

Abstract
Auditory hair cells transduce sound to the brain, and in mammals, these cells reside together with supporting cells in the sensory epithelium of the cochlea, called the organ of Corti. To establish the organ's delicate function during development and differentiation, spatiotemporal gene expression is strictly controlled by chromatin accessibility and cell type–specific transcription factors, jointly representing the regulatory landscape. Bulk sequencing technology and cellular heterogeneity obscured investigations on the interplay between transcription factors and chromatin accessibility in inner ear development. To study the formation of the regulatory landscape in hair cells, we collected single-cell chromatin accessibility profiles accompanied by single-cell RNA data from genetically labeled murine hair cells and supporting cells after birth. Using an integrative approach, we predicted cell type–specific activating and repressing functions of developmental transcription factors. Furthermore, by integrating gene expression and chromatin accessibility data sets, we reconstructed gene regulatory networks. Then, using a comparative approach, 20 hair cell–specific activators and repressors, including putative downstream target genes, were identified. Clustering of target genes resolved groups of related transcription factors and was used to infer their developmental functions. Finally, the heterogeneity in the single-cell data allowed us to spatially reconstruct transcriptional as well as chromatin accessibility trajectories, indicating that gradual changes in the chromatin accessibility landscape are lagging behind the transcriptional identity of hair cells along the organ's longitudinal axis. Overall, this study provides a strategy to spatially reconstruct the formation of a lineage-specific regulatory landscape using a single-cell multi-omics approach.
The mammalian genome was estimated to encode approximately 30,000 protein-coding genes (Shabalina and Spiridonov 2004), and numerous unique combinations of the related gene products account for the molecular variety of the different cell types forming an organism. To orchestrate gene expression and to ensure proper differentiation of a given lineage, a delicate interplay between transcription factors (TFs) and chromatin accessibility controls the developmental program (Klemm et al. 2019). The transcriptional landscape from isolated tissues to whole organisms (Wagner et al. 2018; Packer et al. 2019) has been reconstructed using single-cell whole-transcriptome data and resulted in the development of a variety of bioinformatics algorithms (Bendall et al. 2014; Trapnell et al. 2014; Guo et al. 2017; Qiu et al. 2017b; Ellwanger et al. 2018). More recently, single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) protocols became available to investigate chromatin accessibility at matching resolution (Mezger et al. 2018). The technology identifies accessible chromatin regions, which potentially harbor regulatory elements that can be used to characterize genome-wide DNA/TF interactions (Buenrostro et al. 2013). In combination, single-cell RNA-seq (scRNA-seq) and scATAC-seq technologies offer the potential to resolve dynamic changes in the regulatory landscape (Buenrostro et al. 2018). However, data interpretation remains challenging owing to the sparsity of the scATAC-seq data and biological complexity of the model organism investigated.
The murine organ of Corti is an excellent model to study the development of the regulatory landscape because it consists of only two major cell types, sensory hair cells (HCs) and supporting cells (SCs), that originate from a shared progenitor (Xu et al. 2017). The two major cell types can be subdivided phenotypically (Corti 1851) as well as transcriptionally (Burns et al. 2015; Kolla et al. 2020) into inner HCs (IHCs), outer HCs (OHCs), and a number of different SC types. Tight temporal control is required for development of the organ of Corti, which occurs in waves both molecularly (Lee et al. 2006) and functionally (Lelli et al. 2009), starting at the base and extending toward the apex of the organ. Therefore, isolation of individual cells from the whole organ of Corti at a single developmental time point mirrors a continuum of differentiation, where cells isolated from the base are more mature compared with the cells from the apex. At the transcriptional level, those differences in gene expression were sufficient to reconstruct the organ of Corti in two-dimensional space from single-cell qPCR data (Waldhaus et al. 2015). The aim of our study is to identify the regulatory landscape controlling the differentiation and maturation of the organ of Corti. Here we provide a framework of how to analyze the regulatory landscape of HC differentiation using an integrative single-cell pipeline.
Results
Isolation of organ of Corti HCs and SCs
We generated scATAC-seq profiles from isolated HCs and SCs using fluorescence-activated cell sorting (FACS) in combination with the ATOH1-GFP (Rose et al. 2009)/FGFR3-iCRE (Young et al. 2010)/Ai14-tdTomato (Madisen et al. 2010) mouse line at postnatal day (P) 2 (Fig. 1A). ATOH1-GFP expression labeled sensory HCs, whereas conditional expression of tdTomato delineates two SC types, namely, Pillar cells (PCs) and Deiters’ cells (DCs) (Fig. 1A,B). Apical OHCs showed coexpression of GFP and tdTomato as previously reported (Waldhaus et al. 2015). After microdissection, we divided cochlear ducts into apical and basal compartments, dissociated the tissue to single-cell level, and performed FACS to enrich for HCs and PC/DCs (Fig. 1A,C; Supplemental Fig. S1A). Library preparation of sorted cells was performed using the 10x Genomics scATAC-seq platform, and upon application of stringent quality criteria, we yielded high-quality profiles of 1210 single cells (Fig. 1D). The cells clustered into six populations (Fig. 1E) with a median unique fragment count of 17,048 per cell (Supplemental Fig. S1B). Fragments were enriched at transcriptional start sites (TSSs) (Supplemental Fig. S1C), as well as in distal intergenic regions and introns (Supplemental Fig. S1D). The length of fragments accumulated at 100 bp and 200 bp, indicating nucleosome-free and mononucleosome-bound fragments (Supplemental Fig. S1E). To generate age-matched scRNA-seq data with an identical genetic background, we collected apical and basal compartments from P2 ATOH1-GFP/FGFR3-iCRE/Ai14-tdTomato cochlea for a second time. After single-cell dissociation, we performed FACS and applied less stringent gating criteria compared with the scATAC-seq sorting paradigm to ensure a complete overlap with the previously sorted populations. After library preparation using the 10x Genomics scRNA-seq platform, sequencing, and quality control, we analyzed the transcriptomes of 695 cells and identified 11 clusters (Fig. 1D,F) with a median unique count of 12,343 reads per cell (Supplemental Fig. S1F).
Figure 1.

scATAC-seq and scRNA-seq profiling of isolated HCs and PC/DCs. (A) Schematic representation of the experimental workflow used in this study. (B) Representative fluorescent reporter gene expression of ATOH1-GFP and FGFR3-tdTomato in whole-mount preparations of the organ of Corti at P2. Scale bar, 200 µm. (C) FACS plot and gating strategy to isolate cells expressing GFP and tdTomato. (D) Table summarizing sequencing libraries generated and total number of cells per library after quality control. (E,F) UMAP plots to show the clustering of all organ of Corti cells processed in two aggregated libraries to rule out technical variations for scATAC-seq (E) and scRNA-seq (F) experiments. (G) Expression heat map for 695 organ of Corti scRNA-seq cells (x-axis) and DEGs (y-axis). Shown are the top 100 DEGs for each of the 11 clusters identified. Cluster identities were determined based on DEGs known as canonical markers (also see Supplemental Fig. S2A) and indicated with a color bar at the bottom of the heat map. (H) Accessibility heat map for 1210 scATAC-seq cells. The top 100 DARs for each of the six clusters identified are shown, and cluster IDs are indicated with a color bar at the bottom of the heat map. (I) A Jaccard index similarity matrix reveals relations between scATAC-seq clusters and scRNA-seq clusters based on the overlaps between DEGs and annotated DARs. scATAC-seq cluster annotations were determined by the similarity to scRNA-seq clusters (color-coded as in E and F). (J) Enrichment of chromatin accessibility and expression level of candidate genes corresponding to their clusters. Accumulated scATAC-seq fragments at the individual gene locus (left column) and normalized gene expression levels in violin plots (right column) for the six scATAC-seq clusters. Arrowhead at the bottom of the plot indicates a position of a previously published organ of Corti–specific regulatory element (Wilkerson et al. 2019).
