

Abstract
A rapidly expanding catalog of neurogenetic disorders has encouraged a diagnostic shift towards early clinical whole exome sequencing (WES). Adult primary mitochondrial diseases (PMDs) frequently exhibit neurological manifestations that overlap with other nervous system disorders. However, mitochondrial DNA (mtDNA) is not routinely analyzed in standard clinical WES bioinformatic pipelines. We reanalyzed 11,424 exomes, enriched with neurological diseases, for pathogenic mtDNA variants. Twenty‐four different mtDNA mutations were detected in 64 exomes, 11 of which were considered disease causing based on the associated clinical phenotypes. These findings highlight the diagnostic uplifts gained by analyzing mtDNA from WES data in neurological diseases. ANN NEUROL 2021;89:1240–1247
The rapid expansion in recognized inherited neurological disorders has led to a “genetics first” approach to their diagnosis, which frequently involves the early application of clinical whole exome sequencing (WES). 1 There is also growing evidence that the iterative re‐analysis of exome data significantly improves the diagnostic yield of pathogenic DNA variants in rare diseases, and should therefore be considered in all unresolved cases. 2
Primary mitochondrial diseases (PMDs) have an adult prevalence of 1 in 4,300, 3 thus represent some of the most common neurogenetic disorders, and frequently manifest with nervous system phenotypes that overlap with other neurological disorders. More than 300 mitochondrial proteins, encoded by both nuclear and mitochondrial genomes, are linked with PMDs 4 ; however, mitochondrial DNA (mtDNA) mutations account for the vast majority in adults. 3 Importantly, although mtDNA reads are generated during WES, they are not routinely analyzed. Consequently, standard clinical WES bioinformatic pipelines do not report disease causing mtDNA variants.
We applied a Genome Analysis Toolkit (GATK) Mutect2‐based 5 , 6 bioinformatics pipeline to analyze the mtDNA reads from 11,424 exomes, generated by University College London Queen Square Genomics Facility, and determined the diagnostic uplifts gained by including mtDNA analysis during the iterative reanalysis of neurological disease exomes.
Methods
This study was approved by the Queen Square Research Ethics Committee, London, UK (09/H0716/76).
Ascertainment of Whole Exome Sequencing Data and Cohort Phenotypic Groups
In total, 11,424 DNA samples, extracted from blood, which had undergone WES analysis at the University College London Queen Square Genomics Facility between 2011 and 2019, were included in the study. The cohort was composed of probands with a range of neurological phenotypes, their relatives, and healthy controls. The most common clinical group comprised extrapyramidal and movement disorders (International Classification of Disease‐10 code G20–G26 7 ; 31.5%), followed by systemic atrophies, primarily affecting the central nervous system (G10–G14; 17%), episodic and paroxysmal disorders (G40–G47; 16.4%), diseases of myoneural junction and muscle (G70–G73; 7.5%), other degenerative diseases of the nervous system (G30–G32; 7.4%), polyneuropathies and other disorders of the peripheral nervous system (G60–G64; 3.6%), and mitochondrial metabolism disorders (E88.4; 1.4%). Neurologically normal controls accounted for 15.2% of the exomes.
Mitochondrial Variant Calling and Filtering
Exome capture libraries were generated using SureSelect (Agilent), Nextera (Illumina) or TrueSeq (Illumina) kits, and all samples were sequenced using Illumina instruments generating 100 to 150 bp paired‐end reads. Fastq files were aligned to the GRCh38 genome reference, which includes the revised Cambridge Reference Sequence (rCRS) of human mtDNA, using NovoAlign version 3.08.02 (Novocraft). Alignments were further processed according to the GATK version 4 best practices 5 and variants in mtDNA were called using a somatic variant caller, Mutect2, 6 in tumor‐only mode and filtered using Filter Mutect Calls tool. Only variants in exomes with an average mtDNA coverage of ≥10 times were kept for downstream analysis (Fig 1) and annotated using Variant Effect Predictor, 8 Mitomap, 9 and an in‐house database of mtDNA variants. Variants of interest were defined as those with a “confirmed” pathogenic status in Mitomap. Variants with a read depth ≥10 times and heteroplasmy level of ≥10% in blood were validated and selected for further clinical evaluation.
