

Abstract
Cystic Fibrosis (CF) is a genetic disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. The F508del and G542X are the most common mutations found in US patients, accounting for 86.4% and 4.6% of all mutations, respectively. The F508del causes deletion of the phenylalanine residue at position 508 and is associated with impaired CFTR protein folding. The G542X is a nonsense mutation that introduces a stop codon into the mRNA, thus preventing normal CFTR protein synthesis. Here, we describe the generation of CFTRF508del / F508del and CFTRG542X / G542X lambs using CRISPR/Cas9 and somatic cell nuclear transfer (SCNT). First, we introduced either F508del or G542X mutations into sheep fetal fibroblasts that were subsequently used as nuclear donors for SCNT. The newborn CF lambs develop pathology similar to CFTR −/− sheep and CF patients. Moreover, tracheal epithelial cells from the CFTRF508del / F508del lambs responded to a human CFTR (hCFTR) potentiator and correctors, and those from CFTRG542X / G542X lambs showed modest restoration of CFTR function following inhibition of nonsense‐mediated decay (NMD) and aminoglycoside antibiotic treatments. Thus, the phenotype and electrophysiology of these novel models represent an important advance for testing new CF therapeutics and gene therapy to improve the health of patients with this life‐limiting disorder.
Keywords: animal models, CFTR, cloning, CRISPR/Cas9, human drugs
1. INTRODUCTION
Cystic Fibrosis (CF) is an autosomal recessive genetic disease that affects over 30,000 people in the United States and results from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that disrupt the CFTR synthesis, folding, or function. The CFTR protein is a well‐characterized cAMP‐regulated ATP‐binding cassette (ABC) transporter responsible for regulating anion transport, primarily chloride ions. 1 The CFTR protein is expressed in epithelial cells of multiple organs such as the airways, pancreas, intestine, gallbladder, and male genital ducts. Important features of human CF pathology are early damage to the pancreas, which initiates when pancreatic acini become functional in utero, intestinal obstruction, and persistent lung inflammation associated with infection that causes tissue damage and fibrosis (Reviewed in 2 ). One of the impediments in advancing the understanding of disease mechanisms is the limited ability of most CF animal models to recapitulate some aspects of the disease phenotype observed in humans (reviewed in 3 , 4 ). Of particular importance is the etiology of early lung pathology, which is likely triggered by functional inactivation of CFTR protein early in life. 5
There are over 2000 predicted CFTR mutations that are divided into six classes, depending on how they affect the protein. 6 The F508del mutation is the most frequent in human patients in the United States, followed by G542X. The F508del is identified in 84.7% of patients on at least one allele, whereas the G542X is present at a frequency of 4.6%. 7 The F508del mutation is characterized by loss of a phenylalanine residue at the position 508 (F508) of the CFTR protein and is classified as a Class II alteration since it causes defective processing/trafficking. F508del protein is misfolded and retained in the endoplasmic reticulum (ER) where it is further degraded by proteolytic cleavage. 8 As the most common human CFTR mutation, the F508del CF sheep was considered necessary as a valid ovine model for evaluating lung disease progression in sheep and the development of future therapeutic treatments. Though substantial progress has been made with small molecule approaches to correct this mutant phenotype 9 , 10 (reviewed in 11 ), some patients are poorly responsive to these treatments.
Although effective treatments are now available for CF patients with at least one F508del allele, there is still a great need for therapeutics that target CFTR nonsense mutations (e.g., G542X). These nonsense mutations are Class I alterations, which lack full length of the CFTR protein production and are often targets for nonsense‐mediated decay (NMD) of RNA. Nearly 10% of CF patients carry Class I mutations, the most common being G542X, a G to T substitution in the first nucleotide of the glycine codon (GGA) at position 542 of the protein. This mutation produces a premature termination codon (PTC), which triggers the surveillance pathway and causes mRNA NMD, leading to the lack of CFTR protein production. 12
To the best of our knowledge, no large animal model carrying the G542X mutation has been produced to study in‐depth the nonsense mutations that affect CF patients. Therefore, to better elucidate the pathophysiologic effects of the F508del and G542X mutations, we produced CFTRF508del / F508del and CFTRG542X / G542X sheep using CRISPR/Cas9 ribonucleoprotein (RNP) along with donor single‐stranded oligonucleotide (ssODN) to introduce either the F508del or G542X mutations in the CFTR gene of sheep fetal fibroblasts followed by somatic cell nuclear transfer (SCNT). Here, our goal was to investigate whether F508del and G542X CF sheep models exhibit the same phenotypes as were observed in the CFTR −/− sheep we generated previously. 5 Also it was important to establish the presence of residual CFTR channel activity in F508del CF sheep. We evaluated the effect of CFTR correctors (VX‐661 (Tezacaftor), 13 VX‐809 (Lumacaftor), 14 and VX‐445 (Elexacaftor) 15 ) and potentiator (VX‐770 (Ivacaftor) 16 ) on the restoration of the CFTR channel function in the F508del CF sheep tracheal epithelial (STE) cells by electrophysiology. We also measured the impact of aminoglycoside antibiotics (gentamicin (G418)) 17 and the NMD inhibitor (SMG1‐i (2‐chloro‐N,N‐diethyl‐5‐((4‐(2‐(4‐(3‐methylureido)phenyl)pyridin‐4‐yl)pyrimidin‐2‐yl)amino)benzenesulfonamide)) 18 in G542X CF STE cells. These novel F508del and G542X CF sheep models could become a critical platform for studying the effects of CFTR mutations during fetal development, the mechanisms of onset of lung inflammation, and also provide support for new therapeutic strategies including prenatal therapy toward a genetic cure of the disease.
2. METHODS
2.1. Animals
Domestic Romney sheep (Ovis aries) were used in this study for isolation of sheep fetal fibroblasts (SFFs), and Suffolk and Rambouillet ewes were used as recipients for embryo transfers. All animal studies were approved and monitored by the Institutional Animal Care and Use Committee (IACUC) at Utah State University (IACUC protocol #10089) and conformed to the National Institute of Health guidelines. CFTRF508del / F508del and CFTRG542X / G542X cloned lambs were generated by SCNT using genetically modified SFFs as nucleus donor cells.
2.2. Sheep fetal fibroblasts
Sheep fetal fibroblasts were isolated from male day 45 Romney fetus as previously described and cultured in Dulbecco's modified Eagle's medium ((DMEM) high glucose; HyClone) supplemented with 15% fetal bovine serum (FBS; HyClone) and 100 U/mL penicillin–streptomycin (Life Technologies) at 38.5℃ in an atmosphere of 5% CO2 in air. 19
2.3. Design of guide RNAs/Cas9 and single‐strand oligonucleotides
Based on the sequence results for exons 11 and 12 of the ovine CFTR gene, we designed gRNAs targeting the regions flanking the phenylalanine (F508) site in exon 11 and the Glycine (G542) site in exon 12 using the Benchling software (https://benchling.com/academic) (Figure 1). The gRNA_1 targets the F508 site with the intended “CTT” deletion located within the first and third nucleotide at 3’ end. The gRNA_2 targets the G542 DNA locus with the target nucleotide (Guanine) located within five positions upstream to the CRISPR/Cas9 double‐strand break site. The gRNAs were synthesized by Synthego with chemical modifications (2’‐O‐methyl 3’‐phosphorothioate modifications in the first and last three nucleotides). The TrueCut™ Cas9 V2 protein was purchased from Thermo Fisher Scientific. To introduce the point mutations, we also designed ssODNs carrying either the G542X or F508del mutations as gene‐editing donor oligos with 50 bp homologous arms flanking the mutation sites. The ssODN_1 (5’‐T*G*CTCTCAGTATTCCTGGATCATGCCTGGAACCATTAAAGATAACATCATTGGTGTTTCCTATGATGAATATAGATATAGGAGTGTCATCAAAGCATG*C*C‐3’) carried the F508del mutation (a deletion of three consecutive nucleotides, “CTT”) and the ssODN_2 (5’‐T*T*TGATAATAGGACATCTCCAAGTTTTCAGAGAAAGACAACATAGTTCTTTGAGAAGGTGGAATCACATTGAGTGGAGGTCAGCGAGCAAGAATTTCTT*T*A‐3’) contained the G542X mutation (a “G” nucleotide was replaced with a “T” (bold) leading to the generation of a stop codon, “TGA”). Both ssODNs were synthesized by Integrated DNA Technologies (IDT) containing two phosphorothioate modifications (*) at both 5’ and 3’ ends.
