

Abstract
As antimicrobial resistance becomes an increasing threat, bringing significant economic and health burdens, innovative antimicrobial treatments are urgently needed. While antimicrobial peptides (AMPs) are promising therapeutics, exhibiting high activity against resistant bacterial strains, limited stability and toxicity to mammalian cells has hindered clinical development. Attaching AMPs to polymers provides opportunities to present AMPs in a way that maximizes bacterial killing while enhancing compatibility with mammalian cells, stability, and solubility. Conjugation of an AMP to a linear hydrophilic polymer yields the desired improvements in stability, mammalian cell compatibility, and solubility, yet often markedly reduces bactericidal effects. Non-linear polymer architectures and supramolecular assemblies that accommodate multiple AMPs per polymer chain afford AMP-polymer conjugates that strike a superior balance of antimicrobial activity, mammalian cell compatibility, stability, and solubility. Therefore, we review the design criteria, building blocks, and synthetic strategies for engineering AMP-polymer conjugates, emphasizing the connection between molecular architecture and antimicrobial performance to inspire and enable further innovation to advance this emerging class of biomaterials.
Graphical Abstract
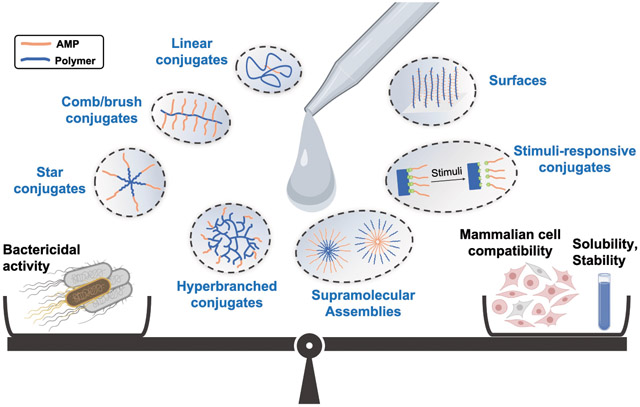
1. Introduction
Antimicrobial peptides (AMPs) are promising candidate therapeutics to counter the increasing threat posed by antibiotic-resistant bacteria. Compared to traditional antibiotics, AMPs are active against a broad spectrum of bacterial species1,2 and are less likely to invoke bacterial resistance.3-5 AMPs combine cationic and hydrophobic components that enable selective interactions with highly anionic bacterial membranes over the more neutral, cholesterol-containing membranes of mammalian cells.6-9 Realizing the promise of AMP-based therapeutic strategies will require fortifying this inherent selectivity, as well as addressing the instability of peptides to proteolytic degradation and their rapid clearance from the blood stream.2,10
Conjugation of AMPs to polymers provides ample opportunities to enhance the therapeutic utility of AMPs.11 Attachment of AMPs to polymers improves AMP solubility, shields peptide constituents from protease degradation, and yields larger conjugates that avoid rapid renal filtration to prolong circulation in the bloodstream. AMP conjugation to neutral hydrophilic polymers can also reduce interactions with eukaryotic cellular membranes, thereby decreasing potential cytotoxic effects against mammalian cells. However, conjugation can also reduce or abrogate AMP-mediated antimicrobial activity, so it is important that AMP-polymer conjugates strike a balance between the cationic, hydrophobic character needed for bacterial killing and the neutral, hydrophilic character that confers biocompatibility. AMP-polymer conjugates can also be engineered to improve antimicrobial activity, control AMP release rate, and modulate host immune responses, ultimately leading to dose sparing (i.e., lower dose levels or frequency).
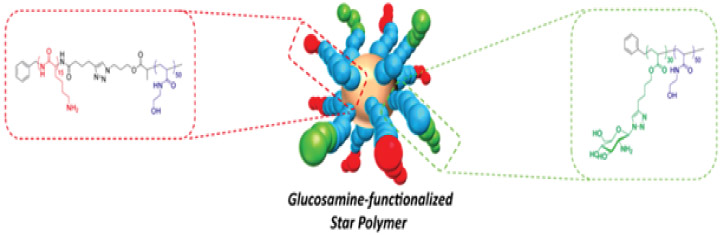
The state-of-the-art in AMP-polymer conjugates builds on advances in polymer chemistry, conjugation chemistry, and molecular design that have propelled polymer-assisted therapeutic delivery.12-16 Polymers can be prepared with well-defined molecular weight, architecture, and composition; these factors determine conjugate size, morphology, and surface charge, and, by extension, antimicrobial performance (Figure 1). Several excellent review articles11,17-21 showcase the compositional diversity and therapeutic potential of AMP-polymer conjugates. Among the various conjugate designs, AMP-polymer conjugates that accommodate multiple AMPs per polymer chain often retain the potent antimicrobial activity of AMPs. Therefore, here we review the use of polymers in conjunction with AMPs towards combating bacteria with a focus on the role of molecular architecture on antimicrobial performance. After a brief discussion of metrics (antimicrobial activity, toxicity to mammalian cells, proteolytic stability) and mechanistic aspects that set design criteria for AMP-polymer conjugates, we describe common polymer and peptide components and discuss synthetic methods for preparing these materials. Thereafter, we dissect the role of polymer architecture on conjugate properties and bactericidal activity. For conjugates with linear, comb/brush, star, and hyperbranched architectures, we compare the antimicrobial activity and mammalian-cell toxicity of diverse conjugates to that of the AMP alone. In addition to molecular AMP-polymer conjugates, we review examples of supramolecular assemblies and surfaces that combine polymers and AMPs. While we focus on in vitro experiments, the implications of conjugating AMPs to polymers certainly extend into antimicrobial performance in vivo.22-25 Finally, since introducing stimuli-responsive functionality to AMP-polymer conjugates can impart on-demand, selective antimicrobial activity, we describe several approaches and examples, many of which involve complex architectures or supramolecular assembly, before concluding with a perspective on challenges and opportunities in the field.
Figure 1.

Molecular engineering of AMP-polymer conjugates to modulate structure, properties, and performance.
2. Antimicrobial performance metrics
Together, the bactericidal activity, safety profile, and stability comprise the overall performance of AMPs and AMP-polymer conjugates. Activity metrics include the minimum inhibitory concentration (MIC, or the minimum concentration of therapeutic that inhibits visible bacterial growth) and the minimum bactericidal concentration (MBC, or the minimum concentration that results in a 3-log10 reduction of bacterial viability), among others and the membrane disruption concentration (MDC, or the minimum concentration required for complete lysis of bacteria).26,27 To elucidate the role of AMP conjugation to polymers on antimicrobial activity, it is particularly useful to adjust MIC, MBC, and MDC values to account for AMP content.28
In general, the antimicrobial activity of a given AMP or conjugate varies against different bacterial species. Particularly pronounced differences are possible between Gram-positive bacteria, e.g., Staphylococcus aureus (S. aureus) and Enterococcus spp, and Gram-negative bacteria, e.g., Escherichia coli (E. coli) and colistin-resistant Pseudomonas aeruginosa (P. aeruginosa), owing to the inherent compositional and structural differences of their cellular envelopes. Gram-positive bacteria have a cytoplasmic membrane that is surrounded by a thick peptidoglycan cell wall embedded with negatively charged teichoic acids, while Gram-negative bacteria have a thin peptidoglycan layer located between a cytoplasmic membrane and an outer membrane with an anionic lipopolysaccharide-presenting exterior leaflet.24,29 The anionic surface charge of Gram-positive and Gram-negative bacteria can draw cationic AMPs to the bacterial cell membrane via electrostatic interactions. In Gram-negative bacteria, the outer membrane provides an adaptable, low-permeability barrier that typically increases resistance to AMPs. Accordingly, higher MIC/MBC values tend to be observed for Gram-negative bacteria relative to Gram-positive organisms.
The safety profile of AMP conjugates includes mammalian cell viability, hemolysis, and immune system engagement, among other factors. Cell viability assays quantify the percentage of live and/or dead cells, and yield an inhibitory concentration (IC), or concentration of AMP (or AMP-polymer conjugate) that reduces the growth rate of cells by a given percentage (e.g., IC50 for 50% inhibition) relative to that in the absence of the AMP.30-34 The hemolytic concentration (HC), or the concentration of AMP or AMP-polymer conjugate that causes lysis of a given percentage of red blood cells (e.g., HC50 for 50% hemolysis), provides another way to gauge the extent to which AMPs and conjugates target mammalian cell membranes.35 The selectivity indices IC50/MIC and HC50/MIC quantify the degree of selectivity of the AMP to bacterial vs. mammalian cells.36 Additionally, a range of assays that measure, among others, platelet activation, complement activation, coagulation, and immune-cell function assess the potential effects of AMP-polymer conjugates on the host immune system.37
Conjugation to polymers can improve AMP resistance to both proteolytic degradation and aggregation. Following incubation of AMPs and conjugates with serum or proteolytic enzymes (e.g., trypsin) to induce degradation and/or aggregation, these changes are measurable with chromatography and scattering, among other techniques.37,38 Additionally, minimal changes to AMP or conjugate bactericidal activity in the presence of proteolytic enzymes and/or serum can also indicate stability.39
3. Mechanism of action of AMPs and AMP-polymer conjugates
While some AMPs have intracellular targets,40 others operate by accumulating and cooperatively disrupting bacterial membranes.2,41-43 As such, it can be helpful to design for and characterize the physical properties of conjugates that impact membrane interaction, like charge, hydrophobicity, size, morphology, and peptide secondary structure. This is highlighted by the abrogation of antibacterial activity by the AMP nisin A upon conjugation to polyethylene glycol (PEG), i.e. ‘PEGylation’.44 On its own, nisin A typically forms pores in bacterial membranes by interaction with lipid II, and the disruption of activity upon PEGylation is attributed to the decrease in hydrophobic character. Yet, conjugation does not always impede membrane interactions: conjugation of multiple polylysine AMPs to chitosan yielded a comb-shaped conjugate that generated large (>30 nm) membrane defects and matched the mechanism of action of polylysine.22,45 However, as highlighted by Hancock and coworkers,43 understanding of the mechanism of action of AMPs is evolving, and there remains much to learn about how AMPs and AMP-polymer conjugates operate so as to guide the design of conjugates that harness the benefits of polymers without compromising bactericidal activity.
4. Constituents of AMP-polymer conjugates
4.1. AMPs
AMP-polymer conjugates feature a plethora of natural and synthetic AMP constituents. Natural AMPs incorporated into AMP-polymer conjugates include anoplin (GLLKRIKTLL),46 a peptide isolated from wasp venom, magainins (e.g. magainin 1 is GIGKFLHSAGKFGKAFVGEIMKS),47 a group of peptides isolated from frogs, and nisin (e.g. nisin A I-Dhb-AI-Dha-LA-Aba-PGAK-Aba-GALMGANMK-Aba-A-Aba-AHASIHV-Dha-K, where Aba is aminobutyric acid, Dha is dehydroalanine, and Dhb is dehydrobutyrine),48-51 a cyclic peptide produced by the bacterium Lactococcus lactis. Modifications to natural AMPs, as well as rationally and computationally designed sequences combining hydrophobic, cationic, and helical character yield synthetic AMPs with high antimicrobial activity, even against multidrug-resistant bacterial isolates.46-51 Advances in solid phase peptide synthesis enable the automated synthesis of AMPs with well-defined sequences exceeding 50 amino acids in length, while ring-opening polymerization of amino acid N-carboxyanhydrides (NCAs) allows the scalable synthesis of polypeptides where precision sequence control is not required, e.g., poly(lysine-co-valine).22,25,58-60,26,27,52-57 While NCA polymerization does not afford well-defined sequences, polypeptide-based AMPs can be synthesized with well-defined molecular weights, compositions, and architectures. Together, inspiration from nature, input from rational design, and advances in peptide chemistry continue to produce structurally diverse, potent AMPs amenable for incorporation into AMP-polymer conjugates.