FIGURE 1.
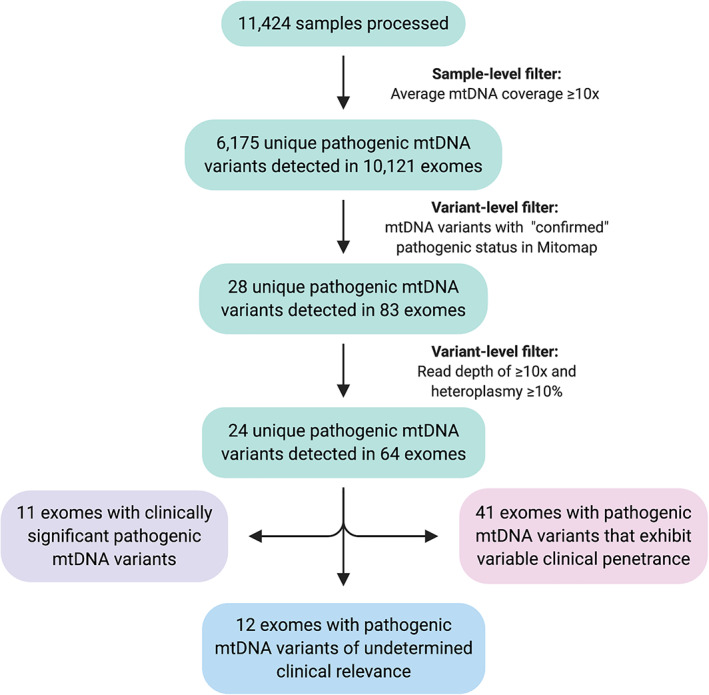
Flow diagram summarizing sample‐level and variant‐level filtering steps applied to 11,424 samples that underwent exome sequencing analysis at the University College London Queen Square Genomics Facility between 2011 and 2019. The figure was created using BioRender.com. mtDNA, mitochondrial DNA. [Color figure can be viewed at www.annalsofneurology.org]
Confirmation of Pathogenic Mitochondrial DNA Variants
All pathogenic mtDNA variants identified from WES data were subsequently confirmed using whole mtDNA next generation sequencing (NGS). Long‐range polymerase chain reaction (PCR) amplification of 2 overlapping fragments covering the entire mtDNA preceded NGS on the MiSeq platform using the Illumina NextEra XT library preparation. Data analysis was performed using an in‐house pipeline aligned to the rCRS, GenBank accession number NC_012920.1. Coverage of the coding region (nucleotides 577–16023) was at a minimum depth of 1,000 times. Variant detection sensitivity was greater than 95% (95% confidence interval).
Classification of Pathogenic Mitochondrial DNA Variants
Pathogenic mtDNA variants were stratified according to their associated neurological presentations as follows: (1) clinically significant pathogenic mtDNA variants, defined as a well‐recognized/reported clinical phenotype and a mutant mtDNA heteroplasmy level of ≥10% in blood; (2) pathogenic mtDNA variants of undetermined clinical relevance, defined as an atypical clinical phenotype and a mutant mtDNA heteroplasmy level of ≥10% in blood; and (3) pathogenic mtDNA variants that exhibit variable clinical penetrance at near homoplasmic or homoplasmic mutant levels and a mutant mtDNA heteroplasmy level of ≥10% in blood (Supplementary Table S1 summarizes a list of pathogenic mtDNA variants that exhibit this phenomenon).
Results
In total, 6,175 mitochondrial variants were identified across 10,121 exomes with sufficient mtDNA coverage (median mtDNA coverage = 43.9 times; see Fig 1). This included 28 pathogenic mtDNA variants in 83 exomes. Twenty‐four variants in 64 exomes met our threshold for further clinical evaluation; specifically, a locus‐specific depth ≥10 times and a heteroplasmy level ≥10% (Tables 1, 2, 3 and Supplementary Table S2).
TABLE 1.