FIGURE 1.
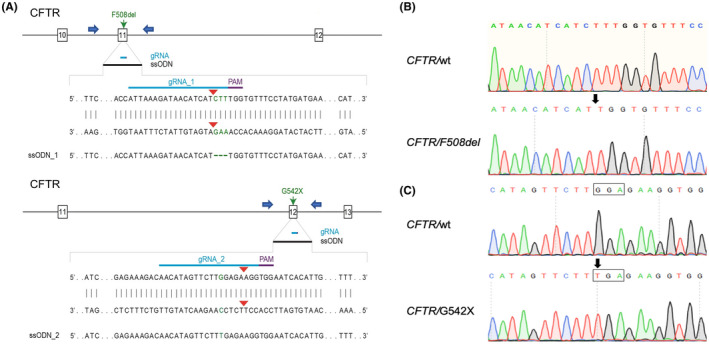
Generation of CFTRF508del / F508del and CFTRG542X / G542X sheep fetal fibroblast cells and lambs. (A) Schematic diagram of CRISPR/Cas9 target sites in exon 11 and 12 for introduction of the F508del and G542X mutations using the ssODN_1 and 2, respectively. The blue arrows represent the primers used to amplify the exon 11 and 12. (B) Representative sequence analysis for cell colonies and lambs containing F508del, similar to the CF patients, the “CTT” nucleotides are deleted between the codons ATC and TTT. Sequencing results indicate that four isolated cell colonies and two cloned lambs contained homozygous F508del mutations. (C) Representative sequence analysis for cell colonies and lambs containing G542X mutation. Similar to the G542X CF patients, Glycine‐encoded codon “GGA” is mutated with a single nucleotide replacement, “G” to “T,” leading to the generation of stop codon, “TGA.” Sequencing results demonstrate that all nine SFF cell colonies and three cloned fetus/lambs present the same homozygous G542X mutation. The arrow represents the point mutation site and the codon sequences are framed
2.4. Transfection and mutation efficiency detection
Nucleofection was performed using an Amaxa Nucleofector System (Program EH‐100, Lonza). Prior to transfection, 3 µl of 100 µM gRNA was incubated with 2 µl of 5 µg/µl Cas9 protein for 10 min at room temperature to form a ribonucleoprotein (RNP) complex. The RNP was then incubated with 2 µl of 200 µM ssODN for 5 min and transfected into 2 × 105 SFFs, in Passage 3, in a 100 µl nucleocuvette system. Two days post‐transfection, cells were harvested for genomic DNA isolation using the DNeasy Blood & Tissue kit (Cat. 69504, Qiagen) following the manufacturer's protocol. Each of the targeted genomic loci was PCR amplified by Phusion High‐fidelity DNA polymerase (Thermo Fisher Scientific) using the genomic DNA isolated from transfected SFFs as templates and the primers (F: 5’‐TGAACTCAGCACCCCATCTCTG‐3’; R: 5’‐TGCAGGCTTCTTATAGCAGGGG‐3’) for F508 locus, and the primers (F: 5’‐GCTGAGATGT GGTGTTCACA‐3’; R: 5’‐GCATCTTCTCTTCC CTGTGC‐3’) for G542 locus. Both PCR products were analyzed by Sanger sequencing before cell colony isolation.
2.5. Isolation of F508del and G542X cell colonies
Three days after gRNA/Cas9/ssODN transfection, cells were subjected to single‐cell colony isolation by serial dilution in 96‐well plates. After 7 days culture in a 96‐well plate, each single cell‐derived colony was allowed to grow to near confluence and then transferred into individual wells of 24‐well plates for 3 more days of culture. Upon reaching confluence, three quarters of the cells were cryopreserved, and one quarter used for DNA extraction. PCR and Sanger sequencing were then performed to identify wild‐type (WT), knock‐out (KO), and knock‐in (KI) colonies.
2.6. Somatic cell nuclear transfer
Sheep SCNT was performed as described by Yang et al. for goats 20 with the minor alteration that the synthetic oviductal fluid (SOF) embryo culture medium was used instead of G1. CFTRF508del / F508del and CFTRG542X / G542X fetal fibroblast cells were grown to 80%–90% confluence and used as nuclear donor cells for SCNT after 24 h of serum starvation (in 0.5% FBS‐containing DMEM medium). The reconstructed embryos were cultured in SOF medium for 10–12 h and then transferred into estrus synchronized recipients as described elsewhere. 19 , 21
2.7. Identification of CFTRF508del/F508del and CFTRG542X/G542X fetuses/lambs
Genomic DNA was extracted from muscle samples collected during the necropsy. Using the set of primers described above, PCR and Sanger sequencing analysis were used for confirmation of the mutations present in the lambs.
2.8. Off‐target analysis
We conducted a BLAST search on the sheep nucleotide sequence database with two gRNA sequences as the queries to find the genomic sequences with the highest homology using Crispor online software (Version 4.97). We selected potential off‐target sites with the highest sequence homology (scores) to each seed sequence. Specific PCR primers were designed to amplify DNA fragments of approximately 500 bp spanning each OT locus. We used PCR and Sanger sequencing for off‐target analysis of the genomic DNA isolated from cloned CFTRF508del / F508del and CFTRG542X / G542X sheep.
2.9. Histopathologic analysis
Necropsy was performed on all lambs that either died or were euthanized within 24 h of birth. The following tissue samples were collected and fixed in 10% neutral buffered formalin for histology: trachea, lung, thyroid glands, adrenal glands, abomasum, intestinal tract, pancreas, liver, kidney, urinary bladder, spleen, heart, testis, and vas deferens, or the area where the vas deferens is expected. Formalin‐fixed tissue sections were processed and embedded in paraffin according to the routine histologic techniques. Sections, 5‐µm thick, were stained with hematoxylin and eosin (H&E), based on standard methods, and examined by light microscopy.
2.10. Tracheal cell isolation, primary culture, and electrophysiological analysis
G542X and F508del CF STE cells were prepared as in a previous study. 5 Briefly, tracheal segments were digested by injecting the subepithelial space with a collagenase solution (type I, 1 mg/mL; Worthington) and incubated at 37℃ for 1 h. After digestion, the epithelial sheets on the luminal surface were collected by scraping, using a plastic coverslip. These sheets were washed and then further digested with Accutase into single cells and small clumps. The cells were then seeded onto permeable supports (Costar 3801) with bronchial epithelial growth medium (BEGM; Lonza) in both upper and lower chambers. Once the cells were post‐confluent for 3 days, the culture medium was switched to PneumaCult‐ALI medium (STEMCELL Technologies) in the lower chamber only for air–liquid interface (ALI) culture. After 6 weeks, the ALI cultures were differentiated into polarized epithelia, which were used for the measurements of transepithelial electrophysiology. The filter inserts were placed in Ussing chambers and bathed on both sides with Krebs‐Ringer bicarbonate solution bubbled with 95% O2/5% CO2 and maintained at 37℃, as described previously. 22 , 23 Transepithelial voltage was measured and clamped to 0 to measure the short‐circuit current. The epithelium was periodically voltage‐clamped to non‐0 values to measure transepithelial resistance. In addition, ciliary beating was observed and recorded in both STE cultures (see, Supporting Information [Link], [Link]).