Identification of organ of Corti cell types using a similarity matrix
Cellular identities of scATAC-seq data were annotated based on similarities between the scATAC-seq and scRNA-seq clusters. First, we identified 11 clusters from scRNA-seq data using Seurat v3 (Stuart et al. 2019). Each of the 11 scRNA-seq clusters were compared with the remaining cells in order to establish a list of differentially expressed genes (DEGs) (Fig. 1G). Overall, we identified a total of 5772 DEGs with a median number of 428 DEGs per cluster. Known markers among the DEGs were used to determine scRNA-seq cluster identities (Supplemental Fig. S2A). Periotic mesenchyme markers like Pou3f4 and Tbx18 (Phippard et al. 1998; Trowe et al. 2008) were among the genes differentially expressed in cluster 1, whereas Fabp7, Mpz, and Egfl8 (Wang et al. 2013; Weiss et al. 2016; Suzuki et al. 2019) in cluster 2 indicated Schwann cell identity. PCs and DCs were represented in cluster 3 as shown by Fgfr3, Prox1, and Hes5 expression (Bermingham-McDonogh et al. 2006; Hartman et al. 2009; Hayashi et al. 2010). Endothelial cells and melanocytes were captured in clusters 4 and 5 as evidenced by the expression of Cldn5, Cdh5, and Sox17 (Gory-Fauré et al. 1999; Morita et al. 1999; Zhou et al. 2015) and of Gsta4, Pmel, and Ptgds (Takeda et al. 2006; Uehara et al. 2009; Hellström et al. 2011), respectively. Atoh1, Pou4f3, and Gfi1 (Xiang et al. 1997; Wallis et al. 2003; Woods et al. 2004) were DEGs of sensory HCs characteristic for cluster 6, whereas lateral SCs in cluster 7 expressed Gata2, Fst, and Hs3st1 (Lilleväli et al. 2004; Hartman et al. 2015; Son et al. 2015). Cluster 8 represented proliferating Schwann cells distinguished by Fabp7, Top2a, and Cdc20 (Fuhrmann et al. 2018; Jessen and Mirsky 2019) expression. Crabp1, Slc12a2, and Atp1b1 (Kolla et al. 2020; Mutai et al. 2020) indicated medial SCs in cluster 9. Cells in cluster 10 represented roof structures consisting of Reissner's membrane and stria vascularis based on Oc90, Otx2, and Cldn8 expression (Kitajiri et al. 2004; Hartman et al. 2015; Vendrell et al. 2015). Finally, immune cells expressed markers like Lyz2 and C1qa (van Schaarenburg et al. 2016; Cochain et al. 2018) in cluster 11. Next, we identified six clusters from scATAC-seq data, and cluster-specific differentially accessible regions (DARs) were determined using SnapATAC (Fig. 1H; Fang et al. 2021). In summary, we identified a total of 67,415 DARs with a median number of 6495 DARs per cluster. Subsequently, a proximity-based approach was used to annotate DARs to genes, which allowed us to compare similarities between DEGs and annotated DARs using a Jaccard index similarity matrix (Fig. 1I). scATAC-seq cluster 1 showed the highest similarity to the scRNA-seq HC cluster, whereas scATAC-seq cluster 2 corresponded to the PC/DC cluster. scATAC-seq clusters 3 through 6 were identified as roof, mesenchyme, endothelial, and immune cells based on their similarities to the scRNA-seq clusters, respectively. Medial and lateral SC-, as well as Schwann cell- and melanocyte-scRNA-seq clusters, did not show high similarities to any of the scATAC-seq clusters, probably owing to the lenient flow sorting strategy applied to the scRNA-seq samples. To validate the accuracy of the similarity-based approach, we integrated scRNA-seq and scATAC-seq for joint alignment analysis using LIGER (Supplemental Fig. S2B,C; Welch et al. 2019). Projecting the similarity-based approach identities onto the LIGER coembedding UMAP revealed high similarity in cell type annotations for both approaches (Supplemental Fig. S2C). Because of the larger number of scATAC-seq cells, the LIGER clustering was dominated by scATAC-seq cells, which potentially obscured the identity of smaller clusters. Therefore, the Jaccard similarity matrix provided an efficient approach to identify and annotate cell types represented by small numbers of cells.
Generally, DARs are likely to be enriched in gene bodies and gene regulatory elements controlling expression of individual transcripts. To test the quality of our scATAC-seq data, we identified five DARs overlapping with previously published organ of Corti–specific enhancer elements (Supplemental Fig. S2D; Wilkerson et al. 2019). Next, we visualized cluster-specific candidate genes contributing to the Jaccard similarity matrix. We plotted chromatin accessibility around the gene body and transcript levels, accordingly (Fig. 1J). Peaks were accumulated and normalized by fragment pileup per million reads for better comparison. Pou4f3 is a known HC-specific marker gene (Xiang et al. 1997), and called peaks at the Pou4f3 locus resolved accessible chromatin in HCs, whereas chromatin in PC/DCs and the other cell types was found in a closed conformation at the same locus. Similarly, the Pou4f3 transcript was only detected in the HC population. Hes5, on the other hand, is a known cochlear PC/DC marker gene (Hartman et al. 2009) and showed cell type–specific chromatin accessibility accompanied by PC/DC-specific expression of the transcript. Cldn8, Tbx18, Cldn5, and C1qa show complementary chromatin accessibilities and transcript expression levels that are known to delineate roof, mesenchymal, endothelial, and immune populations, respectively.
Inference of TF activity in maturating HCs and PC/DCs
HCs and PC/DCs of the organ of Corti develop from a shared progenitor starting around embryonic day (E) 14.5 (Chen et al. 2002). We aimed to identify TFs controlling differentiation of the two cell types and to infer their function in a combinatorial approach from postnatal data. The function of a TF is highly context dependent; nevertheless, it can generally be categorized as a transcriptional activator or repressor. We adopted two assumptions that were previously developed (Berest et al. 2019) to classify TF activity: (1) Upon binding of an activating TF, chromatin accessibility at the regulatory element is increased, resulting in an up-regulation of the respective target gene transcript, and (2) conversely, binding of a repressing TF decreases average chromatin accessibility at the regulatory element and down-regulation of the target gene will occur. Based on the assumptions, we established a classification model tailored toward single-cell experiment workflow.
First, by comparing scRNA-seq data from HC and PC/DC populations, we identified 93 DEGs (P-adjusted < 0.05) encoding TFs such as Lhx3, Gfi1, Sox2, and Tgif1 (Fig. 2A). Next, we calculated TF motif accessibility z-scores using chromVAR (Schep et al. 2017). Briefly, the algorithm quantifies normalized motif accessibility with a z-score by aggregating accessible regions per given TF motif across the whole genome of a single cell. Using TF motifs as features, we visualized clusters in UMAP (Fig. 2B), which faithfully reproduced populations previously identified by SnapATAC. When HC and PC/DC clusters were compared, we identified 353 differentially accessible TF motifs (P-adjusted < 0.05) (Fig. 2C). To infer activating or repressing mode of action, we plotted mRNA fold change versus z-score fold change for the TFs investigated (Fig. 2D). Activating TF function, resulting in chromatin opening upon binding, was inferred if a given TF was differentially expressed in one of the two populations and the corresponding motif was significantly accessible in the same population. A repressor was classified based on its differential expression in combination with a negatively correlated z-score, indicating chromatin closure upon binding. Comparing the HC and PC/DC populations, we identified 56 activators and 23 repressors. To assess the classification performance, we adopted the previously published diffTF algorithm (Berest et al. 2019) and compared the overlap between the two methods (Supplemental Fig. S3A–D). To perform the diffTF analysis, pseudobulk samples corresponding to four biological replicates were generated by aggregating scRNA-seq and scATAC-seq data from the HC and PC/DC clusters, respectively. Using diffTF, we identified 36 activators and 10 repressors. Overall, we found 72% of the diffTF classifications was identical to the current approach. Next, we visualized expression levels and chromatin accessibility for four different HC and PC/DC activators and repressors. TF LHX3 was classified as a transcriptional activator in HCs (Fig. 2D,E), and previously reported differential expression in HCs (Hertzano et al. 2007) was recapitulated, accompanied by differential motif accessibility in HCs as well. In addition, we used the HINT-ATAC algorithm (Li et al. 2019) to generate a footprint from cluster-aggregated scATAC-seq data using HOCOMOCO v10 database (Kulakovskiy et al. 2013), which showed increased chromatin accessibility adjacent to the LHX3 binding site in HCs compared with PC/DCs. Together, these findings support the role of LHX3 as a transcriptional activator in HCs. On the other hand, GFI1 (Fig. 2D,F) is a known zinc-finger transcriptional repressor in HCs (Wallis et al. 2003). Gfi1 transcripts were differentially expressed in HCs; however, the GFI1 z-score, supported by the footprint, indicated significantly lower motif accessibility in HCs compared with PC/DCs, resulting in a repressor classification. TF SOX2 is critical for organ of Corti development (Kiernan et al. 2005) and was identified as a transcriptional activator in PC/DCs based on its positive correlation of mRNA expression and motif accessibility (Fig. 2D,G). Conversely, TGIF1 is a known transcriptional repressor (Shen and Walsh 2005) and was characterized by a negative correlation between mRNA expression level and z-score in developing PC/DCs (Fig. 2D,H). To evaluate the quality of the curated HOCOMOCO v10 motifs, we reran the HINT-ATAC algorithm using the JASPAR 2020 database (Fornes et al. 2020) and obtained virtually identical footprints (Supplemental Fig. S3E–G). In summary, we classified 23 TFs regulating HC development subdivided into six activators and 17 repressors. With respect to PC/DCs, we identified 56 TFs segregating into 50 activators and six repressors.
Figure 2.

Transcriptional activator and repressor classification. (A) Volcano plot of differentially expressed TF genes between the HC and PC/DC clusters (P-adjusted < 0.05). (B) UMAP of TF motif accessibility z-scores calculated from chromVAR. Cells are color-coded based on SnapATAC clusters. (C) Volcano plot of differential TF motif accessibilities calculated from z-scores between the HC and PC/DC clusters (P-adjusted < 0.05). (D) Dot plot of TF classification shown in average log2 fold change (FC) mRNA level and z-scores. The differential expression from scRNA-seq between HCs and PC/DCs is plotted on the x-axis, and the differential accessibility from scATAC-seq is shown on the y-axis. Activators are classified in green, repressors in red, and undetermined TFs in gray. (E–H) TF activities in differentiating HCs and PC/DCs in terms of mRNA expression, chromatin accessibility, and footprints. (E) LHX3, a transcriptional activator in HCs. (First row) Violin plot of RNA expression. Each dot represents a single cell. (Second row) UMAP plot of LHX3 motif accessibility calculated as z-score. Red indicates higher accessibility compared with blue. Each dot represents a single cell. (Third row) LHX3 footprint calculated from scATAC-seq data using HINT-ATAC. Activators are characterized by high scATAC-seq signal in the flanking region of the TF binding sites compared with the control population. Yellow line represents the HC cluster, and orange line represents the PC/DC cluster. (Fourth row) Mouse HOCOMOCO v10 consensus sequence for the LHX3 motif. (F–H) Analogous data representation for (F) GFI1, classified as a HC repressor; (G) SOX2, a PC/DC activator; and (H) TGIF1, a repressor in PC/DCs.