Clinically Significant Pathogenic Mitochondrial DNA Variants Detected in the 11,424 Exomes Analyzed
| Subject | mtDNA variant | Gene | No. ref reads | No. alt reads | Het (%) | Reported associated disease 9 |
|---|---|---|---|---|---|---|
| 1 | m.3697G>A | MT‐ND1 | 7 | 89 | 92 | MELAS/LS/LDYT |
| 2 | m.8851T>C | MT‐ATP6 | 1 | 61 | 98 | BSN/LS |
| 3 | m.8993T>C | MT‐ATP6 | 0 | 65 | 98 | NARP/LS/MILS/other |
| 4 | m.9185T>C | MT‐ATP6 | 1 | 76 | 97 | LS/Ataxia syndromes/NARP‐like disease |
| 5 | m.9185T>C | MT‐ATP6 | 7 | 96 | 92 | LS/Ataxia syndromes/NARP‐like disease |
| 6 | m.10158T>C | MT‐ND3 | 19 | 6 | 25 | LS/MELAS |
| 7 | m.13094T>C | MT‐ND5 | 87 | 55 | 39 | Ataxia and PEO/MELAS, LS, myoclonus, fatigue |
| 8 | m.13513G>A | MT‐ND5 | 47 | 54 | 53 | LS/MELAS/LHON‐MELAS overlap syndrome |
| 9 | m.13513G>A | MT‐ND5 | 19 | 3 | 14 | LS/MELAS/LHON‐MELAS overlap syndrome |
| 10 | m.14459G>A | MT‐ND6 | 1 | 83 | 96 | LDYT/LS |
| 11 | m.14459G>A | MT‐ND6 | 2 | 78 | 97 | LDYT/LS |
BSN = bilateral striatal necrosis; Het = heteroplasmy; LDYT = Leber hereditary optic neuropathy and dystonia; LHON = Leber hereditary optic neuropathy; LS = Leigh syndrome; MELAS = mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes; MILS = maternally inherited Leigh syndrome; mtDNA = mitochondrial DNA; NARP = neurogenic muscle weakness, ataxia, and retinitis pigmentosa; PEO = progressive external ophthalmoplegia.
TABLE 2.
Clinical Features of the Patients with Clinically Significant Pathogenic Mitochondrial DNA Variants in the 11,424 Exomes Analyzed
| Subject | Age, yr/sex | Subject phenotype | Muscle pathology and RCEA | Brain MRI | Family history |
|---|---|---|---|---|---|
| 1 | 3/M | Encephalopathy, lower limb spasticity, generalized tonic–clonic seizures, palatal myoclonus | NA | NA | NA |
| 2 |
82/M Deceased |
Progressive ataxia (onset in 50s), cognitive impairment | NA | Mild supratentorial atrophy with disproportionate cerebellar atrophy and sparing of the brainstem | Identical twin brother with ataxia |
| 3 | 13/M | Developmental delay, spastic‐dystonic gait, ID, axonal neuropathy | NA | NA | One older sibling with developmental delay and ID |
| 4 |
45/M Deceased |
Young adult onset progressive cerebellar ataxia, tremor, optic atrophy, neuropathy, myoclonic epilepsy, urinary frequency, atrial fibrillation | RRF | Basal ganglia changes and cerebellar atrophy | One affected brother (subject 5), mother asymptomatic (47% mutant load in blood), maternal grandmother with epilepsy |
| 5 | 49/M | Young adult onset progressive cerebellar ataxia, pyramidal signs, neuropathy, restless leg syndrome, ADHD | NA | Marked cerebellar atrophy | Less severely affected brother of subject 4 |
| 6 | 29/F | Stroke‐like episodes, cerebellar ataxia, left sided spastic hemiparesis, learning difficulties, seizures, optic atrophy, mild scoliosis, incomplete RBBB |
Mild increase in lipid, no RRF or COX negative fibers Low complex I |
White matter changes and high signal in right basal ganglia and right frontoparietal deep white matter | Mother asymptomatic |
| 7 | 27/M | Encephalopathic episode with bilateral INO and diplopia, nocturnal hypoventilation, reduced visual acuity, LVH |
Normal pathology Normal RCEA |
Symmetrical midbrain, pontine tegmental, brainstem and peduncular T2 signal hyperintensities | Mother and 2 brothers asymptomatic |
| 8 | 26/F | LS, learning disability, limb dystonia, regression of mobility and speech after the age of 9 yr, raised CSF lactate | NA | Basal ganglia lesions | Mother asymptomatic |
| 9 |
20/F Deceased |
Focal epilepsy, mild ID, failure to thrive, dysphagia, constipation, respiratory insufficiency with tracheostomy |
RRF Normal RCEA |
Bilateral and symmetrical T2 high signal in basal ganglia, white matter changes in the periventricular frontal region | NA |