2.11. Effect of CFTR correctors/potentiator on F508del and G542X CF STE cells
F508del STE cell cultures were pretreated with the correctors VX‐809, VX‐661, or VX‐445 at 3.3 μM for 24 h. For the triple combination (as in Trikafta®) VX‐661 and VX‐445 at 3.3 μM were used together for 24 h. At the indicated times, amiloride (100 μM, apical), forskolin (10 μM, basolateral), VX‐770 potentiator (1 μM, apical & basolateral), genistein (30 μM, apical & basolateral), and CFTR inhibitor GlyH‐101 (20 μM, apical) were added for acute treatment. G542X CF STE cells were pretreated with the inhibitor of NMD SMG1‐i (1 μM) 18 plus G418 (50 μM) for 24 h with or without VX‐661 (3.3 μM), followed by acute treatment as described above.
3. RESULTS
3.1. Generation of CFTRF508del/F508del and CFTRG542X/G542X sheep fetal fibroblast colonies using CRISPR/Cas9 RNP and ssODN
Fetal fibroblasts from domestic sheep (O. aries) were used in this study, since they are the preferred cell type for cloning. 24 The cells were isolated as previously described. 21 We designed specific PCR primers according to the sheep CFTR genome sequence (GenBank, NC_019461.2) to amplify exon 11 (F: TGAACTCAGCACCCCATCTCTG, R: TGCAGGCTTCTTATAGCAGGGG, 624 bp) and exon 12 (F: GCTGAGATGTGGTGTTCACA, R: GCATCTTCTCTTCCCTGTGC, 560 bp) and parts of the flanking intron sequences. We designed guide RNA/Cas9 (gRNA/Cas9) targeting sites that were close to the F508 and G542 (gRNA_1: 5’‐ATTAAAGATAACATCATCTT‐3’ and gRNA_2: 5’‐AACATAGTTCTTGGAGAAGG‐3, for exon 11 and 12, respectively), of the CFTR gene. The F508del and G542X mutations were introduced into the corresponding sites of ssODNs in order to induce mutations in SFFs via CRISPR‐mediated homology‐directed repair (HDR) (Figure 1A). SFFs were transfected with gRNA/Cas9 RNP and ssODNs using an electroporation‐based method. After limiting dilution of the transfected cells, we obtained 56 single cell‐derived colonies in the F508del group and 62 colonies in the G542X group. The PCR and Sanger sequencing results demonstrated that mutations, including insertions/deletions (indels) mutations and KIs, were introduced in 53/56 (94.6%) of colonies targeting the F508del site, and four of them (7.1%) contained homozygous F508del mutations (Figure 1B). Whereas a total of mutations were found in 61/62 (98.4%) of colonies in the G542 site, and nine of them (14.5%) contained homozygous G542X mutations (Figure 1C). Four colonies, two from each mutation, were subsequently used for the generation of CFTRF508del / F508del and CFTRG542X / G542X lambs.
3.2. Generation of CFTRF508del/F508del and CFTRG542X/G542X sheep by SCNT
Four SFFs colonies (two with CFTRF508del / F508del and two with CFTRG542X / G542X mutations) were used as donor cells for SCNT. In total, 164 cloned embryos were transferred into 11 estrus‐synchronized recipients. Five pregnancies were initially established, two from the F508del and three from the G542X cells (Table 1). One of the G542X pregnancies was sacrificed around day 40 of gestation and the fetus was used for SFFs G542X / G542X cell rederivation. The remaining pregnancies (n = 4) were allowed to go to term. They resulted in three live offspring and one stillborn lamb. Abdominal ultrasonography confirmed the presence of intestinal obstruction (meconium ileus (MI)) in all F508del and G542X CF offspring. Thus, they were considered not viable due to the MI and were euthanized within a few hours after birth. The sequencing results indicated that all cloned fetuses/lambs carried the same mutations (either F508del or G542X) as those of the donor cells they were derived from (Figure 1B,C). All lambs were submitted for necropsy and tissue collection.
TABLE 1.
Developmental rates following the SCNT using CFTRF508del / F508del and CFTRG542X / G542X fibroblasts as donor cells
| KI colonies (Genotype) | Targeted exon | No of embryos transferred | Pregnancy rate (%) | Term rate (%) | No lambs born alive/ total (%) |
|---|---|---|---|---|---|
| Fd78 (F508del/F508del) | Exon 11 | 47 | 1/3 (33.3) | 1/3 (33.3) | 1/1 (100) |
| Fd92 (F508del/F508del) | Exon 11 | 27 | 1/2 (50.0) | 1/2 (50.0) | 1/1 (100) |
| Gx12 (G542X/G542X) | Exon 12 | 48 | 1/3 (33.3) | 0/3 (0) a | NA |
| Gx44 (G542X/G542X) | Exon 12 | 42 | 2/3 (66.6) | 2/3 (66.6) | 1/2(50) b |
| Total | 164 | 5/11 (45.4) | 4/11 (36.3) | 3/4 (75) |
Fetus collected for isolation of G542X sheep fetus fibroblasts.
The second lamb died about 24 h prior to birth.
3.3. Gross and histologic characterization of the CFTRF508del/F508del and CFTRG542X/G542X sheep
The histopathologic findings of all CFTRF508del / F508del and CFTRG542X / G542X sheep are detailed below and summarized in Table 2.
TABLE 2.
Pathologic findings in CFTR F508del / F508del and CFTR G542X / G542X lambs
| Small intestine | Pancreas | Liver | Gallbladder | Vas deferens | Lung | Kidney | |
|---|---|---|---|---|---|---|---|
| CFTR F508del/F508del (n = 2) | Obstruction, Meconium ileus 2/2 (100%) |
Hypoplasia, mild (1) to severe (1) Acini/duct dilation, minimal inflammation 2/2 (100%) |
Portal ductular reaction, severe, with mild fibrosis Intrahepatic cholestasis, mild (1) to severe (2) 2/2 (100%) |
Hypoplasia 1/2 (50%) | Aplasia or atrophy, bilateral cryptorchidism 2/2 (100%) | No lesions | Hydronephrosis 2/2 (100%) |
| CFTR G542X/G542X (n = 2) | Obstruction, Meconium ileus 2/2 (100%) | Hypoplasia, mild (1) and severe (1) Acini/duct dilation, minimal inflammation 2/2 (100%) |
Portal ductular reaction, mild (1) to severe (1), with mild fibrosis Intrahepatic cholestasis, moderate (1) to severe (1) 2/2 (100%) |
Hypoplasia 2/2 (100%) | Aplasia or atrophy, unilateral cryptorchidism 2/2 (100%) | No lesions | Hydronephrosis 2/2 (100%) |
| CFTR −/− (n = 15) b | Obstruction, Meconium ileus 15/15 (100%) | Hypoplasia or aplasia 11/15 (73.3%) Fibrosis 6/15 (40%) | Portal ductular reaction and fibrosis, mild to severe 11/14 (78.6%), intrahepatic cholestasis, mild to severe 12/14 (85.7%) | Hypoplasia 12/15 (80%) | Aplasia or atrophy 12/12 (100%) | No lesions | Hydronephrosis 13/15 (86.6%) |
Reference CFTR−/− CF sheep data from Fan et al. (2018).
