

Abstract
Thin-film polyimide–metal neuroelectronic interfaces hold the potential to alleviate many neurological disorders. However, their long-term reliability is challenged by an aggressive implant environment that causes delamination and degradation of critical materials, resulting in a degradation or complete loss of implant function. Herein, a rigorous and in-depth analysis is presented on the fabrication and modification of critical materials in these thin-film neural interfaces. Special attention is given to improving the interfacial adhesion between thin films and processing modifications to maximize device reliability. Fundamental material analyses are performed on the polyimide substrate and adhesion-promotion candidates, including amorphous silicon carbide (a-SiC:H), amorphous carbon, and silane coupling agents. Basic fabrication rules are identified to markedly improve polyimide self-adhesion, including optimizing the polyimide-cure profile and maximizing high-energy surface activation. In general, oxide-forming materials are identified as poor adhesive aids to polyimide without targeted modifications. Methods are identified to incorporate effective a-SiC:H interfacial layers to improve metal adherence to polyimide, in addition to examples of alloying between adjacent material layers that can impact the trace resistivity and long-term reliability of the thin-film interfaces. The provided rationale and consequences of key decisions made should promote more reproducible science using robust and reliable neuroelectronic technology.
Keywords: neural interface, adhesion, reliability, thin-film delamination
1. Introduction
Flexible polymer–metal neuroelectronic implants that interface engineered electrodes with neural tissue have been under development for years as therapeutic platforms targeting an array of patient populations with neurological disorders and diseases.[1] Early devices were used to answer fundamental neuroscience questions and as proof-of-concept devices for targeted disease models. More recently, spinal stimulators have shown promise at evoking motor function and alleviating gait deficits,[2,3] electrocorticography arrays have been able to capture neural activity required for brain–machine interfaces,[4] intraneural peripheral implants have been able to improve the control and proprioception of prosthetic limbs,[5,6] and retinal prosthetics have been able to partially restore vision.[7] Next-generation devices are targeting higher channel counts and electrode densities with minimized footprint geometries to further improve therapeutic outcomes. While devices are getting closer to achieving stable performance during chronic implantation in human populations,[5,6] there exist fundamental material and reliability considerations that currently limit their long-term success.
Several polymeric materials have been used as the primary structural and dielectric component of these implanted neural interfaces including polyimides, parylenes, and silicone rubbers that are all suitably flexible, biocompatible, and nontoxic.[1,8] While the choice of the specific material used in a neuroelectronic device depends on the intended application, polyimide-based implants microfabricated from 3,3′,4,4′-biphenyltetracarboxilic dianhydride-p-phenylene diamine (BPDA-PDA) polyamic precursors are some of the most widely used. The fact that BPDA-PDA has excellent chemical resistance and one of the lowest water-uptake ratios[9] of any polyimide, makes it an ideal candidate for the harsh in vivo implant environment.[10] Furthermore, since it has a low coefficient of thermal expansion (CTE, 3–8 ppm K−1),[9] it can be processed at relatively high temperatures (e.g., 450 °C) and integrated into microfabricated devices using many conventional cleanroom deposition, lithography, and etching techniques.[11] The dielectric properties of BPDA-PDA are excellent, with a high dielectric strength (2 MV cm−1), low loss over a wide frequency range, and low relative permittivity (εr = 3.1 at 1 kHz).[9] Furthermore, BPDA-PDA is mechanically robust with an elastic modulus of 9–10 GPa and high toughness allowing it to be formed into flexible, thin layers (e.g., ≈2–20 μm).[8,9]
Although BPDA-PDA polyimide has excellent material properties, thin-film polyimide-metal-based devices can still fail when implanted,[10,12,13] which currently limits their widespread success when used over chronic time periods (i.e., years). The failure of implanted devices can be driven by biotic, abiotic, or a combination biotic and abiotic mechanisms. Due to the complexity of the biological environment, it is difficult to engineer universal methods to limit biotic failure mechanisms (e.g., tissue degradation, fibrotic encapsulation) that depend mainly on the implant modality and device design. On the other hand, abiotic implant failure is typically influenced by parameters that are directly controlled by the specific materials and microfabrication techniques used to manufacture the implant. Common examples include delamination of polyimide–metal and polyimide–polyimide interfaces due to moisture penetration, inflammation-driven chemical degradation, and electrical stimulation during therapeutic sessions.[10,12–14] Understanding the fundamental material science behind the selection of materials and microfabrication process parameters and how they correlate with abiotic failure mitigation offers a pathway to engineer improved chronic reliability for the full spectrum of implanted thin-film polyimide–metal bioelectronic devices, irrespective of implant modality or device design.
The use of micromachining to manufacture neural implants from BPDA-PDA polyimide was first reported in 2000 by the Fraunhofer Institute for Biomedical Engineering.[11] In general, the microfabrication process of thin-film polyimide-based neuroelectronic implants involves a series of steps identified above (Figure 1). Using this general microfabrication process, numerous research groups have developed unique device designs (e.g., transverse intrafascicular multichannel electrode,[15] self-opening intraneural peripheral interface,[16] tissue-engineered electronic neural interface[17]) for a variety of neural targets and disease models. These implantable devices have been successfully tested in proof-of-concept preclinical studies that focused primarily on acute or short term (i.e., months) performance (e.g., biocompatibility, electrophysiology, stimulation efficacy, and histological outcomes). While the general microfabrication process flow is similar, the specific processing variables used often vary widely in terms of the conductive materials (e.g., platinum, gold), adhesion-promotion techniques (e.g., titanium/ chromium, silane coupling agents, oxygen plasma, silicon carbide [SiC]), and polyimide-cure temperatures. The reasonings behind the differing microfabrication processing variables chosen are usually ambiguous and result in neuroelectronic devices with a range of implanted reliability and performance.
Figure 1.
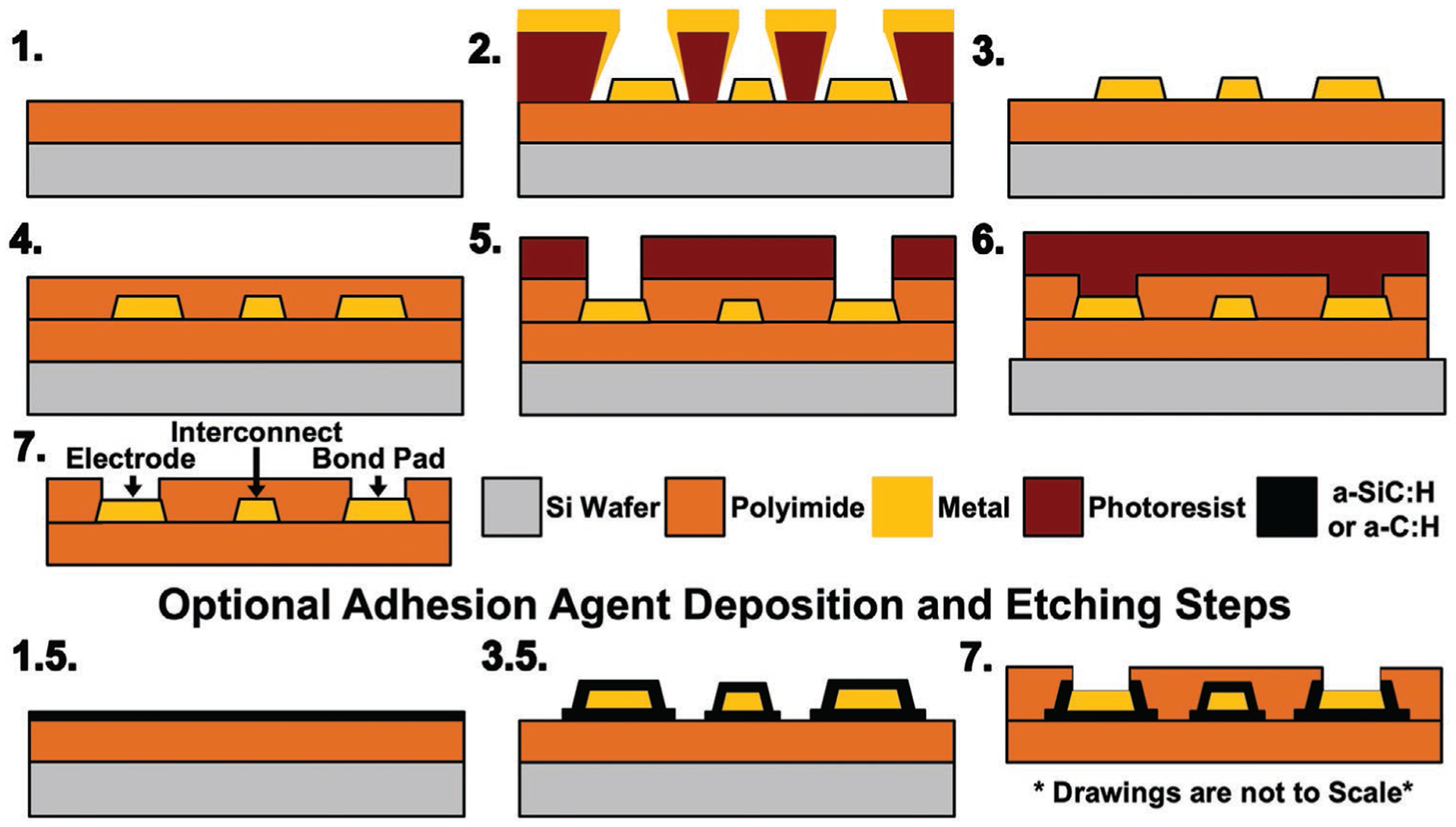
Generic microfabrication process for polyimide-based bioelectronic devices. 1) Deposited and cured bottom polyimide layer, 2) deposited metal layer, 3) patterned metal using a lift-off process, 4) deposited and cured top polyimide layer, 5) etched top layer polyimide to reveal electrodes and bonding sites, 6) etched top and bottom polyimide layers to define final device geometry, and 7) removal of the device from carrier wafer. Optional PECVD deposited materials can also be applied to improve adhesion between polyimide and metal layers including (1.5) deposition of first layer and (3.5) deposition of second layer followed by dry-etch patterning of both layers.
Several important advances to the original polyimide-based microfabrication process have been developed to improve the overall reliability of neural interfaces.[14,18] Notably, amorphous silicon carbide (a-SiC:H) and diamond-like carbon (DLC)/amorphous carbon (a-C:H) were identified as effective adhesion-promotion agents to improve the integrity of the critical polyimide–metal interface (Figure 1). However, a comprehensive analysis of the varying processing parameters used by the community has not been performed, and an examination of fundamental materials science improvements is lacking. Herein, a rigorous and in-depth analysis of both the fabrication and modification of critical materials is presented in the context of polyimide-based neural interfaces. Special attention is given to improving the interfacial adhesion between thin films and chemical/processing modifications that can be performed to maximize device reliability. Detailed descriptions of methodology and related comprehensive discussion sections are provided for key microfabrication process modules used to manufacture reliable polyimide-based neural interfaces, including polyimide curing and adhesion to respective materials, plasma-enhanced chemical vapor deposited (PECVD) adhesion layers (e.g., a-SiC:H and a-C:H), structuring of chemically different metal layers, and thermal alloying between materials. Our objectives are to illuminate the rationale and consequences of key decisions made regarding the microfabrication of polyimide-based neural interfaces and thereby promote a more transparent and reproducible scientific environment to enable the adoption of more robust and reliable neuroelectronic implants.
2. Experimental Section
2.1. Material Deposition and Characterization
2.1.1. Carrier Substrate Cleaning and Photomask Fabrication
All microfabrication steps were performed in a class 100/1000 cleanroom using 100-mm-diameter Si wafers (test grade, P-type, 〈100〉, University Wafer) as carrier substrates. Wafers were first cleaned of residual organics in a 4:1 (v/v) H2SO4:H2O2 solution and coated with hexamethyldisilazane (HMDS) via vapor deposition. Patterned photomasks used in lithography steps were prepared from photoresist-coated blank photomasks (5 in. FeO soda-lime, NanoFilm Microcircuit Technology) that were pattern exposed using a laser writer (DWL 66FS, Heidelberg Instruments GmbH), developed, etched, and stripped of residual photoresist.
2.1.2. Polyimide Spin-Coating, Imidization, and Characterization
To prepare the polyimide base layer, BPDA-PDA polyimide precursor in n-methyl-2-pyrrolidone (NMP) (U-Varnish S, UBE Ind.) was spin-coated (1000–2000 rpm, EC101, Headway Research, Inc.) onto a HMDS wafer and thermally cured on a hotplate (HP1212YH with insulated hood, Wenesco, Inc.) in a N2 atmosphere using a PID temperature controller (EZ-ZONE PM, Watlow Electric Manufacturing Company) with cure profiles modified from UBE, Ind.’s recommendation to a maximum temperature ranging from T1 = 250 to 450 °C (Figure 2). Film thickness was determined using spectral reflectance (F40, Filmetrics, Inc.), bulk chemical functionality was determined by attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR: Nicolet iS50, Thermo Fisher Scientific), and water contact angle was determined using 5 μL droplets of deionized water (DIW, 18.2 MΩ cm) with a custom-made goniometer. To determine the impact of common microfabrication steps on surface functionality, X-ray photoelectron spectroscopy (XPS: 5000 Versa Probe II, ULVAC-PHI, Inc. with low-energy Ar+ and e– charge neutralization) was used to characterize films after the following steps: 1) as-cured, 2) reactive ion etched (RIE) with O2-plasma (Unaxis 790, Plasma-Therm, LLC) at 100 W, 100 mTorr, and 30 sccm O2 for 60 s, 3) RIE with SF6 plasma (Unaxis SLR, Plasma-Therm, LLC) at 50 W, 25 mTorr, 50 sccm SF6 for 60 s, and 4) silanization with aminopropyl triethoxysilane (APTES) organosilane agent (VM-652, HD MicroSystems). APTES was applied after O2-plasma activation, described previously, per the manufacturer’s specifications by spin-coating followed by heating to 120 °C to drive the silanization to completion.