4.2. Polymers
The polymers comprising AMP-polymer conjugates are typically hydrophilic and either charge-neutral or cationic. Charge-neutral, hydrophilic polymers increase solubility and can reduce undesired toxicity to eukaryotic cells.61 Cationic polymers increase solubility and potency, but usually also increase cytotoxicity.22,52,62,63 Poly(ethylene glycol) (PEG) was one of the earliest, and remains one of the most common, polymers conjugated to AMPs. PEG is neutral, non-toxic, highly soluble in water, and approved by the Food and Drug Administration (FDA) for use in humans.64 The commercial availability of PEG with a variety of reactive chain ends enables efficient installation of AMPs to prepare conjugates. Yet, a notable drawback associated with PEG is the generation of anti-PEG antibodies that reduce half-life.64-66 Aside from PEG, conjugation of AMPs to polysaccharides, such as chitosan and dextran, yields a peptidoglycan-mimetic structure. The amine groups of chitosan render it cationic and impart antimicrobial activity,67 while the hydroxyl and amine groups enable AMP conjugation. Chitosan is generally considered safe for human use and employed widely for drug delivery and tissue engineering.68-71 Dextran is a charge-neutral polysaccharide with hydroxyl groups that provide water solubility and enable AMP attachment.72 Since peptidoglycans are a prominent feature of bacteria cell walls, particularly in Gram-positive bacteria, peptidoglycan-mimicking conjugates are postulated to enhance interactions with bacterial membranes.22 Also designed to promote membrane interactions by mimicking cell membrane composition, polyphosphoesters are biodegradable synthetic polymers attractive for biomedical applications, including for conjugation to AMPs.28 Synthetic cationic polymers used previously in AMP-polymer conjugates include poly(ethylene imine) (PEI) and poly(amido amine) (PAMAM). Both are water-soluble and contain multiple amine groups that can initiate ring-opening polymerization (ROP) of amino acid NCAs to form star-shaped conjugates.24,53,73 Other polymers used for AMP conjugation include polylactides, polyacrylamides, and polyacrylates.21,24,74,75
5. Chemistry
Synthetic approaches to prepare AMP-polymer conjugates can be classified as grafting-to, grafting-from, or grafting-through methods, among others (Figure 2).76 In this section, we describe each of these methods and review the chemistry that underlies the molecular engineering of AMP-polymer conjugates. We close the section with a discussion of AMP encapsulation in polymers by physical entrapment and non-covalent interactions.
Figure 2.

Examples of conjugation strategies and possible architectures: (a) grafting-to; (b) grafting-from, illustrated here as a peptide growing from a polymer backbone (polymer can also be grown from a peptide functionalized with an initiator or chain transfer agent); and (c) grafting-through methods. The left column shows conjugation reactions for generating comb/brush polymers, and the right column illustrates other possible architectures that can be realized by these methods.
5.1. Grafting-to approaches
In grafting-to conjugation, AMPs and polymers are prepared separately and then joined (or ‘grafted to’ one another) when a reactive group on the AMP reacts with a complementary reactive group on the polymer. Since steric bulk reduces accessibility to reactive sites on polymers, the grafting-to approach requires highly efficient reactions. Ideally, these reactions should yield no, or easily separable, side products. Therefore, ‘click’ reactions characterized by high yields, mild conditions, and little to no byproduct formation are particularly useful for grafting-to conjugations.77 In cases where the polymer and AMP do not have complementary functional groups, heterobifunctional linkers can be used to prepare conjugates.78 Below, we discuss reactions for preparing AMP-polymer conjugates using the grafting-to approach.
Conjugation reactions involving cysteine thiols on AMPs
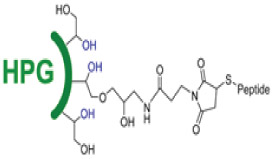
Since thiols participate efficiently and selectively in a variety of radical, nucleophilic substitution, and redox reactions, a common strategy to prepare AMP-polymer conjugates is to append thiol-containing cysteine residues to the N- or C-terminus of AMPs. Using thiol-ene click reactions, thiols on AMPs add to alkenes on polymers via radical addition or nucleophilic substitution.62,72,79-81 In particular, highly electrophilic maleimide alkenes enable conjugation with thiols to proceed readily in aqueous solution at neutral or slightly acidic pH. For example, the reaction of maleimide-functionalized chitosan with the cysteine-terminated peptide HHC10 (CKRWWKWIRW) afforded chitosan-based comb polymers (Figure 3a).81 With less electrophilic acrylate and methacrylate substrates, nucleophilic thiol-ene reactions occur more slowly and, in the case of methacrylates, require catalysts.72,79,80 When alkene substrates are not conjugated to electron-withdrawing groups, radical-based thiol-ene reactions are required.79 While alkenes react with a single cysteine thiol, alkynes can each react with two thiols each in a ‘thiol-yne’ reaction, thereby generating a higher density of AMPs per reactive group.28,82,83 For example, grafting the cysteine-terminated AMP HHC10 to a polyphosphoester with pendant alkyne groups by irradiation at 365 nm in the presence of the radical photoinitiator 2,2-dimethoxy-2-phenylacetophenone (DMPA) furnished AMP-grafted comb polymers (Figure 3b).28 Another reaction used to prepare AMP-polymer conjugates involves electrophilic, thiol-reactive iodoacetamide groups and cysteine thiols.84 Decoration of hyperbranched polyglycerol (HPG) with iodoacetamides enabled conjugation of the cysteine-modified AMP aurein 2.2 (CGLFDIVKKVVGAL) (Figure 3c).85 While this reaction yields iodide salt byproducts, these are easily separated from the AMP-polymer conjugate by dialysis or chromatography. Finally, thiol-containing polymers can form dynamic-covalent conjugation with cysteine-terminated AMPs by oxidation of thiols links, for example, to conjugate AMP (PWKISIHLAAC, derivative of Jelleine-I) with glutathione functionalized chitosan (Figure 3d).86
Figure 3.

AMP-polymer conjugation via grafting-to approaches: a) thiol-ene reaction and example conjugation of a thiol-containing AMP to maleimide-functionalized chitosan, adapted with permission from Pranantyo et al.,81 copyright © 2018 American Chemical Society; b) thiol-yne reaction and example conjugation of a thiol-containing AMP to an alkyne-functionalized polyphosphoester, adapted with permission from Pranantyo et al.,28 copyright © 2016 American Chemical Society; c) iodoacetamide-cysteine reaction and example conjugation of a thiol-containing AMP to an iodoacetamide-functionalized hyperbranched polyglycerol (HPG), adapted with permission from Kumar et al,85 copyright © 2015 American Chemical Society; d) disulfide formation and example conjugation of a thiol-terminated AMP to thiolated chitosan via disulfide linkages, adapted with permission from Costa Petrin et al.,86 copyright © American Chemical Society 2019; e) azide-alkyne reaction and example conjugation of alkyne-terminated AMPs to azide-functionalized chitosan, adapted with permission from Barbosa et al.,87 copyright ® 2017 Elsevier; and f) amidation reactions of amines with carboxylic acids, activated esters, and anhydrides. As an example, we show the conjugation reaction of an amine-terminated AMP to poly(maleic anhydride), followed by capping of acid groups with methyl esters using trimethylsilyldiazomethane, adapted with permission from Liu et al.,88 copyright © 2006 American Chemical Society. In all schemes, R and R’ can represent either polymer or AMP.
Azide-alkyne reactions
The copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) click reaction is frequently applied for grafting-to AMP-polymer conjugation.36,46,87,89 Azide and alkynes form triazole linkages in the presence of a Cu(I) catalyst and stabilizing ligands to accelerate the addition under mild conditions. As an alternative to Cu(I) salts, which are highly susceptible to oxidation during storage and handling, Cu(I) can be produced in situ from a Cu2+ source (e.g., CuCl2 or CuBr2) and a reducing agent (e.g., citric acid or ascorbic acid). In some cases, a weak base is added to deprotonate the alkyne and further accelerate the reaction.90 For example, the alkyne-modified AMP Dhvar-5 (alkyne-LLLFLLKKRKKRKY) was grafted to azide-functionalized chitosan by CuAAC in the presence of: a Cu2+/ascorbic acid pair as the catalyst; 2,6-lutidine as the weak base; tris(3-hydroxypropyltriazolylmethyl)amine as the ligand; and aminoguanidine hydrochloride to avoid modification of arginine side chains that may react with the oxidized form of ascorbic acid (Figure 3e).87 While CuAAC is compatible with a wide range of polymers and peptides, removal of metal catalysts during purification is required to avoid toxicity; this is typically accomplished by washing with Cu chelators (e.g., ethylene diamine tetraacetic acid, EDTA).36,46,87,89,91 Of note, care must be taken when performing azide chemistry, particularly when working with high N:C ratio compounds that present explosion hazards.92
Amidation reactions
The N-terminal primary amines of AMPs and those in lysine side chains also serve as reactive sites for grafting-to conjugation to polymers.38,88,93-95 Primary amines (in the uncharged form) are good nucleophiles and react efficiently with strong electrophiles, such as anhydrides and activated esters (e.g., N-hydroxysuccinimidyl and pentafluorophenyl esters), or with carboxylic acids in the presence of coupling reagents (e.g., carbodiimides) to form amide linkages. For example, the reaction of the N-terminal amine of arginine- and tryptophan-containing AMPs with polymaleic anhydride afforded peptide-grafted comb polymers (Figure 3f).88 Acid groups produced during the reaction were capped with methyl esters using trimethylsilyldiazomethane, and to avoid side-reactions of the amino acid side chains during amidation and capping, side chain protecting groups were removed after completion of these two steps. As another example of amidation, crosslinking poly(lysine-co-alanine) with multi-arm NHS ester-terminated PEG using amine-NHS ester chemistry generated hydrogel networks.93
5.2. Grafting-from approaches
In grafting-from synthesis of AMP-polymer conjugates, AMPs are grown from polymers or polymers are grown from AMPs. Here, we describe the synthesis of AMP-polymer conjugates using ring-opening and controlled radical polymerization, and direct readers to the recent review by Maynard and coworkers for more detail on graft-from methods and applications.16
Most commonly, grafting-from preparation of AMP-polymer conjugates involves ROP of amino acid N-carboxyanhydrides (NCAs) initiated from amines on a synthetic polymer. NCA ROP allows synthesis of polypeptide-based AMPs with limited control over sequence, but fine control over the composition and molecular weight.96-99 Accordingly, this approach is most useful for AMPs in which microbial killing does not require specific amino acid sequences22,24,57-60,100,25-27,52-56 and eliminates the need for ordered, step-by-step peptide synthesis. Polymers with multiple initiating species allow preparation of non-linear AMP-polymer conjugates. For example, copolymerization of lysine and valine NCAs from the primary amines on PAMAM dendrimers yields star-shape conjugates (Figure 4a).26 While NCA ROP is widely applied, challenges include stringent requirements for monomer purity and water-free conditions to prevent chain transfer and other side reactions.96-98,101 A further drawback of NCA polymerization is the use of toxic phosgene derivatives for NCA monomer synthesis.102
Figure 4.

Grafting-from AMP-polymer conjugation: a) NCA ROP scheme and an example ROP of lysine (lys) and valine (val) NCA monomers from PAMAM dendrimers, adapted with permission from Lam et al.,103 copyright © 2016 American Chemical Society; b) RAFT polymerization scheme and an example of grafting poly(2-hydroxyethylmethacrylamide) from an AMP-functionalized chain transfer agent (CTA), adapted with permission from Luppi et al.,104 copyright © 2019 The Royal Society of Chemistry; c) ATRP and d) NMP schemes and sequential polymerization of tert-butyl acrylate and styrene from a resin-bound AMP. Subsequent removal of the conjugates from the resin and removal of tert-butyl groups yielded AMP-poly(acrylic acid)-block-polystyrene conjugates. Adapted with permission from Becker et al.,105 copyright © 2005 American Chemical Society.
The other grafting-from scenario is the growth of polymers from AMPs. Functionalization of AMPs with an initiator or chain transfer agent enables growth of polymers from AMPs using controlled polymerization methods, including reversible addition–fragmentation chain-transfer (RAFT) polymerization, atom transfer radical polymerization (ATRP), and nitroxide-mediated radical polymerization (NMP).16,106,107 In these polymerizations, reversible activation and deactivation of the initiator (a halide-containing compound for ATRP and nitroxides for NMP) or chain transfer agent (CTA, as in RAFT) slows the rate of chain growth relative to initiation to produce polymers with well-defined lengths.
Conducting RAFT polymerization from an AMP-functionalized CTA, typically a trithiocarbonate, yields conjugates with the AMP at one chain end and the trithiocarbonate at the other. If desired, the trithiocarbonate can be cleaved after polymerization, to reveal a thiol for further modification (e.g., with a dye or targeting moiety).106 Of note, when functionalizing AMPs with CTAs, trithiocarbonate-based CTAs are used rather than dithioesters for attachment to the N-terminal amine of AMPs due to their higher stability to aminolysis.16 Semsarilar and coworkers functionalized a carboxylic acid-functionalized trithiocarbonate chain transfer agent to the N-terminal amine of oligolysine during solid phase peptide synthesis to prepare a CTA-modified AMP (Figure 4b).104 Subsequent RAFT polymerization of 2-hydroxypropyl methacrylate from the CTA-modified AMP produced well-defined AMP-polymer conjugates.