3.3.1. Intestinal phenotype
Similar to the CFTR‐null sheep, 5 100% of the CFTRF508del / F508del (n = 2) and CFTRG542X / G542X (n = 2) newborn lambs exhibited intestinal obstruction (MI), (Figure 2; Table 2), a phenotype seen only in ~15% of human CF babies. An abrupt demarcation between a distended oral intestine and small‐diameter aboral small intestine and colon (Figure 2B,C) was present in three out of four lambs. The change in intestinal diameter at the level of the obstruction was less abrupt and extended over an approximately 3 cm long segment in one of F508del lamb. Obstruction was consistently seen in the mid jejunum. The intestine oral to the obstruction point was markedly dilated (ranging from 10 to 40 mm in diameter) and contained large amount of meconium (Figure 2E,F,H,I). The intestine and colon aboral to the obstruction site had a small diameter (4–8 mm) and contained thick mucoid content. Microscopic examination revealed a mucus filled colonic lumen with dilated mucosal glands (Figure 2 K,L,N,O). The gross and microscopic lesions were identical to the ones seen in CFTR −/− lambs 5 and associated with CF disease in humans.
FIGURE 2.
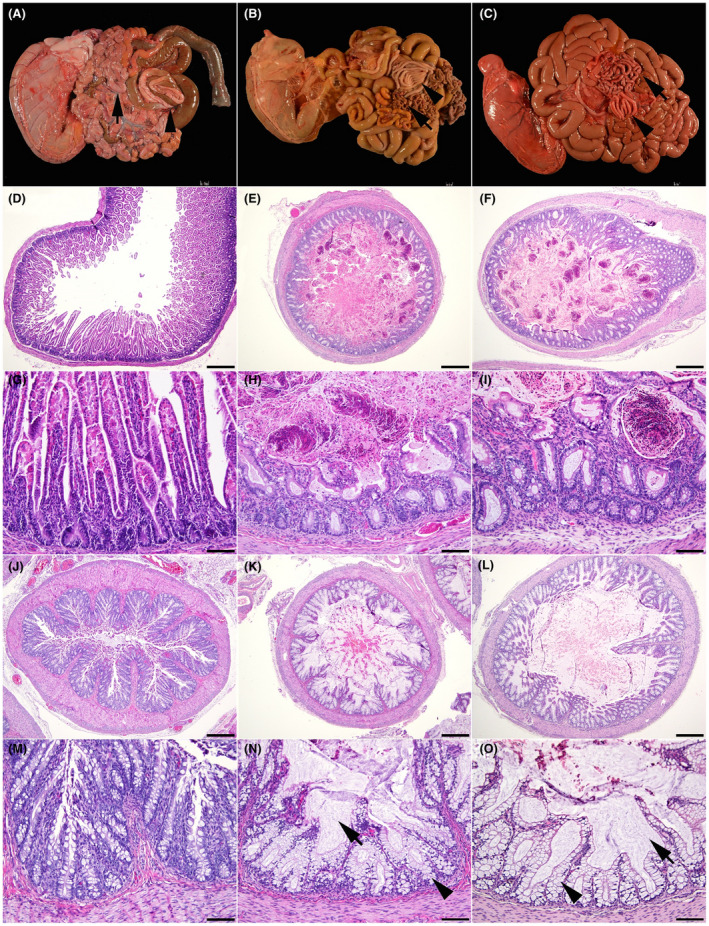
Wild‐type (A, D, G, J, and M), F508del (B, E, H, K, and N), and G542X (C, F, I, L, and O) newborn lamb intestinal tract. (A) Normal intestinal tract in a wild‐type lamb. Note the even intestinal diameter of the small intestine (arrow) and colon (arrowhead). (B) Meconium ileus in an F508del lamb (C) Meconium ileus in a G542X lamb. Note the smaller diameter of the aboral small intestine (arrow), and of the colon (arrowhead). (D) Normal small intestine in a wild‐type lamb. (E) Small intestine filled with meconium in an F508del lamb. (F) Small intestine filled with meconium in a G542X lamb. (G) Higher magnification of the small intestine in a wild‐type lamb. (H) Higher magnification of the small intestine filled with meconium in an F508del lamb. (I) Higher magnification of the small intestine filled with meconium in a G542X lamb. (J) Normal colon in a wild‐type lamb. (K) Colon filled with mucus in an F508del lamb. (L) Colon filled with mucus in a G542X lamb. (M) Higher magnification of the colon in a wild‐type lamb. (N) Higher magnification of the colon in an F508del lamb. (O) Higher magnification of the colon in a G542X lamb. Note the large amount of luminal mucus (arrow) and the goblet cells distended with mucus (arrowhead). Hematoxylin and eosin stain (D–O). 40X (D–F and J–L); bar = 500 μm. 200X (G–I and M–O); bar = 100 μm
3.3.2. Pancreas
The pancreas of all animals was grossly normal in size (Figure 3B,C). However, in all CF lambs, microscopic examination showed variably sized areas of atrophic exocrine pancreas admixed with normal pancreatic tissue (Figure 3E). The atrophic exocrine pancreatic tissue was composed of small acini with sometimes a dilated lumen separated by increased amount of stroma arising from either collapsing of the preexisting stroma or mild fibrosis (Figure 3E,F,H,I). Several acini or ducts with a dilated lumen contained few neutrophils and macrophages. In addition, a few acini and/or ducts had a markedly dilated lumen that contained lightly basophilic flocculent material that stained with alcian blue and periodic acid‐Schiff stain supporting it is likely mucus. These early pancreatic lesions are similar to those reported in humans with CF disease 25 and were also seen in a few CFTR −/− lambs as described by. 5
FIGURE 3.
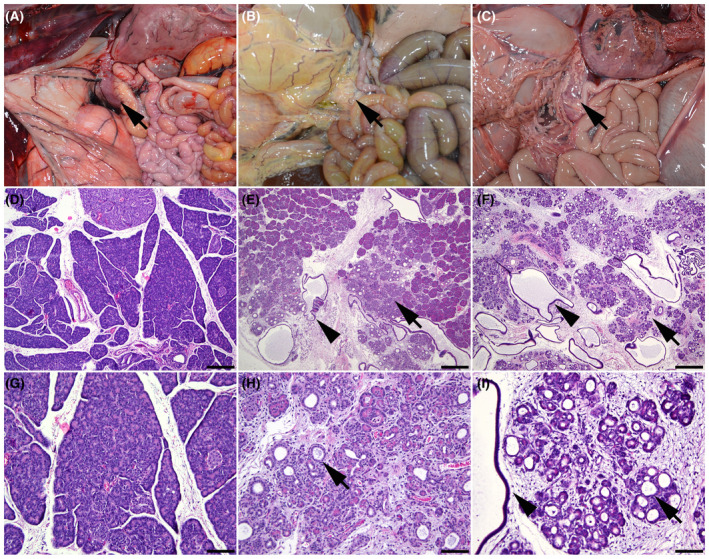
Wild‐type (A, D, and G), F508del (B, E, and H), and G542X (C, F, and I) newborn lamb pancreas. (A) Normal pancreas in a wild‐type lamb (arrow). (B) Pancreas in an F508del lamb (arrow). (C) Pancreas in a G542X lamb (arrow). (D) Pancreas of a wild‐type lamb. (E) Pancreatic acinar atrophy (arrow) and dilatation of acini and ducts (arrowhead) in an F508del lamb. (F) Pancreatic acinar atrophy (arrow) and dilatation of acini and ducts (arrowhead) in a G542X lamb. (G) Higher magnification of the pancreas in a wild‐type lamb. (H) Higher magnification of the pancreatic acinar atrophy and dilatation of acini (arrow) in an F508del lamb. (I) Higher magnification of the pancreatic acinar atrophy and dilatation of acini (arrow) and ducts (arrowhead) in a G542X lamb. Hematoxylin and eosin stain (D–I). 40X (D–F); bar = 500 μm. 200X (G–I); bar = 100 μm
3.3.3. Liver and gallbladder
The liver of all lambs was grossly normal but the gallbladder in three out of four lambs was approximately one quarter of the expected size (Figure 4B,C). In the lamb with a gallbladder of normal size, the cystic duct was tortuous and the gallbladder was found approximately 2 cm more ventral than normal. Microscopic examination revealed moderate to severe bile duct proliferation (ductular reaction) and mild fibrosis in most portal tracts diffusely (biliary fibrosis) in three out of four animals (Figure 4E,F,H,I). Similar lesions but mild were present in the stillborn lamb. Intrahepatic cholestasis was severe in three lambs, and mild in one lamb (Figure 4H,I). These liver lesions were also seen in CFTR −/− sheep 5 and are similar to the phenotype of early onset CF liver disease reported in humans. 26 , 27
FIGURE 4.