Figure 2.
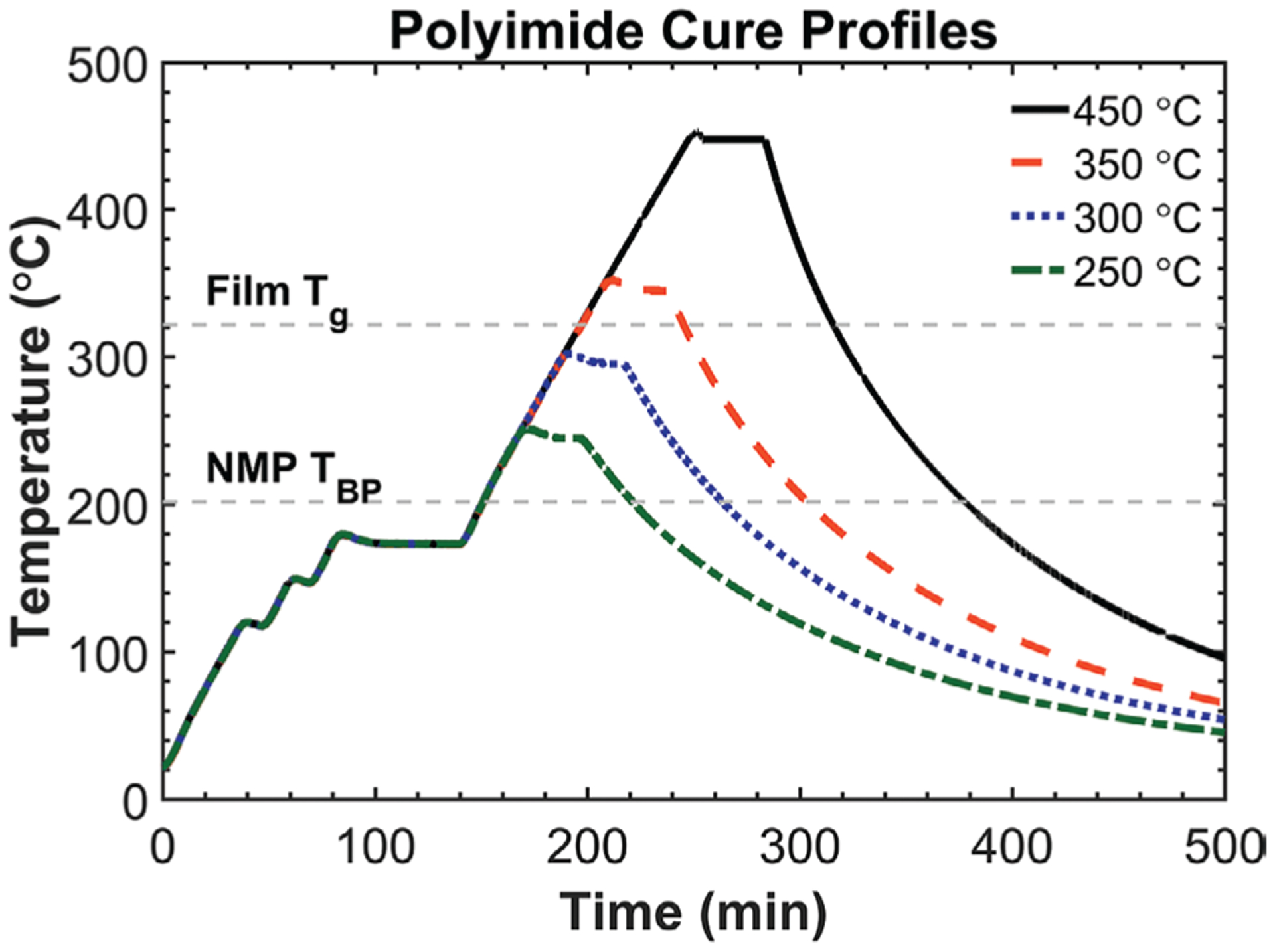
Temperature profiles used to thermally cure BPDA-PDA polyimide. Soft curing occurs from ≈100 to 150 °C, and primary imidization occurs from ≈150 to 250 °C. Maximum hard-cure temperature dictates final film properties (e.g., dielectric loss factor, conductivity, dielectric breakdown field, etc.).[19] Heating rate was fixed at 2.5 °C min−1.
2.1.3. Amorphous, Hydrogenated Silicon Carbide Deposition and Characterization
For a-SiC:H deposition, exposed surfaces (e.g., bare silicon or polyimide) were first cleaned using RIE O2 plasma for 60 s, as described previously. Stoichiometric a-SiC:H (i.e., 1:1 silicon: carbon) was deposited in a PECVD (Unaxis 790, Plasma-Therm, LLC) using a 13.56 MHz RF power supply at 300 °C, 1100 mTorr, 700 sccm He, 300 sccm SiH4 (5% in He), 200 sccm CH4 (nondiluted), and 200 W to a thickness of 250 nm. Thickness of a-SiC:H films was determined using spectral reflectance on bare Si wafers, and film stress was calculated using Stoney’s equation from wafer curvature (Flexxus 2320, KLA-Tencor). Bulk a-SiC:H chemical functionality was determined by transmission FTIR on 500-nm-thick films deposited on both sides of double-sided polished high resistivity wafers (prime grade, P-type, 〈100〉, 10–20 Ω cm, University Wafer).[20,21] Film stoichiometry throughout the bulk was determined using XPS depth-profiling with Ar+ etching, and the impact of common microfabrication steps on surface functionality using XPS was also performed after the same four processes, as described in Section 2.1.2.
2.1.4. Amorphous Hydrogenated Carbon Deposition Development and Characterization
Exploratory depositions of a-C:H using PECVD (Unaxis 790) at 13.56 MHz were performed to optimize film properties and deposition rate. A set of one-way design-of-experiments (DOEs) was performed to assess the impact of modifying deposition temperature, CH4 flow rate, chamber pressure, and deposition power. Deposition conditions of 100 °C, 50 sccm CH4, and 50 mTorr were identified to maximize deposition rate and film properties. Transmission FTIR, XPS, spectral reflectance, and wafer-curvature measurements were performed, as described in Section 2.1.3.
2.2. Film-Adhesion Test-Structure Fabrication and Testing
To quantify film adhesion, modified test structures were designed from ASTM D1876–08: Standard Test Method for Peel Resistance and Adhesives (T-Peel Test) that could be microfabricated on 100-mm-diameter wafers (Figure 3). Specifically, the T-peel test structures allowed quantification of the maximum achievable peel stress, as an analog to adhesive strength, between two polyimide layers and/or different adhesion materials and metals. Each standard coupon was 77.5 mm long and 8.3 mm wide with six replicates/wafer, and each wafer also included two shorter 60-mm-long coupons. All processing steps were chosen to mimic those used to fabricate implantable neuroelectronic devices. However, to isolate the impact of processing parameters and material stacks on adhesion strength, several different versions of the T-peel samples were produced.
Figure 3.
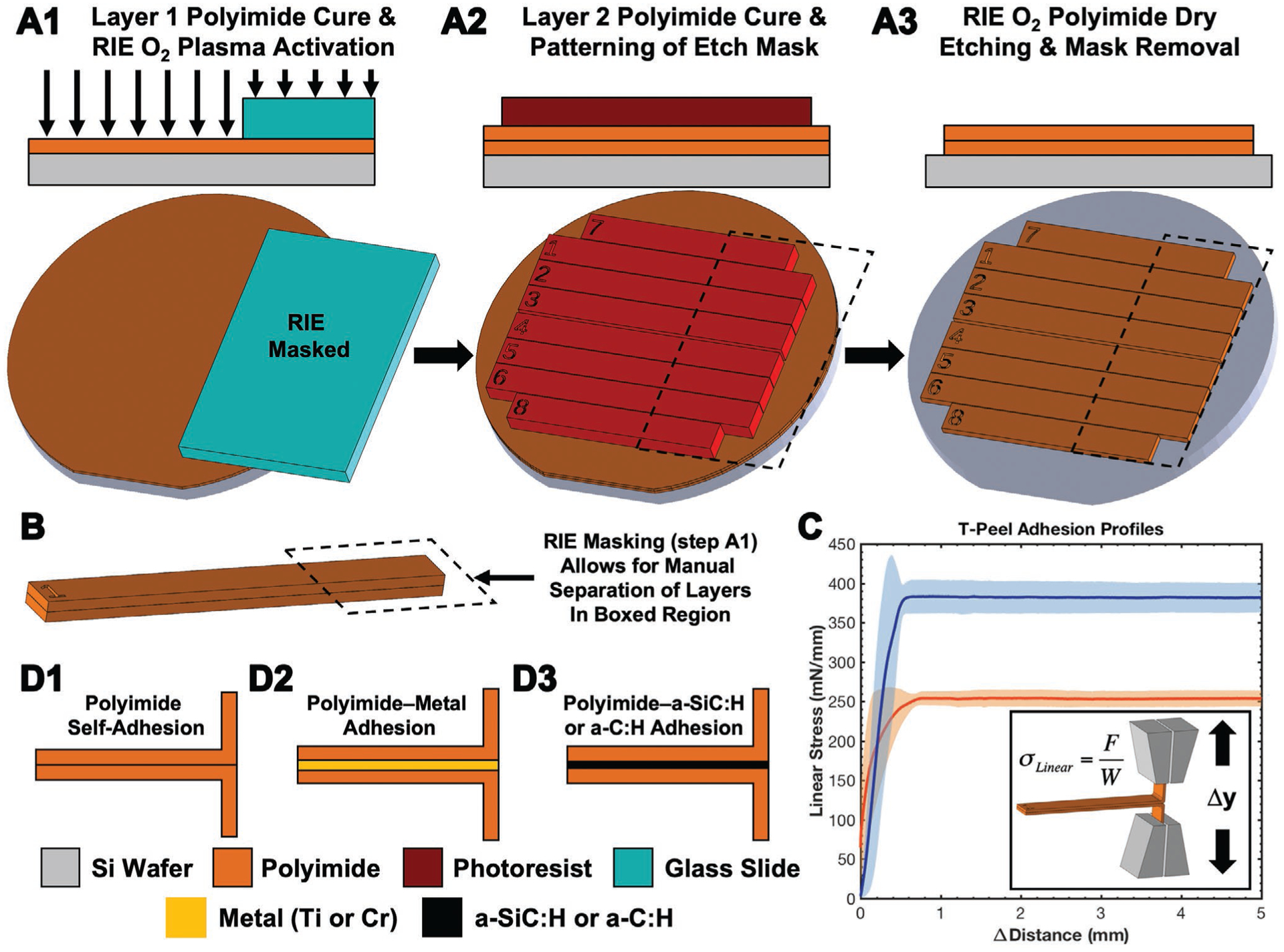
Overview of the fabrication and testing of the T-peel test structures made from two layers of polyimide. A) Wafer-level fabrication process flow highlighting area masked during RIE by a glass slide. B) Highlighted details of an individual T-peel coupon, including the portion that can be peeled apart in order to be secured into a load frame for testing. C) Typical linear stress–distance curves of two sample populations with distinct maximum linear stress values and an inset schematic of the T-peel load grips. Data points represent the arithmetic average (± standard deviation error envelopes). D) Summary of T-peel test structures fabricated to isolate adhesion strength of different material interfaces.
The impact of bottom polyimide curing temperature (T1) on the peel strength between top and bottom polyimide layers was determined by first spin-coating 10 μm of polyimide onto a wafer and curing to a maximum T1 = 250–450 °C (Figure 2). Next, a 50 mm × 75 mm borosilicate glass slide was placed over a portion of the polyimide sample, and an RIE O2 plasma (Unaxis 790) was applied for 60 s, described previously, to chemically activate the exposed polyimide surface (Figure 3A1). A second 10-μm-thick layer of polyimide was then spin-coated on to the first layer, and the layers were cured together at 450 °C (T2). Next, a 25-μm-thick etch-mask layer of photoresist (AZ9260, MicroChemicals GmbH) was spin-coated, rehydrated, exposed (MA6, SUSS MicroTec) through a photomask and developed in 4:1 DIW: AZ400K (MicroChemicals GmbH) (Figure 3A2). Nonmasked polyimide was dry-etched using an RIE O2 plasma through both layers revealing individual test coupons (Figure 3A3). Residual photoresist was removed after etching using PRS3000 (J.T. Baker) at 70 °C.
The two layers of each coupon could be manually separated using tweezers along the region that was masked by the glass slide during step A1 because the interfacial adhesion was greatly reduced without O2-plasma activation. Mechanical T-peel tests were performed in accordance with ASTM D1876–08 using a tensile-load frame (TA.XT.plus, Stable Micro Systems) with a 5 kg load cell at a crosshead translation rate of 0.1 mm s−1. The maximum peel stress was determined by averaging the stable plateau of linear stress–distance plots from 1 to 5 mm with a minimum of 12 replicates from at least two wafers (N = 2, n = 12) (Figure 3C). Coupons that slipped, ripped, or broke during testing were excluded from analysis.
This general fabrication method was modified to determine the impact of other processing parameters and material stacks on peel strength (Figure 3D). Both polyimide layers were cured at T1 = T2 = 450 °C and RIE surface activation was fixed at 60 s during step A1 unless specifically noted otherwise. The effect of RIE activation dosage during step A1 on polyimide–polyimide adhesion was determined by varying O2-plasma time from 0.1 to 500 s (Figure 3D1). The peel strength between metal and polyimide was determined by sputtering different metals onto the bottom polyimide layer during step A1 (Figure 3D2). To pattern metal, a 3-μm-thick layer of negative photoresist (nLOF 2035, MicroChemicals GmbH) was spin-coated, soft-baked, exposed through a photomask, post-exposure baked, and developed (AZ300MIF, MicroChemicals GmbH). The polyimide substrate was cleaned of residual photoresist and chemically activated using a RIE O2 plasma for 45 s, as described previously. It was noted that longer RIE times caused cracking of the nLOF layer. Metal layers including titanium and chromium were sputter-deposited (CMS-18 multisource, Kurt K. Lesker Company) to a thickness of 100 nm. Metal was patterned by lift-off in NMP at 70 °C, and thickness was determined by contact profilometry (Dektak 150 Surface Profiler, Veeco). Subsequent RIE activation of the wafer using the glass-slide mask, top-layer-polyimide curing, and dry-etching processes proceeded, as discussed previously.