Wooley and coworkers generated conjugate nanoparticles by grafting polymers from AMPs using ATRP and NMP (Figure 4c,d).105 First, tert-butyl acrylate and styrene were sequentially polymerized from resin-bound AMPs functionalized with ATRP and NMP initiators. Subsequent acid treatment simultaneously cleaved the conjugates from the resin and the tert-butyl groups on the polymer to afford an amphiphilic AMP-poly(acrylic acid)-block-poly(styrene) conjugate that assembled in aqueous solution into AMP-presenting nanoparticles. With the continued development of biocompatible, synthetically accessible controlled polymerizations, such as metal-free ATRP108 and room-temperature RAFT polymerization,109 these methods are becoming increasingly attractive tools for the synthesis of AMP-polymer conjugates.
5.3. Grafting-through approaches
Grafting-through approaches for AMP-polymer conjugate synthesis involves modifying an AMP with a polymerizable group, followed by polymerization of the resulting monomers into a structure with pendant AMPs on multiple, or even all, repeating units.16,110 For example, free radical polymerization of a lysine- and phenylalanine-containing AMP with a C-terminal polymerizable isobutene group yielded comb/brush polymers with AMPs on each repeating unit (Figure 5a).111 Additionally, UV-initiated, graft-through polymerization of methacrylate-functionalized poly(ethylene glycol)-block-poly(lysine-co-phenylalanine) yielded surfaces decorated with comb/brush AMP-polymer conjugates.112
Figure 5.

Additional approaches to prepare AMP-polymer conjugates: a) Grafting-through synthesis of comb conjugates by conventional radical polymerization of vinyl-terminated lysine-based AMPs.111 b) Cross-linking linear conjugates prepared by RAFT polymerization into stars by chain-extension: example preparation of star conjugates by chain extension of polylysine- and glucosamine-containing linear polymer arms with bisacrylamide. Adapted with permission from Wong et al.,113 copyright © 2016 American Chemical Society.
5.4. Cross-linking linear AMP-polymer conjugates into stars by chain-extension with difunctional monomers
Chan-Park and coworkers bundled AMP- and sugar-containing linear polymer chains into star-shaped conjugates by polymerizing a difunctional monomer from the CTA at the chain ends (Figure 5b).113 This “arm-first” approach allowed precise control of arm length and composition in star-shaped conjugates with multiple types of arms. We note, however, that this strategy provides little control over the number of arms in such core-cross-linked star polymers, which depends on a complex combination of factors, such as concentration, arm molecular weight, and the ratio of difunctional monomer to CTA.114
6. Dissecting the role of polymer architecture on AMP-polymer conjugate properties and antimicrobial performance
Linear AMP-polymer conjugates, in which a single AMP is attached to the end of a linear polymer chain, often improve the stability and biocompatibility of AMPs, but significantly lower antimicrobial activity or abolish it all together. Advances in polymerization chemistry have enabled the synthesis of polymers with well-defined, yet complex architectures. In the context of antimicrobial applications, non-linear polymer architectures can present multiple AMPs on the same polymer chain, which increases the local AMP concentration and, by extension, membrane interactions and bacterial killing. Relative to linear conjugates, the superior balance of high bactericidal activity, low cytotoxicity, and improved stability reported for AMP-polymer conjugates with non-linear architectures continues to motivate the investigation of these more complex structures. As illustrated schematically in Figure 6, this section highlights the impact of molecular architecture on the properties (e.g., surface charge, size, local concentration of AMPs, and hydrophobicity/solubility) and antimicrobial performance of AMP-polymer conjugates, reviewing linear, comb/brush, star, and hyperbranched AMP-polymer conjugates, as well as the effects of varying molecular weight and composition of both the AMP and polymer components within each architecture.
Figure 6.

AMP-polymer conjugates designed in a) linear, b) comb/brush, c) star, and d) hyperbranched architectures.
6.1. Linear AMP-polymer conjugates
Early efforts to improve the therapeutic properties of AMPs by conjugation to biocompatible polymers were first described in the 2000s, and focused on linear architectures (Figure 6a). Conjugation of magainin 2 (GIGKWLHSAKKFGKAFVGEIMNS), tachyplesin I (KWCFRVCYRGICYRRCR, with disulfide bonds connecting cysteine residue 3 to16 and cysteine residue 7 to 12), and nisin A to PEG reduced toxicity to eukaryotic cells, but also markedly decreased antimicrobial activity.44,115,116 For example, while PEGylation of magainin 2 substantially improved viability of mammalian CHO-K1 cells (>95% vs. <1% for the unconjugated peptide at concentrations above the MIC), the conjugates exhibited lower bactericidal activity (MIC = 80 μM) than magainin 2 alone (MIC = 20 μM) against E. coli,116 In this, and similar cases where conjugation to polymers reduces cytotoxic effects, yet severely limits bacterial killing, the peptide is likely buried in the polymer. Later work showed that decreasing PEG length improves retention of AMP antimicrobial activity: conjugates of the AMP CaLL (KWKLFKKIFKRIVQRIKDFLR) to PEG with degree of polymerization (DP) of 44 repeating units exceeded the bactericidal activity of conjugates with longer PEG (DP = 66).117 However, this increase came at the expense of greater cytotoxicity. Table 1 shows that, over multiple examples of linear AMP-polymer conjugates, the trend holds that decreasing PEG length increases both antimicrobial activity and cytotoxic effects. Accordingly, conjugation of the 4.7 kDa AMP cryptin-2 to a 5 kDa PEG chain yielded similar bactericidal activity as AMP alone (equivalent MICs in molar units), while also increasing cell viability, showcasing an ideal outcome of balancing activity and toxicity.39 Moreover, PEGylation of cryptin-2 prolonged bacteria killing in mouse sera, a host environment in which the absorption of proteins may otherwise trigger aggregation and reduce antimicrobial activity.
Table 1.
Linear PEGylated AMPs: decreasing PEG molecular weight increases both antimicrobial activity and mammalian cell toxicity
| Year | AMP | Polymer | Findings | Ref. |
|---|---|---|---|---|
| 2003 | nisin A (3 kDa) | PEG (5 kDa) | Long PEG chain reduced AMP bactericidal activity and toxicity | 44 |
| 2007 | tachyplesin I (2.3 kDa) | PEG (5 kDa) | Long PEG chain reduced AMP bactericidal activity and toxicity | 116 |
| 2007 | magainin 2 (2.5 kDa) | PEG (5 kDa) | Long PEG chain reduced AMP bactericidal activity and toxicity | 115 |
| 2008 | MA (1.6 kDa) | PEG (0.75 and 1.1 kDa) | Decreasing PEG length improved bactericidal activity, but increased toxicity | 118 |
| 2012 | CaLL (2.8 kDa) | PEG (0.67 and 1 kDa) | Conjugation improved cytocompatibility; decreasing PEG length increased conjugate bactericidal activity | 117 |
| 2014 | poly(lysine-co-phenylalanine) (varied from 5.5 to 0.5 kDa) | PEG (5 and 2 kDa) | Decreasing PEG length improved conjugate bactericidal activity, but increased toxicity | 57 |
| 2017 | Peptide 77c (1.8 kDa) | PEG (5 kDa) | Conjugation to PEG reduced bactericidal activity | 37 |
| 2020 | cryptdin-2 (4.7 kDa) | PEG (5 kDa) | Conjugates retained bactericidal activity, reduced toxicity, and improved stability of AMP in murine serum | 39 |
Studies on linear AMP-polymer conjugates highlight the potential value of well-designed AMP conjugation to polymers. An interesting extension is linear amphiphilic conjugates, in which primarily hydrophobic AMPs are conjugated to hydrophilic polymers and vice versa, which can assemble into nanoparticles of well-defined size and shape determined by the molecular weight of each component and the hydrophobic volume fraction, respectively.57,118 Such molecular assemblies can appreciably impact bactericidal activity, and thus we describe these cases in a dedicated ‘supramolecular assembly’ section later in this review.
6.2. Comb/brush AMP-polymer conjugates
Since AMPs tend to operate cooperatively in facilitating bacterial membrane disruption, non-linear polymer architectures that allow conjugation of multiple AMPs119-121 appear to provide more efficient bacterial killing. Comb/brush polymers consist of macromolecular (e.g., polymeric or peptide) pendent groups on a main (backbone) chain that can be either a polymer or a peptide (Figure 6b). The ability to locally concentrate AMPs on a single polymer molecule in comb architecture is conducive to generating potent bactericidal AMP-polymer conjugates. The side chain density, side chain length, and backbone length of comb polymers can all be modulated to improve bactericidal activity and reduce cytotoxic effects by these conjugates.
Increasing AMP side chain density generally increases antimicrobial activity.23,28,46 An informative example by the Chan-Park lab showed tuning the alkyne content in polyphosphoesters to modulate the density of the cysteine-terminated AMP CysHHC10 (H-CKRWWKWIRW-NH2) (Table 2).28 As compared to peptide alone (MIC = 8 μg/mL), conjugates with 41 and 57 weight % peptide exhibited MICs of 16 and 8 μg/mL, respectively against E. coli. These conjugate MICs correspond to 6.6 and 4.6 μg/mL, respectively, of peptide and show the retention and even enhancement of the bactericidal potency of the conjugates relative to the AMP alone. Experiments in Gram-negative, colistin-resistant P. aeruginosa and Gram-positive S. aureus yielded similar results. Moreover, conjugation reduced cytotoxicity: the HC50 values of the conjugates (>4000 μg/mL) exceeded those of the AMP alone (HC50 >1000 μg/mL). Likewise, mammalian-cell viability following exposure to the conjugates (IC50 >160 and >106 μg/mL for conjugates with 41 and 57 wt% peptide) exceeded that for the AMP alone (IC50 >62 μg/mL).
Table 2.
Bactericidal and hemolytic activity of AMP-grafted poly(phosphoester)s; adapted with permission from Pranantyo et al.,28 copyright © 2016 American Chemical Society28
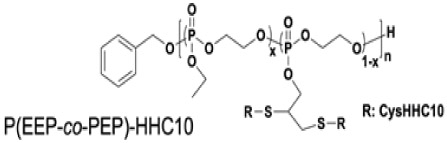
| |||||
|---|---|---|---|---|---|
| MIC (μg/mL) | HC50 (μg/mL) |
IC50 (μg/mL) |
|||
| E. coli | P. aeruginosa | S. aureus | |||
| CysHHC10 | 8 | 16 | 4 | >1000 | >62 |
| P(EEP-co-PEP)-HHC1041% | 16 (6.6)a | 32 (13.1) | 8 (3.3) | >4000 (>1640) | >160 (>65.6) |
| P(EEP-co-PEP)-HHC1057% | 8 (4.6) | 16 (9.1) | 8 (4.6) | >4000 (>2280) | >106 (>60.42) |
Normalized for peptide content conjugate, calculated based on the peptide mass fraction.
In another example, varying the degree of azide substituents on chitosan for reaction with the alkyne-terminated AMP anoplin generated comb conjugates averaging 12, 30, or 40 AMPs per polymer chain with MICs of 256, 128, and 64 μg conjugate/mL, respectively, against Gram-positive S. aureus; this trend extended to Gram-negative E. coli.46 However, no AMP density dependence on MIC was observed against either Gram-positive Enterococcus faecalis (E. faecalis) or Gram-negative, colistin-resistant P. aeruginosa. Circular dichroism spectroscopy, conducted in the presence of membrane-mimicking sodium dodecyl sulfate, showed the helicity of the AMP constituents to increase with AMP density, providing further insight into the AMP density-dependent activity observed in some bacterial species. Additionally, while the hemolytic activity also increased with AMP density, the HC50 values of all conjugates (ranging from 4000 to >30,000 μg/mL) significantly exceeded that of anoplin alone (HC50 = 512 μg/mL).
The above anoplin-chitosan conjugate work highlights another consideration for engineering AMP-polymer conjugates: peptide orientation.46,86 Appending alkynes to either the N-terminus or C-terminus of anoplin enabled variation of the AMP orientation with respect to the polymer backbone.46 Against Gram-positive E. faecalis and Gram-negative, colistin-resistant P. aeruginosa, the conjugates with N-terminally linked anoplin were more active than those with the opposite AMP orientation. Interestingly, these trends appear to be species-specific as conjugates with C-terminally linked anoplin performed as well or better against Gram-positive S. aureus and Gram-negative E. coli.
