrinogen can directly promote the growth of colon cancer cells, we used an in vitro technique to 3D bioprint tumor cells into a fibrin matrix [23]. The 3D bioprinted constructs confer a three-dimensional extracellular microenvironment to in vitro cultured cells. To determine if fibrinogen can directly promote the growth of MC38 colon cancer cells, MC38 cells were cultured in vitro within a hybrid matrix of alginate and fibrinogen for 14 days. Alginate with bovine serum albumin (BSA) was used as a control matrix. We observed a significant increase in MC38 viable tumor cell numbers at days 14 and 21 of culture in 3D constructs with fibrinogen as compared to those with BSA (Fig. 5A).
Figure 5. Fibrinogen in the colonic adenocarcinoma tumor stroma promotes FAK activation.
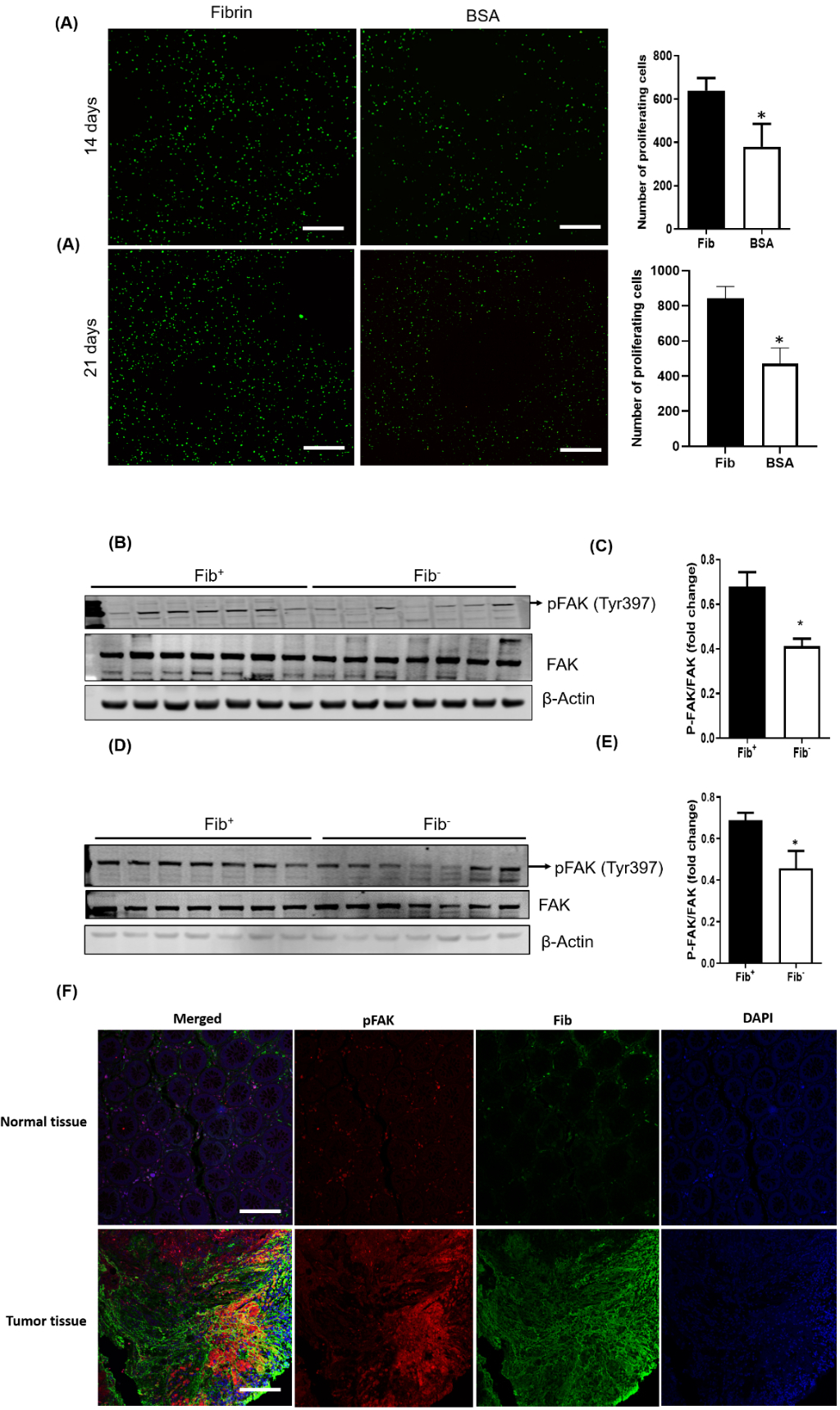
(A) Shown are typical examples of fluorescent micrographs of 3D bioprinted GFP-expressing MC38 tumor models generated using a CELLINK BioX printer (see Methods for details). Significantly greater cellular proliferation was observed after 14 and 21 days in the presence of fibrin versus a BSA control (n = 5 per cohort; *p < 0.05, Mann-Whitney U test). Scale bars represent 500 μm. (B) Western blot analyses for total FAK and pFAK(Tyr397) in MC38 tumor tissues harvested from Fib+ and Fib− mice 20 days after injection. (C) Mean protein levels calculated using densitometric analysis and expressed as a ratio of phospho- to total FAK in Fib+ and Fib− tumors. Beta actin was used as the internal control (n = 7 per cohort, *p < 0.05 Mann Whitney U test). Western blot (D) and densitometric analyses (E) for FAK and pFAK(Tyr397) tumor tissues harvested from Fib+ and Fib− mice 14 days after injection. (n = 7 per cohort, *p < 0.05 Mann Whitney U test) (F) Shown are representative biopsies of normal human colonic tissue and colorectal adenocarcinoma tissue immunofluorescently stained for fibrinogen (green) and pFAK (Tyr397) (red). Nuclei were counterstained with DAPI (blue). Note that areas of intense pFAK staining in colorectal adenocarcinoma samples colocalize with areas of intense fibrinogen staining. Also note that there is very little fibrinogen or pFAK staining in normal colonic tissues. Scale bars represent 100 μm.