TFs controlling HC and SC differentiation
TFs shape the epigenetic landscape of a given cell and direct differentiation through tight transcriptional control of downstream target genes, commonly summarized as regulons. We predicted TF-specific regulons at the whole-transcriptome level by leveraging the scRNA-seq and scATAC-seq data sets. First, coexpression modules of previously classified TFs and their respective downstream targets were identified by analyzing the scRNA-seq data using GENIE3 (Huynh-Thu et al. 2010). To determine activator regulons, we filtered for positively correlated target genes, whereas repressor regulons were constituted from negatively correlated target genes only.
At this point, the list of correlated genes included potential direct and indirect target genes of the respective TFs. Next, we screened the loci of potential target genes for accessible TF binding sites using FIMO (Grant et al. 2011). Links lacking TF binding sites in accessible peaks were considered indirect targets and got removed from the regulon. Once TF-specific regulons were identified, a regulon enrichment score was calculated for each cell using AUCell (Aibar et al. 2017). Overall, regulon activities were visualized for 70 TFs previously categorized as transcriptional activators and repressors (Fig. 3A).
Figure 3.

TFs controlling HC and PC/DC differentiation. (A) AUC enrichment matrix with hierarchical clustering at single-cell resolution revealed the regulon activities during differentiation of HCs and PC/DCs. A regulon summarizes putative downstream target genes as a group of the respective TF. The activity of the regulon is color-coded from blue (depletion) to red (enrichment). Hierarchical clustering reveals similarities between individual cells (x-axis) and between different regulons (y-axis). Color bars on the top and to the side of the heat map indicate library ID, cell type, mode of action, and cell type specificity. (B) Venn diagram of the number of overlapped downstream target genes between the SOX9 regulon and GFI1 regulon. (C,D) Violin plots of Sox9 (C) and S100b (D) expression level. The two genes are representatives of overlapping downstream target genes between the SOX9 and GFI1 regulons. (E,F) Coaccessibility analysis of Sox9 and S100b loci in the PC/DC cluster using Cicero. (E) In PC/DCs, Sox9 TSS is directly and indirectly connected to the accessible sites. They correspond to predicted regulatory elements that contain putative TF binding sites for SOX9 and GFI1, as determined by FIMO motif scanning. (First row) Genome annotation from UCSC Known Genes. (Second row) Coaccessibility plot connects predicted regulatory elements with the TSS. (Third row) Accessible regions aligned with the locus. (Forth row) Putative TF binding sites of SOX9 and GFI1 motifs relative to the accessible regions. TSS position is indicated with a dashed line. (F) Analogous data representation for the S100b locus. (G) Overlapping downstream target genes between known HC transcriptional activators ATOH1 and NHLH1. (H) Zbtb18 is a shared target gene between the ATOH1 and NHLH1 regulons with accessible TF binding sites for both TFs at the TSS in HCs. (I) Immunostaining of ZBTB18 protein expression in IHCs and OHCs in cryosections of the organ of Corti (P2). Arrowhead pointing at the IHC. Bracket delineates OHC location. Scale bar, 20 µm.
Given the lengthy experimental procedure, we aimed to exclude a potential bias of the AUC enrichment matrix toward stress-related regulons. We screened for TFs that were associated with the Gene Ontology term “Stress Response” (GO:0006950) and identified 25 TFs. However, 22 out of the 25 stress-associated genes, like Ils1 (Radde-Gallwitz et al. 2004) and Gata3 (Luo et al. 2013), were previously cited in the context of organ of Corti development as well (Supplemental Table S1). The remaining three genes, namely Atf3 (Maeda et al. 2020), Hif1a (Chung et al. 2004), and Zbtb7a (Yang et al. 2015), were previously published to be differentially expressed after noise exposure and may change their expression in response to the dissection procedure.
To validate the performance of the developed approach, we compared the SOX2 regulon with previously published SOX2 ChIP-seq data from an inner ear cell line (Kwan et al. 2015). Sixty-eight percent and 29% of the SOX2 regulon–associated genes were present in two independently performed ChIP-seq experiments (Supplemental Fig. S4A).
Hierarchical clustering of the AUC enrichment matrix resolved HC and PC/DC clusters (Fig. 3A), and GO term analysis of cluster-specific regulons revealed terms such as auditory receptor differentiation in HCs and positive regulation of cell proliferation in PC/DCs (Supplemental Fig. S4B,C). Clustering also revealed the relations between different TFs with respect to their regulons. The regulons of transcriptional activator SOX9 and repressor GFI1 were closely related with an overlap of 71 target genes (Fig. 3B). All 71 overlapping genes were confirmed to be differentially expressed in the PC/DC cluster (Fig. 1G) and thus represented a portion of the PC/DC-specific transcriptome to be repressed in Gfi1-positive HCs. For further analysis, we focused on S100b and Sox9 genes, which were differentially expressed in PC/DCs and were among the shared targets between the SOX9 and GFI1 regulons (Fig. 3B–D). Using Cicero (Pliner et al. 2018), the cis-regulatory landscape of Sox9 in the PC/DC cluster was reconstructed, and motif scanning identified accessible SOX9 binding sites in the predicted Sox9 regulatory elements, suggesting autoregulation (Fig. 3E). SOX9 motifs were also found in the regulatory elements of S100b (Fig. 3F). In the absence of the repressor GFI1 in PC/DCs, GFI1 motifs in the regulatory elements of S100b and Sox9 were accessible. Conversely, expression of Gfi1 in HCs rendered its binding sites and flanking regions inaccessible to prevent the expression of the PC/DC-specific genes in HCs (Supplemental Fig. S4D,E). Similarly, regulatory elements containing SOX9 motifs were not accessible in HCs.
As the AUC enrichment matrix successfully resolved TFs contributing to the regulatory landscape of HC and PC/DC differentiation, the matrix was used as a discovery tool. The clustering allowed categorization of TFs in groups controlling a similar set of target genes. Nhlh1, for example, was previously identified in an RNA-based screening to be expressed in developing HCs (Scheffer et al. 2015). The functional context of TF NHLH1 was not investigated. However, the NHLH1 regulon was closely related to the ATOH1 regulon with an overlap of 665 target genes (Fig. 3A,G), suggesting synergistic effects in early HC differentiation. Conversely, the ZBTB18 regulon clustered further away from the ATOH1 regulon and clustered more similarly to the LHX3 regulon, which is expressed later during HC development (Fig. 3A; Hertzano et al. 2007). ATOH1 and NHLH1 TF binding sites at the Zbtb18 TSS indicate that the gene is a shared target (Fig. 3H). Protein expression of ZBTB18 was confirmed (Fig. 3I), and regulon-associated genes such as Cdh23, Cib2, Espn, Myo7a, Pcdh15, Tmie, Ush2a, and Whrn (Fettiplace 2017), among others, suggest a role for Zbtb18 in hair bundle formation. In summary, the AUC enrichment matrix was used to visualize regulon activities for 16 HC-associated and 54 PC/DC-associated TFs at single-cell resolution.
Spatial reconstruction of HCs from scATAC-seq and scRNA-seq data
Organ of Corti development proceeds in gradients, in which cells located in the base of the organ are more mature compared with the cells in the apex (Chen et al. 2002; Lee et al. 2006). To further analyze the effects of differentiation on the regulatory landscape of HCs, we aimed to reconstruct individual HC's anatomical position from scRNA-seq and scATAC-seq data. We followed the conceptual idea previously published for reconstruction of the mouse organ of Corti from single-cell qPCR data (Waldhaus et al. 2015). During the process of dissection (Fig. 4A), flow sorting (Fig. 4B), and library preparation, the apical and basal compartments of the organ of Corti were processed separately in both scRNA-seq and scATAC-seq experiments (Fig. 4C,D). In total, 427 DEGs (P-value < 0.005) (Fig. 4E) and 147 DARs (P-value < 0.001) (Fig. 4F; Supplemental Fig. S5A,B) between the apical and basal compartments were identified. Using the DEGs and DARs as features, we were able to project the HCs in a one-dimensional (1D) PCA along the y-axis (Fig. 4G,H). The distribution along the x-axis reflects random jitter for better visualization. HCs were plotted according to their rank order, which resolved the relative position of each individual cell along the apex-to-base axis for both scRNA-seq and scATAC-seq data. For validation of the spatial reconstruction, library IDs were plotted onto the 1D spatial reconstruction maps. Pkhd1l1 was among the DEGs and annotated DARs with expression and accessibility gradients predicted to be significantly higher in basal HCs compared with the apical counterparts. Graded expression of Pkhd1l1 transcript was previously reported (Wu et al. 2019), and was reproduced by RNA staining (Fig. 4I,J).