Chan-Park and coworkers studied the effects of pendent AMP length and cationic-hydrophobic balance, as well as backbone length on bactericidal activity and toxicity with a series of lysine (K)-grafted chitosan comb conjugates (Table 3).22 Conjugation to chitosan markedly improved antimicrobial activity of the AMPs, as K1-K25 alone all exhibited MICs > 1000 μg/mL. At constant AMP density on the conjugates (i.e., molar ratio of AMP-containing side chains), increasing AMP side chain length improved activity. Comb conjugates with graft lengths of 1 (CS-g-K1) and 25 (CS-g-K25) exhibited MICs of 160 and 10 μg conjugate/mL, respectively, corresponding to 73 and 4.6 μg peptide/mL respectively, against Gram-positive S. aureus; this trend was similar for Gram-negative E. coli. In this case, increasing AMP length also increased charge. Holding graft length constant at 25 and incorporating 50% hydrophobic phenylalanine residues (i.e., CS-g-K12.5F12.5) substantially reduced activity and increased hemolysis. At constant AMP length, composition, and graft density, conjugates with shorter chitosan backbones exhibited superior activity: trilysine-grafted chitosan (CS-g-K3) with molecular weights of 13,500 g/mol (CS-g-K3) and 290,000 g/mol (CS-g-K3-HMW) yielded MICs of 40 and 160 μg conjugate/mL (29 and 150 μg peptide/mL), respectively, against S. aureus; experiments against P. aeruginosa and E. coli produced similar results. The authors attributed the lower bactericidal activity of the higher molecular weight conjugates to reduced passage through the cell wall, and thus, decreased disruption of the cytoplasmic membrane. A similar effect of backbone length on bactericidal activity was also reported for lysine- and phenylalanine-grafted dextran72, and for ε-polylysine-grafted chitosan,23 where conjugates with the lowest molecular weight examined showed the greatest antimicrobial activity.
Table 3.
Antimicrobial and hemolytic activity of polylysine-grafted chitosan; adapted with permission from Li et al.,22 copyright © WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
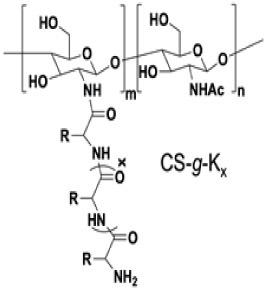
| |||||||
|---|---|---|---|---|---|---|---|
| MIC (μg/mL) | HC50 (μg/mL) |
Selectivityb (HC50/MIC) |
|||||
| E. coli |
P.
aeruginosa |
S.
aureus |
|||||
| Increasing AMP length and charge |
|
CS-g-K1 | 80 (37)a | 80 (37) | 160 (73) | >50000 | >625 |
| CS-g-K3 | 20 (15) | 20 (15) | 40 (29) | >50000 | >2500 | ||
| CS-g-K9 | 10 (8.8) | 20 (17) | 20 (17) | >50000 | >5000 | ||
| CS-g-K16 | 10 (9.3) | 20 (19) | 10 (9.3) | >100000 | >10000 | ||
| CS-g-K25 | 10 (9.5) | 40 (38) | 10 (4.6) | >50000 | >5000 | ||
| Increasing AMP hydrophobicity; decreasing charge |
|
CS-g-K16 | 10 (9.3) | 20 (19) | 10 (9.3) | >100000 | >10000 |
| CS-g-K8F8 | >2500 (>2308) | >2500 (>2308) | 2500 (2308) | 12500 | <5 | ||
|
|
CS-g-K25 | 10 (9.5) | 40 (38) | 10 (4.6) | >50000 | >5000 | |
| CS-g-K12.5F12.5 | 630 (598) | 1250 (1187) | 1250 (1187) | 7500 | 12 | ||
| Increasing backbone length |
|
CS-g-K3 | 20 (15) | 20 (15) | 40 (29) | >50000 | >2500 |
| CS-g-K3-HMW | 80 (56) | 310 (218) | 160 (152) | >25000 | >313 | ||
Normalized for peptide content conjugate, calculated based on the peptide mass fraction determined from the peptide degree of polymerization and chitosan deacetylation degree.
Calculated from MIC against E.coli
Table 4 lists additional examples of comb/brush AMP-polymer conjugates, which largely corroborate the findings discussed in this section. Overall, these studies indicate that increasing AMP side chain length and/or density increases activity, while increasing backbone length decreases activity. Generally, conjugation of AMPs to comb/brush polymers provide an avenue to reduce cytotoxicity, while retaining, or even enhancing, bactericidal activity. These outcomes provide an immense benefit towards antimicrobial therapy.
Table 4.
Other examples of comb/brush AMP-polymer conjugates.
| Year | AMP | Polymer | Findings | Ref. |
|---|---|---|---|---|
| 2006 | Acetyl-RRWW-NH2 Acetyl-RWRW-NH2 |
poly(maleic anhydride) | Conjugate exhibited increased antimicrobial and hemolytic activity compared to the AMP alone | 88 |
| 2014 | ε-polylysine | chitosan | Conjugates permeabilized bacterial membranes | 46 |
| 2014 | nisin | hyaluronic acid | Increasing AMP graft density increased antimicrobial activity | 51 |
| 2017 | ε-polylysine | chitosan | Bactericidal activity increased with AMP density and decreased with greater backbone length | 23 |
| 2017 | pentalysine (KKKKK) | chitosan | Grafting pentalysine AMPs from short chitosan chains elevated activity above that of linear polylysine with higher charge density; result attributed to conjugate assembly into nanoparticles | 52 |
| 2018 | HHC10 (KRWWKWIRW) | chitosan | Conjugation reduced hemolytic activity and mammalian cell toxicity of the AMP; grafting AMPs to chitosan hydroxyls to leave amines available for membrane interactions improved bactericidal activity | 81 |
| 2019 | RWAAC-NH2; CAAWR-NH2; PWKISIHLAAC-NH2 | chitosan | Conjugation improved bactericidal activity; conjugates exhibited minimal cytotoxicity near MIC | 86 |
| 2020 | poly(lysine-co-valine) | chitosan | Grafting of AMPs improved bactericidal activity of chitosan | 100 |
6.3. Star-shaped AMP-polymer conjugates
Star-shaped polymers feature multiple polymer chains (arms) emanating from a central core (Figure 6c). The arm number, arm length, and composition all impact the balance between the bactericidal activity, mammalian-cell toxicity, protease resistance, and aggregation of star-shaped AMP-polymer conjugates. Since star-shaped conjugates are, in most cases, prepared by ROP of amino acid NCAs from initiating groups on a small molecule or polymer, AMP arm length is proportional to the NCA monomer:initiator ratio, while the number of initiating groups on the core determines arm number. The commercially available small molecules 1,1,1-tris(hydroxymethyl) propane; pentaerythritol; dipentaerythritol; and tripentaerythritol are used as cores to generate star polymers with 3, 4, 6, and 8 arms, respectively.25 To synthesize stars with a higher number of arms, hyperbranched polymers or dendrimers, precision hyperbranched polymers with discrete molecular weights, are used as the core.122,123 For example, ROP of lysine and valine NCAs from PAMAM dendrimers with 16 and 32 peripheral primary amines generated 16- and 32-arm star-shaped poly(lysine-co-valine)s, respectively.
Since star polymers present multiple AMPs per molecule, the high local concentration of AMPs typically translates to potent antimicrobial activity. This is especially evident when comparing linear and star polymers with similar compositions and AMP:polymer ratios. In one example, Reynolds, Qiao, and coworkers showed a linear and a 16-arm star poly(lysine-co-valine), with similar arm lengths, to yield MBCs of 29.5 and 0.72 μM, respectively, against Gram-negative E. coli. Since every star polymer carries 16 AMPs, normalizing for peptide concentration by multiplying the MBC of the star conjugate by 16 gives 11.52 μM, still less than half the MBC of the linear polymer. Moreover, both the linear and star polymer had hemolytic concentrations above the bactericidal concentrations, with HC50 = 675 and 58 μM, respectively.26 In another example, bacterial killing experiments with linear and 8-arm star poly(l-lysine) (PLL) with comparable molecular weights showed the star polymers to exhibit superior activity against two Gram-positive and two Gram-negative bacterial species.24 Experiments and molecular simulations on these materials showed the surface charge of the star-shaped conjugates to exceed that of the linear PLL, indicating a higher density of cationic lysine residues. The higher charge density and electrostatic driving force for interaction with, and disruption of, bacterial membranes is consistent with the more potent bactericidal activity of the star polymers. Additionally, the star polymer demonstrated greater resistance to trypsin-mediated degradation, which may also result from the higher lysine density of the star polymer.
The bactericidal activity of star AMP-polymer conjugates generally increases with both the AMP number and length.25-27 Holding AMP length constant at ca. 15 repeating units on poly(lysine-co-valine)-grafted PAMAM dendrimers, the 4-, 8- and 16-arm star conjugates exhibited MICs of 23.2, 7.5, and 5.2 μg conjugate/mL, respectively, against E. coli.27 Converting the MICs to molar values, and correcting for AMP content by multiplying the molar MICs by the arm number (since the 16-arm star contains 4 times more AMPs per conjugate than the 4-arm star) highlights the increase in the inherent activity of the conjugates with higher arm numbers (Table 5), likely due to a higher local AMP concentration. This series of star polymers provided selective antimicrobial activity, with selectivity indices (SI = IC50/MIC) ranging from 5-9. With the same materials system, 4-arm star polymers with AMP lengths of 12, 19, and 26 amino acids per arm yielded MICs of 23.2, 7.9, and 7.7 μg/mL, respectively. While activity improved with arm length, the plateau in this trend was attributed to a reduction in the local AMP concentration at the outermost surface of the star polymers (i.e., due to the greater distance between AMPs with increasing arm length).
Table 5.
The effect of arm number on the antimicrobial activity and selectivity of star-shaped poly(lysine-co-valine)-poly(amidoamine)s; reprinted with permission from Shirbin et al.,27 copyright © 2018, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
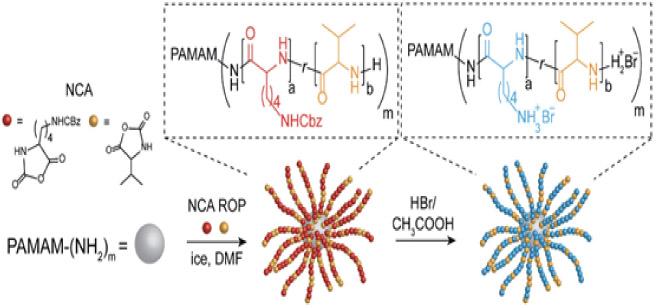
| |||||
|---|---|---|---|---|---|
| Arm number |
AMP length (amino acids) |
MIC (μg/mL) |
MIC (μM) |
MIC x arm Number (μM)a |
TI (IC50/ MIC)b |
| 4 | 12 | 23.2 | 2.636 | 10.544 | 5.6 |
| 8 | 16 | 7.5 | 0.322 | 2.576 | 6.1 |
| 16 | 14 | 5.2 | 0.127 | 2.032 | 8.6 |
Since 1 mol of the 16-arm star conjugate contains 4x more AMPs than the 4-arm star conjugate, we normalize for these effects by multiplying the molar MIC by arm number.
Therapeutic index (TI) = IC50 against H4IIE rat hepatoma cells / MIC against E. coli
In addition to modulating arm length and arm number, versatile synthetic methods enable variation of the polymer and AMP composition of the arms that comprise a star polymer.26,27,54,89,103,113 Incorporating 20% hydrophobic indole groups into star-shaped PLL enhanced antimicrobial activity and reduced toxicity to lung and kidney cells as compared to the unmodified PLL star; a result attributed to a reduction in overall charge and hydrophilicity of the conjugate.25 Instead of uniformly varying the composition of all arms, Chan-Park and coworkers prepared star-shaped polymers with a combination of cationic PLL arms and sugar-containing polymer arms linked to a core via neutral hydrophilic polymer linkers (Table 6).113 Zeta potential and antimicrobial activity increased with PLL arm content, consistent with a higher charge density and local AMP concentration yielding more potent bacterial killing. Comparing compatibility with human aortic smooth muscle cells to MIC, the conjugates containing 75% PLL arms provided the highest selectivity (IC50/MIC) = 7. Inclusion of at least 25% glycopolymer arms was crucial in mitigating the toxicity of PLL, as the selectivity of conjugates without glycopolymer arms approached 0. These star conjugates did not induce appreciable hemolytic activity (HC50 > 10,000 μg/mL), which the authors attributed to the lack of hydrophobic groups.
Table 6.