These in vitro data suggest that fibrinogen in the tumor microenvironment could promote tumor growth via a tumor cell intrinsic mechanism. Focal adhesion kinase (FAK) is a cytoplasmic tyrosine kinase located in focal adhesion complexes that regulates integrin-mediated cell spreading, migration, and signaling events. Focal adhesion complexes are known to regulate cancer cell growth, and FAK activation has been specifically shown to regulate p53 [24]. We hypothesized that FAK is a key mediator between tumor cell integrin(s) and fibrin(ogen) in the MC38 tumor stroma. To test this hypothesis, we assessed the activation of FAK in Fib+ and Fib− tumors by quantifying phosphorylation of FAK at Tyrosine 397, an established indicator of FAK activation, in tumors harvested 21 days after inoculation. At this later timepoint, there was significantly less FAK activation in tumor tissue harvested from Fib− mice relative to controls (Figs. 5B & C; supplementary Figure 2A & B). To look at an earlier timepoint when the fibrinogen-dependent differences in tumor growth are less apparent, we analyzed tumors 14 days after inoculation, an early timepoint when tumor size was relatively small and similar between genotypes (supplementary Fig. 3). A significant fibrinogen-dependent difference in FAK activation was seen at this early time point (Fig. 5D & E), suggesting that fibrinogen-mediated FAK activation occurs prior to significant differences in tumor growth.
Together these data suggest that fibrinogen in the TME activates FAK, leading to destabilization of p53 through MDM2. Decreased p53 results in diminished expression of 14-3-3σ and p21, which prevents cell senescence and contributes to the acceleration of cell proliferation. This view is further supported by analyses of immunofluorescently stained human colonic adenocarcinoma biopsies where we found abundant deposition of fibrin(ogen) in areas staining positive for FAK activation as compared to adjacent normal colonic tissues (Fig. 5F).
Earlier studies from our group showed that prothrombin supports the growth and local invasion of colonic adenocarcinoma in mice.[6] To determine if FAK signaling is dependent on thrombin functions, we analyzed FAK activation and 14-3-3σ expression in tumor tissues collected from mice with low circulating prothrombin levels and controls. Consistent with our findings that loss of thrombin-mediated fibrin polymer formation (FibAEK) and deletion of FXIIIA had not impact on MC38 tumor growth, we did not observe any prothrombin-dependent changes in the activation of FAK and 14-3-3σ expression. (Supplementary Figure 4 A &B). These findings suggest that thrombin promotes colonic adenocarcinoma growth through fibrinogen-independent mechanisms.
Fibrinogen in the tumor stroma promotes metabolic reprogramming
14-3-3σ and p53 are also known to regulate cancer cell metabolism [18, 25]. We used NMR-based metabolomics to determine the influence of fibrinogen on cancer cell metabolism in vivo. A total of 54 metabolites were quantified in the polar extracts of whole tumor tissues from Fib+ and Fib− mice (Fig. 6A & supplementary table 3). As speculated, 39 out of 54 metabolites were significantly different in tumors from Fib+ mice as compared to tumors from Fib− mice (supplementary table 3). The key metabolites that were found to be significantly decreased in Fib− tumors relative to Fib+ tumors were pyruvate, lactate, glutamate, NAD+ and ATP (Figs. 6B–E). These metabolites are involved in aerobic glycolysis and are essential for tumor growth. In order to confirm that metabolic activity is reduced in the absence of fibrinogen, we analyzed the relative expression levels of key genes involved in glycolysis by qPCR. We observed significant down-regulation of pyruvate kinase isozyme M2 (PKM2), Enolase-1, lactate dehydrogenase (LDHA), hexokinase-2 (HK2), and pyruvate dehydrogenase kinase isoform 2 (PDK2) in Fib− tumors relative to Fib+ tumors (Fig. 6F). Notably, PDK2 enhances glycolysis and lactic acid production, essential functions for robust tumor growth (Fig. 6F). Likewise, the expression of c-Myc target genes involved in glutaminolysis [Slc38a3 (Sn1)] and mitochondrial related genes (ND-1 & TFAM) were decreased in Fib− tumors (Fig. 6F). To define the role of fibrinogen in tumor metabolism at an earlier timepoint, we performed a similar comparison of tumors harvested from Fib− mice and controls 14 days after inoculation, prior to any significant genotype dependent difference in tumor growth (supplementary Figure 5A). We observed significant changes in multiple metabolites including ADP, ATP, succinate, creatine, Sn-glycero-3-phosphocholine, taurine and different amino acids (glycine, threonine, isoleucine, valine, methionine, proline and serine) (supplementary Figure 5B). These results suggest that fibrinogen/FAK interactions promote metabolic reprogramming needed for robust tumor growth by limiting 14-3-3σ expression.
Figure 6. Fibrinogen promotes tumor metabolism.