The adhesion between polyimide and either a-SiC:H or a-C:H layers was determined by: 1) O2 activating the bottom polyimide layer, with the glass slide masking as before, 2) PECVD depositing the respective film with the glass slide mask still in place, using deposition methods described in Sections 2.1.3 and 2.1.4, and 3) O2 activating the PECVD film’s surface (Figure 3D3). For a-SiC:H containing samples, variations of this general protocol were further made to determine the impact of the APTES adhesion promoter between the a-SiC:H and polyimide layers, deposited as described previously. Encapsulation in the top polyimide layer proceeded as described previously followed by dry etching to reveal individual coupons. As the a-SiC:H layer is not appreciably etched in an O2 plasma during step A3, the RIE gas flow (Unaxis 790) was changed to 30 sccm CF4 and 6 sccm O2 at 40 mTorr once the a-SiC:H layer was reached. After the a-SiC:H layer was etched through, the gas flow was returned to previous levels to etch the remaining polyimide.
2.3. Metal Alloying and XPS Depth Profiling
Depth profiling of polyimide/a-SiC:H/metal/a-SiC:H stacks was performed to identify alloying and diffusion between layers due to thermal processing experienced during device microfabrication. Each test substrate consisted of a Si wafer with 5 μm of 450 °C cured polyimide + 250 nm stoichiometric a-SiC:H + metal stack + 250 nm a-SiC:H. Samples were then heated to 450 °C to simulate a full-stack polyimide cure. Four different metal-stack compositions were characterized (Table 1). Depth profiling was performed by alternating between XPS analysis and Ar+ ion-beam sputter etching. Etching was stopped once the polyimide layer was reached. Each cycle consisted of four high-resolution XPS sweeps of C1s, Si2p, O1s, Ti2p, Pt4f, and Au4f binding energies with a 0.4 eV step size followed by 2 min of Ar+ sputter-etching at 4 kV over a 2 × 2 mm2 area with Zolar rotation to maximize etch uniformity. Total time to etch through the full stack was ≈310 min with 155 XPS/sputter cycles.
Table 1.
Deposited metal stacks used for XPS depth profiling. Stacks were encapsulated between two 250 nm a-SiC:H layers on 5 μm polyimide and annealed at 450 °C.
| Metal stack identifier | Bottom adhesion | Metal core | Top adhesion |
|---|---|---|---|
| Stack A | 50 nm Ti | 300 nm Pt | 50 nm Ti |
| Stack B | 50 nm Ti | 300 nm Au | 50 nm Ti |
| Stack C | 50 nm Ti | 100 nm Pt + 100 nm Au + 100 nm Pt | 50 nm Ti |
| Stack D | 50 nm Ti | 100 nm Pt + 100 nm Au + 100 nm Pt | None |
2.4. Metal Resistivity Measurements
Four-point probe metal resistivity test structures (Figure S1, Supporting Information) were fabricated using procedures described previously and consisted of stacks of 5 μm polyimide + a-SiC:H/metal/a-SiC:H + 5 μm polyimide according to Table 1. Micropositioner probe tips were used to contact each of the four contact pads, and metal resistivity was determined by dividing the voltage drop measured across the inner two contact pads by the dc current from a power supply (E3611A, Agilent Technologies) delivered through the outer two pads. The voltage drop was measured directly across the two inner pads with a multimeter. Current was determined by dividing the voltage drop across a precision 10 kΩ resistor, which was measured by a second multimeter, by its resistance value.
3. Results and Discussion
3.1. Polyimide Curing and Surface Modification
The polyamic acid precursor used to fabricate BPDA-PDA polyimide starts to thermally imidize at ≈125–150 °C and reaches maximal percent imidization at ≈250 °C as both the water formed during imide-ring formation and the NMP solvent evaporate.[19] Tracking changes in the functional groups during this process with FTIR spectroscopy can determine relative imidization (Figure S2, Supporting Information).[19] The maximum cure temperature is linked to bulk film properties such as dielectric loss factor, dc conductivity, and dielectric breakdown field.[19] After thermal curing, the final polyimide film is relatively inert, chemically resistant, and near impossible to thermally reprocess (e.g., by extrusion, thermoforming). Although these robust properties make BPDA-PDA polyimide an excellent material choice for neural implants, its relative resistance to thermal and chemical reprocessing can make forming chronically stable and robust implants difficult due to weak adhesion at material interfaces. To address this challenge, it is important to understand the surface properties and how different materials (e.g., additional polyimide, conductive metal, a-SiC:H, etc.) can be adhered to the cured polyimide films.
Tracking changes in polyimide surface properties during microfabrication and their influence on subsequent adhesion is critical to create reliable devices. Surface changes of cured BPDA-PDA films were characterized using XPS after a variety of common processing steps used during device fabrication including: 1) as-cured, 2) RIE O2 plasma (used to improve adhesion, remove organic contaminates, and etch polyimide), 3) RIE SF6 plasma (used to etch metals, oxides, and ceramics including a-SiC:H), and 4) silanization with APTES (a common adhesion promotion agent for oxide-forming materials) (Table 2 and Figure 4). Survey scans of as-cured polyimide identified carbon, oxygen, and nitrogen, and high-resolution peak fitting resolved constituent C—C/C—H, C=O, and C—N bonds as expected from the BPDA-PDA backbone chemistry. The O2 plasma oxidized surface carbon as indicated by a decrease in C% and increase in O% with corresponding appearances of C—O peaks within both the C1s and O1s atomic envelopes arising from alcohol and carboxyl groups. These new hydrophilic functional groups lowered the water contact angle of BPDA-PDA films from 68 ± 2° to 3 ± 2° with as little as 10 s of O2-plasma activation, and this value did not appreciably change with longer activation times. The activated functional groups added to the surface can be used to form covalent linkages with a second polyimide layer, react with various adhesion agents (e.g., APTES), and improve adhesion with deposited metals, semiconductors, and ceramics.
Table 2.
Atomic composition of the polyimide surface after various processing steps obtained using XPS survey scans. Data shown include the arithmetic average (+/− standard deviation), n = 3.
| Process step | Carbon (%) | Oxygen (%) | Nitrogen (%) | Fluorine (%) | Silicon (%) |
|---|---|---|---|---|---|
| As-cured | 75.4 (2.1) | 18.1 (1.4) | 6.4 (2.3) | 0.0 (0.0) | 0.0 (0.0) |
| O2-RIE activated | 67.7 (2.5) | 25.8 (2.2) | 6.4 (2.4) | 0.0 (0.0) | 0.0 (0.0) |
| SF6-RIE exposed | 50.8 (1.9) | 11.0 (2.7) | 5.0 (1.2) | 33.3 (3.4) | 0.0 (0.0) |
| APTES silanized | 49.1 (3.9) | 34.1 (3.2) | 6.8 (2.2) | 0.0 (0.0) | 10.0 (1.1) |
Figure 4.
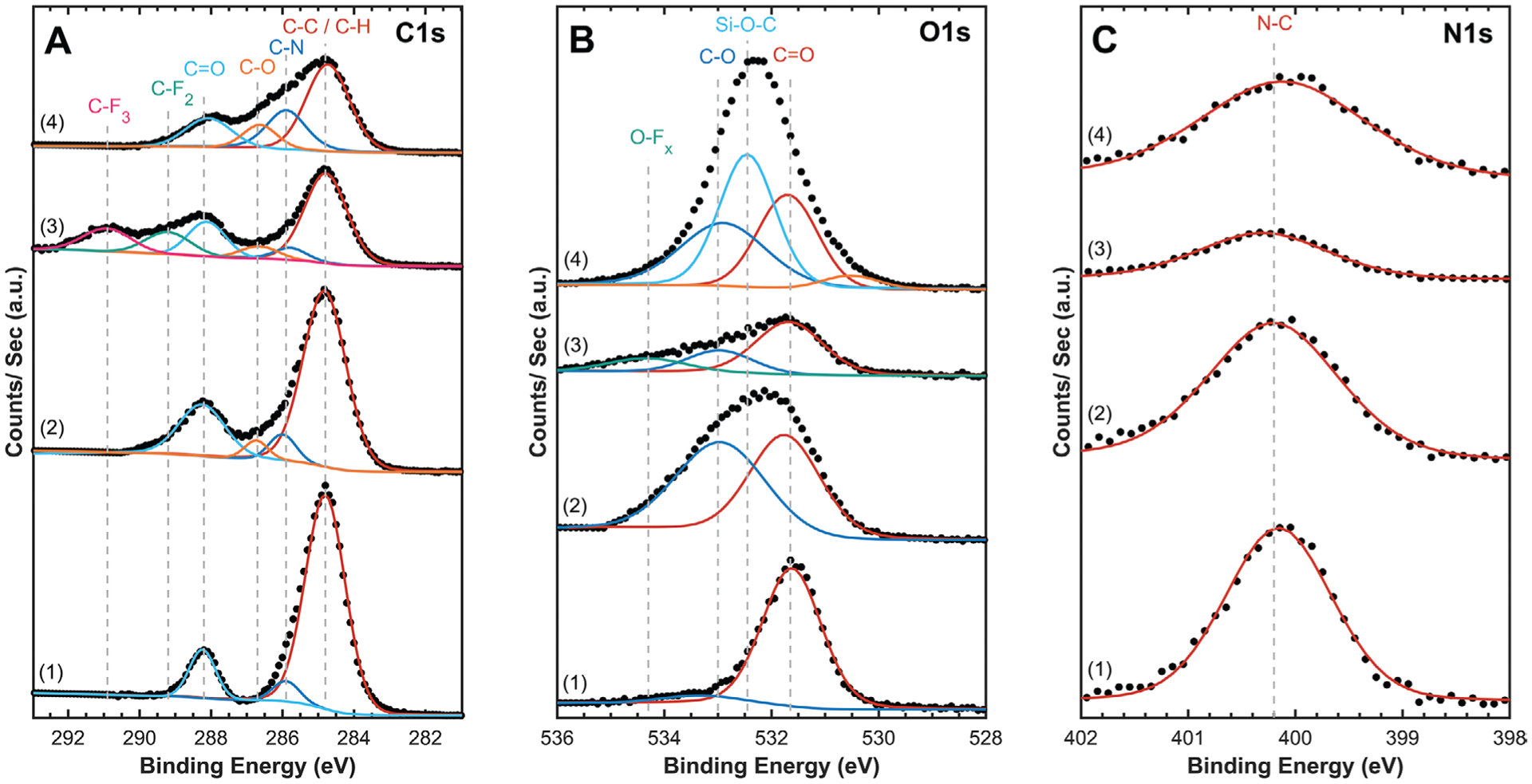
Representative high-resolution XPS analysis of A) carbon, B) oxygen, and C) nitrogen 1s orbitals for BPDA-PDA polyimide films after various processing steps. 1) as-cured, 2) O2 RIE activated, 3) SF6 RIE exposed, and 4) O2 RIE activated and APTES silanized. Spectra were peak-shift corrected to 284.8 eV C—C peak to account for charging, and each spectra is shifted along the Y-axis to allow for comparison. Experimental data points are overlaid with fitted peaks showing identified bonding environments.
During thin-film device microfabrication, an SF6 plasma can be used to etch a variety of oxides (e.g., SiO2), ceramics (e.g., a-SiC:H), and metals (e.g., titanium) that are not significantly etched by an O2 plasma. Although an SF6 plasma does not appreciably etch polyimide, fluorinated species can become imbedded in the polyimide surface and form a variety of fluorinated compounds that can hinder adhesion to subsequent layers. As shown in Table 2, SF6 plasma causes the atomic composition of the surface of polyimide to become ≈33% fluorine, which bonds to carbon and oxygen in the polyimide. This problematic contamination can be reduced by using a subsequent RIE Ar plasma or fully removed by etching 100–200 nm of the polyimide surface using a subsequent O2 plasma.
Silane agents are commonly used to form covalent linkages between surface oxides (e.g., SiO2, oxidized metals, oxidized a-SiC:H) and polymers. Specifically, APTES is used with polyimides because its primary amine head (R—NH2) can react with carboxylic acid groups (R—C=O—OH) in either the polyamic acid precursor or on O2-plasma activated polyimide to form amide bonds (R1—C=O—NH—R2) at >100 °C. The triethoxy silane tail of APTES can react with oxides forming silanols (R1—Si—O—R2) through condensation of ethanol. An XPS analysis of O2-activated polyimide that was silanized with APTES revealed 10% of the surface atomic composition was Si, and there was an increase in C—O, C—N, and Si—O—C bonding character indicating successful silanization. The water contact angle correspondingly increased from 3 ± 2° to 49 ± 1° as the hydrophilic functional groups on the O2-activated polyimide were capped with APTES. While both the C1s and O1s orbital peaks were able to accurately track changes in surface chemistry, the N1s peak remained consistent for all different surfaces measured. None of the plasma-processing steps are expected to significantly alter the N—C bonds, and the weak N1s signal was insensitive to distinctions between the primary amine of APTES and the nitrogen within the imide bond of the polyimide.