Zeta potential and selectivity of glucosamine-functionalized star polymers. In the schematic, red = polylysine, green = sugar-containing polymer, blue = neutral hydrophilic poly(hydroxylethylacrylamide). Adapted with permission from Wong et al.,113 copyright © 2016 American Chemical Society.
| |||
|---|---|---|---|
| PLL arm fraction (%) on conjugate |
Zeta potential (mV) |
MIC against S. aureus (μg/mL) |
Selectivity: IC50/MIC |
| 100 | 44.4 | 16-32 | ~0 |
| 75 | 33.6 | 32-64 | 7.0 |
| 50 | 33.4 | 64-128 | 3.8 |
| 35 | 23.2 | 256 | 2.4 |
6.4. Hyperbranched AMP-polymer conjugates
With highly branched three-dimensional structures, hyperbranched polymers offer a high density of peripheral functional groups for AMP conjugation (Figure 6d). In one such example, the peripheral groups of hyperbranched polyglycerol (HPG) were substituted with the AMP 77c (RLWDIVRRVWGWL) (Table 7).37 . Although conjugation reduced antimicrobial activity (MICs of 50-325 μg conjugate/mL against S. aureus) as compared to the AMP alone (MIC = 8 μg/mL), it reduced toxicity to mammalian cells and improved compatibility with blood components. Indeed, while AMP 77c was highly hemolytic and toxic to fibroblasts, the conjugate was much better tolerated (hemolysis <10%; cell viability >80%) at bactericidal concentrations (highest concentration tested = 250 μg conjugate/mL). Proteolytic stability of the AMP also improved upon conjugation, as noted by the lack of fragmentation observed by mass spectrometry after incubation with trypsin as compared to the fragmented spectra of AMP 77c alone. As observed for AMP-polymer conjugates with other architectures, decreasing HPG molecular weight was associated with increased antimicrobial activity. Although there are still relatively few studies to date on hyperbranched AMP-polymer conjugates, this example and several others56,73,85,124,125 showcase the potential of hyperbranched architectures to balance bactericidal activity, mammalian-cell compatibility, and proteolytic stability by varying the composition, degree of branching, molecular weight, and end group functionality.
Table 7.
AMP-hyperbranched polyglycerol conjugates with similar peptide content, adapted with permission from Kumar et al., copyright © 2017 American Chemical Society37
| ||
|---|---|---|
| AMPs per polymer (by NMR spectroscopy) |
MIC against S. aureus (μg/mL) |
|
| peptide 77c | - | 8 |
| 22k HPG-peptide 77c | 7 ± 1 | 50 |
| 44k HPG-peptide 77c | 11 ± 3 | 100 |
| 105k HPG-peptide 77c | 20 ± 5 | 325 |
7. Supramolecular assembly of AMP-polymer conjugates
Amphiphilic AMP-polymer conjugates can assemble in aqueous solution into higher order structures, such as micelles and nanosheets (Figure 7).56,105,111 Such supramolecular assemblies can both increase the local concentration of AMPs to improve bactericidal activity and provide protection against proteolytic degradation and aggregation. 36,58,59,62,104,126 One strategy is the attachment of a hydrophilic AMP to a hydrophobic polymer, yielding nanostructures in solution with the AMP presented on the outer surface.57,59,104,105 Wooley and coworkers conjugated an AMP to the hydrophilic poly(acrylic acid) end of the amphiphilic block copolymer poly(acrylic acid)-b-poly(styrene).105 In aqueous solution, the conjugates assembled into ~50 nm micelles with a hydrophobic polystyrene core, a hydrophilic poly(acrylic acid) shell, and the AMP on the outer surface (Figure 7a). The micelles, containing 10% AMP by mass, exhibited an MIC of 13 μg conjugate/mL against both S. aureus and E. coli, which was superior to the performance of the unconjugated AMP (MIC = 17 μg/mL against S. aureus and 33 μg/mL against E. coli). Presentation of the AMP on micelles also tempered the toxicity of the AMP against mouse myeloma B cells.
Figure 7.

AMP-polymer conjugate assemblies in aqueous solution: a) AMP-functionalized poly(acrylic acid)-block-polystyrene assembles into micelles with polystyrene cores, polyacrylic acid shells, and the AMP on the outermost surface, adapted with permission from Becker et al.,105 copyright © 2005 American Chemical Society; 105 b) comb polymer with pendant AMPs assembles into nanoparticles postulated to kill bacteria by membrane disruption, adapted with permission from Zhen et al.,111 copyright © 2019 Royal Society of Chemistry; and c) hyperbranched AMP-polymer conjugates form nanosheets that disrupt bacterial membranes, adapted with permission from Gao et al., copyright © 2016 American Chemical Society.56
In contrast to nanoparticles with AMPs on the surface, AMP-conjugates can also be engineered to form micelles with AMPs located within the core. Conjugation of hydrophobic AMPs to PEG results in assembly in aqueous solution of micelles with a hydrophilic PEG shell and hydrophobic peptide core.118,126,127 Sequestering AMPs into micelle cores can mask cytotoxicity and provide protection against AMP degradation and aggregation. For example, the MIC of the PEGylated AMP MA (GLLALILWIKRKR) against S. aureus increased only slightly, from 12 to 24 μM, in the presence of serum; in contrast, the MIC of the unconjugated AMP increased from 12 μM to 163 μM.118 As we discuss in the following section on ‘stimuli-responsive conjugates’, assemblies with AMPs within the core are often designed as responsive systems to mitigate cytotoxicity and to reveal the AMPs on-demand at sites of infection.
MICs exceeding the aggregation concentrations of assembling AMP-polymer conjugates indicate that the supramolecular assemblies, rather than the individual conjugates, are responsible for bactericidal activity. Yang and coworkers generated amphiphilic comb polymers by free radical polymerization of a vinyl-functionalized AMPs consisting of hydrophobic phenylalanine and leucine residues, as well as cationic lysines (Figure 7b).111 Assembly in aqueous solution above 12 μg/mL generated cationic nanoparticles. Scanning electron microscopy showed the nanoparticles to be ~100 nm in diameter, and dynamic light scattering measured slightly larger hydrodynamic diameters of ~150 nm. Against six bacterial species, the nanoparticles afforded MICs of 16 μg/mL, remarkably close to the aforementioned critical aggregation concentration. Mammalian-cell viability exceeded 80% after incubation with 40 μg/mL of the nanoparticles for 24 h, revealing low cytotoxicity at bactericidal concentrations. Amphiphilic, hyperbranched AMP-polymer conjugates formed nanosheets in aqueous solution (Figure 7c).56 Transmission electron microscopy and atomic force microscopy revealed ca. 10 nm thick nanosheets > 50 μm2 in area. These nanosheets exhibited an MIC value (16 μg/mL) just above the critical aggregation concentration (15 μg/mL) against both Gram-negative E. coli and Gram-positive S. aureus. Despite the low surface charge (zeta potential ~+6 mV), these nanosheets displayed potent antimicrobial activity attributed to a “wrap and penetrate” mechanism, whereby the high surface area of the nanosheets provides high contact area with bacterial membranes. Overall, supramolecular AMP-polymer conjugate nanostructures that concentrate AMPs, provide protection from degradation and aggregation, and interact with membranes in distinct ways from unassembled conjugates are promising materials for antimicrobial applications and warrant further investigation.
8. Stimuli-responsive systems
Stimuli-responsive AMP-polymer conjugates can reduce undesired side effects by selectively inducing antimicrobial activity at target sites. Both the elevated concentrations of the natural reducing agent glutathione associated with bacteria and the acidic pH at infection sites (reaching as low as 5.5) can cause AMP-polymer conjugates to reveal or release AMPs.63,128-130 Additionally, applying light at the infection site can trigger AMP release from photo-responsive conjugates or activate photothermal materials to produce synergistic therapeutic effects.131-133 Examples of stimuli-responsive AMP-polymer systems include those featuring degradable linkages,38,40,74,133 membrane-induced AMP conformational changes,36 and therapeutic compounds for combination therapies63,130,131,133-135, among others40,74,136 (Figure 8).
Figure 8.

Schematic illustrations of stimuli-responsive AMP-polymer conjugate systems. a) Redox-responsive poly(2-ethyl-2-oxazoline) (PEtOx)-cyclic peptide nanotube (CPNT) conjugates are activated by intracellular glutathione-triggered disulfide reduction, cleaving the PEtOx polymer to reveal bactericidal CPNTs.38 b) Conjugates of the AMP LK13 and PEGylated chitosan form nanospheres in aqueous environments. The neutral surface charge of the nanospheres allows for diffusion into the negatively charged extracellular matrix surrounding P. aeruginosa. Interaction with the bacterial membrane causes disassembly of the nanospheres and conversion of LK13 from random coil to a bactericidal α-helical conformation.36 c) Emulsion-templated fabrication of nanoparticles from pH-responsive linear triblock copolymers composed of poly(d,l-lactic-co-glycolic acid) (PLGA), poly(ethylene glycol) (PEG), and poly(l-histidine) (PLH) furnished pH-responsive nanoparticles encapsulating vancomycin. At physiological pH, the nanoparticles are anionic. Lowering the pH to 6.0-6.5 protonates the imidazole groups on PLH, rendering the nanoparticles cationic for increased interaction with bacterial membranes and subsequent vancomycin-induced bacteria killing.130 d) Capping lysine amines of the AMP KLA on PEG-KLA conjugates with pH-responsive 2,3-dimethylmaleic anhydride (DA) yields anionic conjugates at physiological pH. In acidic environments (pH 5.5), the DA caps are cleaved to reveal cationic lysines that target bacterial membranes. Encapsulation of PDT agents in the PEG-KLA conjugates enables light-triggered bacteria killing.131
Degradable Linkages
The introduction of degradable linkages, such as hydrolysis-labile esters and redox-cleavable disulfides, between AMPs and polymers provides the opportunity to increase selectivity by facilitating controlled release of AMPs. For example, PEGylation of AMP Bac7(1-35) through an ester linkage enables peptide release by hydrolysis or upon exposure to esterases present in human serum and plasma40. Compared to analogous conjugates with more stable linkages, the cleavable conjugate yielded higher bactericidal activity, thus highlighting the opportunity to leverage natural degradation mechanisms to enhance the performance of Bac7(1-35).
The elevated concentration of the natural reducing agent glutathione (GSH) in E. Coli (> 10 mM) compared to that in eukaryotic cells (1-2 mM) has motivated the design of reducible, disulfide-linked conjugates for GSH-triggered release of AMPs.38,128,129,133,137 Perrier and coworkers increased the selectivity of membrane pore-forming cyclic AMP nanotubes (cyclo(l-Trp-d-Leu-l-Lys-d-Leu-l-Trp-d-Leu-l-Lys-d-Leu)) (CPNTs) through conjugation to biocompatible poly(2-ethyl-2-oxazoline) (PEtOx) (Figure 8a).38,138 PEtOx disperses the CPNTs in aqueous solution and limits off-target toxicity prior to disulfide cleavage in reductive environments. Upon exposure to 10 mM GSH, to cleave PEtOx, dynamic light scattering measurements showed CPNT size increases indicative of nanotube formation and conducive to membrane interactions. Hemolysis assays and calcein leakage studies showed PEtOx to reduce the hemolytic and membrane pore-forming activity of the CPNTs prior to removal.
Membrane-induced AMP conformational changes
The hydrophobic environment within bacterial membranes can induce conformational changes in AMPs that cause membrane pore formation and/or disruption. AMPs and conjugates that undergo these conformational switches can remain benign prior to membrane interaction. For instance, the cationic AMP LK13 (LKLLKKLLKKLKK) adopts a random-coil conformation in aqueous environments. Exposure of LK13 to the hydrophobic environment within P. aeruginosa membranes induces a conformational change to α-helical, initiating antimicrobial activity through membrane pore formation.41,139 While this conformational change confers selective antimicrobial activity to LK13 alone (HC10/MIC = 800 and 1,600 against E. coli and P. aeruginosa, respectively), conjugation to PEGylated chitosan (CS-PEG-LK13) further improved selectivity (>2,000 and >4,000 against E. coli and P. aeruginosa, respectively). Moreover, CS-PEG-LK13 conjugates formed neutrally charged nanospheres in aqueous solution that facilitated diffusion of the AMP through the highly anionic extracellular matrix surrounding P. aeruginosa (Figure 8b).36 In this case, the polymer assisted with both transport through the extracellular infection environment and bacterial membrane targeting.