| 10 | 52/F | Cognitive abnormalities, slowly progressive gait changes with onset at 30 yr | NA | NA | One affected brother (subject 11) |
| 11 |
45/M Deceased |
Gait changes with onset at 20 yr | NA | NA | One affected sister (subject 10) |
ADHD = attention deficit hyperactivity disorder; COX = cytochrome c oxidase; CSF = cerebrospinal fluid; ID = intellectual disability; INO = intra‐nuclear ophthalmoplegia; LS = Leigh syndrome; LVH = left ventricular hypertrophy; MRI = magnetic resonance imaging; NA = not available; RBBB = right bundle branch block; RCEA = respiratory chain enzyme activity; RRF = ragged red fibers.
TABLE 3.
Pathogenic Mitochondrial DNA Variants of Undetermined Clinical Relevance in the 11,424 Exomes Analyzed
| Subject | Age, yr /sex | mtDNA variant | Gene | No. ref reads | No. alt reads | Het (%) | Reported associated disease 9 | Subject phenotype/ investigations |
|---|---|---|---|---|---|---|---|---|
| 12 | 77/F | m.3243A>G | MT‐TL1 | 0 | 17 | 98 | MELAS/LS/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/cardiac + multi‐organ dysfunction |
IBM diagnosed 2012 Onset of symptoms aged 40 yr with grasp and proximal lower limb weakness Muscle biopsy; endomysial inflammatory cell infiltrate with invasion of non‐necrotic muscle fibers, fibers with rimmed vacuoles, ragged red and COX negative fibers |
| 13 | NA | m.3243A>G | MT‐TL1 | 10 | 4 | 29 | MELAS/LS/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/Cardiac and multi‐organ dysfunction | Adult subject with brain calcifications, no family history |
| 14 | 3/f | m.3260A>G | MT‐TL1 | 7 | 16 | 68 | MMC/MELAS |
Hypotonia since birth, severe intellectual disability, stereotyped hand movements, choreic movements, dystonic postures, anarthria MRI; thin corpus callosum with delayed myelination Confirmed heterozygous VAMP2 mutation |
| 15 | NA | m.3271T>C | MT‐TL1 | 51 | 20 | 30 | MELAS/DM | MSA; pathologically confirmed |
| 16 | NA | m.7497G>A | MT‐TS1 | 32 | 26 | 45 | MM/EXIT | MSA; pathologically confirmed |
| 17 | NA | m.8344A>G | MT‐TK | 54 | 107 | 65 | MERRF; Other ‐ LS/depressive mood disorder/Leukoencephalopathy/HCM | MSA; pathologically confirmed |
| 18 | NA | m.8851T>C | MT‐ATP6 | 22 | 5 | 21 | BSN/LS | Unaffected subject |
| 19 | NA | m.8993T>G | MT‐ATP6 | 20 | 28 | 58 | NARP/Leigh Disease/MILS/Other | PD |
| 20 | NA | m.9176T>C | MT‐ATP6 | 17 | 15 | 47 | FBSN/Leigh disease | Unaffected subject |
| 21 | NA | m.9185T>C | MT‐ATP6 | 16 | 5 | 25 | Leigh disease/Ataxia syndromes/NARP‐like disease | Extrapyramidal disorder |
| 22 | NA | m.11778G>A | MT‐ND4 | 4 | 11 | 73 | LHON/progressive dystonia | PD |
| 23 | 21/m | m.13513G>A | MT‐ND5 | 69 | 27 | 28 | Leigh disease/MELAS/LHON‐MELAS overlap syndrome | Cyclic vomiting syndrome |
ASD = autistic spectrum disorder; BSN = bilateral striatal necrosis; CPEO = chronic progressive external ophthalmoplegia; DM = diabetes mellitus; DMDF = diabetes deafness; EXIT = exercise intolerance; FBSN = familial bilateral striatal necrosis; FSGS = focal segmental glomerulosclerosis; HCM = hypertrophic cardiomyopathy; Het = heteroplasmy; IBM = inclusion body myositis; LHON = Leber hereditary optic neuropathy; LDYT = LHON and dystonia; LS = Leigh syndrome; MELAS = mitochondrial encephalomyopathy with lactic acidosis and stroke‐like episodes; MERRF = Myoclonic epilepsy with ragged‐red fibers; MIDD = Maternally inherited diabetes and deafness; MILS = maternally inherited Leigh syndrome; MM = mitochondrial myopathy; MMC = maternal myopathy and cardiomyopathy; MSA = multi‐system atrophy; mtDNA = mitochondrial DNA; NA = not available; NARP = neurogenic muscle weakness, ataxia, and retinitis pigmentosa; No. = number; PD = Parkinson's disease; SNHL = sensorineural hearing loss.