3.2. a-SiC:H Deposition and Surface Modification
Crystalline and semicrystalline SiC polymorphs (e.g., 3C-SiC, 6C-SiC, and 4H-SiC) are commonly used materials in a variety of industries due to their chemical inertness, physical strength, and semiconducting properties.[22] Although SiC is typically produced by high-temperature (≥1500 °C) firing or sintering, which is common for many ceramics, such high-temperature fabrication methods are not compatible with polymeric neural-interface fabrication. Instead, SiC can be deposited by relatively low-temperature (e.g., 100–350 °C) PECVD, which results in an amorphous dielectric film composed of silicon, carbon, and a significant amount of hydrogen (e.g., ≈27%)[23] that originates from incomplete dissociation of the precursors (e.g., SiH4 and CH4). This type of PECVD-deposited SiC is designated as amorphous hydrogenated SiC (a-SiC:H) to distinguish it from the high-temperature polymorphs.
Thin films of a-SiC:H are “biocompatible,” do not cause an exaggerated inflammatory/tissue response,[24] and are resistant to dissolution in biological solutions.[25] Thin films of a-SiC:H have been used in a variety of application areas,[24] including as an adhesive aid between polyimide and metal,[1,14,18,26,27] as a dielectric encapsulant around stiff probes,[28] and as the primary dielectric and/or structural material for thin-film probes.[29,30] Its use as an adhesive aid is of most relevance within the context of this work. The primary factors important for incorporation of thin-film a-SiC:H into reliable polyimide devices are its intrinsic film stress, stoichiometry, and chemical and surface characteristics. Stoichiometric films are targeted so that there is an equal probability of forming a carbide or silicide bond with adjacent material layers (e.g., polyimide, titanium, platinum). The analysis presented here does not focus on intrinsic film stress that is typically controlled primarily by the deposition conditions and postdeposition thermal annealing. It is understood that excessive film stress should be mitigated as much as possible. Instead, the analysis focuses on more fundamentally applicable chemical and surface properties of deposited films and how these material properties impact the integration of a-SiC:H into functional thin-film devices.
Depth profiling of a-SiC:H throughout the entire film thickness using XPS confirmed film stoichiometry with only trace oxygen (e.g., <1%) (Figure S3, Supporting Information); however, it is important to note that XPS cannot determine the hydrogen content within the film. Transmission FTIR confirmed the chemical-bonding environment within the deposited film was in agreement with literature sources,[20,21,23,28] which confirms that a-SiC:H was the primary phase deposited instead of other possible phases, such as hydrogenated amorphous silicon, hydrogenated amorphous carbon (a-C:H), or SiO2 (Figure S3, Supporting Information). Surface changes of stoichiometric a-SiC:H films were monitored with XPS after a variety of processing steps (Table 3 and Figure 5), in a fashion similar to the analysis discussed previously for polyimide. Survey scans of as-deposited a-SiC:H identified both carbon and silicon, as expected, as well as significant oxygen due to a native-oxide layer. High-resolution XPS of the C1s envelope confirmed that Si—C is the primary carbon bond with a minority of C—C/C—H bonds from incomplete dissociation of the CH4 precursor. Analysis of the Si2p envelope identified a majority of Si—C bonds as well as several oxidation states of silicon oxycarbide (SixOyCz)[31] and SiO2 confirming that the native oxide is formed primarily on silicon atoms as opposed to carbon.
Table 3.
Atomic surface composition of stoichiometric a-SiC:H films after various processing steps obtained using XPS survey scans. Data shown include the arithmetic average (± standard deviation), n = 3.
| Process step | Carbon (%) | Silicon (%) | Oxygen (%) | Nitrogen (%) | Argon (%) |
|---|---|---|---|---|---|
| As-deposited | 28.3 (3.1) | 43.6 (2.1) | 28.1 (2.8) | 0.0 (0.0) | 0.0 (0.0) |
| Ar+ etched | 49.5 (1.8) | 48.1 (2.9) | 0.5 (0.3) | 0.0 (0.0) | 1.9 (0.7) |
| O2-RIE cleaned | 9.3 (2.8) | 32.6 (1.9) | 58.1 (2.2) | 0.0 (0.0) | 0.0 (0.0) |
| APTES agent | 28.5 (2.2) | 20.9 (4.1) | 44.2 (3.9) | 6.3 (1.2) | 0.0 (0.0) |
Figure 5.
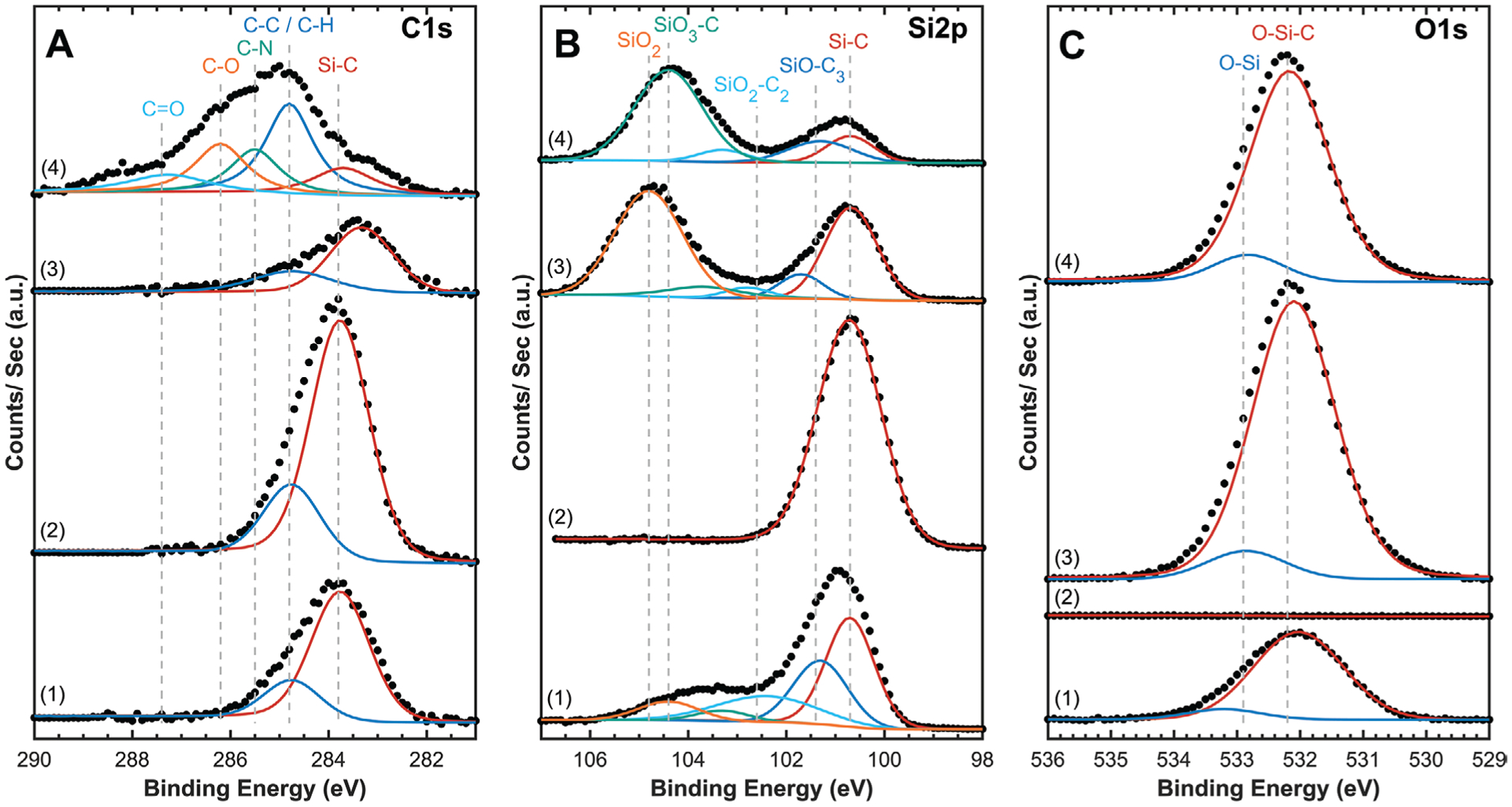
Representative high-resolution XPS analysis of A) C1s, B) Si2p, and C) O1s orbitals for stoichiometric a-SiC:H films. 1) As-deposited with native oxide, 2) after native oxide removed with Ar+ etching, 3) O2-RIE cleaned with increased O2-bond density, and 4) O2-RIE cleaned and APTES silanized. Spectra were peak-shift corrected to 284.8 eV C—C peak to account for charging, and spectra are shifted along the Y-axis to allow for comparison. Experimental data points are overlaid with fitted peaks showing identified bonding environments.
The bulk a-SiC:H film can be analyzed by removing the thin native oxide layer using Ar+ sputtering to etch the first few atomic layers of the film while within the ultrahigh vacuum XPS chamber. Survey scans of Ar+-sputtered films confirmed the a-SiC:H was stoichiometric with <1% O, and high-resolution XPS of the respective elemental envelopes was dominated by Si—C and C—C/C—H bonding with no appreciable Si—O bonds. As to be expected, sputtering with Ar+ does result in minor argon implantation into the film surface. Cleaning with O2 plasma is a common processing step used to activate surfaces and remove residual contaminants (e.g., photoresist, fluorinated species) to maximize adhesion between different material layers. Cleaning the surface of a-SiC:H with RIE O2 plasma oxidizes carbon bonds, reducing the carbon content and increasing the oxygen content through the formation of SiO2. The dominant SiO2 character of the a-SiC:H surface will dictate adhesion to subsequent layers, especially to polyimide that is spin-coated and cured onto the a-SiC:H, discussed in detail in Section 3.4.3. However, the SiO2 moieties can be functionalized with silane adhesion agents (e.g., APTES) to control the adhesion of a-SiC:H with subsequent materials. Films of a-SiC:H cleaned with O2 plasma were silanized with APTES, which resulted in a relative increase in the carbon content and incorporation of nitrogen into the film from APTES’s primary amine. High-resolution analysis of the C1s orbital confirmed successful APTES silanization with the appearance of both C—N and C—O peaks. Furthermore, the Si2p orbital demonstrated a strong SiO3—C peak from the silanol tail and a complete absence of SiO2 indicating that the majority of the a-SiC:H surface was properly silanized.
3.3. a-C:H Deposition and Film Properties
Diamond-like carbon (DLC) is typically described as a meta-stable form of amorphous carbon that contains a significant portion of sp3 bonds compared to graphitic sp2 bonds.[32] The sp3 character of the disordered isotropic DLC film produces material properties similar to diamond (e.g., increased hardness, elastic modulus, and thermal conductivity) which allow DLC films to be used in a wide array of applications.[32] However, DLC is a catch-all name used to describe films that can be deposited by various methods (e.g., pulsed laser deposition, sputtering, cathodic arc, PECVD),[32] can be formed into an array of different polymorphs (e.g., glassy carbon, a-C, ta-C), and can contain a varying amount hydrogen and sp3 bonding that can result in disparate film properties. Due to this high DLC-film variability, it is very challenging to make direct comparisons between different literature sources on DLC films unless explicit material properties and deposition conditions are provided. In the context of polyimide-based neural implants, PECVD deposited DLC has been reported to form C—C bonds[26] with polyimide that provide more robust adhesion than a-SiC:H. Based on this encouraging result, DLC of an unknown polymorph was used as a tie layer between a-SiC:H and polyimide in place of APTES.[18] Unfortunately, reproducing the reported DLC results remains difficult since few experimental details were given (e.g., deposition conditions) and key film properties (e.g., bulk and surface chemistry) were not provided.
Herein, the development of an amorphous carbon film is reported with extensive details that can be used within a polyimide microfabrication process for neural devices (Figure 6). For the sake of clarity and accuracy, all carbon films deposited via PECVD will be reported as amorphous hydrogenated carbon (a-C:H) to distinguish them from the many other popular polymorphs. Films of a-C:H were deposited using 13.56 MHz RF PECVD with CH4 gas. Deposition conditions were varied using one-way DOEs to isolate the effect of CH4 flow rate, chamber pressure, deposition power, and temperature (Figure 6A–C). CH4 is relatively difficult to ionize, compared to other carbon sources like acetylene or benzene, and capacitive coupling at 13.56 MHz is relatively inefficient due to low plasma density.[32] At 100 °C, the deposition rate was maximized at low gas flow rates (e.g., 50 sccm) that maximized ionized gas residence time within the chamber[33–35] and at low pressures (e.g., 50 mTorr) that maximized the ion-radical fraction in the plasma.[32–36] However, depositions performed below 35 mTorr or 25 sccm CH4 either failed to ignite a plasma or resulted in no deposited film. At higher temperatures of 300 °C, deposition was only successful at these optimized conditions of 50 sccm CH4 and 50 mTorr while higher values resulted in a failure to ignite a plasma, no significant deposition, or inhomogeneous surface coverage. Introducing helium into the chamber had minimal impact on film deposition.
Figure 6.