Figure 4.
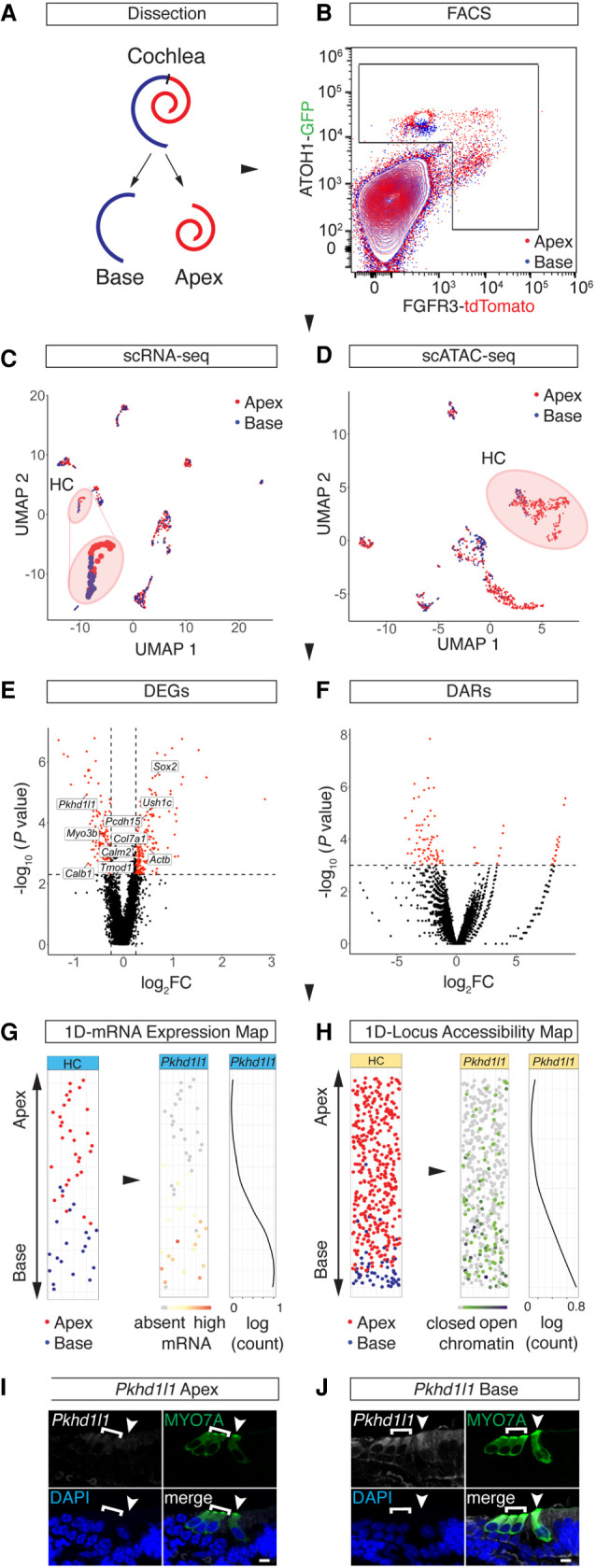
Spatial reconstruction of HC origins along the longitudinal axis. (A) Schematic representation of the sampling strategy used in this study. Color code is as follows: apex, red; base, blue. (B) FACS plot and gating strategy to isolate GFP- and tdTomato-expressing cells. Color code identifies compartmental identities (color code same as in A). (C,D) UMAP projections of all cells analyzed in the scRNA-seq (C) and scATAC-seq (D) experiments with color code for library ID (color code as in A and B). HC populations are highlighted with a circle and are magnified for better visibility in C. Dots correspond to single cells. (E,F) Volcano plots of DEGs (E) and DARs (F) comparing apical and basal compartments. scRNA-seq cutoff: P < 0.005 and absolute value of log2FC > 0.25. scATAC-seq cutoff: P < 0.001. (G,H) 1D spatial reconstruction of single-cell transcript expression levels and chromatin accessibilities. (G) 1D HC expression map. (Left) 1D PCA based on the DEGs shown in E. y-Axis resolves predicted apex (top) to base (bottom) axis. Data points are randomly spread along x-axis for better visibility. Dots correspond to single cells. Color code depicts library ID (same as in A–D). (Middle) Gene expression level of Pkhd1l1 projected onto the 1D expression map. (Right) Pkhd1l1 expression fitted into a regression line. y-Axis corresponds to the apex-to-base axis; x-axis, to expression level shown in log counts. (H) Analogous data representation as in G, showing 1D accessibility map with library ID and Pkhd1l1 accessibility projected. (I,J) RNAscope staining of Pkhd1l1 transcript comparing HCs of apical (I) and basal (J) origin. HCs were counter-stained with anti-MYO7A and DAPI nuclear stain. IHC (arrowhead) and OHC (bracket) staining using identical imaging settings. Scale bar, 10 µm.
Chromatin dynamics during HC differentiation
Around birth, segregation into IHCs and OHCs is distinct, based on anatomical position and differential gene expression (Kolla et al. 2020). IHC and OHC subclusters, with respect to the scRNA-seq data, were identified based on a set of previously published marker genes (Fig. 5A; Waldhaus et al. 2015). Differential gene expression analysis revealed 169 IHC-specific and 115 OHC-specific genes (P < 0.01) (Fig. 5B). Plotting cluster ID, library ID, and DEGs like Fgf8 and Cdh1 onto the 1D spatial expression map allowed for visualization of cell type–specific transcriptomes spanning the longitudinal axis of the developing organ of Corti (Fig. 5C). However, at the chromatin level further subclustering using z-scores appeared to be driven by the original position along the apex-to-base axis rather than IHC and OHC identities. To visualize this observation, we projected DAR-based rank order, library ID, and z-scores of the OHC-specific TF INSM1 (Wiwatpanit et al. 2018) onto the UMAP (Fig. 5D; Supplemental Fig. S6A,B). To test if IHC- and OHC-specific chromatin remodeling occurs along the tonotopic axis around birth, we reconstructed a HC-specific trajectory based on z-scores (Fig. 5E; Supplemental Fig. S6C). Using CellTrails (Ellwanger et al. 2018), single HCs were aligned on a Y-shaped trajectory representing four individual states. To validate the trajectory prediction, Slingshot (Street et al. 2018) and Monocle (Trapnell et al. 2014; Qiu et al. 2017a,b) were used (Supplemental Fig. S6D,E). Projecting DAR-based rank order onto the CellTrails map (Fig. 5F) resolved apical identity for state S4, which corresponded to the smaller of the two subclusters seen in the UMAP projection (Fig. 5D; Supplemental Fig. S6C). Basal rank order identities related to states S2 and S3. Similarly, library IDs were distributed asymmetrically along the trajectory (Supplemental Fig. S6F). States S1 and S4 were constituted by cells dissected from the apex only, whereas S2 and S3 contained both apical and basal HCs. Given the gradual differentiation along the apex-to-base axis, we hypothesized that S4 may correspond to an apical, immature HC state and that S2 and S3 may represent more mature IHC and OHC states at the base. To test this hypothesis, we first visualized differential Atoh1 mRNA expression increasing from the base toward the apex (Fig. 5G). Next, differential z-scores between the four states were determined in pairwise comparisons using Wilcoxon sum rank tests. In support of the hypothesis that states S4 corresponds to the apical end of the organ of Corti, we found that both the ATOH1 z-score and the footprint were significantly more accessible in state S4 compared with S2 and S3. To further validate our hypothesis that S2 and S3 may correspond to basal IHCs and OHCs, we focused on the TF INSM1, a transcriptional repressor expressed in OHCs (Fig. 5H; Wiwatpanit et al. 2018). Differential expression of Insm1 mRNA was confirmed. Consistent with the repressive function of INSM1, state S3 showed lower z-scores compared with S2; and the footprint was less accessible in state S3 compared with S2 as well. Together, these findings imply OHC identity for cells representing state S3. Aiming to test if state S2 represented IHCs, we visualized the IHC marker Hivep2’s mRNA levels and motif accessibility (Fig. 5I). Although the function of HIVEP2 in HC development remains to be determined, differential expression has been reported for IHCs and OHCs (Li et al. 2016). The current study confirms the differential mRNA expression in IHCs and confirms that the HIVEP2 motif was significantly more accessible in state S2 compared with S3. These findings suggested HIVEP2 may function as a transcriptional activator in IHCs. In summary, the data presented support the hypothesis that state S4 represented apical immature HCs whereas S2 and S3 corresponded to IHCs and OHCs, respectively.
Figure 5.