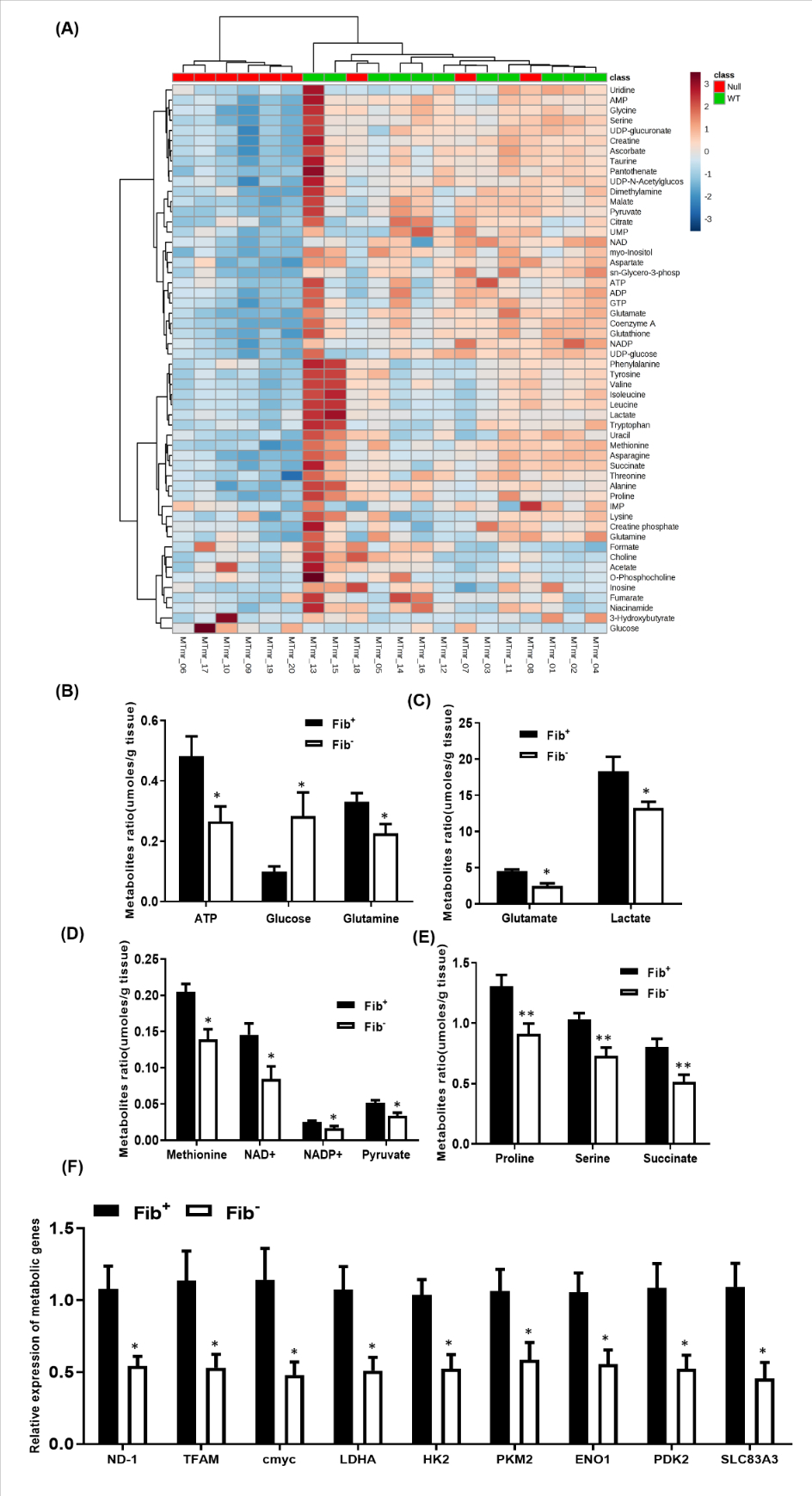
(A) Heat map showing the patterns of different metabolites in Fib− vs Fib+ tumor tissues. (B) Bar graphs showing significant fibrinogen-dependent differences in multiple key metabolites. Metabolite concentrations (umoles) are normalized to tissue mass. (Fib+, n=9; Fib−,n=11; *p < 0.05, **p < 0.001 Kruskal–Wallis test) (C) Bar graph showing the transcript levels of key metabolic genes in tumor tissues harvested from Fib+ and Fib− mice. Bars and error bars represent mean and SEMs (n=6 per cohort, Mann-Whitney U test, *p < 0.05).
DISCUSSION
Colonic adenocarcinoma appears to be at least one important malignancy whereby fibrinogen contributes to tumorigenesis and primary tumor growth. The studies presented here point to a previously unrecognized mechanism by which fibrinogen in the TME promotes cancer growth. The data presented support the conclusion that fibrinogen in the TME activates focal adhesion kinase (FAK), which in turn promotes the ubiquitination of the tumor suppressor, p53. This leads to a decrease in key p53 downstream targets, including p21 and 14-3-3σ, which eventually results in tumor cell metabolic reprogramming, acceleration of tumor cell proliferation, and inhibition of senescence (Fig. 7).
Figure 7: Proposed model explaining fibrinogen-driven colon cancer growth.

Fibrinogen/integrin interactions in the TME leads to FAK activation, ultimately leading to the degradation of p53 and downregulation of downstream p53 targets (14-3-3σ and p21), thereby promoting cell proliferation and survival. In the absence of fibrinogen, p53 is stabilized leading to increased expression of its downstream targets, resulting in cell senescence and growth inhibition.