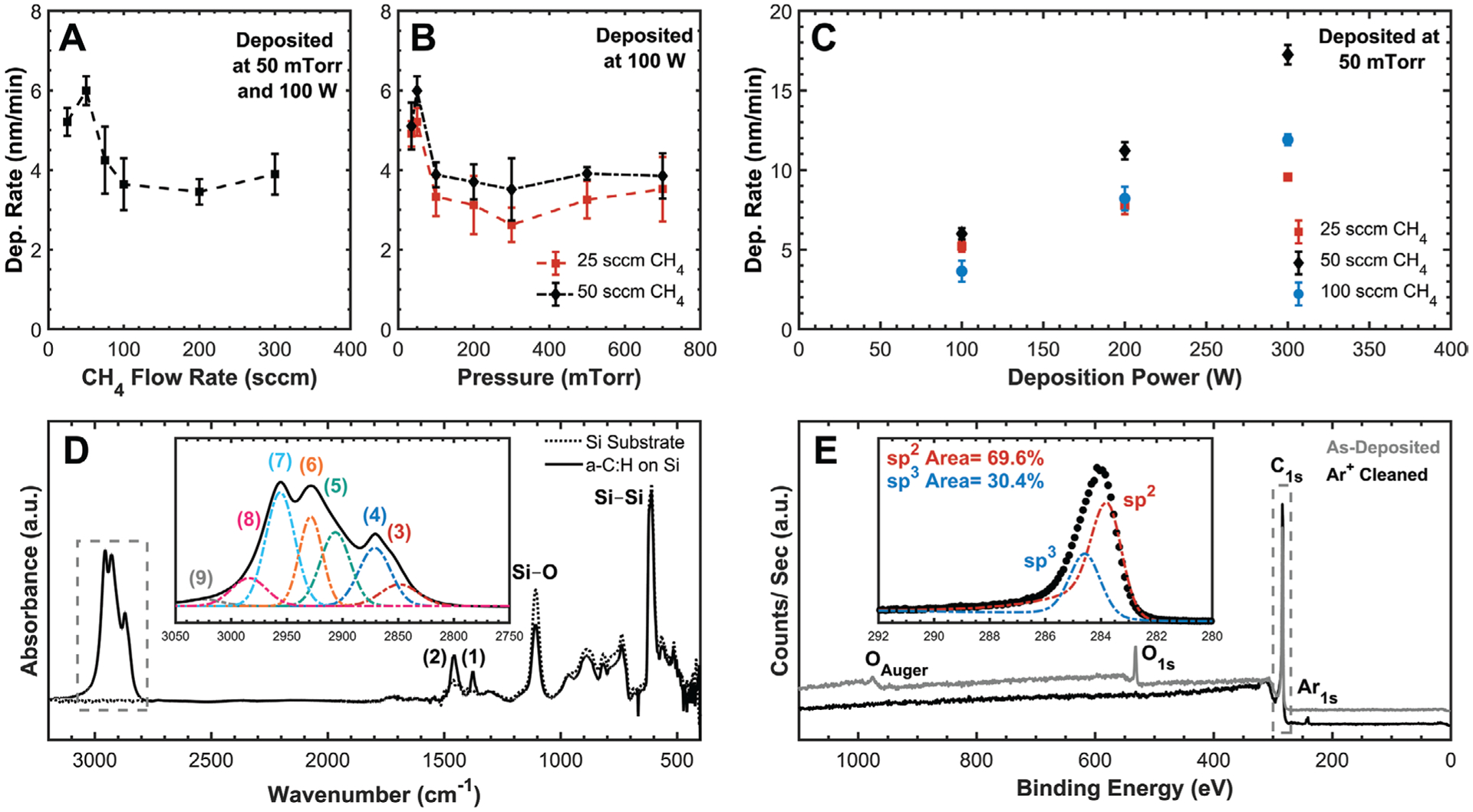
Summary of a-C:H deposition development and resultant film properties. One-way DOEs at 100 °C showing the influencing effect of deposition A) flow rate, B) pressure, and C) power. Deposition rate and surface coverage were maximized at 50 sccm CH4 and 50 mTorr with a linear increase with deposition power. Data points represent the arithmetic average with error bars representing one standard deviation, n = 3, N = 5, and dashed lines are simply to aid visualization. D) Transmission FTIR spectra of a-C:H film with included inset of the C—H stretching vibration region (boxed) showing a dominant sp3 character. Si—Si and Si—O peaks are from the Si wafer substrate, and a-C:H peak fitted IDs are included in Table 4. E) XPS survey spectra of a-C:H with included inset of the peak fitted C1s photoemission envelope from Ar+ cleaned surface (boxed) with no detectable oxygen. Spectra were peak-shift corrected to 284 eV C—C to account for charging, and spectra are shifted along the Y-axis to allow for comparison.
Films were characterized using both FTIR and XPS to determine chemical bonding within the film including their general sp2/sp3 character (Figure 6D,E). Peak fitting of FTIR spectra identified a majority of sp3 carbon–hydrogen bonds with no evidence of ordered aromatics (e.g., graphite) and only weak peaks corresponding to a minority of sp2 bonded alkene carbon (Table 4). Survey XPS scans of as-deposited a-C:H identified oxygen contamination (12.1%) on the surface that could be removed using Ar+ sputtering (Figure 6E), and RIE etching the surface with an O2 plasma increased the oxygen content (21.9%). However, high-resolution analysis of the C1s photoemission envelope did not detect any changes in peak shape with or without the presence of oxygen suggesting that it is likely weakly adsorbed atmospheric O2. Peak fitting of the C1s envelope identified both asymmetric sp2 (284 eV) and symmetric sp3 (284.8 eV) peaks, but the relative ratio between the two is highly susceptible to both the peak-fitting parameters used and their relative positions and should not be considered as ground truth. Using both FTIR and XPS, efforts were made to maximize the sp3 (i.e., diamond-like) character through varying deposition parameters, but no significant changes in the chemical bonding state were identified through the full range of conditions tested (e.g., temperature, pressure, power, flow-rate, etc.). It is likely that an RF restricted PECVD without additional power supplies, deposition frequencies, carrier gases (e.g., argon), or deposition gases (e.g., acetylene) limits the tunability of film properties. Additional studies are required using more advanced PECVD tools or different deposition methods (e.g., pulsed laser deposition).
Table 4.
| Peak identification | Position [cm−1] | Vibrational mode assignment |
|---|---|---|
| 1 | 1374 | sp3 C—H3 (symmetric bend) |
| 2 | 1455 | sp3 C—H2 (scissor) |
| 3 | 2848 | sp3 C—H2 (symmetric stretch) |
| 4 | 2871 | sp3 C—H3 (symmetric stretch) |
| 5 | 2907 | sp3 C—H (stretch) |
| 6 | 2929 | sp3 C—H2 (asymmetric stretch) |
| 7 | 2956 | sp3 C—H3 (asymmetric stretch) |
| 8 | 2984 | sp2 C—H (stretch) |
| 9 | 3028 | sp2 C—H2 (stretch) |
3.4. Polyimide Adhesion
A common failure mechanism experienced by polyimide-based thin-film neuroelectronic devices both in vitro and in vivo is delamination of dielectric layers (e.g., polyimide, a-SiC:H) and/or conductive metal traces resulting in penetration of body fluid and loss of electrical integrity.[10,12–14] These critical interfaces were investigated using adhesion T-peel tests to determine fabrication strategies and materials that would maximize interfacial integrity. Improving these weak points within thin-film devices should improve their resistance to failure and overall chronic reliability. Mechanical loading of T-peel coupons causes delamination (i.e., fracture/cracking at a bimaterial interface) due to a stress concentration between two layers that can be quantified for comparative purposes. A primary advantage of such tests is the ability to measure adhesion between multiple material interfaces (e.g., polyimide–metal–polyimide) to identify the weakest interface.
3.4.1. Polyimide–Polyimide Self-Adhesion
Maintaining robust adhesion between laminated polyimide layers that comprise the majority of the mechanical and dielectric integrity of a neuroelectronic device is of paramount importance. Two different sets of T-peel test coupons were fabricated to isolate the impact of fabrication parameters on polyimide–polyimide adhesion: 1) effect of T1 cure temperature and 2) RIE O2-plasma activation time (Figure 3D1). Although manufacturer specifications recommend curing the BPDA-PPD polyimide to 450 °C to maximize imidization and mechanical/dielectric properties, these specifications do not consider polyimide–polyimide adhesive strength. Therefore, the effect of curing the bottom polyimide layer at lower T1 temperatures while still curing the final polyimide stack at T2 = 450 °C was investigated (Figure 7A). The specific T1 values were chosen to be above the boiling point of NMP (202 °C) with two temperatures below the final cured film’s glass transition temperature (Tg = 322–324 °C) and two above.
Figure 7.
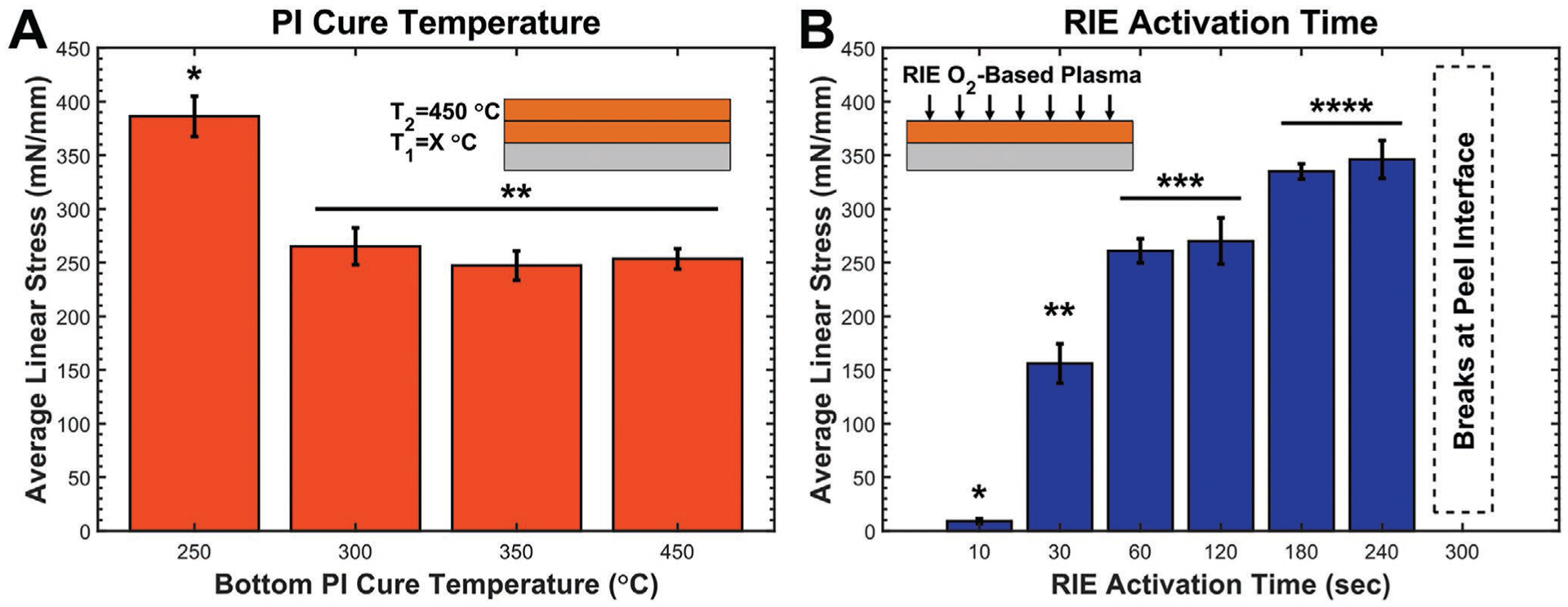
Polyimide–polyimide T-peel summary highlighting the impact of different fabrication parameters. A) Curing the bottom polyimide layer at different T1 temperatures using a standard O2-RIE-activation time of 60 s shows that a lower T1 = 250 °C improves adhesion. B) The effect of O2-RIE-activation time using a standard T1 = T2 = 450 °C PI cure shows that increasing activation time improves adhesion. T-peel coupons activated for 300 s or longer were nontestable due to consistent breakage of the peel arms at the peel interface; therefore, the upper limit of this trend is unknown. Data points represent the arithmetic average with standard deviation error bars, N = 2, n = 12. Asterisks indicate statistically homogenous subsets, 1-way ANOVA, α = 0.05, Tukey’s post hoc analysis.
A T1 = 250 °C significantly increased the linear peel stress between the polyimide layers compared to curing at T1 = 300 to 450 °C. Similar results were reported for PMDA-ODA[39,40] polyimide that showed increased peel strength with lower T1 cure temperatures. Analysis with XPS and forward recoil spectroscopy revealed that lowering T1 resulted in increased interdiffusion between polyimide layers due to increased polymer chain mobility.[39,40] Furthermore, maximizing the differential between T1 and T2 resulted in maximum peel strength.[39] The majority of the imidization of BPDA-PDA occurs between 175 and 250 °C as the NMP solvent and condensed water is removed with a stable plateau around 90% imidization at temperatures >250 °C,[19] and this is supported by the fact that there were no significant chemical changes between films cured at 250 or 450 °C (Figure S2, Supporting Information). While film thickness decreased by 31.5% from 120 to 250 °C as the solvent was removed and the film densifies, there was only an additional 2.0% decrease when curing to 450 °C. It is important to note that all T-peel tests reported were between layers that were O2-plasma activated for 60 s to improve adhesion, and areas that were not activated could be easily separated using tweezers.
Surface activation with high-energy O2 plasma is a widely used method to improve adhesion between various materials and polymers.[41,42] By oxidizing C—C bonds along the cured polyimide backbone, the plasma creates a variety of surface functional groups (e.g., carboxylic acid, alcohol, Figure 4) that can react with functional groups within the subsequently deposited polyamic acid precursor to form covalent linkages between the two layers during curing. The plasma also etches and effectively roughens the surface, which leads to increased surface area that can improve the physical interlocking to subsequently deposited materials.[43] Within the context of adhesive strength between two polyimide layers, maximizing both the quantity of surface functional groups and surface roughness will maximize adhesion strength. Using T-peel coupons, increasing RIE time from 10 to 240 s resulted in a measurable increase in peel strength between the polyimide layers (Figure 7B).
Longer RIE times (i.e., 300–600 s) were also tested, but all T-peel coupons fractured at the polyimide–polyimide interface before delaminating suggesting that the localized peak stress experienced was near the yield stress of the polyimide. For T-peel samples, the localized stress concentration at the interface can be significantly higher than the actual peel stress due to sharp geometric changes in film thickness.[44] During O2-plasma activation, the polyimide is etched at a rate of ≈87 nm min−1 resulting in an abrupt and non-insignificant step change in the thickness of the bottom polyimide layer as glass-slide masks are used to prevent some regions from being exposed to the plasma. As a consequence, these T-peel test results do not indicate that the peel interface was stronger than the bulk material, but rather that the interface was no longer the lowest energy path for crack propagation.[39] Although attempts were made to fabricate thicker T-peel coupons (e.g., using 20-μm-thick polyimide layers) that could potentially resist fracture during testing, the extreme film thickness resulted in the formation of large bubbles that typically caused the film to delaminate from the wafer during the microfabrication process. Bubbles likely originated from residual solvent that was unable to completely diffuse out of the thicker films during curing.