Combination therapies enabled by pH-responsive AMP-polymer conjugate
The ability of AMPs to interact with microbial membranes renders AMP-polymer conjugates particularly well suited for delivering secondary antimicrobial agents in combination therapy. A majority of these systems involve pH-responsive polymers and/or AMPs that leverage the pH difference between normal extracellular conditions (pH 7.4) and infection sites (pH ~ 5.5-6.5) for selective treatment.63,131,133 For example, emulsion-templated synthesis allowed encapsulation of the antibiotic vancomycin in nanoparticles composed of poly(d,l-lactic-co-glycolic acid)-b-poly(l-histidine)-b-poly(ethylene glycol) (PLGA-PLH-PEG) triblock copolymers (Figure 8c).130 At physiological pH these nanoparticles display a negative surface charge; lowering the pH to 5.5-6.5 protonates the imidazole groups on the poly(l-histidine) AMPs to facilitate vancomycin delivery and bactericidal activity. In another example, Wen, Quan, and coworkers designed a creative modification of antimicrobial star-polylysine, in which bifunctional linear PEG was used to cross-link PLL stars under dilute conditions, yielding cyclic polylysine-polyethylene glycol constructs. The protonatable amines of PLL confer inherent pH-responsive behavior, and these conjugates facilitated pH-dependent encapsulation and release of recombinant human interferon α-2b, an anionic cytokine involved in antimicrobial host responses.54 Another example comes from Park and coworkers, who functionalized cationic amine moieties of α-poly(l)lysine side-grafted onto a chitosan backbone (CS-PLL) with anionic citraconyl amide (CA) to form nanostructures at physiological pH.135 CA hydrolysis leads to disassembly at acidic pH (<6) found in bacterial infections. TEM and DLS were able to show disassembly of the nanostructures after incubation at pH 5, leading to both a decrease in hydrodynamic diameter and an increase in zeta potential. The CS-PLL conjugates had an MIC of 8-32 μg conjugate/mL against E. Coli, P. aeruginosa, S. aureus, S. epidermis, and MRSA bacteria; however, the CA-capped conjugates showed no antimicrobial activity up to 1024 μg/mL. Incubation of the CA-capped conjugates at pH 5.0 for 1 day yielded comparable MICs to the CS-PLL conjugates alone, while incubation at pH 7.4 produced no change in the low activity of the capped conjugates. The linear PLL, CS-PLL conjugates and the protected CA-capped conjugates showed no HC50 value up to 1024 μg/mL. Capping lysine amines with CA significantly reduced toxicity of the conjugates: whereas linear PLL and CS-PLL conjugates yielded 10 and 28 μg/mL in mouse 3T3 fibroblast cells, respectively, the CA-capped conjugates showed no appreciable toxicity up to 1024 μg/mL.
In a dual-stimuli responsive system, lysine-rich AMPs were functionalized with acid-cleavable groups and conjugated to PEG for the delivery of photodynamic therapy (PDT) antimicrobial agents. Specifically, the PEGylated cationic AMP KLA (KLAKLAKKLAKLAK) facilitated delivery of the PDT agents α-cyclodextrin conjugated nitric oxide (α-CD-NO) and chlorin e6 (α-CD-Ce6) into S. aureus biofilms.131 To conceal the positive charge until encountering the acidic pH in S. aureus biofilms (pH. 5.5), anionic 2,3-dimethylmaleic anhydride (DA) was attached to the lysine residues of KLA (Figure 8d). Confocal microscopy showed infiltration of the pH-responsive conjugates into S. aureus biofilms, while, in contrast, permanently anionic control conjugates displayed relatively low biofilm infiltration. In the presence of the conjugate at bactericidal concentrations, NIH 3T3 fibroblasts remained >90% viable. Irradiating biofilms infused with the pH-responsive conjugates encapsulating PDT at 660 nm to initiate photodynamic therapy decreased the number of live bacteria in biofilms by more than 90%, while the permanently anionic conjugates encapsulating PDT agents decreased the live bacteria by only 50%. These examples highlight the utility of introducing stimuli-responsive functionality into AMP-polymer conjugates to facilitate selective antimicrobial activity and enabling potent combination therapies.
9. Surfaces
In addition to envisioned applications of AMP -polymer conjugates as injectable, oral, or topical antimicrobial therapeutics, they are also expected to serve as anti-infective coatings, for example on high-touch hospital surfaces and implantable devices (e.g., grafts, stents, and catheters). Ideally, coatings should exhibit antimicrobial activity while also preventing bacterial adhesion and biofilm formation. These criteria present a biomaterials design challenge as bacterial killing is generally associated with hydrophobic, positively charged materials, while anti-fouling surfaces, those that prevent the adhesion of bacteria and proteins, are typically hydrophilic and charge-neutral. AMP-polymer conjugates are well-positioned to address these contrasting requirements by combining cationic AMPs with neutral polymers, such as PEG, polysaccharides, or zwitterionic polymers that generate highly-hydrated surfaces.140,141 The studies highlighted in this section tune AMP-polymer conjugate composition and architecture towards realizing dual bactericidal and anti-fouling and bactericidal character.
Polymerization of AMP-containing monomers from surfaces strike a particularly good balance of antimicrobial and antifouling activity. In one example, post-polymerization attachment of the cationic AMPs E6 (KRWRIRVRVIRKC) and Tet 20 (KRWRIRVRVIRKC) to reactive side chains of hydrophilic polymers grown from nanoparticles generated AMP-comb polymer conjugate surfaces.78,142 Varying the polymer constituents of these surfaces showed that conjugates formed from the protonatable poly(dimethyl acrylamide) (DMA) to exhibit superior antimicrobial activity and antifouling performance relative to conjugates prepared from the zwitterionic polymers poly(2-[(methacryloyl)oxy]ethyl]-phosphorylcholine) and poly(sulfobetaine methacrylamide). For the polyDMA-based surfaces, increasing AMP density improved antimicrobial activity.143,144,145 Another example shows the effect of PEG length on antifouling and antimicrobial activity. Polymerization of methacrylate-functionalized poly(ethylene glycol)-block-poly(lysine-co-phenylalanine) from poly(dimethylsiloxane) produced surfaces with AMP-polymer ‘bottlebrushes’ (Figure 9).112 Increasing the length of the PEG linker separating the AMP from the polymer backbone marginally reduced bacterial killing, with conjugates having 10, 45, and 90 PEG repeating units yielding >99%, >99%, and 92-95% dead bacteria, respectively. Varying the PEG linker length within these AMP-polymer conjugates minimally impacted the antifouling character of the surface.
Figure 9.

UV-induced polymerization of PEG-AMP from polydimethylsiloxane (PDMS) surfaces to generate tethered bottlebrush AMP-polymer conjugates. Adapted with permission from Gao et al.,112 copyright © 2017 Elsevier.
In addition to comb/brush conjugates, functionalization of silicone rubber surfaces with the AMP ILPWRWPWWPWRR and cell adhesive peptide RGD conjugated to linear triblock copolymers of poly(propylene oxide) flanked by two poly(ethylene oxide) blocks has been reported to kill bacteria, limit bacterial adhesion, and promote integration into host tissues.145 Varying the amount of the AMP-functionalized polymer added to silicone showed that both bactericidal activity and adhesion increase with AMP content. Incorporation of even small amounts (≤10%) of triblock polymers functionalized with the cell-adhesive peptide RGD increased surface coverage by healthy host cells, reflecting of the ability of these surfaces to integrate with healthy tissue.
10. Hydrogels
Immobilizing AMPs to polymeric hydrogels as side chains or cross-linkers combines the antimicrobial properties of AMPs with the biocompatibility, tissue-mimetic mechanics, and stimuli-responsive properties of polymeric hydrogels to yield advanced biomaterials well suited for preventing and treating bacterial infections.82,94,146-148 For example, conjugating thiol-terminated AMPs to alkyne-modified hydrogels imparted bactericidal properties, markedly reducing growth of both Gram-positive and Gram-negative bacterial species relative to the unmodified hydrogel.82 In another example, cross-linking antimicrobial polylysine stars with the natural, small-molecule cross-linker genipin formed a hydrogel that allowed the loading of vascular endothelial growth factor, thereby yielding an effective combinational wound treatment system.146
11. Concluding remarks
The conjugation of AMPs to biocompatible polymers can markedly influence the bactericidal activity, mammalian cell compatibility, and proteolytic stability of AMPs. Linear conjugation of AMPs to polymers towards reducing cytotoxic effects can simultaneously reduce bactericidal activity, particularly in cases when the polymer surrounds the AMP, thus shielding it from interactions with both mammalian and bacterial membranes. This tradeoff is perhaps most evident in the PEGylation of AMPs, where increasing PEG molecular weight reduces both toxicity and bactericidal activity. Enhancement in AMP activity is often seen in cases where multiple AMPs are conjugated to a single polymer or when supramolecular assembly of AMP-polymer conjugates into nanoparticles locally concentrates AMPs. There is still much to learn about optimizing the bactericidal and off-target effects of more complex non-linear conjugates and supramolecular assemblies; to this end, several elegant studies reviewed in this work provide a strong foundation for future investigations.
By expanding tool sets to tailor both AMP and polymer constituents, guided by many of the design rules presented here, we can realize potent antibiotic formulations that address the growing challenge of antimicrobial resistance. For example, advances in controlled polymerization have enabled the preparation of a range of intricate molecular architectures. Going forward, systematic studies on AMP-polymer conjugates with similar compositions, but with varying molecular architectures will further elucidate design rules for enhancing bacterial killing and minimizing mammalian-cell toxicity. Investigating potential changes in the bactericidal mechanism-of-action upon varying molecular architecture will also provide essential insight for engineering synergistic combinations of AMPs and polymers. Other promising future directions include the introduction of new peptide and polymer compositions, varying the orientation of AMPs with respect to the polymer, and imparting stimuli-responsive functionality to release or reveal AMPs at microbial infection sites. We hope that this review will serve to update researchers with diverse backgrounds, from infectious disease to polymer chemistry, of the advancements in AMP-functionalized polymers towards tackling the mounting challenges posed by antimicrobial-resistant bacterial pathogens.
Acknowledgements:
The authors gratefully acknowledge support from the National Institutes of Health (NIH R01AI150941, NIH R21AI139947), the Global Infectious Diseases Institute, the Ivy Biomedical Innovation Fund, and the University of Virginia. Additionally, V.P.G. acknowledges support from the UVA Biotechnology Training Program (NIH 1T32GM136615) and M.S.B. acknowledges support from a UVA Engineering Dean’s Scholar Fellowship.