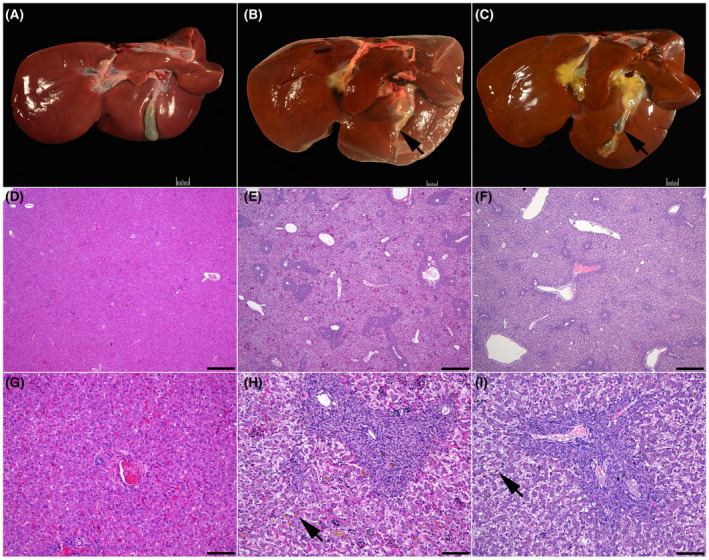
Wild‐type (A, D, and G), F508del (B, E, and H), and G542X (C, F, and I) newborn lamb liver. (A) Normal liver wild‐type lamb. Note the well‐developed gallbladder. (B) Gallbladder hypoplasia in an F508del lamb. (C) Gallbladder hypoplasia in a G542X lamb. Note the small gallbladder (arrow). (D) Normal liver in a wild‐type lamb. (E) Biliary fibrosis in an F508del lamb. (F) Biliary fibrosis in a G542X lamb. (G) Higher magnification of the liver in a wild‐type lamb. (H) Higher magnification of the liver with biliary fibrosis in an F508del lamb. (I) Higher magnification of the liver with biliary fibrosis in a G542X lamb. Note the intrahepatic cholestasis (arrow) in image H and I. Hematoxylin and eosin stain (D–I). 40X (D–F); bar = 500 μm. 200X (G–I); bar = 100 μm
3.3.4. Epididymis and vas deferens
None of the lambs had a grossly visible or microscopically detectable vas deferens. Epididymis was present in all animals. However, bilateral cryptorchidism, was present in both F508del CF lambs, and unilateral cryptorchidism was present in the G542X CF lambs (one right and one left cryptorchid). Cryptorchidism, an absence of at least one testis from the scrotum, is not a CF‐associated lesion and was not previously described in our CFTR‐null sheep, 5 also it may occur due to various factors, such as birth weight, absence of appendix testis, as well as maternal and environmental factors that disrupt hormones and physical changes. 28
3.3.5. Lungs
The lungs of the F508del and G542X CF lambs were grossly and microscopically unremarkable.
3.3.6. Kidney
Hydronephrosis, a common pathology in cloned sheep 29 , 30 and cattle 31 , 32 associated with SCNT, was evident in all four CF lambs.
3.3.7. Other organs
One lamb (G542X CF) had a moderately rounded heart due to right ventricular dilation, whereas all other animals had no heart lesions. Three lambs had a small (± 20 mL) to moderate (± 100 mL) amount of translucent watery fluid in the abdominal cavity, while the stillborn lamb had the thoracic and abdominal cavities filled with dark red watery fluid. These lesions were most likely associated with SCNT. 33 No microscopic lesions were detected in the other evaluated tissues including thyroid gland, adrenal glands, trachea, thymus, spleen, umbilicus, skeletal muscle, forestomach, urinary bladder, and brain obtained from either F508del or G542X CF lambs.
3.4. Functional rescue of mutant ovCFTR using hCFTR therapeutic compounds
In order to evaluate whether the Vertex compounds, which achieve partial functional rescue of mutations in human CFTR (hCFTR), are also effective on equivalent mutations in ovine CFTR (ovCFTR), we treated STE cells with the relevant reagents and monitored CFTR channel activity in Ussing chambers. Treatment with VX‐661, 13 VX‐809, 14 and VX‐445, 15 all three of which promote maturation/correct folding of F508del hCFTR, increased the function of F508del ovCFTR in STE cells (Figure 5A). Following activation of F508del ovCFTR with the VX‐770 potentiator of hCFTR, 16 the increased short‐circuit current caused by the corrector was VX‐445 > VX‐661 > VX‐809 (Figure 5A). The most potent activation was observed with the triple combination of simultaneous treatment with VX‐445 and VX‐661, followed by VX‐770 (Figure S1). In the G542X STE cells, the function of ovCFTR G542X was moderately restored by SMG1‐i 18 and G418 17 treatments (Figure 5B). Furthermore, exposure to G418 with VX‐661 alone did not improve the CFTR function (not shown).
FIGURE 5.