These two strategies were combined by fabricating T-peel coupons with a T1 = 250 °C and RIE activation time of 240 s; however, the coupons fractured at the interface indicating that these effects are likely additive resulting in an even higher average linear stress to fracture. Overall, these results indicate that to maximize polyimide–polyimide self-adhesion, the first polyimide layer should be cured to T1 = 250 °C with maximal O2-plasma-RIE activation before applying and curing the second polyimide layer to T2 = 450 °C. Owing to the lowered T1, all subsequent processes (e.g., PECVD) performed prior to curing the top polyimide layer should be performed at T < 250 °C so as not to impact the polyimide structure.
3.4.2. Polyimide–Metal Adhesion
The polyimide acts as the primary dielectric layer between conductive metal traces and protects sensitive electrode sites on the neuroelectronic device by maintaining electrical isolation between all channels while limiting parasitic capacitance between adjacent traces. Failure of the metal–polyimide interface allows for fluid ingress that leads to electrical shorting between traces and a decrease in electrode performance, and failure is often exacerbated during electrical stimulation.[45] Furthermore, delaminated metal structures are free to move within the polyimide and can act as a stiff “blade” that mechanically forces additional delamination. Unfortunately, it can be challenging to form a robust adhesive interface since polymers traditionally form only relatively weak interactions with metals through Van der Waals, ionic, oxide, and/or limited carbide bonds.[46–48] Titanium and chromium are traditionally used as adhesive tie layers between polyimide and either gold or platinum as they can readily form carbide bonds and are highly reactive with oxides while both platinum and gold do not form carbides at the temperatures used for neural device fabrication.
T-peel coupons made with titanium or chromium were used to investigate the critical metal–polyimide interface (Figure 3D2). The thickness of metal tie layers is typically kept thin (≈15–50 nm) to provide the chemical functionality needed while minimizing the impact of film-stress and CTE mismatch. As the tie layer is typically applied to both the top and bottom of the core noble metals (e.g., platinum, gold), 100 nm was chosen for these studies to equate to two 50-nm-thick films. The peel strength of polyimide-titanium-polyimide samples was extremely weak, with an average peel strength of 3 N m−1 compared to the polyimide–polyimide control interface (Table 5). Coupons with the titanium layer were easily peeled apart by hand and partially delaminated when simply placing test samples onto the load frame. As the measured peel strength was near the lower limit of the load-cell detection range, the experimentally obtained value should be considered only an approximation of this weak interface.
Table 5.
Average linear peel stress of T-peel coupons using a variety of adhesion materials between two 10-μm-thick polyimide layers. Data points represent the arithmetic average (+/− standard deviation) from at least 12 replicates between two wafers.
| Adhesion identifier | Treatment of bottom polyimidea) | Adhesive tie-layer | Treatment prior to top polyimidea) | Average linear stress [N m−1] |
|---|---|---|---|---|
| Polyimide (control) | N/A | N/A | O2 RIE (60 s) | 253 (10) |
| Titanium | O2 RIE (45 s)b) | Titanium | O2 RIE (60 s) | 3 (6) |
| Chromium | O2 RIE (45 s)b) | Chromium | O2 RIE (60 s) | 194 (18) |
| a-SiC:H | O2 RIE (60 s) | a-SiC:H | O2 RIE (60 s) | 4 (5) |
| a-SiC:H + APTES | O2 RIE (60 s) | a-SiC:H | O2 RIE (60 s) + APTES silanization | Cohesive fracture |
| APTES + a-SiC:H+APTES | O2 RIE (60 s) + APTES silanization | a-SiC:H | O2 RIE (60 s) + APTES silanization | Cohesive fracture |
| APTES | N/A | N/A | O2 RIE (60 s) + APTES silanization | 200 (47) |
| a-C:H | O2 RIE (60 s) | a-C:H | O2 RIE (60 s) | 32 (21) |
All polyimide layers were cured at T1 = T2 = 450 °C, and all O2-plasma RIE was performed at 100 W, 100 mTorr, and 30 sccm O2 for the time indicated;
Longer RIE times would damage the nLOF photoresist used to pattern the metal layers.
While delamination failure was clearly observed between the titanium and the top cured polyimide, it was not detected between the titanium and the underlying previously cured polyimide. An underlying cause of these results could be because of the fact that titanium rapidly forms a surface oxide when exposed to the atmosphere or an O2 plasma. Curing polyimide on top of oxide materials (e.g., SiO2, Al2O3, TiO2, etc.) makes use of only weak ionic interactions, which are formed at the interface primarily by charged carboxylic acid groups in the polyamic acid precursor and result in poor adhesive bonds that can be further weakened by water and aging.[49] It is likely a titanium-oxide surface layer promoted adhesive failure to the top polyimide layer. Conversely, depositing titanium in vacuum onto the bottom polyimide surface prevents any oxide formation. In addition, sputtering is known to improve adhesion owing to the bombardment of high-energy particles that can result in higher packing densities and surface coverage.[50] Detailed XPS studies indicated that the first atomic layers of sputtered titanium can react with carbonyl groups in the cured polyimide through charge transfer and form an initial Ti—O bond with a second transfer reaction forming Ti—C bonds.[47,51,52] Despite the presence of these bonds, the maximum peel strength of a Ti–PMDA–ODA interface from 90° peel studies was only 28.1 N m−1, and it further weakened during thermal aging experiments (85 °C, 85% relative humidity).[52] Importantly, previously published XPS-informed adhesion studies only analyzed the impact of depositing metal onto previously cured polyimide and were unable to investigate the bonds formed when instead curing polyimide onto previously deposited metal. Although these results indicate that titanium alone does not act as an effective adhesion-promotion agent with polyimide due to the presence of a significant oxide layer that prevents carbide formation, in later sections the use of titanium when combined with a-SiC:H will be discussed.
On the other hand, T-peel coupons containing chromium formed a relatively robust interface that was only modestly weaker than the polyimide–polyimide control interface (Table 5). Various studies have shown that chromium adhesion to polyimide can be improved by activating the polyimide surface with reactive plasmas including O2,[48,53] N2,[54] and atmospheric He/O2/NF3 [55] prior to metal deposition, with increased peel strength at higher plasma powers and longer plasma doses. Studies performed with XPS indicate that the first two monolayers of Cr deposited onto PMDA-ODA are oxidized by the pendant carbonyl oxygen along the polyimide backbone[56,57] forming Cr—O and Cr—C bonds in a fashion similar to titanium.[47,51,52] Others have argued that chromium–polyimide interactions are instead driven by π-arene ring complexation.[58,59] Whichever case it may be, the failure of T-peel coupons was again relegated to the top-cured polyimide–metal interface, indicating that the adhesion of polyimide onto chromium with its native oxide is weaker than the interface between chromium sputtered onto polyimide. As chromium and its oxide forms a comparatively stronger bond to BPDA-PDA than titanium, chromium is the recommended adhesion agent to use with polyimide if no additional agents are available. However, given that chromium ions (e.g., Cr6+) can be toxic and damaging to tissue,[60,61] it is not typically used for chronic implants if there is a risk of ion formation.
3.4.3. Polyimide–a-SiC:H and a-C:H Adhesion
It is clear from metal T-peel testing and published literature that BPDA-PDA polyimide does not naturally form robust interfaces when cured on top of titanium. This is a serious drawback for polyimide neural interface devices that hope to one day be used in humans for chronic timeframes. However, a-SiC:H, which has been reported to be an effective adhesion agent for polyimide-based neural implants, could alleviate this problem due to its ability to form carbide bonds with both polyimide and titanium and silicide bonds with platinum.[1,18,27] The use of thin layers of a-SiC:H has been shown to decrease delamination failure in polyimide-based devices in aggressive in vitro reactive accelerated aging studies[14] and in devices undergoing significant electrical stimulation during in vivo studies.[10]
Unfortunately, the peel strength of polyimide–a-SiC:H–polyimide samples without additional treatments was extremely weak compared to the standard polyimide–polyimide control interface (Table 5). This weak interface behaved in a manner similar to the titanium samples, with the primary failure interface between the a-SiC:H and polyimide cured on top. The use of XPS analysis on a-SiC:H identified the formation of a surface SiO2 layer that becomes more pronounced after O2-plasma activation (Figure 5). It is apparent that the thin SiO2 layer prevents the formation of covalent C—C bonds between the polyimide and a-SiC:H. On the other hand, the interface formed between a-SiC:H deposited onto previously cured polyimide did not delaminate, and this was attributed to the fact that PECVD deposition uses high-energy ions that can form strong bonds with the deposition surface and the lack of oxygen during deposition prevents SiO2 formation, allowing for the formation of robust carbide bonds between a-SiC:H and polyimide.
The troublesome surface SiO2 on a-SiC:H can be removed prior to top polyimide spin-coating/curing (Figure 1, step 4) using a variety of methods, including an HF buffered-oxide wet etch or dry etching in Ar+, SF6, or CF4 plasma. However, the oxide is expected to reform in minutes, likely before the top polyimide layer can be applied. In addition, all of these oxide-removal methods would also impact the exposed polyimide surface that could drastically inhibit its adhesion to subsequent polyimide layers (e.g., through fluorine contamination). Furthermore, as O2-plasma activation of the polyimide surface is absolutely required to form robust interfaces with subsequent polyimide, oxidizing the a-SiC:H cannot be avoided. There are two primary methods to overcome this problem and create robust polyimide–a-SiC:H–polyimide interfaces: 1) use of a silane adhesion agent (e.g., APTES) after RIE O2-plasma activation or 2) depositing an a-C:H[1,18] tie layer on top of the a-SiC:H.
APTES bridges this interface by coupling with the oxide on the a-SiC:H surface via covalent silanol bonds (Si—O—Si) and with the polyimide via covalent amide bonds, and silanization of both a-SiC:H and polyimide has already been discussed in detail (Sections 3.1 and 3.2). T-peel coupons using APTES silanized a-SiC:H exhibited a qualitatively stronger interface than samples without (Table 5). This interface could not be easily peeled apart by hand, but the exact adhesive peel strength could also not be accurately quantified because the interface exhibited cohesive fracture of the a-SiC:H layer during testing as opposed to adhesive delamination from the polyimide. Optical inspection of fractured surfaces identified broken islands of a-SiC:H on both polyimide layers. It is apparent that the use of APTES can increase adhesion strength between polyimide and a-SiC:H to a value above the fracture strength of a-SiC:H. Similar cohesive fracture occurred when the bottom polyimide was also silanized, prior to a-SiC:H deposition, in addition to the top a-SiC:H layer (Table 5). However, while APTES improves adhesion with a-SiC:H, it unfortunately weakens polyimide–polyimide bonding by ≈20% (Table 5), which is likely due to the fact that silanol groups do not significantly react with polyimide.
T-peel coupons using 250 nm a-C:H films as an adhesion aid between polyimide layers exhibited a relatively weak peel strength compared to the polyimide control (Table 5), and qualitatively, the a-C:H films formed weaker interfaces to polyimide than the APTES a-SiC:H films. Thinner a-C:H layers ranging in thickness from 50 to 250 nm and films deposited over the full range of conditions tested (Figure 6A–C) were also investigated with similar peel-strength results. Although it is unclear exactly how hydrogen content or the sp2/sp3 character of the film impacts adhesion to polyimide, it is apparent that using the deposition conditions reported here does not produce films that form strong chemical bonds with BPDA-PDA polyimide. Efforts failed to repeat the success reported for DLC films,[1,18] but as detailed material properties of the film were not reported, we cannot firmly state if carbon-based films in general are not suitable for polyimide-based neural implants or if the deposition tool or deposition conditions employed were simply insufficient. Although the theoretical chemical benefits of transitioning from a-SiC:H to a carbon film to polyimide are profound, additional testing and research is required to identify an optimal carbon film.
3.5. Metal-Patterning and Annealed-Film Properties
Noble metals (e.g., platinum, gold) are typically used in polyimide neural devices as the primary electrically conductive component to record bioelectric action potentials from firing neurons, electrically stimulate neural tissue near electrode sites, and conduct electrical signals through the microfabricated implant. Although “exotic” conductive/semiconductive materials (e.g., glassy carbon,[62–64] doped SiC[30]) have recently garnered increased interest, more research is needed to determine if they can outperform traditional metallic devices. The choice of gold, platinum, or both depends on the fabrication options available, the final application of the neural device, and their impact on reliability. The lower melting point of gold (Tm,Au = 1064 °C) compared to platinum (Tm,Pt = 1768 °C) makes it easier to deposit via thermal evaporation. Gold also has a lower electrical resistivity and is more ductile than platinum, which allows it to reduce trace resistance and corresponding power loss as well as more easily tolerate large mechanical strains without cracking. On the other hand, platinum has a larger charge-injection capacity that makes it more suited for electrical stimulation.
While noble metals are resistant to common corrosion mechanisms reported for other metals (e.g., steel, copper[65]), they typically require either titanium or chromium tie layers to improve adhesion, that can themselves corrode. Warnings of galvanic corrosion of such bimetallic junctions have been given,[1] but no significant corrosion related failure has been widely reported for polyimide–metal devices. Numerous examples of polyimide-based neural devices exist that use Ti—Pt,[66,67] Ti—Au,[68–70] Cr—Au,[71,72] etc. In addition, a multilayer stack of Pt/Au/Pt (i.e., platinum layers above and below a gold core) has been suggested to combine the improved stimulation and electrochemical behavior of platinum electrodes with the low resistance and power loss of mostly gold-based traces.[18] While the benefits of chromium as a more effective tie layer than titanium and the added benefits from the use of a-SiC:H has been discussed in detail previously, the pros/cons of using different noble metal stacks on device performance and reliability have not been provided. Furthermore, the addition of a-SiC:H creates a new material layer that could react with and diffuse into the metal layers causing changes in electrical and other material properties. These omissions are discussed in detail now.