Earlier studies suggested that downregulation of 14-3-3σ expression is crucial for the progression of multiple cancers [26–29]. 14-3-3σ is required to prevent mitotic catastrophe in response to DNA damage and guard genomic stability.[30] 14-3-3σ expression is upregulated by p53, a critical tumor-suppressor gene which works together with 14-3-3σ in the setting of DNA damage to promote cellular senescence and limit proliferation [30]. Previous reports have established a positive correlation between 14-3-3σ and p53 stabilization [14, 31–33]. A study by Yang et al demonstrated that p53 and 14-3-3σ interact with each other, and 14-3-3σ stabilizes p53 when there is DNA damage [14]. Previous studies also showed that p53 expression increased in response to increases in 14-3-3σ, indicating that 14-3-3σ has a positive impact on p53 stability [14, 33]. In the present study, 14-3-3σ and p53 were found to be significantly upregulated in Fib− tumor tissues as compared to Fib+ tumors and correlated with significantly diminished tumor size in Fib− mice, suggesting that tumor growth is suppressed by 14-3-3σ and p53 in the absence of fibrinogen. Consistent with previous reports showing that 14-3-3σ enhances p53 transcriptional activity [14], the current study also suggests that 14-3-3σ enhances p53 transcriptional activity towards an endogenous target gene, namely p21. 14-3-3σ can stabilize p53 by multiple mechanisms. In this context, we observed enhanced ubiquitination of MDM2 in Fib− tumors. Moreover, increased acetylated p53 was observed in Fib− tumors, and prior studies have shown that acetylated p53 cannot be targeted by MDM2 [34, 35]. Overall these analyses imply that there is less interaction of MDM2 with p53 in the absence of fibrinogen which leads to the stabilization of p53, which drives cell senescence. This is consistent with the increased cell senescence observed in tumors from Fib− mice.
In order to find a mechanistic link between fibrinogen, an extracellular matrix protein, and the 14-3-3σ/p53 axis, we assessed the potential of fibrinogen in the TME to activate FAK. Our analyses showed reduced FAK activation in Fib− tumor tissues compared to Fib+ counterparts, suggesting that fibrinogen in the TME activates tumor cell associated FAK. Earlier studies have shown that once FAK is activated it can trigger the ubiquitination of p53 [12, 13, 24]. In cell fraction analyses for p53 and 14-3-3σ, we found accumulation of these molecules in the nuclear fraction of tumors from Fib− mice, indicating increased nuclear translocation of stabilized p53 in the absence of fibrinogen. Stabilization of p53 in the absence of fibrinogen then leads to decreased tumor cell proliferation through multiple mechanisms, including upregulation of p21 and 14-3-3σ. Given the apparent broad importance of FAK activation in cancer growth, it remains unclear why fibrinogen promotes the growth of some tumors but not others. The fact that other C57BL/6 derived tumor cell lines grew robustly in the same Fib− mice used in these studies strongly suggest that these animals are not mounting an adaptive immune response to C57BL/6-derived tumors, consistent with the fact that they are heavily inbred onto a C57BL/6 background. Other matrix proteins capable of FAK activation (fibronectin, thrombospondin, etc.) could be the critical factor over fibrinogen in the extracellular matrix of other tumors. Alternatively, other tumors may have other mechanisms for downregulating p53, p21 and 14-3-3σ. Understanding these other mechanisms will be an important future goal.
Multiple integrins are capable of FAK activation and determining precisely how fibrinogen in the TME activates FAK is an important future direction. Our data indicates that fibrinogen mediated FAK activation is independent of the fibrinogen γ chain binding motifs for αMβ2 and αIIbβ3. Tumor growth was unaffected by a mutation in the fibrinogen α chain that prevents polymer formation (FibAEK), as well as complete deficiency of FXIII, the transglutaminase responsible for fibrin crosslinking. This would suggest that immobilized fibrinogen in the TME is sufficient to activate FAK and promote tumor growth in this context in the absence of fibrin polymerization. This is not entirely surprising given that immobilized fibrinogen has been shown to bind to multiple integrins. [9–11] However, it is conceivable that thrombin-mediated cleavage of the intact fibrinogen β chain cleavage site could alter fibrinogen structure in a manner that supports colon cancer growth in this context. It is notable that FibAEK mice demonstrated no evidence of anything resembling fibrin polymer in vivo [20]. Definitively determining whether immobilized fibrinogen is the form of the molecule driving tumor growth in this context will require further study. It is also notable that previous studies have shown that colon cancer growth, including the colon cancer cells used in these studies, is strongly dependent on thrombin function [6]. The finding that fibrinogen also promotes colon cancer growth in mice, but without the need for stable fibrin polymer formation, suggests that thrombin promotes colon cancer growth through mechanism(s) independent of fibrin polymer formation. Consistent with this view, analyses of tumors harvested from mice with low circulating prothrombin showed no changes in the activation of FAK and 14-3-3σ as compared to WT controls despite manifesting significantly slower tumor growth. Thrombin has over a dozen recognized substrates [36, 37]. In addition to fibrin polymerization, thrombin can trigger platelet activation, initiate regulatory pathways that both promote and suppress coagulation, activate factor XI, protein C, thrombin-activated fibrinolysis inhibitor (TAFI), and activate three G protein-coupled protease activated receptors (PAR-1, −3, and −4) [38, 39]. Determining the fibrinogen-independent mechanisms coupling thrombin to colon cancer growth will also be an important future direction.
Previous studies from our laboratory have shown that fibrin(ogen) promotes colonic tumorigenesis in the context of inflammatory colitis by a mechanism related to engagement of the leukocyte integrin αMβ2 [5]. In those previous studies, mice carrying a mutant form of fibrinogen lacking the αMβ2 binding motif (Fibγ390−396A) were significantly protected from colitis-driven adenoma formation [5]. Notably, imposition of the Fibγ390–396A fibrinogen variant had no impact on the growth of established colonic adenocarcinoma in the current studies, suggesting that the mechanism by which fibrinogen activates FAK in this context does not involve αMβ2. A limitation of the current study is the fact that the murine colon cancer cell line was implanted into the skin of the dorsal subcutis, as hemostasis related concerns make orthotopic injection directly into the colon problematic. Nevertheless, the previous and current findings suggest that the role of fibrin(ogen) in the TME may change over time as colon cancer moves from the early tumorigenesis phase, to fully transformed adenocarcinoma. In the pre-cancerous niche, fibrin(ogen)-leukocyte interactions mediated by αMβ2 appear to promote inflammatory events that drive tumorigenesis in the context of colitis. The data presented here suggests that at some point the inflammatory events mediated by fibrin(ogen)-αMβ2 interactions are no longer required to support the growth of fully transformed colonic adenocarcinoma. However, fibrinogen-mediated FAK activation triggered by another integrin promotes tumor growth. Whether fibrin(ogen)-mediated FAK activation and subsequent downregulation of p53, p21 and 14-3-3σ play a role in early events important in colonic tumorigenesis remains to be determined.