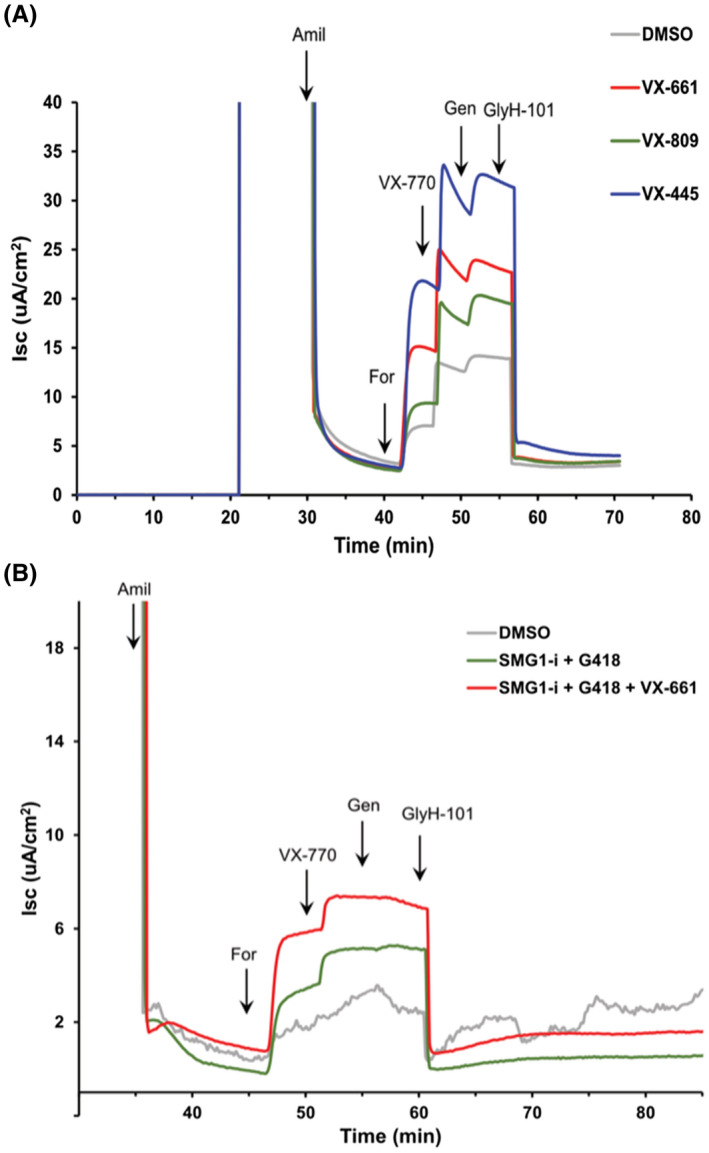
Representative traces of short‐circuit current in CFTR F508del and G542X sheep tracheal epithelial cell cultures. Primary cultures of sheep tracheal epithelial cells were seeded onto permeable filter supports and maintained at the ALI for 6 weeks. At the time of measurement, the filter inserts were placed in Ussing chambers and bathed on both sides with Krebs‐Ringer bicarbonate solution bubbled with 95% O2/5% CO2 and maintained at 37℃. (A) Effect of CFTR correctors on F508del cells. Cultures were pretreated with VX‐809, VX‐661, or VX‐445 at 3.3 μM for 24 h. At the indicated times, amiloride (Amil, 100 μM, apical), forskolin (For, 10 μM, basolateral), VX‐770 (1 μM, apical & basolateral), genistein (Gen, 30 μM, apical & basolateral), and GlyH‐101 (20 μM, apical) were added for acute treatment. (B) Effect of G418 and SMG1‐i on G542 cells. Cells were pretreated with SMG1‐i (1 μM) plus G418 (50 μM) for 24 h with or without VX‐661 (3.3 μM), followed by acute treatment as described above
3.5. Evaluation of potential introduction of mutations in off‐target sites by CRISPR/Cas9
To examine whether unintended off‐targeting (OT) events occurred in CFTRF508del / F508del or CFTRG542X / G542X sheep, we conducted a blast search of the sheep nucleotide sequence database with CFTR targeting sequences as the queries to find the genomic sequences with the highest homologies. We chose 10 potential OT sites related to the gRNA_1 and 11 OT sites to the gRNA_2 in the sheep genome with the highest sequence homology (scores >1) for the OT analysis (Table S1). Specific PCR primers were designed to amplify DNA fragments ranging from 300 to 800 bp spanning each potential OT locus (Table S2). Genomic DNA isolated from two F508del and three G542X cloned sheep were used as the templates for PCR amplification. Sequencing results verified that none of the animals had mutations in the analyzed potential OT sites (Figure S2).
4. DISCUSSION
One impediment to advancing the understanding of CF disease mechanisms is the limited ability of most CF animal models to recapitulate some aspects of humans’ disease phenotype (reviewed in 34 ). Also, the CFTR‐null models do not represent the same molecular mechanisms seen in patients with the most common mutations, F508del and G542X, as the random disruption of the gene may lead to unpredictable effects on protein synthesis. Currently, a few F508del and G542X CF animal models have been established, including pig, 35 rat, 36 , 37 and mouse 38 for the F508del mutation, and mouse 39 and rat 40 for the G542X mutation. The F508del mutants have similar abnormalities as seen in their respective CFTR‐null models of the same species, except with less severe damage in some organs due to residual CFTR function 35 , 36 (reviewed in 41 ). The availability of sheep models of the F508del and G542X mutations provides an extremely valuable resource, not only to reveal critical events in the early pathology of CF disease, which are not accessible in humans, but also to evaluate new therapeutic approaches. Similar to our observations on the efficacy of the Vertex therapeutics to restore the function of ovine F508del CFTR protein, primary nasal cultures of F508del rats demonstrated that the combination of VX‐809 and VX‐770, human therapeutics, restored CFTR function, and improved the Cl− anion transport up to 37%. 36 Thus these therapeutics may be effective in multiple species as the molecular mechanism of the CFTR synthesis seems to be similar. Of note, ongoing efforts to optimize CFTR correctors, which although they correct the folding process do not restore the reduced open state probability of the F508del CFTR channel, 42 may also benefit from additional animal model systems for in vivo studies. Nonetheless, even with these most promising postnatal treatments, they cannot fully correct all the deficits arising from the absence of appropriate CFTR levels during fetal development. For this reason, the F508del CF sheep models may also be an excellent candidate to test not only postnatal but also prenatal therapeutic approaches. Sheep, like humans, frequently have one to two offspring, which provides an advantage for prenatal treatment. CF pig models, on the other hand, require a minimum of four offsprings to maintain the pregnancy, 43 and normally have 6–10 offspring per pregnancy.
G542X CF mice and rats were generated recently 39 , 40 and exhibit a similar phenotype to their CFTR‐null models with no residual CFTR functionality. However, organoids isolated from the intestine of G542X CF mice and epithelial cells isolated from the trachea of G542X CF rats indicated that CFTR protein synthesis could be rescued by G418 antibiotic treatment, thus, partially restoring the CFTR channel. Also, McHugh et al. observed that the intestinal organoids from the same G542X CF mouse, when treated with PTC124, showed no enhancement of CFTR gene transcription. 39 More recently, McHugh et al. tested the efficacy of a combination of read‐through agents and NMD inhibitors to restore the CFTR function in primary airway epithelial (PAE) cells from a G542X CF mouse model. Their analysis strongly indicates that the SMG1‐i and G418 act in synergy to restore the CFTR channel in PAE cell cultures. 44 Furthermore, several studies 45 , 46 , 47 , 48 have demonstrated that drugs like G418 and PTC124 enable ribosomes to read through nonsense mutation sites of PTC by inserting a near‐cognate aminoacyl tRNA that inserts a range of alternative amino acids to allow translation of the full‐length protein; thus, preventing early PTC. However, these compounds cause side effects or provide no significant enhancement, that is, gentamicin is nephro and ototoxic, whereas PTC124 has little impact on clinical disease in CF patients in Phase 3 clinical trials. Therefore, currently, there are no adequate and safe therapies available to recover the production of the CFTR gene for nonsense mutations. 12,39 Again, the G542X sheep model may serve as not only an excellent candidate for the discovery of new corrective drugs but also prenatal therapeutic approaches.
Among all etiologies associated with CF, one of particular importance is the early lung pathology, which is likely triggered by functional inactivation of the CFTR protein early in life. Current models of human lung development heavily rely on seminal work in the sheep lung, carried out many decades ago (reviewed in 49 ). 50 Also, the developmental time course of CFTR gene expression in the sheep lung appears similar to limited observations in humans. 51 , 52 , 53 , 54 Hence, it is evident that the availability of CF sheep models could be a valuable asset to reveal key events in the human CF lung, which are not accessible for study in vivo in humans. 55 Similar to human CF patients, newborn CF sheep do not exhibit any histological lung lesions. However, further molecular characterization of the lung tissue derived from these animals could provide novel insights into CF‐associated deficits occurring prior to observations of clinical lesions. Due to meconium ileus, our current models only survive 24–48 h after birth, and therefore, the short life span prevents further studies. To overcome this limitation, we intend to genetically correct the intestinal phenotype by expressing a transgene under regulation of an intestinal cell‐specific promoter, similarly to what was previously accomplished in mice, ferrets, and pigs. 56 , 57 , 58 On these aspects, CF sheep models carrying the most common mutations (F508del and G542X) seem necessary in order to advance the research on the CF disease.