3.5.1. Metal Deposition and Lift-Off
Four different metal stacks were sputtered, according to Table 1, directly onto lift-off photoresist patterned and O2-plasma RIE activated polyimide without an a-SiC:H tie layer. Sporadic delamination of ≈5–10% of the patterned metal features during lift-off was evidenced (Figure S4, Supporting Information), likely due to a combination of film stress and poor chemical adhesion between titanium and the underlying polyimide substrate. Deposited film stress for metal stack C ranged from −185 to −225 MPa (compressive), and there was no significant qualitative difference in the number of lost features between the different metal stacks using titanium tie layers. Conversely, analogous experiments using a chromium tie layer exhibited no sporadic delamination during lift-off, due to the stronger adhesive strength between chromium and polyimide (Table 5). As described above, the relatively weak titanium-to-polyimide adhesion can be improved significantly by adding a thin film of a-SiC:H between the polyimide and titanium layers. With these results in mind, the metal depositions were repeated on polyimide films that were first coated with 250 nm of stoichiometric a-SiC:H. As expected, no metal delamination from the a-SiC:H layer was observed during lift-off. However, T-peel tests were not performed to directly study the a-SiC:H–titanium interface owing to challenges with the experimental setup. Specifically, fabricating a metal or a-SiC:H film thick enough to allow for physical manipulation and peeling (≈10 μm) would be prohibitively expensive. However, accelerated soak tests of functional devices in 10–20 × 10−3 m H2O2 in PBS at 87 °C confirmed that the a-SiC:H–metal interface was incredibly robust and no delamination was observed.[14]
Although the use of a-SiC:H significantly improves metal adherence during fabrication, there are certain technicalities that must be considered. As discussed previously, a-SiC:H films have a native oxide surface that is exacerbated during O2-plasma cleaning. As both gold and platinum are known to adhere poorly to SiO2 surfaces, they are prone to delaminate from a-SiC:H during fabrication. Initial attempts to replicate work from published sources[18] by depositing platinum directly onto a-SiC:H without a tie layer resulted in widespread failure during lift-off, but this failure was avoided by using a 50-nm-thick titanium tie layer. If the titanium layer cannot be used for any specific reason, this problem can be overcome by first removing the a-SiC:H oxide using Ar+ plasma within the deposition tool and then depositing metal without breaking vacuum, thereby preventing a new oxide layer from forming. It is important to note that this method was only confirmed when depositing platinum and not gold. Although Pt—Si bonds that form between the deposited platinum and a-SiC:H provide robust adhesion, diffusion and alloying between these layers can also occur when curing subsequent polyimide layers, discussed in detail in Section 3.5.2.
3.5.2. Thermally Driven Metallic Interdiffusion and Alloying
The final properties of the metallic stack (e.g., chemistry, resistivity, stress) and how the metal interacts with tie layers (e.g., a-SiC:H) can have a significant impact on the performance and reliability of the neural-interface device. However, measuring film properties after deposition and lift-off does not capture the final properties present during implantation. Instead, characterization should ideally be performed after thermally annealing the full material stacks using the same temperature profile used to cure the final polyimide layer. Materials that diffuse and alloy together when provided sufficient time and thermal energy can experience significant changes in their film properties. Experimentally, four different metal stacks (Table 1) were deposited between two 250-nm-thick a-SiC:H layers, annealed at 450 °C, and XPS depth profiled to identify interdiffusion and alloying between materials (Figure 8).
Figure 8.
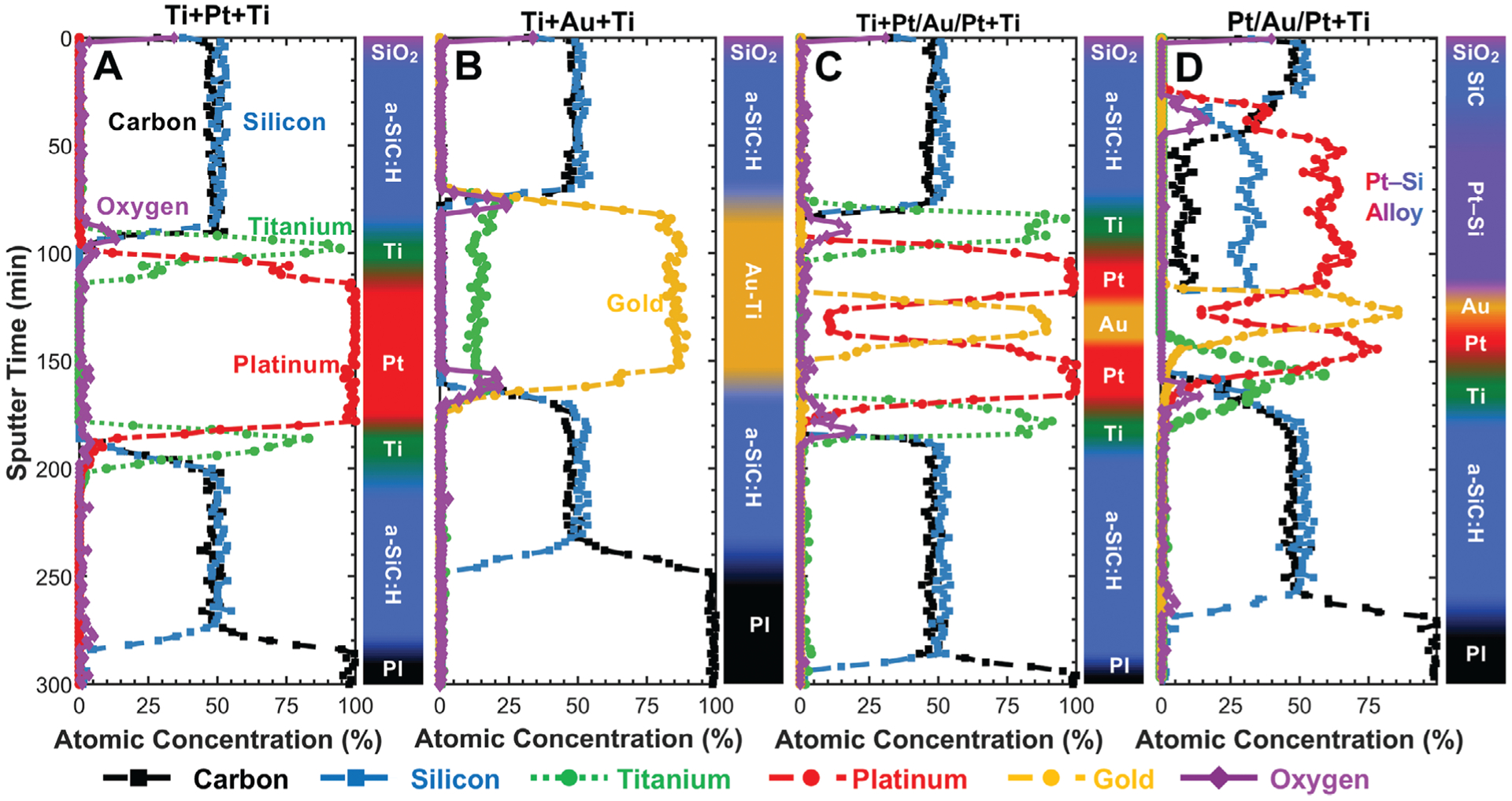
XPS depth profiles of different a-SiC:H/metal/a-SiC:H stacks deposited onto polyimide (PI) and annealed at 450 °C. The top of each plot represents the initial surface of the material stack before sputter etching occurs. The plot then shows the atomic composition as the Ar+-beam-based sputter etch progresses downward through the materials to finally reach the polyimide layer. A) The Ti+Pt+Ti film shows distinct titanium bands sandwiching the platinum layer with no significant alloying. B) The Ti+Au+Ti film shows complete alloying of the titanium layer into the gold layer as evidenced by the lack of distinct titanium bands and the stable titanium signal throughout the entirety of the gold layer. C) The Ti+Pt/Au/Pt+Ti film shows distinct bands for all deposited metal, which indicates that the platinum acts as an effective diffusion barrier between the titanium and gold layers. D) As the Pt/Au/Pt+Ti stack does not have a top titanium layer, Pt—Si forms as platinum diffuses into and alloys with the top a-SiC:H layer. The bottom titanium layer is an effective diffusion barrier between platinum and the lower a-SiC:H layer, preventing the formation of Pt—Si there.
For metal stack A (Ti+Pt+Ti), each metal layer was readily distinguishable, and no significant alloying was identified between either the adjacent a-SiC:H layers or between the metals (Figure 8A). This analysis method was also able to detect the surface oxide that formed on the top titanium layer when the sample was removed from the sputter chamber and transferred to the PECVD for a-SiC:H deposition. In addition, the method detected a minor peak at the bottom titanium–a-SiC:H interface, which indicates that a surface oxide formed on the a-SiC:H before sputtering, as expected. Titanium has a strong affinity for oxygen and thus forms strong bonds to oxidized substrates such as a-SiC:H. For metal stack B (Ti+Au+Ti), the metals completely diffused together, resulting in an Au—Ti alloy composed of ≈85% Au and ≈15% Ti (Figure 8B). Significant Au—Ti interdiffusion in thin films has been observed to occur at temperatures as low as 200 °C, with complete alloying at 350 °C.[73,74] The surface oxide peaks at the top and bottom of the metal stack are identifiable and appear to demarcate the interface between a-SiC:H and the Au—Ti alloy. No long-range a-SiC:H diffusion into the metal was identified.
For metal stack C (Ti+Pt/Au/Pt+Ti), only minor diffusion of Pt into the Au layer was evidenced (i.e., the Au layer became an ≈90% Au and 10% Pt alloy), and no reverse diffusion of Au into the Pt layer was identified. (Figure 8C). Furthermore, the 100-nm-thick Pt layers acted as diffusion barriers that prevented the formation of Au—Ti alloys, and all of the transitions between metals were relatively sharp. The oxide layers were also identifiable at the Ti–a-SiC:H interfaces, which is consistent with the results for metal stacks A-B.
For metal stack D (Pt/Au/Pt+Ti) without the top Ti layer, significant interdiffusion between the top platinum and a-SiC:H layers occurred, which resulted in the formation of a platinum silicide (Pt—Si) layer composed of ≈60% platinum, 30% silicon, and ≈10% carbon (Figure 8D). However, the platinum did not diffuse through the entire 250-nm-thick a-SiC:H layer as seen by the residual stoichiometric a-SiC:H film over the first ≈25 min of profiling (estimated thickness of 75–100 nm). The final thickness of platinum silicide was likely limited by the original thickness of the platinum layer and the final stoichiometry of the silicide. The a-SiC:H–platinum silicide interface contains a depleted silicon and platinum transition zone with a corresponding local maximum in carbon and oxygen. The presence of the interfacial oxide peak is interesting because the a-SiC:H bulk does not contain any detectable oxygen. Therefore, it is likely that the oxygen originated from the surface oxide on the sputtered platinum that was subsequently excluded during the formation of platinum silicide and forced into the a-SiC:H–platinum silicide interface. It is unlikely that the oxygen originated from the upper a-SiC:H surface, as no detectable oxygen diffusion through a-SiC:H was identified for metal stacks A–C. Annealing thin-film platinum deposited directly onto Si wafers results in diffusion-limited platinum-silicide formation that starts at ≈200 °C and progresses from Pt3Si→Pt2Si→PtSi.[75,76] While direct comparisons cannot be made between films formed using Si wafers or a-SiC:H as the source of silicon, it is apparent that annealing films to 450 °C causes a complete depletion of the platinum film in favor of silicide formation and cannot be avoided unless a diffusion barrier such as titanium is used.
3.5.3. Metal Resistivity
Electrical resistivity of the metal traces used for neural devices contributes to power loss experienced within the device. Resistivity was calculated for each metal stack after thermal annealing to determine the impact of chemistry and alloying on the electrical characteristics of the traces (Figure 9). A set of four-point-probe resistance measurements were performed on test-structure traces, and resistivity was calculated using the as-fabricated trace geometries (Figure S1, Supporting Information). The metal traces were fully encapsulated between a-SiC:H and polyimide (except for contact pad sites) and cured to 450 °C. Platinum in metal stack A had an average resistivity of 27.2 μΩ cm, which is more than double the 10.06 value of bulk platinum.[77] However, this result is not unexpected as thin films often have higher resistivities than bulk materials due to size-induced surface and grain-boundary scattering effects and higher defect density from low-temperature sputtering that all inhibit electron transport.[78,79] It was expected that metal stack B would have a significantly lower resistivity compared to the platinum stack due to the lower resistivity of gold. However, the annealed Au—Ti alloy had a resistivity of 59.8 μΩ cm, which is 24 times higher than bulk gold (i.e., 2.44 μΩ cm)[77] and 1.4 times higher than bulk titanium (i.e., 42.0 μΩ cm),[77] which suggests that the titanium dominates the resistivity. The increased resistivity and resultant power consumption should be considered when designing neural implants that use gold with titanium tie layers if the devices need to be heated above ≈200 °C during fabrication.
Figure 9.