Developmental bifurcation of IHCs and OHCs. (A) UMAP projection of scRNA-seq HCs delineates IHCs and OHCs subclusters. A single dot represents a single cell. (B) Volcano plot of DEGs (P < 0.01) between IHC and OHC clusters. (C) 1D spatial reconstruction map with cell identity (first column) and library ID (second column) projected. Expression levels of IHC-specific gene Fgf8 (third column) and OHC-specific gene Cdh1 (fourth column) were projected onto the 1D spatial reconstruction map. Dashed line delineates IHCs (left) from OHCs (right). (D) UMAP plot of HC cluster from scATAC-seq data with projection of spatial rank order as determined by 1D spatial reconstruction map. (E,F) Trajectory reconstruction based on scATAC-seq z-scores using CellTrails. CellTrails states (E) and DAR-based rank order (F) projected onto the trajectory. (G,I) Comparative analysis of selected TFs in terms of mRNA expression, motif accessibility, and footprints. (G, top left column) Atoh1 mRNA expression projected onto the 1D spatial reconstruction map. Dashed line delineates IHCs (left) from OHCs (right). (Bottom left column) Violin plots with mRNA levels for IHCs and OHCs. (Top right column) Contour plot of ATOH1 z-scores with CellTrails trajectory in the background. (Bottom right column) ATOH1 footprint from scATAC-seq data for selected CellTrails states (same color code as in E). ATOH1 consensus sequence is depicted at the bottom left of the footprint plot. (H,I) Analogous data representation for INSM1 (H) and HIVEP2 (I).
TFs controlling IHC and OHC differentiation
Using the AUC enrichment matrix, we aimed to identify novel IHC- and OHC-specific TFs in order to add to our knowledge of the regulatory landscape during HC maturation. A total of seven differentially expressed TFs (P-adjusted < 0.05) were identified (Fig. 6A). Given the small number of IHCs identified from the scRNA-seq data, we confirmed robustness of the seven TFs identified by comparison with previously published bulk RNA-seq data from perinatal and adult IHCs and OHCs (Supplemental Fig. S6G,H; Li et al. 2018; Wiwatpanit et al. 2018). Next, 254 differential z-scores (P-adjusted < 0.05) were calculated (Fig. 6B) from scATAC-seq data. Correlation of expression level with motif accessibility correctly annotated INSM1 as an OHC repressor and HIVEP2 as an IHC activator (Fig. 6C). In addition, one OHC activator (TCF4) and two IHC activators (FOXO4 and GLIS3) were categorized and visualized in the AUC enrichment matrix (Fig. 6D). Projecting mRNA levels and z-scores of OHC activator TCF4 revealed differential expression between IHCs and OHCs, and a major gradient in TCF4 motif accessibility was observed along the apex-to-base axis (Fig. 6E). TCF4-antibody staining revealed qualitative differences, in the nucleus as well as the cytoplasm, between IHCs and OHCs (Fig. 6F). Foxo4 mRNA was differentially expressed in IHCs, and the FOXO4 motif was differentially accessible in state S2 compared with S3 (Fig. 6G). FOXO4-antibody staining revealed the presence of FOXO4 in the nucleus and the cytoplasm of IHCs and OHCs (Fig. 6H). Glis3 mRNA was differentially expressed in IHCs, and the GLIS3 motif was differentially accessible in state S2 compared with the OHC state S3 (Fig. 6I). Nuclear staining was present in IHCs with GLIS3 antibody (Fig. 6J). Together, these findings reveal dynamic changes in the regulatory landscape of IHCs and OHCs along the apex-to-base axis.
Figure 6.

TFs controlling IHC and OHC differentiation. (A) Volcano plot of differentially expressed TF genes (P-adjusted < 0.05) between IHCs and OHCs. (B) Volcano plot of differentially accessible TF motifs (P-adjusted < 0.05) comparing IHC and OHC clusters. (C) Dot plot of TF classification shown in average log2FC mRNA transcripts and z-scores. The differential expression between IHCs and OHCs is plotted on the x-axis, and differential accessibility is shown on the y-axis. Activators are classified in green, repressors in red, and undetermined TFs in gray. (D) AUC enrichment matrix of TF regulons contributing to IHC/OHC segregation. Color bars on the top and to the side of the heat map indicate library ID, cell type as determined based on DEGs, and mode of action. (E,G,I) Comparative analysis of selected TFs in terms of mRNA expression, motif accessibility, and footprints. Analogous data representation as in Figure 5, F and G. (E) OHC activator TCF4. (F) Anti-TCF4 staining localizes to OHC nuclei and cytoplasm. HCs are counter-stained with anti-MYO7A and DAPI nuclear stain. Arrowhead points at IHC nucleus; bracket highlights OHC region. Scale bar, 10 µm. (G) IHC activator FOXO4. (H) Anti-FOXO4 staining in IHC and OHC cytoplasm and nuclei. Counter-stain, scale bar, and labeling analogous to F. (I) IHC activator GLIS3. (J) Anti-GLIS3 in IHC and OHC cytoplasm and nuclei. Counter stain, scale bar, and labeling analogous to F.
Discussion
Current single-cell-based RNA-seq and ATAC-seq protocols resolve thousands of genes and accessible chromatin regions per individual cell. This is in stark contrast to many classic developmental tool sets focusing on gene expression and regulatory networks such as ChIP-seq, knockout, or overexpression studies, resolving one candidate gene at a time. The amount of data generated using novel sequencing protocols provides information at steadily increasing resolution. Therefore, there is a need for novel data analysis strategies to provide a biological context for the accumulating information. The AUC enrichment matrix presented in this study identified 20 regulons contributing to the differentiation of sensory HCs. Previous whole-transcriptome-based studies (Burns et al. 2015) resolved each of the 20 TFs to be expressed in the developing organ of Corti as well. However, based on previous study design, their roles in inner ear development remained elusive. Here we present an integrative approach, combining scRNA-seq and scATAC-seq technology, to compare cell types that develop from shared progenitors. Analyzing differentially expressed TFs that showed differential motif accessibility allows us to robustly resolve how multiple TFs function in concert to activate or repress cell type–specific target genes at the whole-transcriptome level. The organ of Corti was used as a model owing to its developmental gradient along the longitudinal axis. The same analytical framework can be applied to other organ systems in which the cells analyzed share a common progenitor.
The scRNA-seq and scATAC-seq data were generated from age-matched, genetically labeled HCs and PC/DCs of the organ of Corti. To integrate both data sets, an AUC enrichment matrix was computed in a sequential workflow. We leveraged existing algorithms and developed novel elements to study HC and PC/DC differentiation and started by predicting activating and repressing function of differentially expressed TF genes. Existing algorithms like diffTF (Berest et al. 2019) were developed for bulk-based experiments comparing two groups with multiple biological replicates using Pearson's correlation analysis. We tested this approach with pseudobulk samples from single-cell data and identified a number of activator and repressor TFs in each cell type. However, diffTF failed to classify key TFs previously described for inner ear development, likely owing to the limitations of cell numbers and sample numbers. To overcome this challenge, we adopted the hypothesis that upon binding of an activator, the flanking regions of the TF binding sites would open, and conversely, binding of a repressor would decrease the average accessibility at the regulatory elements controlled by the TF. Therefore, we compared fold changes in TF expression levels with TF motif accessibility. This approach successfully identified known TF activators and repressors, like LHX3 and GFI1, with remarkable robustness and helped us classify 79 known and unknown TFs contributing to organ of Corti development. Although this classification algorithm virtually considers all TFs expressed in the scRNA-seq data set, it is limited by the motif database used to calculate motif accessibilities using chromVAR. Specifically, previously published TFs like POU4F3 and IKZF2 (Xiang et al. 1997; Chessum et al. 2018) were not annotated in the mouse HOCOMOCO v10 database and therefore were not considered in this study.
To calculate the AUC enrichment matrix, general concepts from the SCENIC algorithm were adopted (Aibar et al. 2017), and we integrated information regarding TF classification and motif accessibility from the scATAC-seq experiment. First, a coexpression matrix was computed using GENIE3 (Huynh-Thu et al. 2010), considering both positively and negatively correlated TF–target gene links by integrating activator and repressor predictions, respectively. Next, indirect target genes were excluded by leveraging scATAC-seq data. Finally, the regulon activities for each single cell were calculated with AUCell (Aibar et al. 2017) and projected to the AUC heat map. Integration of data from independently acquired scRNA-seq and scATAC-seq experiments robustly identified developmentally relevant activating and repressing TFs and provided insights into TF function by resolving direct target genes. This approach was not conceived to replace classic knockout and overexpression experiments but to provide an alternative approach accessing the whole regulatory network in a single experiment. For example, ChIP-seq experiments require considerably larger amounts of input material compared with the single-cell technology, explaining limited numbers of inner ear–specific ChIP-seq data in the literature (Cai et al. 2015; Kwan et al. 2015; Stojanova et al. 2016; Li et al. 2020a; Menendez et al. 2020). Together, this illustrates why the current approach is specifically useful for populations with limited cell numbers, like IHCs in mice, in which only about 800 cells exist per inner ear (Ehret and Frankenreiter 1977). Finally, the current approach may prove less sensitive compared with ChIP-seq technology; however, the overlap between both methods aiming to identify SOX2 target genes underlined its relevance in understanding gene regulation during organ of Corti differentiation.