Clinically Significant Pathogenic Mitochondrial DNA Variants
Clinically significant pathogenic mtDNA variants were detected in 11 exomes from 9 unrelated families (mean age = 36 years; age range = 3–82 years; 36% female; see Tables 1 and 2). The variants broadly resided within 2 groups of genes: 7 pathogenic variants were detected in mitochondrial complex I subunit genes (MT‐ND1, MT‐ND3, MT‐ND5, and MT‐ND6); and 4 pathogenic variants resided in MT‐ATP6, encoding the ATP6 subunit of mitochondrial ATP synthase (complex V). Mutant levels of the MT‐ATP6 variants were uniformly high in blood (92–98%). This corresponded to predominantly central nervous system (CNS) phenotypes, including Leigh syndrome (LS) and Leigh‐like syndrome, which typically occur when the mutant load is ≥90%. 10 One individual survived into the 9th decade despite harboring 98% m.8851T>C levels, a mutant load previously linked with severe pediatric phenotypes. 11 , 12 Mutant mtDNA levels in exomes with pathogenic complex I subunit variants were more variable in blood (14–97%). There was also a less obvious correlation between the clinical phenotype and the underlying mutant load, given that all patients had severe CNS phenotypes (LS/Leigh‐like syndrome and mitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodes [MELAS]), despite some harboring relatively low heteroplasmy levels in blood. Variable neurological manifestations have previously been reported with low mutant MT‐ND5 levels, 13 thus reaffirming that pathogenic mtDNA variants measured peripherally (even in post‐mitotic tissues) do not always accurately reflect CNS levels.
Pathogenic Mitochondrial DNA Variants of Undetermined Clinical Relevance
Pathogenic mtDNA variants with undetermined clinical relevance were detected in 12 exomes from 12 unrelated families (see Table 3). The variants were located in mitochondrial tRNA genes (MT‐TL1, MT‐TS1, and MT‐TK), MT‐ATP6 and complex I subunit genes (MT‐ND4 and MT‐ND5). The range of mutant mtDNA levels in blood were as follows: mitochondrial tRNA genes (29–98%); MT‐ATP6 (21–58%); and complex I subunit genes (28–73%). None of the clinical manifestations reported were typical for the well‐recognized phenotypes associated with the detected pathogenic mtDNA variants, and, in some instances, an alternative diagnosis was confirmed pathologically (subjects 15, 16, and 17) or genetically (subject 14). Furthermore, one patient (subject 12) had a clinicopathological diagnosis of inclusion body myositis, despite harboring 98% mutant levels of the pathogenic m.3243A>G variant in MT‐TL1 associated with MELAS, and we are currently investigating the potential protective mechanisms preventing PMDs from manifesting in this individual.