In conclusion, the current study demonstrates a key role for fibrinogen in tumor growth. Our studies suggest that in the context shown here, fibrinogen in the extracellular matrix interacts with integrin receptor(s) on the tumor cell triggering the downstream effector molecule (FAK) on the cytosolic segment of focal adhesion complexes. The phosphorylation of FAK triggers a signaling cascade that ultimately results in the ubiquitination of p53, targeting it towards degradation by nuclear proteasomes. The decrease in p53 results in the destabilization of 14-3-3σ and arrest of p21 transcription. The absence/deactivation of these three key cell cycle regulators results in unchecked cell division and tumor proliferation (Fig. 7). This implies that there may be contexts where inhibiting fibrinogen/FAK interactions in the tumor stroma could limit tumor progression. Small molecules capable of disrupting the interaction of fibrinogen with FAK could be exploited for therapeutic use without any concern for bleeding. Future studies will be focused on identifying the specific integrin responsible for the interaction of fibrinogen with FAK, as well as determining the potential relevance of fibrin(ogen)-mediated FAK activation on human colonic adenocarcinoma.
MATERIALS AND METHODS
Gene-targeted mice and murine tumor growth analyses
Mice with fibrinogen (Fib−/−) and FXIIIA (FXIII−/−) deficiency have been described [21] [40]. The following mice carrying mutant forms of fibrinogen have also been described: Fibγ390−396A mice carry a form of fibrinogen lacking the γ chain binding motif for the leukocyte integrin αMβ2; FibγΔ5 mice lack the γ chain binding motif for the platelet integrin αIIbβ3; FibAEK mice carry a mutation in the α chain that prevents thrombin-mediated cleavage of fibrinopeptide A, “locking” fibrinogen in a soluble state [9, 19, 20]. All mice were backcrossed onto a C57BL/6 background at least 8 generations. Age and sex-matched WT and homozygous mutant mice ages 8 to 12 weeks were used in all experiments. All mouse protocols were in compliance with the National Institutes of Health guidelines for animal care and use and approved by Cincinnati Children’s Hospital Research Foundation Institutional Animal Care and Use Committee.
MC38 tumor cells (Edith Janssen, University of Cincinnati) were grown in culture and injected into the dorsal subcutis (2.5 × 105 cells/mouse) as previously described [6]. Tumor volumes were estimated by serial caliper measurements as previously described.
Analyses of Human colorectal adenocarcinoma samples
Formalin-fixed, paraffin-embedded cases of colorectal cancer and the adjacent normal colon samples from 20 patients were procured from the Biospecimen Core of the University of Cincinnati Medical Center. This biorepository was approved by the IRB of the UC Medical Center, and informed consent was obtained prior to procurement for the biorepository. All specimens were reviewed by pathologists at UC Medical Center. Sections with >50% lesion from each case were used for immunostaining and analysis. Fibrin(ogen) and FAK immunostaining was performed as previously described [41, 42].
Immunohistochemistry and β-galactosidase staining of murine tumor tissues
For immunohistochemistry (IHC), tumor tissues sections were deparaffinized in xylene and antigen retrieval was performed with 0.01 M citrate buffer (pH-6.1) for 15 minutes in a microwave. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 10 min, followed by incubation with PBST containing 10% normal goat serum for 60 minutes. For detection of Ki67 protein expression, specimens were incubated overnight at 4°C with Ki67 mAb (#Ki-67 (D2H10) Rabbit mAb (IHC Specific) #9027 Cell Signaling Technology) at a dilution of 1:200. As a negative control, the primary antibody was replaced by normal, nonimmune goat serum. Hematoxylin and eosin (H&E) staining was performed on all the formalin-fixed tissues. Senescence β-Galactosidase staining was done using a commercially available kit (Cell Signaling Technology) following the manufacturer’s instructions.
RNA preparation, cDNA synthesis, and quantitative PCR
Trizol Reagent (#15596–026; Life Technologies) was used to isolate total RNA. Total RNA (2 ug) from each sample was reverse transcribed into cDNA using High Capacity RNA-to-cDNA kit (Applied Biosystems). Quantitative PCR (qPCR) analysis was performed using PowerUp™ SYBR™ Green Master Mix (#A25776; Thermo Fisher Scientific). The qPCR primers used are provided in Supplementary Table 1.
RNA Seq analysis
Following removal of primers and barcodes, raw reads were aligned to the Hg38 human genome with annotations provided by UCSC, using the TopHat and Cufflinks from the Tuxedo pipeline, which quantifies transcript abundances of high-throughput sequencing reads with fragments per kilobase per million reads (FPKM) as the output. All further processing was performed in GeneSpring 14.9. Each transcript was log2-normalized and baselined to the median across all samples. Between-genotype significance (Fib− vs Fib+) was assessed using a moderated t-test, with an FDR-corrected significance cutoff of 0.05 and a fold change requirement of 1.25 (n=213 genes). Ontological analyses of differentially regulated genes was performed using the ToppGene suite (toppgene.cchmc.org and toppcluster.cchmc.org) and the ClueGo app in Cytoscape.