References:
- 1.Lei J, Sun LC, Huang S, Zhu C, Li P, He J, Mackey V, Coy DH and He QY, Am. J. Transl. Res, 2019, 11, 3919–3931. [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar P, Kizhakkedathu JN and Straus SK, Biomolecules, 2018, 8, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Som A, Vemparala S, Ivanov I and Tew GN, Biopolym. - Pept. Sci. Sect, 2008, 90, 83–93. [DOI] [PubMed] [Google Scholar]
- 4.Defraine V, Schuermans J, Grymonprez B, Govers SK, Aertsen A, Fauvart M, Michiels J, Lavigne R and Briers Y, Antimicrob. Agents Chemother, 2016, 60, 3480–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lazzaro BP, Zasloff M and Rolff J, Science (80-. ), 2020, 368, eaau5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahlapuu M, Håkansson J, Ringstad L and Björn C, Front. Cell. Infect. Microbiol, 2016, 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohammed I, Said DG and Dua HS, Prog. Retin. Eye Res, 2017, 61, 1–22. [DOI] [PubMed] [Google Scholar]
- 8.Chen CH and Lu TK, Antibiotics, 2020, 9, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mookherjee N, Anderson MA, Haagsman HP and Davidson DJ, Nat. Rev. Drug Discov, 2020, 19, 311–332. [DOI] [PubMed] [Google Scholar]
- 10.Andersson DI, Hughes D and Kubicek-Sutherland JZ, Drug Resist. Updat, 2016, 26, 43–57. [DOI] [PubMed] [Google Scholar]
- 11.Sun H, Hong Y, Xi Y, Zou Y, Gao J and Du J, Biomacromolecules, 2018, 19, 1701–1720. [DOI] [PubMed] [Google Scholar]
- 12.Gauthier MA and Klok HA, Chem. Commun, 2008, 2591–2611. [DOI] [PubMed] [Google Scholar]
- 13.Shu JY, Panganiban B and Xu T, Annu. Rev. Phys. Chem, 2013, 64, 631–657. [DOI] [PubMed] [Google Scholar]
- 14.Ekladious I, Colson YL and Grinstaff MW, Nat. Rev. Drug Discov, 2019, 18, 273–294. [DOI] [PubMed] [Google Scholar]
- 15.Chen C, Ng DYW and Weil T, Prog. Polym. Sci, 2020, 105, 1–40. [Google Scholar]
- 16.Messina MS, Messina KMM, Bhattacharya A, Montgomery HR and Maynard HD, Prog. Polym. Sci, 2020, 100, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mukhopadhyay S, Bharath Prasad AS, Mehta CH and Nayak UY, World J. Microbiol. Biotechnol, 2020, 36, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Bai H, Yang Y, Yoon J, Wang S and Zhang X, Adv. Mater, 2019, 31. [DOI] [PubMed] [Google Scholar]
- 19.Martin-Serrano Á, Gómez R, Ortega P and La Mata FJD, Pharmaceutics, 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faya M, Kalhapure RS, Kumalo HM, Waddad AY, Omolo C and Govender T, J. Drug Deliv. Sci. Technol, 2018, 44, 153–171. [Google Scholar]
- 21.Li W, Separovic F, O’Brien-Simpson NM and Wade JD, Chem. Soc. Rev, 2021, 50, 4932–4973. [DOI] [PubMed] [Google Scholar]
- 22.Li P, Zhou C, Rayatpisheh S, Ye K, Poon YF, Hammond PT, Duan H and Chan-Park MB, Adv. Mater, 2012, 24, 4130–4137. [DOI] [PubMed] [Google Scholar]
- 23.Su Y, Tian L, Yu M, Gao Q, Wang D, Xi Y, Yang P, Lei B, Ma PX and Li P, Polym. Chem, 2017, 8, 3788–3800. [Google Scholar]
- 24.Lu C, Quan G, Su M, Nimmagadda A, Chen W, Pan M, Teng P, Yu F, Liu X, Jiang L, Du W, Hu W, Yao F, Pan X, Wu C, Liu D and Cai J, Adv. Ther, 2019, 2, 1–11. [Google Scholar]
- 25.Chen YF, Da Lai Y, Chang CH, Tsai YC, Tang CC and Jan JS, Nanoscale, 2019, 11, 11696–11708. [DOI] [PubMed] [Google Scholar]
- 26.Lam SJ, O’Brien-Simpson NM, Pantarat N, Sulistio A, Wong EHH, Chen YY, Lenzo JC, Holden JA, Blencowe A, Reynolds EC and Qiao GG, Nat. Microbiol, 2016, 1, 1–11. [DOI] [PubMed] [Google Scholar]
- 27.Shirbin SJ, Insua I, Holden JA, Lenzo JC, Reynolds EC, O’Brien-Simpson NM and Qiao GG, Adv. Healthc. Mater, 2018, 7, 1800627. [DOI] [PubMed] [Google Scholar]
- 28.Pranantyo D, Xu LQ, Kang ET, Mya MK and Chan-Park MB, Biomacromolecules, 2016, 17, 4037–4044. [DOI] [PubMed] [Google Scholar]
- 29.Silhavy TJ, Kahne D and Walker S, Cold Spring Harb. Perspect. Biol, 2010, 2, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strober W, Curr. Protoc. Immunol, 2015, 111, A3.B.1–A3.B.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosmann T, J. lmmunological Methods, 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- 32.Repetto G, Del Peso A and Zurita JL, Nat. Protoc, 2008, 3, 1125–1131. [DOI] [PubMed] [Google Scholar]
- 33.Riss TL, Moravec RA, Niles AL, Duellman S, Benink HA, Worzella TJ and Minor L, Cell Viability Assays, Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2013. [PubMed] [Google Scholar]
- 34.Tominaga H, Ishiyama M, Ohseto F, Sasamoto K, Hamamoto T, Suzuki K and Watanabe M, Anal. Commun, 1999, 36, 47–50. [Google Scholar]
- 35.Ganewatta MS and Tang C, Polymer (Guildf)., 2015, 63, A1–A29. [Google Scholar]
- 36.Ju X, Chen J, Zhou M, Zhu M, Li Z, Gao S, Ou J, Xu D, Wu M, Jiang S, Hu Y, Tian Y and Niu Z, ACS Appl. Mater. Interfaces, 2020, 12, 13731–13738. [DOI] [PubMed] [Google Scholar]
- 37.Kumar P, Takayesu A, Abbasi U, Kalathottukaren MT, Abbina S, Kizhakkedathu JN and Straus SK, ACS Appl. Mater. Interfaces, 2017, 9, 37575–37586. [DOI] [PubMed] [Google Scholar]
- 38.Hartlieb M, Catrouillet S, Kuroki A, Sanchez-Cano C, Peltier R and Perrier S, Chem. Sci, 2019, 10, 5476–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaur N, Dilawari R, Kaur A, Sahni G and Rishi P, Sci. Rep, 2020, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benincasa M, Zahariev S, Pelillo C, Milan A, Gennaro R and Scocchi M, Eur. J. Med. Chem, 2015, 95, 210–219. [DOI] [PubMed] [Google Scholar]
- 41.Brogden KA, Nat. Rev. Microbiol, 2005, 3, 238–250. [DOI] [PubMed] [Google Scholar]
- 42.Li J, Koh JJ, Liu S, Lakshminarayanan R, Verma CS and Beuerman RW, Front. Neurosci, 2017, 11, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haney EF, Straus SK and Hancock REW, Front. Chem, 2019, 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guiotto A, Pozzobon M, Canevari M, Manganelli R, Scarin M and Veronese FM, Farmaco, 2003, 58, 45–50. [DOI] [PubMed] [Google Scholar]
- 45.Hyldgaard M, Mygind T, Vad BS, Stenvang M, Otzen DE and Meyer RL, Appl. Environ. Microbiol, 2014, 80, 7758–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sahariah P, Sørensen KK, Hjálmarsdóttir MA, Sigurjónsson ÓE, Jensen KJ, Másson M and Thygesen MB, Chem. Commun, 2015, 51, 11611–11614. [DOI] [PubMed] [Google Scholar]
- 47.Blin T, Purohit V, Leprince J, Jouenne T and Glinel K, Biomacromolecules, 2011, 12, 1259–1264. [DOI] [PubMed] [Google Scholar]
- 48.Breukink E, Wiedemann I, Van Kraaij C, Kuipers OP, Sahl HG and De Kruijff B, Science (80-. ), 1999, 286, 2361–2364. [DOI] [PubMed] [Google Scholar]
- 49.Da Silva IM, Boelter JF, Da Silveira NP and Brandelli A, J. Nanoparticle Res, , DOI: 10.1007/s11051-014-2479-y. [DOI] [Google Scholar]
- 50.Biswaro LS, Sousa M. G. d. C., Rezende TMB, Dias SC and Franco OL, Front. Microbiol, 2018, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lequeux I, Ducasse E, Jouenne T and Thebault P, Eur. Polym. J, 2014, 51, 182–190. [Google Scholar]
- 52.Hou Z, Shankar YV, Liu Y, Ding F, Subramanion JL, Ravikumar V, Zamudio-Vázquez R, Keogh D, Lim H, Tay MYF, Bhattacharjya S, Rice SA, Shi J, Duan H, Liu XW, Mu Y, Tan NS, Tam KC, Pethe K and Chan-Park MB, ACS Appl. Mater. Interfaces, 2017, 9, 38288–38303. [DOI] [PubMed] [Google Scholar]
- 53.Zhao J, Dong Z, Cui H, Jin H and Wang C, ACS Appl. Mater. Interfaces, 2018, 10, 42058–42067. [DOI] [PubMed] [Google Scholar]
- 54.Lu C, Wen T, Zheng M, Liu D, Quan G, Pan X and Wu C, Pharmaceutics, , DOI: 10.3390/pharmaceutics12010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thappeta KRV, Vikhe YS, Yong AMH, Chan-Park MB and Kline KA, ACS Infect. Dis, 2020, 6, 1228–1237. [DOI] [PubMed] [Google Scholar]
- 56.Gao J, Wang M, Wang F and Du J, Biomacromolecules, 2016, 17, 2080–2086. [DOI] [PubMed] [Google Scholar]
- 57.Costanza F, Padhee S, Wu H, Wang Y, Revenis J, Cao C, Li Q and Cai J, RSC Adv., 2014, 4, 2089–2095. [Google Scholar]
- 58.Bin Zhen J, Kang PW, Zhao MH and Yang KW, Bioconjug. Chem, 2020, 31, 51–63. [DOI] [PubMed] [Google Scholar]
- 59.Xi Y, Song T, Tang S, Wang N and Du J, Biomacromolecules, 2016, 17, 3922–3930. [DOI] [PubMed] [Google Scholar]
- 60.Liu B, Yao T, Ren L, Zhao Y and Yuan X, Colloids Surfaces B Biointerfaces, 2018, 172, 330–337. [DOI] [PubMed] [Google Scholar]
- 61.Sun H, Hong Y, Xi Y, Zou Y, Gao J and Du J, Biomacromolecules, 2018, 19, 1701–1720. [DOI] [PubMed] [Google Scholar]
- 62.Bin Qi G, Zhang D, Liu FH, Qiao ZY and Wang H, Adv. Mater, 2017, 29, 1–10. [DOI] [PubMed] [Google Scholar]
- 63.Su YR, Yu SH, Chao AC, Wu JY, Lin YF, Lu KY and Mi FL, Colloids Surfaces A Physicochem. Eng. Asp, 2016, 494, 9–20. [Google Scholar]
- 64.Joralemon MJ, McRae S and Emrick T, Chem. Commun, 2010, 46, 1377–1393. [DOI] [PubMed] [Google Scholar]
- 65.Yang Q and Lai SK, Wiley Interdiscip. Rev. Nanomedicine Nanobiotechnology, 2015, 7, 655–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thanh T, Thi H, Pilkington EH, Nguyen DH and Lee JS, Polymers (Basel). [Google Scholar]
- 67.Mellegård H, Strand SP, Christensen BE, Granum PE and Hardy SP, Int. J. Food Microbiol, 2011, 148, 48–54. [DOI] [PubMed] [Google Scholar]
- 68.Yaghoubi A, Ghojazadeh M, Abolhasani S, Alikhah H and Khaki-Khatibi F, J. Cardiovasc. Thorac. Res, 2015, 7, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sultankulov B, Berillo D, Sultankulova K, Tokay T and Saparov A, Biomolecules, 2019, 9, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zielinski BA and Aebischer P, Biomaterials, 1994, 15, 1049–1056. [DOI] [PubMed] [Google Scholar]
- 71.Bernkop-Schnürch A and Dünnhaupt S, Eur. J. Pharm. Biopharm, 2012, 81, 463–469. [DOI] [PubMed] [Google Scholar]
- 72.Chen Y, Yu L, Zhang B, Feng W, Xu M, Gao L, Liu N, Wang Q, Huang X, Li P and Huang W, Biomacromolecules, 2019, 20, 2230–2240. [DOI] [PubMed] [Google Scholar]
- 73.Labena A, Kabel KI and Farag RK, Mater. Sci. Eng. C, 2016, 58, 1150–1159. [DOI] [PubMed] [Google Scholar]