Pathogenic Mitochondrial DNA Variants That Exhibit Variable Clinical Penetrance
Pathogenic mtDNA variants that exhibit variable clinical penetrance were detected in 41 exomes. This group comprised mtDNA mutations associated with Leber hereditary optic neuropathy (LHON; n = 12), deafness and/or aminoglycoside‐induced deafness (n = 27), and maternally inherited cardiomyopathy (n = 2; see Supplementary Table S2). None of the patients (apart from subject 50) exhibited visual failure, deafness, or cardiomyopathy, despite many carrying near homoplasmic or homoplasmic mutant mtDNA levels. Subject 50 had bilateral sensorineural hearing loss associated with a complex neurological phenotype that would not be accounted for by the m.1555A>G mtDNA mutation alone. The absence of clinical manifestations, despite many patients harboring near homoplasmic or homoplasmic mutant mtDNA levels, is in keeping with the variable penetrance characteristic of these recurrent mtDNA variants. 14
Discussion
Re‐analysis of 11,424 exomes identified 24 unique pathogenic mtDNA variants across 64 exomes. Clinically significant pathogenic mtDNA variants were detected in 11 exomes from 9 unrelated families, thus confirming an mtDNA‐related PMD diagnosis in these patients. Of these 11 individuals, PMD had been considered in 6 (subjects 4–9), but mtDNA analysis was not completed prior to WES. This was partly due to the absence of characteristic mitochondrial abnormalities observed in muscle (subjects 6 and 7), but also the limited access to diagnostic mtDNA NGS at the local recruiting centers. In 5 of the 11 patients, a PMD diagnosis was not contemplated prior to WES despite phenotypes that, in hindsight, were compatible with the underlying pathogenic mtDNA variants subsequently detected. These findings highlight the major challenges faced by neurologists when evaluating patients with PMDs, which results from the clinical overlap that exists between PMD phenotypes and other neurological disorders. There has also been a shift toward a “genetics first” approach to diagnosing genetic neurological diseases, through the application of large nuclear gene panels and WES. However, mtDNA analysis is not routinely included in standard clinical WES bioinformatic pipelines, despite pathogenic mtDNA (rather than nuclear DNA) variants accounting for the majority of adult PMDs. 3 Consequently, including mtDNA analysis when evaluating neurological disease exomes, represents an opportunity to capture genetically undiagnosed mtDNA‐related PMDs. Importantly, mtDNA NGS of a post‐mitotic tissue (eg, skeletal muscle) remains the “gold standard” molecular approach for excluding pathogenic mtDNA variants.
Muscle histopathology was available in 4 patients in whom PMD had been considered prior to WES; 2 patients (subjects 4 and 9) demonstrated typical pathological hallmarks of PMD (ragged red fibers), 1 patient exhibited a mild increase in lipid deposition only, albeit associated with reduced mitochondrial respiratory chain complex I enzyme activity (subject 6), and 1 patient was normal (subject 7). The most common brain magnetic resonance imaging (MRI) findings in patients with clinically significant pathogenic mtDNA variants included signal abnormalities of the basal ganglia and brainstem, in patients with Leigh/Leigh‐like syndrome (subjects 4, 7, 8, and 9) and MELAS (subject 6), and cerebellar atrophy (subjects 2, 4, and 5). Diffuse white matter changes were also observed in 2 patients (subjects 6 and 9).
Pathogenic mtDNA variants of undetermined clinical relevance were detected in 12 exomes from 12 unrelated families, whereas 41 exomes contained pathogenic mtDNA variants known to exhibit variable penetrance, despite near homoplasmic or homoplasmic mutant levels. The latter included pathogenic mtDNA variants associated with LHON (n = 12), deafness and/or aminoglycoside‐induced deafness (n = 27), and cardiomyopathy (n = 2). There was no evidence to suggest an underlying PMD in these individuals, and mutant mtDNA levels for some variants were below the threshold necessary for clinical symptoms to manifest (with the caveat that they were all detected in blood). However, there remains the possibility that disease onset could occur later in life or that subclinical manifestations are present (eg, cardiomyopathy or cardiac conduction defects). These findings have therefore been confirmed by diagnostic mtDNA NGS, with further analysis of a second tissue to assess risk, where appropriate. Reporting of secondary actionable findings (ie, genetic variants causing risk of an unrelated disease that would affect patient management) in large‐scale genetic testing is not straightforward, and mtDNA encoded genes are not currently included in the American College of Medical Genetics and Genomics (ACMG) medically actionable genes list. 15 , 16 However, the finding of a pathogenic mtDNA variant that potentially explains part or all of a clinical phenotype is within the remit of a primary finding, given it addresses the medical basis for requesting the test. There is no uniform definition of what is “medically actionable.” 17 Consequently, when a pathogenic mtDNA variant did not clearly contribute toward the disease (see Table 3), the recruiting clinician was informed to determine whether it was actionable for that individual and whether the patient's consent supported reporting of secondary findings.