Immunoblotting
Whole protein lysates were prepared using RIPA lysis buffer (#89901; Thermo Fisher Scientific) supplemented with a complete Mini Protease Inhibitor Tablet (#11836153001; Roche Life Science). For Western blot analysis, 30 ug protein was separated on NuPAGE precast gels (Invitrogen/Life Technologies), transferred using an XCell II Blot module (#090707–098; Invitrogen/Life Technologies) onto Immobilon-FL membranes (#IPFL00010; EMD Millipore, Billerica, MA, USA), and probed with specific primary antibodies: 14-3-3-sigma (#ab14123; abcam); p53 (#SC-126;Santa Cruz); FAK (#SC-271126; Santa Cruz); p-FAK (#SC-81493;Santa Cruz); p21(SC-6246; Santa cruz); Ubiquitin (SC-47721 Santa cruz);acetylated p53 (SC-2570; Cell signaling Technology); Cleaved caspase-3 (9664; Cell signaling Technology); MDM2(# ab226939; abcam); Lamin A + C (#ab108922; Abcam); and b-actin (#A5441; Sigma-Aldrich). All the antibodies were used at a dilution of 1:1000 except acetylated p53, which was used at 1:500 dilution and b-actin at 1:10,000 dilution. The following IRDye-conjugated secondary antibodies were used: donkey anti-mouse IRDye 800CW (#926–32212), and donkey anti-rabbit IRDye 800CW (#926–32213; both from Li-Cor Biosciences). Li-Cor Biosciences Odyssey Classic Imager was used to detect Western blots. For immunoprecipitation assays (MDM2/Ub), 600 ug protein from whole-cell lysates and Protein A/G Plus-agarose immunoprecipitation reagent (sc-2003; Santa Cruz Biotechnology) were used.
Nuclear and cytoplasmic protein tumor fractions were prepared using the nuclear extraction kit (#10009277; Cayman Chemicals).
3D-Bioprinting
Sterilized alginate hydrogel (20 % w/v) was prepared by adding the required amount of sodium alginate (Sigma Aldrich, USA) slowly in deionized water at 40 °C on a magnetic stirrer at 300 RPM, followed by autoclaving. The cell encapsulated hydrogel (bioink) was prepared by encapsulating MC38 cells (cell density, 4×106 cells/ml) in 20% (w/v) sodium alginate hydrogel with or without fibrinogen (Molecular Innovations (MFBGN-1mg/mL). Self-standing cell laden constructs were printed using BioX 3D bioprinter (CELLINK). The 3D bio-printing was done at 280C using a pneumatic print head by dispensing the bioink through a plastic 410 μm diameter nozzle using pneumatic pressure of 100–150 kPa and a deposition speed of 7 mm/min into a 3 layered 1mm ×1mm square structure with 15% infill density in a rectilinear fashion and a layer height of 41%. Printed MC38 cells encapsulated in alginate constructs were crosslinked with sterile filtered 100 mM CaCl2 under sterile conditions. The constructs were cultured in DMEM (Hyclone) supplemented with 10% FBS (Gibco), 5% penicillin/streptomycin (Gibco) and incubated at 370C, 5% CO2.
LIVE/DEAD® Viability/Cytotoxicity Kit (Invitrogen) was used for cell viability following the manufacture’s instructions. The bioprinted alginate incubated for 30 min in the live/dead assay solution at 37° C and visualized by Nikon A1R LUN-V. The number of live and dead cells were counted by Image J software (NIH).
NMR based metabolites analysis
Extraction solvent volumes were added based on tissues weights. Modified Bligh and Dyer extraction was used to obtain polar metabolites.[43, 44] Briefly, cold methanol and water were added to the samples and homogenized for 30 s at 5000 rpm 2.8mm ceramic beads. Cold chloroform and water were used to bring the final methanol:chloroform:water ratio to 2:2:1.8. The polar phase was vacuum dried and resuspended in 220 uL of NMR buffer containing 100mM phosphate buffer, pH7.3, 1mM TMSP (3-Trimethylsilyl 2,2,3,3-d4 propinoate), 1mg/mL sodium azide prepared in D2O.
One-dimensional 1H NMR spectra were acquired on a Bruker Avance II 600 MHz spectrometer with 5mm, BBO Prodigy probe. Data were collected at 298 Kelvin using the noesygppr1d pulse sequence in the Bruker pulse sequence library. Topspin 3.6 software (Bruker Analytik) was used for data analysis. For a representative sample, two dimensional data 1H-1H total correlation spectroscopy (TOCSY) and 2D 1H-13C heteronuclear single quantum coherence (HSQC) were collected for metabolites assignment.
Chemical shifts were assigned to metabolites based on 1D 1H, 2D TOCSY and HSQC NMR experiments with reference spectra found in databases, Human Metabolome Database (HMDB)[ and Chenomx® NMR Suite profiling software (Chenomx Inc).[45] The concentrations of metabolites in polar extracts were calculated using Chenomx software based on internal standard, TMSP. 54 polar metabolites were studied. Concentrations were normalized to tissue weights. R studio and MetaboAnalyst were used for analyses.[46]
Statistical analysis
Statistical analyses were performed using Prism GraphPad software. Unless otherwise noted, data are presented as mean ± standard error of mean (SEM) and statistical analyses were performed using the Mann–Whitney U test.
Supplementary Material
ESSENTIALS.
Elevated plasma fibrinogen correlates with poor prognosis in colorectal cancer (CRC).
Fibrinogen regulates the expression of genes involved in cell cycle regulation and metabolism.
Fibrinogen in the tumor microenvironment activates FAK, leading to destabilization of p53.
Decreased p53 diminishes 14-3-3σ and p21, accelerating proliferation and limiting senescence.
Acknowledgments
Grant Support
This work was funded by grants from the National Institutes of Health (J. S. Palumbo, R01 CA204058 and CA207503).