The AUC enrichment matrix identified 20 TFs, including regulons contributing to the differentiation of sensory HCs. Many of the TFs like ZBTB18 were known to cause sensorineural hearing loss (SNHL) (Aleksiūnienė et al. 2017), but their role in inner ear development remained elusive. This is of particular interest, as a limited set of only four TFs, namely, SIX1, ATOH1, POU4F3, and GFI1, is sufficient to convert fibroblasts into induced HC-like cells (Menendez et al. 2020). Together, these findings illustrate that a limited number of TFs play key roles in controlling the regulatory landscape of HC differentiation; nevertheless, a significantly larger array of TFs is necessary to allow for differentiation of functional HCs. Generally, after undergoing terminal mitosis, HCs develop in three overlapping phases. Initially, the regulatory landscape is represented by TFs, like ATOH1, LHX3, and GFI1 (Wallis et al. 2003; Woods et al. 2004; Hertzano et al. 2007), establishing an early HC fate by delineating HCs from PC/DCs. Later around birth, the developmental bifurcation into IHCs and OHCs is controlled by TFs like INSM1 and IKZF2 (Chessum et al. 2018; Wiwatpanit et al. 2018). In parallel, genes contributing to functions like mechanotransduction and synaptic transmission are up-regulated to complete the developmental progression (Fettiplace 2017). The 20 TFs identified contribute across the entire continuum of HC development. Comparing NHLH1 and ZBTB18 regulons, for example, allowed us to annotate their roles in initial differentiation and functional maturation, respectively. Likewise, when comparing IHC with OHC regulatory landscapes, five differentially active TFs were identified. Among the five TFs, INSM1 was previously published to control OHC development (Wiwatpanit et al. 2018), supporting our findings. Except for HIVEP2, the remaining four TFs, namely, INSM1, FOXO4, TCF4, and GLIS3, were either directly or indirectly associated with SNHL (Hishiya et al. 2006; Dimitri et al. 2011; de Winter et al. 2016). Although clustering of previously published marker transcripts allowed for IHC and OHC separation in scRNA-seq data, cellular identities were less prominent when analyzing scATAC-seq data. Based on the trajectory reconstruction, we were able to visualize differential motif accessibility for the TFs INSM1, FOXO4, TCF4, GLIS3, and HIVEP2 between basal IHCs and OHCs. The trajectories were used to visualize differences between transcriptome- and chromatin-based identities of HCs during differentiation. Together, our findings support the observation that during development, dynamic changes of the chromatin accessibility landscape on average lag behind changes of the transcriptional landscape (Hu et al. 2018; Li et al. 2020b).
In conclusion, this study provides a framework to analyze dynamic changes in the regulatory landscape of various cellular lineages that develop from a shared progenitor. With respect to the development of auditory HCs, we reconstructed a regulatory landscape featuring 20 individual TFs. Given the observation that several of those TFs have been linked to SNHL, the data provided by this study will help to further the knowledge regarding sensory HC differentiation and maturation in the mammalian inner ear.
Methods
Single-cell isolation and flow sorting
At P2, the cochlear ducts of FGFR3-iCre;Ai14-tdTomato;ATOH1-GFP pups were processed as previously described (Durruthy-Durruthy et al. 2014). To enrich for HCs and PC/DCs before sequencing, cells were purified with FACS. These samples were then used for standard 10x Genomics preparations for scRNA-seq or scATAC-seq experiments. For details, see Supplemental Methods. Critical commercial assays are listed in the Supplemental Key Resources Table.
RNAscope and immunofluorescence staining
Cochlear samples from P2 neonatal mice were fixed in 4% paraformaldehyde (Electron Microscopy Sciences 15710) and subjected to cryosectioning. For RNAscope and immunofluorescence staining, we followed the manufacturer's instructions with modifications. Details can be found in the Supplemental Methods. Antibodies, reagents, sources, and corresponding IDs are listed in the Supplemental Key Resources Table.
scATAC-seq analysis
scATAC-seq libraries were subject to SnapATAC clustering analysis as previously described (Fang et al. 2021). Details can be found in the Supplemental Methods.
Quality control of scATAC-seq data set
Ataqv (Orchard et al. 2020), an ATAC-seq QC and visualization tool, was used to measure the scATAC-seq data quality. Details can be found in the Supplemental Methods.
scRNA-seq analysis
The scRNA-seq data set was analyzed using Seurat v3 pipeline (Stuart et al. 2019). Details can be found in the Supplemental Methods.
Cell-type identification in scATAC-seq clusters using Jaccard index similarity matrix
A Jaccard index similarity matrix was generated to annotate scATAC-seq clusters by calculating the overlaps between DEGs and annotated DARs from the scRNA-seq and scATAC-seq data. Details can be found in the Supplemental Methods.
LIGER multi-omics integration
LIGER (Welch et al. 2019), a joint alignment algorithm, was adopted to jointly define cell identities from scRNA-seq and scATAC-seq data sets. Details can be found in the Supplemental Methods.
TF motif activity estimation
The cell-by-peak matrix, generated by SnapATAC, was subjected to chromVAR (Schep et al. 2017) with the mouse HOCOMOCO v10 database. Details can be found in the Supplemental Methods.
TF classification into activators and repressors
An integrative approach, which considers the associations between TF gene expression and chromatin accessibility, was developed to classify TF mode of action into activators and repressors. Details can be found in the Supplemental Methods.
TF footprint identification
HINT-ATAC (Li et al. 2019) was used to identify TF binding sites with footprints for different populations. Details can be found in the Supplemental Methods.
Gene regulatory network inference
A three-step pipeline was developed to reconstruct gene regulatory networks. The first step is to identify coexpression modules from scRNA-seq data using GENIE3 (Huynh-Thu et al. 2010). The second step is to identify direct target genes of TFs and to set up a quality control for GENIE3 by scanning putative TF binding sites within accessible regions and removing the links lacking TF binding sites. The third step is to calculate regulon enrichment scores for each individual cell using AUCell (Aibar et al. 2017). Details can be found in the Supplemental Methods.
1D spatial reconstruction of HCs
Individual HC's anatomical position from scRNA-seq and scATAC-seq data was determined using a 1D-PCA (Waldhaus et al. 2015). Details can be found in the Supplemental Methods.
Prediction of cis-regulatory interactions
Cicero (Pliner et al. 2018) was performed to calculate peak-to-peak coaccessibility from scATAC-seq data for different clusters separately. Details can be found in the Supplemental Methods.
IHC and OHC identification using scRNA-seq data
Seven previously published marker genes were leveraged to identify IHC and OHC subpopulations from HC cluster in scRNA-seq data. Details can be found in the Supplemental Methods.
HC chromatin accessibility trajectory inference
CellTrails (Ellwanger et al. 2018) was applied to reconstruct HC developmental trajectory using z-scores from scATAC-seq data. Slingshot (Street et al. 2018) and Monocle (Trapnell et al. 2014; Qiu et al. 2017a,b) were adopted to validate the trajectory inference from CellTrails. Details can be found in the Supplemental Methods.
Bulk RNA-seq analysis
Two previously published bulk RNA-seq data sets (Li et al. 2018; Wiwatpanit et al. 2018) were leveraged to validate differential expression analysis between IHC and OHC owing to the limited cell numbers. Details can be found in the Supplemental Methods.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE157398. The analytical code is available as Supplemental Code and at GitHub (https://github.com/shuzwang/P2_cochlea).
Supplementary Material
Acknowledgments
We thank the staff at University of Michigan Advanced Genomics Core for their help and support and thank members of the J.W. laboratory and Yujuan Fu for the discussion of the manuscript. This work was supported by National Institute on Deafness and Other Communication Disorders/National Institutes of Health grant (R21DC015861) to J.W.
Author contributions: J.W. and S.W. conceived the study, and J.W. and J.L. supervised the project. S.W., S.J., and M.P.L. acquired and analyzed data. S.W. and J.W. wrote the manuscript with the help of J.L., M.P.L., and S.J.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.271080.120.
Competing interest statement
The authors declare no competing interests.