Here, we generated two models of human CF mutations in sheep that develop pathologies similar to what is seen in human patients. Analogous to the F508del CF rat and G542X CF rat and mouse, our F508del and G542X CF sheep cells are very responsive to the CFTR correctors (VX‐661, VX‐809, and VX‐445) and potentiator (VX‐770) in F508del STE cells, as well as for SMG1‐i and G418 with and without corrector (VX‐661) in G542X STE cells, respectively. As was shown earlier, the ovCFTR channel is more active than the human channel and the F508del mutation in sheep has a reduced impact in vitro on channel gating when compared to hCFTR protein. 59 Thus, our F508del CF sheep model may contribute substantially to the discovery of new therapeutics. G542X CF models, on the other hand, remain poorly understood as only one mouse model has recently been generated, and does not recapitulate all aspects of CF in humans as reviewed in. 39 Our G542X CF model will likely provide a robust platform to study in‐depth the effects of nonsense mutations in the CFTR gene and provide an opportunity to enhance the in vivo development of therapeutics that overcome the PTC. Moreover, with the rapid advancement of CRISPR/Cas9 technologies as molecular tools, both CF models may provide excellent platforms to observe the correction of the CFTR gene through gene editing and gene therapy approaches.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
AUTHOR CONTRIBUTIONS
AH, KLW, and IAP conceived and managed the study. IVP and ZW designed and/or carried out the CRISPR/Cas9 and ssODN designs. YL, ZF, JK, and MR conducted SCNT experiments. AVW carried out histopathological analysis of the CF lambs. SHL established tracheal epithelial cell cultures and MDW, KMC, SNM, and TJK performed electrophysiological analysis of these cells. IVP, ZF, AVW, SHL, AH, KLW, and IAP wrote the manuscript. All authors reviewed the manuscript.
Supporting information
Supplementary Material
Video S1
Video S2
ACKNOWLEDGEMENTS
The authors thank Drs. Rusty Stott, Holy Mason, and Alexis Sweat for performing embryo transfers procedures, and David Forrester, Taylor Martin, and Angie Robinson for excellent assistance with animal care. Also, the authors acknowledge the Epithelial Cell Core at the CWRU CF Center (Cystic Fibrosis Foundation RDP R447‐CR11) and Dr. R. Bridges of Rosalind Franklin University for generous donation of SMG1‐i, through the CFFT compound distribution program. The authors also acknowledge Amanda Wilheim and Sherry Iodice for preparation of the histology slides. This work was partially supported by US Department of Agriculture Multistate Project W‐4171 (IAP) and the Utah Agricultural Experiment Station (Project 1343).
Viotti Perisse I, Fan Z, Van Wettere A, et al. Sheep models of F508del and G542X cystic fibrosis mutations show cellular responses to human therapeutics. FASEB BioAdvances. 2021;3:841–854. 10.1096/fba.2021-00043
Contributor Information
Ann Harris, Email: ann.harris@case.edu.
Irina A. Polejaeva, Email: irina.polejaeva@usu.edu.
REFERENCES
- 1. Hwang T‐C, Kirk KL. The CFTR ion channel: gating, regulation, and anion permeation. Cold Spring Harb Perspect Med. 2013;3(1):a009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ratjen F, Grasemann H. New therapies in cystic fibrosis. Curr Pharm Des. 2012;18(5):614‐627. [DOI] [PubMed] [Google Scholar]
- 3. McCarron A, Parsons D, Donnelley M. Animal and cell culture models for cystic fibrosis: which model is right for your application? Am J Pathol. 2021;191(2):228‐242. [DOI] [PubMed] [Google Scholar]
- 4. Rosen BH, Chanson M, Gawenis LR, et al. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. 2018;17(2):S28‐S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fan ZQ, Perisse IV, Cotton CU, et al. A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. Jci Insight. 2018;3(19). 10.1172/jci.insight.123529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marangi M, Pistritto G. Innovative therapeutic strategies for cystic fibrosis: moving forward to CRISPR technique. Front Pharmacol. 2018;9:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cystic Fibrosis Foundation . Cystic Fibrosis Foundation Patient Registry 2018 Annual Data Report. 2019. [Google Scholar]
- 8. Chung WY, Song M, Park J, et al. Generation of ΔF508‐CFTR T84 cell lines by CRISPR/Cas9‐mediated genome editing. Biotech Lett. 2016;38(12):2023‐2034. [DOI] [PubMed] [Google Scholar]
- 9. Ren HY, Grove DE, De La Rosa O, et al. VX‐809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane‐spanning domain 1. Mol Biol Cell. 2013;24(19):3016‐3024. 10.1091/mbc.E13-05-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wainwright CE, Elborn JS, Ramsey BW. Lumacaftor‐Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(18):1783‐1784. 10.1056/NEJMc1510466 [DOI] [PubMed] [Google Scholar]
- 11. Strug LJ, Stephenson AL, Panjwani N, Harris A. Recent advances in developing therapeutics for cystic fibrosis. Hum Mol Genet. 2018;27(R2):R173‐R186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yeh JT, Hwang TC. Positional effects of premature termination codons on the biochemical and biophysical properties of CFTR. J Physiol. 2020;598(3):517‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pettit RS, Fellner C. CFTR modulators for the treatment of cystic fibrosis. Pharm Ther. 2014;39(7):500. [PMC free article] [PubMed] [Google Scholar]
- 14. Ren HY, Grove DE, De La Rosa O, et al. VX‐809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane‐spanning domain 1. Mol Biol Cell. 2013;24(19):3016‐3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Keating D, Marigowda G, Burr L, et al. VX‐445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612‐1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Accurso FJ, Rowe SM, Clancy J, et al. Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation. N Engl J Med. 2010;363(21):1991‐2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eustice DC, Wilhelm JM. Mechanisms of action of aminoglycoside antibiotics in eucaryotic protein synthesis. Antimicrob Agents Chemother. 1984;26(1):53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gopalsamy A, Bennett EM, Shi M, Zhang W‐G, Bard J, Yu K. Identification of pyrimidine derivatives as hSMG‐1 inhibitors. Bioorg Med Chem Lett. 2012;22(21):6636‐6641. [DOI] [PubMed] [Google Scholar]
- 19. Yang M, Perisse I, Fan Z, Regouski M, Meyer‐Ficca M, Polejaeva IA. Increased pregnancy losses following serial somatic cell nuclear transfer in goats. Reprod Fertil Dev. 2018;30(11):1443. 10.1071/RD17323 [DOI] [PubMed] [Google Scholar]
- 20. Yang M, Hall J, Fan Z, et al. Oocytes from small and large follicles exhibit similar development competence following goat cloning despite their differences in meiotic and cytoplasmic maturation. Theriogenology. 2016;86(9):2302‐2311. 10.1016/j.theriogenology.2016.07.026 [DOI] [PubMed] [Google Scholar]
- 21. Fan Z, Yang M, Regouski M, Polejaeva IA. Gene knockouts in goats using CRISPR/Cas9 system and somatic cell nuclear transfer. Methods Mol Biol. 2019;1874:373‐390. 10.1007/978-1-4939-8831-0_22 [DOI] [PubMed] [Google Scholar]
- 22. Davis PB, Silski CL, Kercsmar CM, Infeld M. Beta‐adrenergic receptors on human tracheal epithelial cells in primary culture. Am J Physiol‐Cell Physiol. 1990;258(1):C71‐C76. [DOI] [PubMed] [Google Scholar]