Abbreviations:
- Fib
fibrinogen
- FAK
focal adhesion kinase
- CRC
colorectal cancer
Footnotes
Disclosures
Nothing to disclose.
REFERENCES
- 1.Flick MJ, Palumbo JS. Platelets couple inflammation to tumorigenesis, a bridge too far. J Thromb Haemost. 2018; 16: 759–61. 10.1111/jth.13967. [DOI] [PubMed] [Google Scholar]
- 2.Kawai K, Watanabe T. Colorectal cancer and hypercoagulability. Surg Today. 2014; 44: 797–803. 10.1007/s00595-013-0606-5. [DOI] [PubMed] [Google Scholar]
- 3.Pedrazzani C, Mantovani G, Salvagno GL, Baldiotti E, Ruzzenente A, Iacono C, Lippi G, Guglielmi A. Elevated fibrinogen plasma level is not an independent predictor of poor prognosis in a large cohort of Western patients undergoing surgery for colorectal cancer. World J Gastroenterol. 2016; 22: 9994–10001. 10.3748/wjg.v22.i45.9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vossen CY, Hoffmeister M, Chang-Claude JC, Rosendaal FR, Brenner H. Clotting factor gene polymorphisms and colorectal cancer risk. J Clin Oncol. 2011; 29: 1722–7. 10.1200/jco.2010.31.8873. [DOI] [PubMed] [Google Scholar]
- 5.Steinbrecher KA, Horowitz NA, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, Finkelman FD, Flick MJ, Pinkerton MD, Talmage KE, Kombrinck KW, Witte DP, Palumbo JS. Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin alpha(M)beta(2) engagement of fibrinogen. Cancer Res. 2010; 70: 2634–43. 10.1158/0008-5472.Can-09-3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adams GN, Rosenfeldt L, Frederick M, Miller W, Waltz D, Kombrinck K, McElhinney KE, Flick MJ, Monia BP, Revenko AS, Palumbo JS. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Res. 2015; 75: 4235–43. 10.1158/0008-5472.Can-15-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol. 2018; 15: 366–81. 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019; 79: 4557–66. 10.1158/0008-5472.CAN-18-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004; 113: 1596–606. 10.1172/jci20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez J, Ferber A, Bach TL, Yaen CH. Interaction of fibrin with VE-cadherin. Ann N Y Acad Sci. 2001; 936: 386–405. 10.1111/j.1749-6632.2001.tb03524.x. [DOI] [PubMed] [Google Scholar]
- 11.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001; 167: 2887–94. 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 12.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005; 280: 25008–21. 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 13.Golubovskaya VM, Finch R, Zheng M, Kurenova EV, Cance WG. The 7-amino-acid site in the proline-rich region of the N-terminal domain of p53 is involved in the interaction with FAK and is critical for p53 functioning. Biochem J. 2008; 411: 151–60. 10.1042/BJ20071657. [DOI] [PubMed] [Google Scholar]
- 14.Yang HY, Wen YY, Chen CH, Lozano G, Lee MH. 14-3-3 sigma positively regulates p53 and suppresses tumor growth. Mol Cell Biol. 2003; 23: 7096–107. 10.1128/mcb.23.20.7096-7107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu H, Subramanian RR, Masters SC. 14-3-3 proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000; 40: 617–47. 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- 16.Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996; 84: 889–97. 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 17.Ling C, Su VM, Zuo D, Muller WJ. Loss of the 14-3-3sigma tumor suppressor is a critical event in ErbB2-mediated tumor progression. Cancer Discov. 2012; 2: 68–81. 10.1158/2159-8290.Cd-11-0189. [DOI] [PubMed] [Google Scholar]
- 18.Phan L, Chou PC, Velazquez-Torres G, Samudio I, Parreno K, Huang Y, Tseng C, Vu T, Gully C, Su CH, Wang E, Chen J, Choi HH, Fuentes-Mattei E, Shin JH, Shiang C, Grabiner B, Blonska M, Skerl S, Shao Y, Cody D, Delacerda J, Kingsley C, Webb D, Carlock C, Zhou Z, Hsieh YC, Lee J, Elliott A, Ramirez M, Bankson J, Hazle J, Wang Y, Li L, Weng S, Rizk N, Wen YY, Lin X, Wang H, Wang H, Zhang A, Xia X, Wu Y, Habra M, Yang W, Pusztai L, Yeung SC, Lee MH. The cell cycle regulator 14-3-3 sigma opposes and reverses cancer metabolic reprogramming. Nat Commun. 2015; 6: 7530. 10.1038/ncomms8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmback K, Danton MJ, Suh TT, Daugherty CC, Degen JL. Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin alpha IIb beta 3. Embo j. 1996; 15: 5760–71. [PMC free article] [PubMed] [Google Scholar]
- 20.Prasad JM, Gorkun OV, Raghu H, Thornton S, Mullins ES, Palumbo JS, Ko YP, Hook M, David T, Coughlin SR, Degen JL, Flick MJ. Mice expressing a mutant form of fibrinogen that cannot support fibrin formation exhibit compromised antimicrobial host defense. Blood. 2015; 126: 2047–58. 10.1182/blood-2015-04-639849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palumbo JS, Barney KA, Blevins EA, Shaw MA, Mishra A, Flick MJ, Kombrinck KW, Talmage KE, Souri M, Ichinose A, Degen JL. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost. 2008; 6: 812–9. 10.1111/j.1538-7836.2008.02938.x. [DOI] [PubMed] [Google Scholar]
- 22.Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, Yao TP. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo j. 2001; 20: 1331–40. 10.1093/emboj/20.6.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang T, Munguia-Lopez JG, Gu K, Bavoux MM, Flores-Torres S, Kort-Mascort J, Grant J, Vijayakumar S, De Leon-Rodriguez A, Ehrlicher AJ, Kinsella JM. Engineering bioprintable alginate/gelatin composite hydrogels with tunable mechanical and cell adhesive properties to modulate tumor spheroid growth kinetics. Biofabrication. 2019; 12: 015024. 10.1088/1758-5090/ab3a5c. [DOI] [PubMed] [Google Scholar]