References
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. 2017. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14: 1083–1086. 10.1038/nmeth.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksiūnienė B, Matulevičiūtė R, Matulevičienė A, Burnytė B, Krasovskaja N, Ambrozaitytė L, Mikštienė V, Dirsė V, Utkus A, Kučinskas V. 2017. Opposite chromosome constitutions due to a familial translocation t(1;21)(q43;q22) in two cousins with development delay and congenital anomalies: a case report. Medicine (Baltimore) 96: e6521. 10.1097/MD.0000000000006521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir e-A, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe'er D. 2014. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157: 714–725. 10.1016/j.cell.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berest I, Arnold C, Reyes-Palomares A, Palla G, Rasmussen KD, Giles H, Bruch PM, Huber W, Dietrich S, Helin K, et al. 2019. Quantification of differential transcription factor activity and multiomics-based classification into activators and repressors: diffTF. Cell Rep 29: 3147–3159.e12. 10.1016/j.celrep.2019.10.106 [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh O, Oesterle EC, Stone JS, Hume CR, Huynh HM, Hayashi T. 2006. Expression of Prox1 during mouse cochlear development. J Comp Neurol 496: 172–186. 10.1002/cne.20944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. 10.1038/nmeth.2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, Greenleaf WJ. 2018. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173: 1535–1548.e16. 10.1016/j.cell.2018.03.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JC, Kelly MC, Hoa M, Morell RJ, Kelley MW. 2015. Single-cell RNA-seq resolves cellular complexity in sensory organs from the neonatal inner ear. Nat Commun 6: 8557. 10.1038/ncomms9557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Jen HI, Kang H, Klisch TJ, Zoghbi HY, Groves AK. 2015. Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. J Neurosci 35: 5870–5883. 10.1523/JNEUROSCI.5083-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Johnson JE, Zoghbi HY, Segil N. 2002. The role of Math1 in inner ear development: uncoupling the establishment of the sensory primordium from hair cell fate determination. Development 129: 2495–2505. 10.1242/dev.129.10.2495 [DOI] [PubMed] [Google Scholar]
- Chessum L, Matern MS, Kelly MC, Johnson SL, Ogawa Y, Milon B, McMurray M, Driver EC, Parker A, Song Y, et al. 2018. Helios is a key transcriptional regulator of outer hair cell maturation. Nature 563: 696–700. 10.1038/s41586-018-0728-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JW, Kang HH, Shin JE, Kim JU. 2004. Accumulation of hypoxia-inducible factor-1α in mouse inner ear by noise stimulation. Neuroreport 15: 2353–2356. 10.1097/00001756-200410250-00010 [DOI] [PubMed] [Google Scholar]
- Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, Zernecke A. 2018. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 122: 1661–1674. 10.1161/CIRCRESAHA.117.312509 [DOI] [PubMed] [Google Scholar]
- Corti A. 1851. Recherches sur l'organe de l'ouïe des mammifères. Z Wiss Zool 3: 109–169. [Google Scholar]
- de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RC. 2016. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis 11: 37. 10.1186/s13023-016-0422-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitri P, Warner JT, Minton JA, Patch AM, Ellard S, Hattersley AT, Barr S, Hawkes D, Wales JK, Gregory JW. 2011. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol 164: 437–443. 10.1530/EJE-10-0893 [DOI] [PubMed] [Google Scholar]
- Durruthy-Durruthy R, Gottlieb A, Hartman BH, Waldhaus J, Laske RD, Altman R, Heller S. 2014. Reconstruction of the mouse otocyst and early neuroblast lineage at single-cell resolution. Cell 157: 964–978. 10.1016/j.cell.2014.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret G, Frankenreiter M. 1977. Quantitative analysis of cochlear structures in the house mouse in relation to mechanisms of acoustical information processing. J Comp Physiol 122: 65–85. [Google Scholar]
- Ellwanger DC, Scheibinger M, Dumont RA, Barr-Gillespie PG, Heller S. 2018. Transcriptional dynamics of hair-bundle morphogenesis revealed with cellTrails. Cell Rep 23: 2901–2914.e13. 10.1016/j.celrep.2018.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. 2021. Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat Commun 12: 1337. 10.1038/s41467-021-21583-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace R. 2017. Hair cell transduction, tuning, and synaptic transmission in the mammalian cochlea. Compr Physiol 7: 1197–1227. 10.1002/cphy.c160049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranašić D, et al. 2020. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48: D87–D92. 10.1093/nar/gkz1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann D, Mernberger M, Nist A, Stiewe T, Elsässer HP. 2018. Miz1 controls Schwann cell proliferation via H3K36me2 demethylase Kdm8 to prevent peripheral nerve demyelination. J Neurosci 38: 858–877. 10.1523/JNEUROSCI.0843-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gory-Fauré S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. 1999. Role of vascular endothelial-cadherin in vascular morphogenesis. Development 126: 2093–2102. 10.1242/dev.126.10.2093 [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Bao EL, Wagner M, Whitsett JA, Xu Y. 2017. SLICE: determining cell differentiation and lineage based on single cell entropy. Nucleic Acids Res 45: e54. 10.1093/nar/gkw1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman BH, Basak O, Nelson BR, Taylor V, Bermingham-McDonogh O, Reh TA. 2009. Hes5 expression in the postnatal and adult mouse inner ear and the drug-damaged cochlea. J Assoc Res Otolaryngol 10: 321–340. 10.1007/s10162-009-0162-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman BH, Durruthy-Durruthy R, Laske RD, Losorelli S, Heller S. 2015. Identification and characterization of mouse otic sensory lineage genes. Front Cell Neurosci 9: 79. 10.3389/fncel.2015.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Ray CA, Younkins C, Bermingham-McDonogh O. 2010. Expression patterns of FGF receptors in the developing mammalian cochlea. Dev Dyn 239: 1019–1026. 10.1002/dvdy.22236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström AR, Watt B, Fard SS, Tenza D, Mannström P, Narfström K, Ekesten B, Ito S, Wakamatsu K, Larsson J, et al. 2011. Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet 7: e1002285. 10.1371/journal.pgen.1002285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzano R, Dror AA, Montcouquiol M, Ahmed ZM, Ellsworth B, Camper S, Friedman TB, Kelley MW, Avraham KB. 2007. Lhx3, a LIM domain transcription factor, is regulated by Pou4f3 in the auditory but not in the vestibular system. Eur J Neurosci 25: 999–1005. 10.1111/j.1460-9568.2007.05332.x [DOI] [PubMed] [Google Scholar]
- Hishiya A, Iemura S, Natsume T, Takayama S, Ikeda K, Watanabe K. 2006. A novel ubiquitin-binding protein ZNF216 functioning in muscle atrophy. EMBO J 25: 554–564. 10.1038/sj.emboj.7600945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, Cui K, Fang D, Hirose S, Wang X, Wangsa D, Jin W, Ried T, Liu P, Zhu J, et al. 2018. Transformation of accessible chromatin and 3D nucleome underlies lineage commitment of early T cells. Immunity 48: 227–242.e8. 10.1016/j.immuni.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. 2010. Inferring regulatory networks from expression data using tree-based methods. PLoS One 5: e12776. 10.1371/journal.pone.0012776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. 2019. Schwann cell precursors; multipotent glial cells in embryonic nerves. Front Mol Neurosci 12: 69. 10.3389/fnmol.2019.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. 2005. Sox2 is required for sensory organ development in the mammalian inner ear. Nature 434: 1031–1035. 10.1038/nature03487 [DOI] [PubMed] [Google Scholar]
- Kitajiri SI, Furuse M, Morita K, Saishin-Kiuchi Y, Kido H, Ito J, Tsukita S. 2004. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear Res 187: 25–34. 10.1016/S0378-5955(03)00338-1 [DOI] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, Greenleaf WJ. 2019. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20: 207–220. 10.1038/s41576-018-0089-8 [DOI] [PubMed] [Google Scholar]
- Kolla L, Kelly MC, Mann ZF, Anaya-Rocha A, Ellis K, Lemons A, Palermo AT, So KS, Mays JC, Orvis J, et al. 2020. Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat Commun 11: 2389. 10.1038/s41467-020-16113-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, Makeev VJ. 2013. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res 41: D195–D202. 10.1093/nar/gks1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Shen J, Corey DP. 2015. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports 4: 47–60. 10.1016/j.stemcr.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Liu F, Segil N. 2006. A morphogenetic wave of p27Kip1 transcription directs cell cycle exit during organ of Corti development. Development 133: 2817–2826. 10.1242/dev.02453 [DOI] [PubMed] [Google Scholar]
- Lelli A, Asai Y, Forge A, Holt JR, Géléoc GS. 2009. Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol 101: 2961–2973. 10.1152/jn.00136.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu H, Barta CL, Judge PD, Zhao L, Zhang WJ, Gong S, Beisel KW, He DZ. 2016. Transcription factors expressed in mouse cochlear inner and outer hair cells. PLoS One 11: e0151291. 10.1371/journal.pone.0151291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu H, Giffen KP, Chen L, Beisel KW, He DZZ. 2018. Transcriptomes of cochlear inner and outer hair cells from adult mice. Sci Data 5: 180199. 10.1038/sdata.2018.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Schulz MH, Look T, Begemann M, Zenke M, Costa IG. 2019. Identification of transcription factor binding sites using ATAC-seq. Genome Biol 20: 45. 10.1186/s13059-019-1642-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang T, Ramakrishnan A, Fritzsch B, Xu J, Wong EYM, Loh YE, Ding J, Shen L, Xu PX. 2020a. Dynamic changes in cis-regulatory occupancy by Six1 and its cooperative interactions with distinct cofactors drive lineage-specific gene expression programs during progressive differentiation of the auditory sensory epithelium. Nucleic Acids Res 48: 2880–2896. 10.1093/nar/gkaa012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu Y, Yang H, Zhang T, Naruse K, Tu Q. 2020b. Dynamic transcriptional and chromatin accessibility landscape of medaka embryogenesis. Genome Res 30: 924–937. 10.1101/gr.258871.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilleväli K, Matilainen T, Karis A, Salminen M. 2004. Partially overlapping expression of Gata2 and Gata3 during inner ear development. Dev Dyn 231: 775–781. 10.1002/dvdy.20185 [DOI] [PubMed] [Google Scholar]