- 23. Veizis EI, Carlin CR, Cotton CU. Decreased amiloride‐sensitive Na+ absorption in collecting duct principal cells isolated from BPK ARPKD mice. Am J Physiol‐Renal Physiol. 2004;286(2):F244‐F254. [DOI] [PubMed] [Google Scholar]
- 24. Schnieke AE, Kind AJ, Ritchie WA, et al. Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts. Science. 1997;278(5346):2130‐2133. 10.1126/Science.278.5346.2130 [DOI] [PubMed] [Google Scholar]
- 25. Reid GJ, Hyde K, Ho SB, Harris A, Weatherall D. Cystic fibrosis of the pancreas: involvement of MUC6 mucin in obstruction of pancreatic ducts. Mol Med. 1997;3(6):403‐411. [PMC free article] [PubMed] [Google Scholar]
- 26. Colombo C. Liver disease in cystic fibrosis. Curr Opin Pulm Med. 2007;13(6):529‐536. [DOI] [PubMed] [Google Scholar]
- 27. Debray D, Narkewicz MR, Bodewes FAJA, et al. Cystic fibrosis–related liver disease: research challenges and future perspectives. J Pediatr Gastroenterol Nutr. 2017;65(4):443‐448. [DOI] [PubMed] [Google Scholar]
- 28. Leslie SW, Sajjad H, Villanueva CA. Cryptorchidism. StatPearls. StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC.; 2021. [Google Scholar]
- 29. Dawson AJ, King TJ, Wilmut I, Harkness LM, Kelly BG, Rhind SM. Immunohistochemical characterization of cloned lamb nephropathy. J Histochem Cytochem. 2004;52(12):1657‐1664. [DOI] [PubMed] [Google Scholar]
- 30. Rhind SM, King TJ, Harkness LM, et al. Cloned lambs—lessons from pathology. Nat Biotechnol. 2003;21(7):744‐745. [DOI] [PubMed] [Google Scholar]
- 31. Chavatte‐Palmer P, Heyman Y, Richard C, et al. Clinical, hormonal, and hematologic characteristics of bovine calves derived from nuclei from somatic cells. Biol Reprod. 2002;66(6):1596‐1603. [DOI] [PubMed] [Google Scholar]
- 32. Kato Y, Tani T, Tsunoda Y. Cloning of calves from various somatic cell types of male and female adult, newborn and fetal cows. J Reprod Fertil. 2000;120(2):231‐238. [PubMed] [Google Scholar]
- 33. Hill JR. Incidence of abnormal offspring from cloning and other assisted reproductive technologies. Annu Rev Anim Biosci. 2014;2(1):307‐321. [DOI] [PubMed] [Google Scholar]
- 34. Lavelle GM, White MM, Browne N, McElvaney NG, Reeves EP. Animal models of cystic fibrosis pathology: phenotypic parallels and divergences. Biomed Res Int. 2016;2016:5258727. 10.1155/2016/5258727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ostedgaard LS, Rogers CS, Dong Q, et al. Processing and function of CFTR‐ΔF508 are species‐dependent. Proc Natl Acad Sci. 2007;104(39):15370‐15375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dreano E, Bacchetta M, Simonin J, et al. Characterization of two rat models of cystic fibrosis—KO and F508del CFTR—Generated by Crispr‐Cas9. Anim Models Exp Med. 2019;2(4):297‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McCarron A, Cmielewski P, Reyne N, et al. Phenotypic characterization and comparison of cystic fibrosis Rat models generated using CRISPR/Cas9 gene editing. Am J Pathol. 2020;190(5):977‐993. [DOI] [PubMed] [Google Scholar]
- 38. Colledge WH, Abella BS, Southern KW, et al. Generation and characterization of a ΔF508 cystic fibrosis mouse model. Nat Genet. 1995;10(4):445‐452. [DOI] [PubMed] [Google Scholar]
- 39. McHugh DR, Steele MS, Valerio DM, et al. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS One. 2018;13(6):e0199573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sharma J, Abbott J, Klaskala L, Zhao G, Birket SE, Rowe SM. A novel G542X CFTR rat model of cystic fibrosis is sensitive to nonsense mediated decay. Front Physiol. 2020;11:611294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Semaniakou A, Croll RP, Chappe V. Animal models in the pathophysiology of cystic fibrosis. Front Pharmacol. 2019;9:1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ostedgaard LS, Meyerholz DK, Chen J‐H, et al. The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis–like disease in pigs. Sci Transl Med. 2011;3(74):74ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polejaeva IA, Chen S‐H, Vaught TD, et al. Cloned pigs produced by nuclear transfer from adult somatic cells. Nature. 2000;407(6800):86‐90. [DOI] [PubMed] [Google Scholar]
- 44. McHugh DR, Cotton CU, Hodges CA. Synergy between readthrough and nonsense mediated decay inhibition in a murine model of cystic fibrosis nonsense mutations. Int J Mol Sci. 2021;22(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bukowy‐Bieryllo Z, Dabrowski M, Witt M, Zietkiewicz E. Aminoglycoside‐stimulated readthrough of premature termination codons in selected genes involved in primary ciliary dyskinesia. RNA Biol. 2016;13(10):1041‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dabrowski M, Bukowy‐Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug‐stimulated translational readthrough of premature termination codons. Mol Med. 2018;24(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bolze F, Mocek S, Zimmermann A, Klingenspor M. Aminoglycosides, but not PTC124 (Ataluren), rescue nonsense mutations in the leptin receptor and in luciferase reporter genes. Sci Rep. 2017;7(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moosajee M, Tracey‐White D, Smart M, et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC‐414 in human choroideremia fibroblasts and the nonsense‐mediated zebrafish model. Hum Mol Genet. 2016;25(16):3416‐3431. [DOI] [PubMed] [Google Scholar]
- 49. Thorburn GD, Harding R. Textbook of fetal physiology. Oxford: Oxford University Press; 1994. [Google Scholar]
- 50. Cotton CU, Lawson EE, Boucher RC, Gatzy JT. Bioelectric properties and ion transport of airways excised from adult and fetal sheep. J Appl Physiol Respir Environ Exerc Physiol. 1983;55(5):1542‐1549. 10.1152/jappl.1983.55.5.1542 [DOI] [PubMed] [Google Scholar]
- 51. Broackes‐Carter FC, Mouchel N, Gill D, Hyde S, Bassett J, Harris A. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum Mol Genet. 2002;11(2):125‐131. 10.1093/Hmg/11.2.125 [DOI] [PubMed] [Google Scholar]
- 52. Harris A, Chalkley G, Goodman S, Coleman L. Expression of the cystic fibrosis gene in human development. Development. 1991;113(1):305‐310. [DOI] [PubMed] [Google Scholar]
- 53. Mouchel N, Broackes‐Carter F, Harris A. Alternative 5’ exons of the CFTR gene show developmental regulation. Hum Mol Genet. 2003;12(7):759‐769. [DOI] [PubMed] [Google Scholar]
- 54. Trezise AE, Chambers JA, Wardle CJ, Gould S, Harris A. Expression of the cystic fibrosis gene in human foetal tissues. Hum Mol Genet. 1993;2(3):213‐218. [DOI] [PubMed] [Google Scholar]
- 55. Harris A. Towards an ovine model of cystic fibrosis. Hum Mol Genet. 1997;6(13):2191‐2193. 10.1093/Hmg/6.13.2191 [DOI] [PubMed] [Google Scholar]
- 56. Stoltz DA, Rokhlina T, Ernst SE, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Investig. 2013;123(6):2685‐2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013;3(9):a009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun X, Sui H, Fisher JT, et al. Disease phenotype of a ferret CFTR‐knockout model of cystic fibrosis. J Clin Investig. 2010;120(9):3149‐3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cai Z, Palmai‐Pallag T, Khuituan P, et al. Impact of the F508del mutation on ovine CFTR, a Cl− channel with enhanced conductance and ATP‐dependent gating. J Physiol. 2015;593(11):2427‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Video S1
Video S2