One limitation of mtDNA analysis using WES data is the depth of mtDNA coverage achieved. Unlike clinical diagnostic laboratories, which use dedicated deep sequencing of mtDNA, 18 WES achieves comparatively low coverage of extracted mtDNA reads. Consequently, low‐level heteroplasmic mtDNA variants, particularly in blood, might remain undetected, despite the presence of higher, clinically relevant levels in post‐mitotic tissues. A second potential limitation is the spurious identification of a variant within a nuclear pseudogene that closely resembles an mtDNA gene – a so called “nuclear mitochondrial DNA segments” or “NUMTs.” 19 However, we simultaneously aligned reads to nuclear and mitochondrial genomes and confirmed pathogenic mtDNA variants by sequencing long‐range PCR enriched mtDNA, thus the risk of such errors is extremely low. 20
In conclusion, exome re‐analysis for pathogenic mtDNA variants in our cohort achieved a diagnostic uplift in 11 patients, thus highlighting the importance of including mtDNA analysis during the evaluation of neurological disease exomes.
Author Contributions
O.V.P., D.M., E.B., C.E.W., J.V., M.G.H., A.P., and R.D.S.P. contributed to study concept and design. O.V.P., C.P., D.M., M.F., W.L.M., E.B., C.E.W., R.L., S.E., V.S., V.C., R.K., R.M., A.A.A., A.G., S.J.K., H.J., N.W., H.H., J.V., M.G.H., A.P., and R.D.S.P. contributed to data acquisition and analysis. O.V.P., C.P., D.M., M.F., W.L.M., E.B., J.V., M.G.H., A.P., and R.D.S.P. drafted the text and figures.
The members of the SYNaPS Study Group are included in the Supplementary online Table S3.
Potential Conflicts of Interest
The authors declared no conflicts of interest.
Supporting information
Supplementary Table S1: Recurrent pathogenic mitochondrial DNA variants that exhibit variable clinical penetrance at near homoplasmic or homoplasmic mutant levels
Supplementary Table S2: Pathogenic mitochondrial DNA variants that exhibit variable clinical penetrance at near homoplasmic or homoplasmic mutant levels detected in the 11,424 exomes analyzed
Supplementary Table S3: Members of the SYNaPS Study Group
Acknowledgments
This research was conducted as part of the Queen Square Genomics Group at University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The University College London Hospitals/University College London Queen Square Institute of Neurology sequencing facility receives a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. The clinical and diagnostic “Rare Mitochondrial Disorders” Service in London is funded by the UK NHS Highly Specialized Commissioners. R.D.S.P. and M.F. are supported by a Medical Research Council (UK) Clinician Scientist Fellowship (MR/S002065/1). R.D.S.P., M.G.H., H.H., J.V., H.J., and W.L.M. receive funding from a Medical Research Council (UK) strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD) (MR/S005021/1). J.V. holds a fellowship from the Health Education England Genomics Education Programme. V.C. is funded by the Association of British Neurologists/Multiple System Atrophy Trust Clinical Research Training Fellowship. H.H. is grateful to the Medical Research Council (UK), the Wellcome Trust Synaptopathies Award, Ataxia UK, Rosetrees Trust, Brain Research UK, University College London Official Development Assistance and Low and Middle Income Country award, The Multiple System Atrophy Trust, Muscular Dystrophy UK, and Muscular Dystrophy Association.