- 74.Danial M, Tran CMN, Jolliffe KA and Perrier S, J. Am. Chem. Soc, 2014, 136, 8018–8026. [DOI] [PubMed] [Google Scholar]
- 75.Song Q, Yang J, Hall SCL, Gurnani P and Perrier S, ACS Macro Lett., 2019, 8, 1347–1352. [DOI] [PubMed] [Google Scholar]
- 76.Feng C, Li Y, Yang D, Hu J, Zhang X and Huang X, Chem. Soc. Rev, 2011, 40, 1282–1295. [DOI] [PubMed] [Google Scholar]
- 77.Arslan M, Acik G and Tasdelen MA, Polym. Chem, 2019, 10, 3806–3821. [Google Scholar]
- 78.Yu K, Lo JCY, Mei Y, Haney EF, Siren E, Kalathottukaren MT, Hancock REW, Lange D and Kizhakkedathu JN, ACS Appl. Mater. Interfaces, 2015, 7, 28591–28605. [DOI] [PubMed] [Google Scholar]
- 79.Lowe AB, Polym. Chem, 2014, 5, 4820–4870. [Google Scholar]
- 80.Hoyle CE, Lowe AB and Bowman CN, Chem. Soc. Rev, 2010, 39, 1355–1387. [DOI] [PubMed] [Google Scholar]
- 81.Pranantyo D, Xu LQ, Kang ET and Chan-Park MB, Biomacromolecules, 2018, 19, 2156–2165. [DOI] [PubMed] [Google Scholar]
- 82.Cai XY, Li JZ, Li NN, Chen JC, Kang ET and Xu LQ, Biomater. Sci, 2016, 4, 1663–1672. [DOI] [PubMed] [Google Scholar]
- 83.Panja S, Bharti R, Dey G, Lynd NA and Chattopadhyay S, ACS Appl. Mater. Interfaces, 2019, 11, 33599–33611. [DOI] [PubMed] [Google Scholar]
- 84.Sechi S and Chait BT, Anal. Chem, 1998, 70, 5150–5158. [DOI] [PubMed] [Google Scholar]
- 85.Kumar P, Shenoi RA, Lai BFL, Nguyen M, Kizhakkedathu JN and Straus SK, Biomacromolecules, 2015, 16, 913–923. [DOI] [PubMed] [Google Scholar]
- 86.Petrin THC, Fadel V, Martins DB, Dias SA, Cruz A, Sergio LM, Arcisio-Miranda M, Castanho MARB and Dos Santos Cabrera MP, Biomacromolecules, 2019, 20, 2743–2753. [DOI] [PubMed] [Google Scholar]
- 87.Barbosa M, Vale N, Costa FMTA, Martins MCL and Gomes P, Carbohydr. Polym, 2017, 165, 384–393. [DOI] [PubMed] [Google Scholar]
- 88.Liu Z, Deshazer H, Rice AJ, Chen K, Zhou C and Kallenbach NR, J. Med. Chem, 2006, 49, 3436–3439. [DOI] [PubMed] [Google Scholar]
- 89.Pranantyo D, Xu LQ, Hou Z, Kang ET and Chan-Park MB, Polym. Chem, 2017, 8, 3364–3373. [Google Scholar]
- 90.Liang L and Astruc D, Coord. Chem. Rev, 2011, 255, 2933–2945. [Google Scholar]
- 91.Amirah MN, Afiza AS, Faizal WIW, Nurliyana MH and Laili S, J. Environ. Pollut. Hum. Heal, 2013, 1, 1–5. [Google Scholar]
- 92.Bräse S, Gil C, Knepper K and Zimmermann V, Angew. Chemie - Int. Ed, 2005, 44, 5188–5240. [DOI] [PubMed] [Google Scholar]
- 93.Song A, Rane AA and Christman KL, Acta Biomater., 2012, 8, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin Z, Wu T, Wang W, Li B, Wang M, Chen L, Xia H and Zhang T, Int. J. Biol. Macromol, 2019, 140, 330–342. [DOI] [PubMed] [Google Scholar]
- 95.Zhu J, Han H, Li F, Wang X, Yu J, Qin X and Wu D, Chem. Mater, 2019, 31, 4436–4450. [Google Scholar]
- 96.Kricheldorf HR, Angew. Chemie - Int. Ed, 2006, 45, 5752–5784. [DOI] [PubMed] [Google Scholar]
- 97.Hadjichristidis N, Iatrou H, Pitsikalis M and Sakellariou G, Chem. Rev, 2009, 109, 5528–5578. [DOI] [PubMed] [Google Scholar]
- 98.Deming TJ, Chem. Rev, 2016, 116, 786–808. [DOI] [PubMed] [Google Scholar]
- 99.Rasines Mazo A, Allison-Logan S, Karimi F, Chan NJA, Qiu W, Duan W, O’Brien-Simpson NM and Qiao GG, Chem. Soc. Rev, 2020, 49, 4737–4834. [DOI] [PubMed] [Google Scholar]
- 100.Dong Z, Wang Y, Wang C, Meng H, Li Y and Wang C, Adv. Healthc. Mater, 2020, 9, 1–9. [DOI] [PubMed] [Google Scholar]
- 101.Semple JE, Sullivan B and Sill KN, Synth. Commun, 2017, 47, 53–61. [Google Scholar]
- 102.Environmental Protection Agency US, Toxicological Review of Phosgene, 2005. [Google Scholar]
- 103.Lam SJ, Wong EHH, O’Brien-Simpson NM, Pantarat N, Blencowe A, Reynolds EC and Qiao GG, ACS Appl. Mater. Interfaces, 2016, 8, 33446–33456. [DOI] [PubMed] [Google Scholar]
- 104.Luppi L, Babut T, Petit E, Rolland M, Quemener D, Soussan L, Moradi MA and Semsarilar M, Polym. Chem, 2019, 10, 336–344. [Google Scholar]
- 105.Becker ML, Liu J and Wooley KL, Biomacromolecules, 2005, 6, 220–228. [DOI] [PubMed] [Google Scholar]
- 106.Perrier S, Macromolecules, 2017, 50, 7433–7447. [Google Scholar]
- 107.Corrigan N, Jung K, Moad G, Hawker CJ, Matyjaszewski K and Boyer C, Prog. Polym. Sci, 2020, 111, 101311. [Google Scholar]
- 108.Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, De Alaniz JR, Fors BP and Hawker CJ, J Am Chem Soc, 2014, 134, 16096–16101. [DOI] [PubMed] [Google Scholar]
- 109.Convertine AJ, Ayres N, Scales CW, Lowe AB and Mccormick CL, Biomacromolecules, 2004, 5, 1177–1180. [DOI] [PubMed] [Google Scholar]
- 110.Sun H, Yang L, Thompson MP, Schara S, Cao W, Choi W, Hu Z, Zang N, Tan W and Gianneschi NC, Bioconjug. Chem, 2019, 30, 1889–1904. [DOI] [PubMed] [Google Scholar]
- 111.Bin Zhen J, Zhao MH, Ge Y, Liu Y, Xu LW, Chen C, Gong YK and Yang KW, Biomater. Sci, 2019, 7, 4142–4152. [DOI] [PubMed] [Google Scholar]
- 112.Gao Q, Yu M, Su Y, Xie M, Zhao X, Li P and Ma PX, Acta Biomater., 2017, 51, 112–124. [DOI] [PubMed] [Google Scholar]
- 113.Wong EHH, Khin MM, Ravikumar V, Si Z, Rice SA and Chan-Park MB, Biomacromolecules, 2016, 17, 1170–1178. [DOI] [PubMed] [Google Scholar]
- 114.Blencowe A, Tan JF, Goh TK and Qiao GG, Polymer (Guildf)., 2009, 50, 5–32. [Google Scholar]
- 115.Imura Y, Nishida M and Matsuzaki K, Biochim. Biophys. Acta - Biomembr, 2007, 1768, 2578–2585. [DOI] [PubMed] [Google Scholar]
- 116.Imura Y, Nishida M, Ogawa Y, Takakura Y and Matsuzaki K, Biochim. Biophys. Acta - Biomembr, 2007, 1768, 1160–1169. [DOI] [PubMed] [Google Scholar]
- 117.Morris CJ, Beck K, Fox MA, Ulaeto D, Clark GC and Gumbleton M, Antimicrob. Agents Chemother, 2012, 56, 3298–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang G, Han B, Lin X, Wu X and Yan H, J. Biochem, 2008, 144, 781–788. [DOI] [PubMed] [Google Scholar]
- 119.Liu Z, Young AW, Hu P, Rice AJ, Zhou C, Zhang Y and Kallenbach NR, ChemBioChem, 2007, 8, 2063–2065. [DOI] [PubMed] [Google Scholar]
- 120.Liu SP, Zhou L, Lakshminarayanan R and Beuerman RW, Int. J. Pept. Res. Ther, 2010, 16, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chamorro C, Boerman MA, Arnusch CJ, Breukink E and Pieters RJ, Biochim. Biophys. Acta - Biomembr, 2012, 1818, 2171–2174. [DOI] [PubMed] [Google Scholar]
- 122.Gillies ER and Fréchet JMJ, Drug Discov. Today, 2005, 10, 35–43. [DOI] [PubMed] [Google Scholar]
- 123.Mintzer MA and Grinstaff MW, Chem. Soc. Rev, 2011, 40, 173–190. [DOI] [PubMed] [Google Scholar]
- 124.Kabel KI, Labena A, Keshawy M and Hozzein WN, Materials (Basel)., 2020, 13, 2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Demircan D and Zhang B, Carbohydr. Polym, 2017, 157, 1913–1921. [DOI] [PubMed] [Google Scholar]
- 126.Gao Y, Wang J, Hu D, Deng Y, Chen T, Jin Q and Ji J, Macromol. Rapid Commun, 2019, 40, 1–6. [DOI] [PubMed] [Google Scholar]
- 127.Joshi PR, McGuire J and Neff JA, J. Biomed. Mater. Res. - Part B Appl. Biomater, 2009, 91, 128–134. [DOI] [PubMed] [Google Scholar]
- 128.Ferguson GP and Booth IR, J. Bacteriol, 1998, 180, 4314–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Forman HJ, Zhang H and Rinna A, Mol. Aspects Med, 2009, 30, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Radovic-Moreno AF, Lu TK, Puscasu VA, Yoon CJ, Langer R and Farokhzad OC, ACS Nano, 2012, 6, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hu D, Deng Y, Jia F, Jin Q and Ji J, ACS Nano, 2020, 14, 347–359. [DOI] [PubMed] [Google Scholar]
- 132.Wang Y, Li S, Liu L and Feng L, ACS Appl. Bio Mater, 2018, 1, 27–32. [Google Scholar]
- 133.Park SC, Kim YM, Lee JK, Kim NH, Kim EJ, Heo H, Lee MY, Lee JR and Jang MK, J. Control. Release, 2017, 256, 46–55. [DOI] [PubMed] [Google Scholar]
- 134.Garton M, Nim S, Stone TA, Wang KE, Deber CM and Kim PM, Proc. Natl. Acad. Sci, 2018, 115, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pranantyo D, Kang E-T and Chan-Park MB, Biomater. Sci, 2021, 9, 1627–1638. [DOI] [PubMed] [Google Scholar]
- 136.Pu Y, Khin MM and Chan-Park MB, Mater. Lett, 2018, 223, 69–72. [Google Scholar]
- 137.Lee J, Oh ET, Lee H, Kim J, Kim HG, Park HJ and Kim C, Bioconjug. Chem, 2020, 31, 43–50. [DOI] [PubMed] [Google Scholar]
- 138.Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM and Ghadiri MR, Nature, 2001, 412, 452–455. [DOI] [PubMed] [Google Scholar]
- 139.Wiradharma N, Khoe U, Hauser CAE, Seow SV, Zhang S and Yang YY, Biomaterials, 2011, 32, 2204–2212. [DOI] [PubMed] [Google Scholar]
- 140.Hadjesfandiari N, Yu K, Mei Y and Kizhakkedathu JN, J. Mater. Chem. B, 2014, 2, 4968–4978. [DOI] [PubMed] [Google Scholar]
- 141.Salwiczek M, Qu Y, Gardiner J, Strugnell RA, Lithgow T, McLean KM and Thissen H, Trends Biotechnol., 2014, 32, 82–90. [DOI] [PubMed] [Google Scholar]
- 142.Gao G, Lange D, Hilpert K, Kindrachuk J, Zou Y, Cheng JTJ, Kazemzadeh-Narbat M, Yu K, Wang R, Straus SK, Brooks DE, Chew BH, Hancock REW and Kizhakkedathu JN, Biomaterials, 2011, 32, 3899–3909. [DOI] [PubMed] [Google Scholar]
- 143.Hilpert K, Elliott M, Jenssen H, Kindrachuk J, Fjell CD, Körner J, Winkler DFH, Weaver LL, Henklein P, Ulrich AS, Chiang SHY, Farmer SW, Pante N, Volkmer R and Hancock REW, Chem. Biol, 2009, 16, 58–69. [DOI] [PubMed] [Google Scholar]
- 144.Gao G, Yu K, Kindrachuk J, Brooks DE, Hancock REW and Kizhakkedathu JN, Biomacromolecules, 2011, 12, 3715–3727. [DOI] [PubMed] [Google Scholar]
- 145.Muszanska AK, Rochford ETJ, Gruszka A, Bastian AA, Busscher HJ, Norde W, Van Der Mei HC and Herrmann A, Biomacromolecules, 2014, 15, 2019–2026. [DOI] [PubMed] [Google Scholar]
- 146.Hsu FM, Hu MH, Jiang YS, Lin BY, Hu JJ and Jan JS, Mater. Sci. Eng. C, 2020, 112, 110923. [DOI] [PubMed] [Google Scholar]
- 147.Annabi N, Rana D, Shirzaei Sani E, Portillo-Lara R, Gifford JL, Fares MM, Mithieux SM and Weiss AS, Biomaterials, 2017, 139, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xie Z, Aphale NV, Kadapure TD, Wadajkar AS, Orr S, Gyawali D, Qian G, Nguyen KT and Yang J, J. Biomed. Mater. Res. - Part A, 2015, 103, 3907–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]