- 24.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM- enhanced p53 degradation. Mol Cell. 2008; 29: 9–22. 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, Deng Y. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014; 8: 1461–74. 10.1016/j.celrep.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad GL, Valverius EM, McDuffie E, Cooper HL. Complementary DNA cloning of a novel epithelial cell marker protein, HME1, that may be down-regulated in neoplastic mammary cells. Cell Growth Differ. 1992; 3: 507–13. [PubMed] [Google Scholar]
- 27.Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA, Stampfer MR, Sukumar S. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A. 2000; 97: 6049–54. 10.1073/pnas.100566997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwata N, Yamamoto H, Sasaki S, Itoh F, Suzuki H, Kikuchi T, Kaneto H, Iku S, Ozeki I, Karino Y, Satoh T, Toyota J, Satoh M, Endo T, Imai K. Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3 sigma gene in human hepatocellular carcinoma. Oncogene. 2000; 19: 5298–302. 10.1038/sj.onc.1203898. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Itoh F, Toyota M, Kikuchi T, Kakiuchi H, Imai K. Inactivation of the 14-3-3 sigma gene is associated with 5’ CpG island hypermethylation in human cancers. Cancer Res. 2000; 60: 4353–7. [PubMed] [Google Scholar]
- 30.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999; 401: 616–20. 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 31.Su CH, Zhao R, Zhang F, Qu C, Chen B, Feng YH, Phan L, Chen J, Wang H, Wang H, Yeung SC, Lee MH. 14-3-3sigma exerts tumor-suppressor activity mediated by regulation of COP1 stability. Cancer Res. 2011; 71: 884–94. 10.1158/0008-5472.Can-10-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, Wen YY, Zhao R, Lin YL, Fournier K, Yang HY, Qiu Y, Diaz J, Laronga C, Lee MH. DNA damage-induced protein 14-3-3 sigma inhibits protein kinase B/Akt activation and suppresses Akt-activated cancer. Cancer Res. 2006; 66: 3096–105. 10.1158/0008-5472.Can-05-3620. [DOI] [PubMed] [Google Scholar]
- 33.Yang H, Zhao R, Lee MH. 14-3-3sigma, a p53 regulator, suppresses tumor growth of nasopharyngeal carcinoma. Mol Cancer Ther. 2006; 5: 253–60. 10.1158/1535-7163.Mct-05-0395. [DOI] [PubMed] [Google Scholar]
- 34.Kobet E, Zeng X, Zhu Y, Keller D, Lu H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci U S A. 2000; 97: 12547–52. 10.1073/pnas.97.23.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem. 2002; 277: 50607–11. 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 36.Remiker AS, Palumbo JS. Mechanisms coupling thrombin to metastasis and tumorigenesis. Thromb Res. 2018; 164 Suppl 1: S29–S33. 10.1016/j.thromres.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Cantrell R, Palumbo JS. The thrombin-inflammation axis in cancer progression. Thromb Res. 2020; 191 Suppl 1: S117–S22. 10.1016/S0049-3848(20)30408-4. [DOI] [PubMed] [Google Scholar]
- 38.Nesheim M, Wang W, Boffa M, Nagashima M, Morser J, Bajzar L. Thrombin, thrombomodulin and TAFI in the molecular link between coagulation and fibrinolysis. Thromb Haemost. 1997; 78: 386–91. [PubMed] [Google Scholar]
- 39.Adams GN, Sharma BK, Rosenfeldt L, Frederick M, Flick MJ, Witte DP, Mosnier LO, Harmel-Laws E, Steinbrecher KA, Palumbo JS. Protease-activated receptor-1 impedes prostate and intestinal tumor progression in mice. J Thromb Haemost. 2018; 16: 2258–69. 10.1111/jth.14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, Simon DI, Potter S, Degen JL. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995; 9: 2020–33. 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]
- 41.Kopec AK, Joshi N, Cline-Fedewa H, Wojcicki AV, Ray JL, Sullivan BP, Froehlich JE, Johnson BF, Flick MJ, Luyendyk JP. Fibrin(ogen) drives repair after acetaminophen-induced liver injury via leukocyte alphaMbeta2 integrin-dependent upregulation of Mmp12. J Hepatol. 2017; 66: 787–97. 10.1016/j.jhep.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X, Cance WG. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003; 9: 215–22. [PubMed] [Google Scholar]
- 43.Wu H, Southam AD, Hines A, Viant MR. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem. 2008; 372: 204–12. 10.1016/j.ab.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Watanabe M, Meyer KA, Jackson TM, Schock TB, Johnson WE, Bearden DW. Application of NMR-based metabolomics for environmental assessment in the Great Lakes using zebra mussel (Dreissena polymorpha). Metabolomics. 2015; 11: 1302–15. 10.1007/s11306-015-0789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, Fung C, Nikolai L, Lewis M, Coutouly MA, Forsythe I, Tang P, Shrivastava S, Jeroncic K, Stothard P, Amegbey G, Block D, Hau DD, Wagner J, Miniaci J, Clements M, Gebremedhin M, Guo N, Zhang Y, Duggan GE, Macinnis GD, Weljie AM, Dowlatabadi R, Bamforth F, Clive D, Greiner R, Li L, Marrie T, Sykes BD, Vogel HJ, Querengesser L. HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007; 35: D521–6. 10.1093/nar/gkl923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chong J, Wishart DS, Xia J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr Protoc Bioinformatics. 2019; 68: e86. 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.