- Luo XJ, Deng M, Xie X, Huang L, Wang H, Jiang L, Liang G, Hu F, Tieu R, Chen R, et al. 2013. GATA3 controls the specification of prosensory domain and neuronal survival in the mouse cochlea. Hum Mol Genet 22: 3609–3623. 10.1093/hmg/ddt212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Kariya S, Uraguchi K, Takahara J, Fujimoto S, Sugaya A, Nishizaki K. 2020. Immediate changes in transcription factors and synaptic transmission in the cochlea following acoustic trauma: a gene transcriptome study. Neurosci Res 165: 6–13. 10.1016/j.neures.2020.05.001 [DOI] [PubMed] [Google Scholar]
- Menendez L, Trecek T, Gopalakrishnan S, Tao L, Markowitz AL, Yu HV, Wang X, Llamas J, Huang C, Lee J, et al. 2020. Generation of inner ear hair cells by direct lineage conversion of primary somatic cells. eLife 9: e55249. 10.7554/eLife.55249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, Farmer A, Fordyce P, Linnarsson S, Greenleaf W. 2018. High-throughput chromatin accessibility profiling at single-cell resolution. Nat Commun 9: 3647. 10.1038/s41467-018-05887-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita K, Sasaki H, Furuse M, Tsukita S. 1999. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147: 185–194. 10.1083/jcb.147.1.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutai H, Wasano K, Momozawa Y, Kamatani Y, Miya F, Masuda S, Morimoto N, Nara K, Takahashi S, Tsunoda T, et al. 2020. Variants encoding a restricted carboxy-terminal domain of SLC12A2 cause hereditary hearing loss in humans. PLoS Genet 16: e1008643. 10.1371/journal.pgen.1008643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard P, Kyono Y, Hensley J, Kitzman JO, Parker SCJ. 2020. Quantification, dynamic visualization, and validation of bias in ATAC-Seq data with ataqv. Cell Syst 10: 298–306.e4. 10.1016/j.cels.2020.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer JS, Zhu Q, Huynh C, Sivaramakrishnan P, Preston E, Dueck H, Stefanik D, Tan K, Trapnell C, Kim J, et al. 2019. A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science 365: eaax1971. 10.1126/science.aax1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phippard D, Heydemann A, Lechner M, Lu L, Lee D, Kyin T, Crenshaw EB. 1998. Changes in the subcellular localization of the Brn4 gene product precede mesenchymal remodeling of the otic capsule. Hear Res 120: 77–85. 10.1016/S0378-5955(98)00059-8 [DOI] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. 2018. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol Cell 71: 858–871.e8. 10.1016/j.molcel.2018.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C. 2017a. Single-cell mRNA quantification and differential analysis with census. Nat Methods 14: 309–315. 10.1038/nmeth.4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C. 2017b. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14: 979–982. 10.1038/nmeth.4402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde-Gallwitz K, Pan L, Gan L, Lin X, Segil N, Chen P. 2004. Expression of Islet1 marks the sensory and neuronal lineages in the mammalian inner ear. J Comp Neurol 477: 412–421. 10.1002/cne.20257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, Zoghbi HY. 2009. Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64: 341–354. 10.1016/j.neuron.2009.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer DI, Shen J, Corey DP, Chen ZY. 2015. Gene expression by mouse inner ear hair cells during development. J Neurosci 35: 6366–6380. 10.1523/JNEUROSCI.5126-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. 2017. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14: 975–978. 10.1038/nmeth.4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Spiridonov NA. 2004. The mammalian transcriptome and the function of non-coding DNA sequences. Genome Biol 5: 105. 10.1186/gb-2004-5-4-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Walsh CA. 2005. Targeted disruption of Tgif, the mouse ortholog of a human holoprosencephaly gene, does not result in holoprosencephaly in mice. Mol Cell Biol 25: 3639–3647. 10.1128/MCB.25.9.3639-3647.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son EJ, Ma JH, Ankamreddy H, Shin JO, Choi JY, Wu DK, Bok J. 2015. Conserved role of sonic hedgehog in tonotopic organization of the avian basilar papilla and mammalian cochlea. Proc Natl Acad Sci 112: 3746–3751. 10.1073/pnas.1417856112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanova ZP, Kwan T, Segil N. 2016. Epigenetic regulation of Atoh1 guides hair cell development in the mammalian cochlea. Development 143: 1632. 10.1242/dev.137976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S. 2018. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19: 477. 10.1186/s12864-018-4772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. 2019. Comprehensive integration of single-cell data. Cell 177: 1888–1902.e21. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Inada H, Han C, Kim MJ, Kimura R, Takata Y, Honkura Y, Owada Y, Kawase T, Katori Y, et al. 2019. “Passenger gene” problem in transgenic C57BL/6 mice used in hearing research. Neurosci Res 158: 6–15. 10.1016/j.neures.2019.10.007 [DOI] [PubMed] [Google Scholar]
- Takeda K, Yokoyama S, Aburatani H, Masuda T, Han F, Yoshizawa M, Yamaki N, Yamamoto H, Eguchi N, Urade Y, et al. 2006. Lipocalin-type prostaglandin D synthase as a melanocyte marker regulated by MITF. Biochem Biophys Res Commun 339: 1098–1106. 10.1016/j.bbrc.2005.11.125 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 32: 381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowe MO, Maier H, Schweizer M, Kispert A. 2008. Deafness in mice lacking the T-box transcription factor Tbx18 in otic fibrocytes. Development 135: 1725–1734. 10.1242/dev.014043 [DOI] [PubMed] [Google Scholar]
- Uehara S, Izumi Y, Kubo Y, Wang CC, Mineta K, Ikeo K, Gojobori T, Tachibana M, Kikuchi T, Kobayashi T, et al. 2009. Specific expression of Gsta4 in mouse cochlear melanocytes: a novel role for hearing and melanocyte differentiation. Pigment Cell Melanoma Res 22: 111–119. 10.1111/j.1755-148X.2008.00513.x [DOI] [PubMed] [Google Scholar]
- van Schaarenburg RA, Suurmond J, Habets KL, Brouwer MC, Wouters D, Kurreeman FA, Huizinga TW, Toes RE, Trouw LA. 2016. The production and secretion of complement component C1q by human mast cells. Mol Immunol 78: 164–170. 10.1016/j.molimm.2016.09.001 [DOI] [PubMed] [Google Scholar]
- Vendrell V, López-Hernández I, Durán Alonso MB, Feijoo-Redondo A, Abello G, Gálvez H, Giráldez F, Lamonerie T, Schimmang T. 2015. Otx2 is a target of N-myc and acts as a suppressor of sensory development in the mammalian cochlea. Development 142: 2792–2800. [DOI] [PubMed] [Google Scholar]
- Wagner DE, Weinreb C, Collins ZM, Briggs JA, Megason SG, Klein AM. 2018. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360: 981–987. 10.1126/science.aar4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhaus J, Durruthy-Durruthy R, Heller S. 2015. Quantitative high-resolution cellular map of the organ of Corti. Cell Rep 11: 1385–1399. 10.1016/j.celrep.2015.04.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, Zoghbi HY, Orkin SH, Bellen HJ. 2003. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130: 221–232. 10.1242/dev.00190 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang B, Jiang H, Zhang L, Liu D, Xiao X, Ma H, Luo X, Bojrab D, Hu Z. 2013. Myelination of the postnatal mouse cochlear nerve at the peripheral-central nervous system transitional zone. Front Pediatr 1: 43. 10.3389/fped.2013.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Taschner-Mandl S, Bileck A, Slany A, Kromp F, Rifatbegovic F, Frech C, Windhager R, Kitzinger H, Tzou CH, et al. 2016. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 64: 2133–2153. 10.1002/glia.23045 [DOI] [PubMed] [Google Scholar]
- Welch JD, Kozareva V, Ferreira A, Vanderburg C, Martin C, Macosko EZ. 2019. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177: 1873–1887.e17. 10.1016/j.cell.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson BA, Chitsazan AD, VandenBosch LS, Wilken MS, Reh TA, Bermingham-McDonogh O. 2019. Open chromatin dynamics in prosensory cells of the embryonic mouse cochlea. Sci Rep 9: 9060. 10.1038/s41598-019-45515-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiwatpanit T, Lorenzen SM, Cantú JA, Foo CZ, Hogan AK, Márquez F, Clancy JC, Schipma MJ, Cheatham MA, Duggan A, et al. 2018. Trans-differentiation of outer hair cells into inner hair cells in the absence of INSM1. Nature 563: 691–695. 10.1038/s41586-018-0570-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C, Montcouquiol M, Kelley MW. 2004. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci 7: 1310–1318. 10.1038/nn1349 [DOI] [PubMed] [Google Scholar]
- Wu X, Ivanchenko MV, Al Jandal H, Cicconet M, Indzhykulian AA, Corey DP. 2019. PKHD1L1 is a coat protein of hair-cell stereocilia and is required for normal hearing. Nat Commun 10: 3801. 10.1038/s41467-019-11712-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Gan L, Li D, Chen ZY, Zhou L, O'Malley BW, Klein W, Nathans J. 1997. Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci 94: 9445–9450. 10.1073/pnas.94.17.9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ueno H, Xu CY, Chen B, Weissman IL, Xu PX. 2017. Identification of mouse cochlear progenitors that develop hair and supporting cells in the organ of Corti. Nat Commun 8: 15046. 10.1038/ncomms15046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Cai Q, Bard J, Jamison J, Wang J, Yang W, Hu BH. 2015. Variation analysis of transcriptome changes reveals cochlear genes and their associated functions in cochlear susceptibility to acoustic overstimulation. Hear Res 330: 78–89. 10.1016/j.heares.2015.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KM, Mitsumori T, Pringle N, Grist M, Kessaris N, Richardson WD. 2010. An Fgfr3-iCreERT2 transgenic mouse line for studies of neural stem cells and astrocytes. Glia 58: 943–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Williams J, Smallwood PM, Nathans J. 2015. Sox7, Sox17, and Sox18 cooperatively regulate vascular development in the mouse retina. PLoS One 10: e0143650. 10.1371/journal.pone.0143650 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.