Contributor Information
Robert D.S. Pitceathly, Email: r.pitceathly@ucl.ac.uk.
the SYNaPS Study Group:
Issam Alkhawaja, Selina Banu, Maria Bonsignore, Marianthi Breza, Gabriella Di Rosa, Morteza Heidari, Georgios Koutsis, Arn M.J.M. van den Maagdenberg, Alfons Macaya, Alexander Münchau, Carmela Scuderi, and Nazira Zharkinbekova
References
- 1. Macken WL, Vandrovcova J, Hanna MG, Pitceathly RDS. Applying genomics and trascnritpomic advances to mitochondrial medicine. Nat Rev Neurol 2021. 10.1038/s41582-021-00455-2. Online ahead of print. [DOI] [PubMed] [Google Scholar]
- 2. Liu P, Meng L, Normand EA, et al. Reanalysis of clinical exome sequencing data. N Engl J Med 2019;380:2478–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol 2015;77:753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stenton SL, Prokisch H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem 2018;62:399–408. [DOI] [PubMed] [Google Scholar]
- 5. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.0.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benjamin D, Sato T, Cibulskis K, et al. Calling somatic SNVs and indels with Mutect2. bioRxiv. 2019.
- 7. World Health Organization . International Statistical Classification of Diseases and Related Health Problems. Tenth Revision. Vol. 1: Tabular List (1992); Vol. 2: Instruction Manual (1993); Vol. 3: Index (1994). Geneva: WHO, 1992. [Google Scholar]
- 8. McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics 2010;26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brandon MC, Lott MT, Nguyen KC, et al. MITOMAP: a human mitochondrial genome database—2004 update. Nucleic Acids Res 2005;33:D611–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA‐associated Leigh syndrome and NARP. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. Seattle, WA: GeneReviews((R)), 1993. [PubMed] [Google Scholar]
- 11. De Meirleir L, Seneca S, Lissens W, et al. Bilateral striatal necrosis with a novel point mutation in the mitochondrial ATPase 6 gene. Pediatr Neurol 1995;13:242–246. [DOI] [PubMed] [Google Scholar]
- 12. Honzik T, Tesarova M, Vinsova K, et al. Different laboratory and muscle biopsy findings in a family with an m.8851T>C mutation in the mitochondrial MTATP6 gene. Mol Genet Metab 2013;108:102–105. [DOI] [PubMed] [Google Scholar]
- 13. Ng YS, Lax NZ, Maddison P, et al. MT‐ND5 mutation exhibits highly variable neurological manifestations at low mutant load. EBioMedicine 2018;30:86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giordano C, Iommarini L, Giordano L, et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber's hereditary optic neuropathy. Brain 2014;137:335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:249–255. [DOI] [PubMed] [Google Scholar]
- 16. Saelaert M, Mertes H, De Baere E, Devisch I. Incidental or secondary findings: an integrative and patient‐inclusive approach to the current debate. Eur J Hum Genet 2018;26:1424–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Royal College of Physicians, Royal College of Pathologists and British Society for Genetic Medicine . Consent and Confidentiality in Genomic Medicine: Guidance on the Use of Genetic and Genomic Information in the Clinic. Report of the Joint Committee on Genomics in Medicine. 3rd ed. RCP, RCPath and BSGM: London, 2019. [Google Scholar]
- 18. Zhang W, Cui H, Wong LJ. Comprehensive one‐step molecular analyses of mitochondrial genome by massively parallel sequencing. Clin Chem 2012;58:1322–1331. [DOI] [PubMed] [Google Scholar]
- 19. Samuels DC, Han L, Li J, et al. Finding the lost treasures in exome sequencing data. Trends Genet 2013;29:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Santibanez‐Koref M, Griffin H, Turnbull DM, et al. Assessing mitochondrial heteroplasmy using next generation sequencing: a note of caution. Mitochondrion 2019;46:302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Recurrent pathogenic mitochondrial DNA variants that exhibit variable clinical penetrance at near homoplasmic or homoplasmic mutant levels
Supplementary Table S2: Pathogenic mitochondrial DNA variants that exhibit variable clinical penetrance at near homoplasmic or homoplasmic mutant levels detected in the 11,424 exomes analyzed
Supplementary Table S3: Members of the SYNaPS Study Group