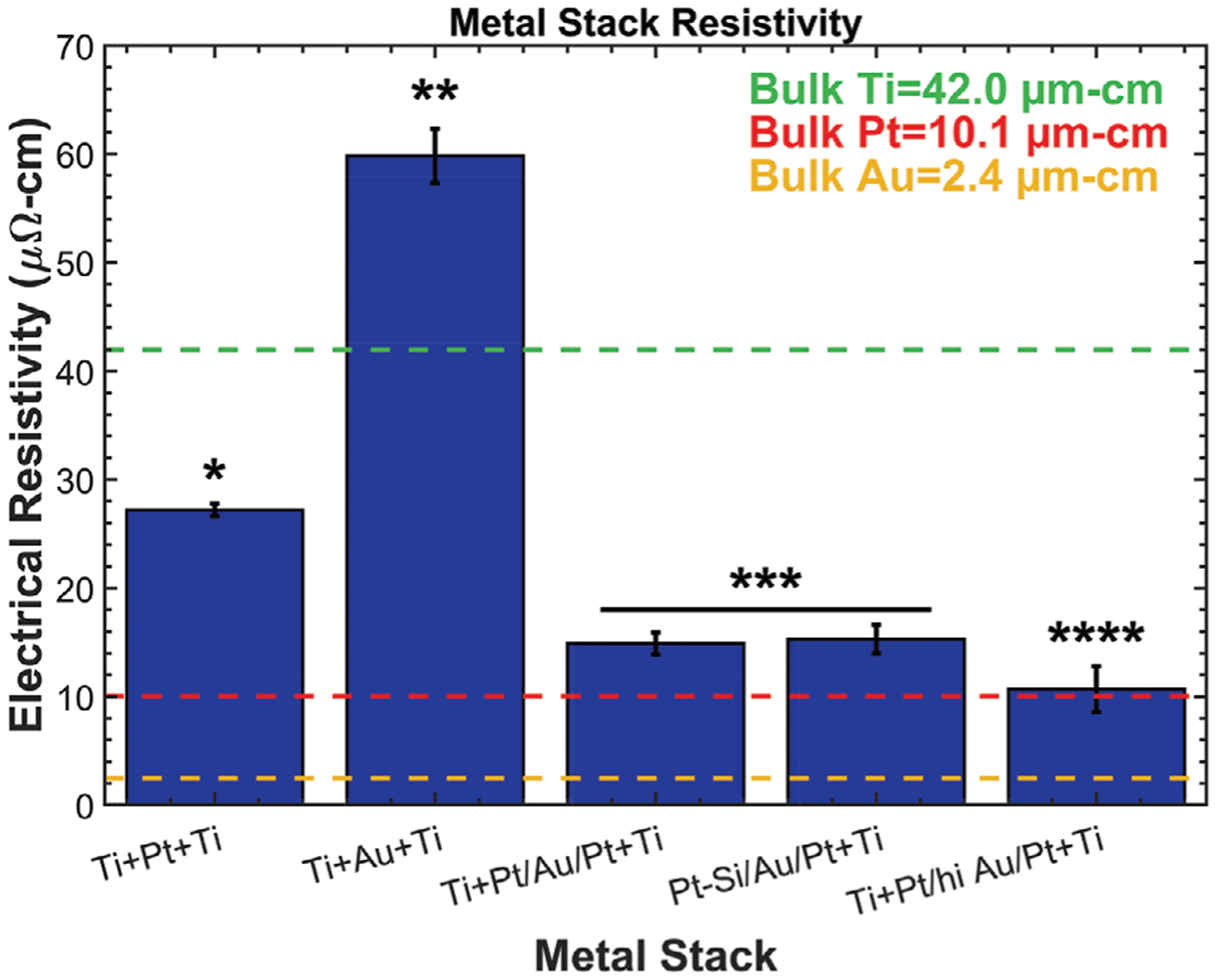
Metal resistivity for a set of metal stacks described in Table 1. The addition of non-alloyed gold lowered the resistivity compared to platinum-titanium stacks, but gold-titanium alloys had significantly higher resistivity. Platinum silicide formation had no impact on resistivity. Asterisks indicate statistically homogenous subsets, one-way ANOVA, and Tukey’s post hoc analysis.
The gold core of the hybrid metal stack C (Pt/Au/Pt) stack lowered the resistivity to 14.9 μΩ cm, which is only a modest increase compared to bulk platinum and superior to both metal stacks A and B. The gold served its purpose of lowering resistivity when it did not alloy with titanium, owing to the platinum diffusion barrier. Interestingly, metal stack D (Pt—Si/Au/Pt) had a comparable resistivity, which indicates that the platinum silicide does not strongly impact the electrical resistivity of the trace. A fifth metal stack modified from stack C was also tested to verify the trend of decreasing resistivity with increasing gold content (without the formation of the gold-titanium alloy). This stack was fabricated with the ratio of gold to platinum increased from 0.5:1 to 5:1 by using a Ti (50 nm) + Pt (25 nm)/Au (250 nm)/Pt (25 nm) + Ti (50 nm) metal stack. The increase in gold content significantly lowered the trace resistivity to 10.7 μΩ cm, which is similar to bulk platinum.
4. Conclusions
Fabrication of reliable polyimide-based neural interfaces that can withstand the harsh implant environment is a difficult challenge that must be overcome to unlock their full therapeutic potential. A critical, material-science investigation was performed in an effort to provide research groups and industry with the tools and materials foundation needed to fabricate more reliable and robust devices. Polyimide and possible adhesion-promotion aids (e.g., a-SiC:H, a-C:H) were analyzed using a variety of techniques including FTIR and XPS to provide fundamental understanding of the bulk and surface chemical changes that each material undergoes during different microfabrication steps. This information in conjunction with T-peel adhesion testing was used to identify and improve upon critical interfacial weaknesses of different material stacks. Basic fabrication rules were identified to markedly improve polyimide self-adhesion including modifying the T1 cure temperature to 250 °C and maximizing RIE O2-plasma activation (>≈300 s). Effective a-SiC:H adhesive aids between titanium-based metal stacks and polyimide surfaces were identified; however, the use of an additional APTES tie layer was needed to provide robust adhesion with subsequently applied polyimide layers. Efforts failed to identify a-C:H deposition methods that could produce films to eliminate reliance on APTES and aid polyimide–polyimide adhesion. Quantitative adhesion and qualitative delamination studies identified chromium as an effective tie layer if a-SiC:H–titanium is unavailable. Depth profiling of a-SiC:H–metal stacks identified examples of alloying between adjacent material layers (e.g., gold/titanium and platinum/a-SiC:H) that could impact the bulk trace resistivity and possibly their long-term reliability. While there is undoubtedly more progress to be made, the efforts reported herein should enable readers to fully understand the rationale and consequences of key decisions made regarding the microfabrication of polyimide-based neural interfaces and thereby promote a more transparent and reproducible scientific environment in this field to enable the adoption of more robust and reliable fabrication methodologies.
Supplementary Material
Acknowledgements
This work was sponsored by the Defense Advanced Research Projects Agency (DARPA): Biological Technology Office Electrical Prescriptions (ElectRx) program, under the auspices of Dr. Doug Weber and Dr. Eric Van Gieson through the DARPA contracts Management Office, Pacific Cooperative Agreement No. HR0011-15-2-0030 and by the National Institutes of Health: National Institute of Neurological Disorders and Stroke under the auspices of Dr. Sahana N. Kukke (Agreement No. R01 NS111518).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Contributor Information
Cary A. Kuliasha, Department of Electrical and Computer Engineering, University of Florida, Gainesville, FL 32611, USA Nanoscience Institute for Medical and Engineering Technology, University of Florida, Gainesville, FL 32611, USA.
Jack W. Judy, Department of Electrical and Computer Engineering, University of Florida, Gainesville, FL 32611, USA Nanoscience Institute for Medical and Engineering Technology, University of Florida, Gainesville, FL 32611, USA.
References
- [1].Ordonez J, Schuettler M, Boehler C, Boretius T, Stieglitz T, MRS Bull. 2012, 37, 590. [Google Scholar]
- [2].Capogrosso M, Milekovic T, Borton D, Wagner F, Moraud EM, Mignardot JB, Buse N, Gandar J, Barraud Q, Xing D, Rey E, Duis S, Jianzhong Y, Ko WKD, Li Q, Detemple P, Denison T, Micera S, Bezard E, Bloch J, Courtine G, Nature 2016, 539, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wenger N, Moraud EM, Gandar J, Musienko P, Capogrosso M, Baud L, Le Goff CG, Barraud Q, Pavlova N, Dominici N, Minev IR, Asboth L, Hirsch A, Duis S, Kreider J, Mortera A, Haverbeck O, Kraus S, Schmitz F, DiGiovanna J, Van Den Brand R, Bloch J, Detemple P, Lacour SP, Bézard E, Micera S, Courtine G, Nat. Med 2016, 22, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chiang CH, Won SM, Orsborn AL, Yu KJ, Trumpis M, Bent B, Wang C, Xue Y, Min S, Woods V, Yu C, Kim BH, Kim SB, Huq R, Li J, Seo KJ, Vitale F, Richardson A, Fang H, Huang Y, Shepard K, Pesaran B, Rogers JA, Viventi J, Sci. Transl. Med 2020, 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Petrini FM, Valle G, Strauss I, Granata G, Di Iorio R, D’Anna E, Čvančara P, Mueller M, Carpaneto J, Clemente F, Controzzi M, Bisoni L, Carboni C, Barbaro M, Iodice F, Andreu D, Hiairrassary A, Divoux JL, Cipriani C, Guiraud D, Raffo L, Fernandez E, Stieglitz T, Raspopovic S, Rossini PM, Micera S, Ann. Neurol 2019, 85, 137. [DOI] [PubMed] [Google Scholar]
- [6].Petrini FM, Bumbasirevic M, Valle G, Ilic V, Mijović P, Čvančara P, Barberi F, Katic N, Bortolotti D, Andreu D, Lechler K, Lesic A, Mazic S, Mijović B, Guiraud D, Stieglitz T, Alexandersson A, Micera S, Raspopovic S, Nat. Med 2019, 25, 1356. [DOI] [PubMed] [Google Scholar]
- [7].Bloch E, Luo Y, da Cruz L, Ther. Adv. Ophthalmol 2019, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hassler C, Boretius T, Stieglitz T, J. Polym. Sci., Part B: Polym. Phys 2011, 49, 18. [Google Scholar]
- [9].Polyimides: Fundamentals and Applications (Eds: Ghosh MK, Mittal KL), Marcel Dekker, New York: 1996. [Google Scholar]
- [10].Čvančara P, Boretius T, López-Álvarez VM, Maciejasz P, Andreu D, Raspopovic S, Petrini F, Micera S, Granata G, Fernandez E, Rossini PM, Yoshida K, Jensen W, Divoux J-L, Guiraud D, Navarro X, Stieglitz T, J. Neural Eng 2020, 17, 046006. [DOI] [PubMed] [Google Scholar]
- [11].Stieglitz T, Beutel H, Schuettler M, Meyer JU, Biomed. Microde-vices 2000, 2, 283. [Google Scholar]
- [12].Schuettler M, Koch KP, Stieglitz T, in Proc. 8th Annual Int. Conf. Int. Functional Electrical Stimulation Society, 2003, pp. 306–310. [Google Scholar]
- [13].Lee SM, Byeon HJ, Kim BH, Lee J, Jeong JY, Lee JH, Moon JH, Park C, Choi H, Lee SH, Lee KH, BioChip J. 2017, 11, 153. [Google Scholar]
- [14].Kuliasha CA, Judy JW, in 40th Annual Int. Conf. IEEE Engineering in Medical and Biology Society, IEEE, Honolulu, Hawaii: 2018, pp. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Boretius T, Badia J, Pascual-Font A, Schuettler M, Navarro X, Yoshida K, Stieglitz T, Biosens. Bioelectron 2010, 26, 62. [DOI] [PubMed] [Google Scholar]
- [16].Cutrone A, Del Valle J, Santos D, Badia J, Filippeschi C, Micera S, Navarro X, Bossi S, J. Neural Eng 2015, 12, 016016. [DOI] [PubMed] [Google Scholar]
- [17].Kuliasha CA, Spearman BS, Atkinson EW, Rustogi P, Furniturewalla AS, Nunamaker EA, Otto KJ, Schmidt CE, Judy JW, in 2018 Solid-State Sensors Actuators Microsystems Workshop Hilton Head 2018, 2018, p. 46. [Google Scholar]
- [18].Ordonez JS, Boehler C, Schuettler M, Stieglitz T, in 2012 Annual Int. Conf. IEEE EMBS, IEEE, San Diego, CA: 2012, p. 5134. [DOI] [PubMed] [Google Scholar]
- [19].Diaham S, Locatelli ML, Lebey T, Malec D, Thin Solid Films 2011, 519, 1851. [Google Scholar]
- [20].Gat E, El Khakani MA, Chaker M, Jean A, Boily S, Pépin H, Kieffer JC, Durand J, Cros B, Rousseaux F, Gujrathi S, J. Mater. Res 1992, 7, 2478. [Google Scholar]
- [21].Cogan SF, Edell DJ, Guzelian AA, Liu YP, Edell R, J. Biomed. Mater. Res 2003, 67A, 856. [DOI] [PubMed] [Google Scholar]
- [22].Park YS, SiC Materials and Devices, Vol. 52, Academic, San Diego, CA: 1998. [Google Scholar]
- [23].Jean A, Chaker M, Diawara Y, Leung PK, Gat E, Mercier PP, Pépin H, Gujrathi S, Ross GG, Kieffer JC, J. Appl. Phys 1992, 72, 3110. [Google Scholar]
- [24].Knaack GL, Charkhkar H, Cogan SF, Pancrazio JJ, Silicon Carbide Biotechnology: A Biocompatible Semiconductor Advanced Biomedical Devices and Applications, 2nd ed., Elsevier, Oxford: 2016, p. 249. [Google Scholar]
- [25].Lei X, Kane S, Cogan S, Lorach H, Galambos L, Huie P, Mathieson K, Kamins T, Harris J, Palanker D, J. Neural Eng 2016, 13, 046016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ordonez JS, Boehler C, Schuettler M, Stieglitz T, Mater. Res. Soc. Symp. Proc 2012, 1466, 61. [Google Scholar]
- [27].Cogan SF, Ehrlich J, Plante TD, Gingerich MD, Shire DB, IEEE Trans. Biomed. Eng 2010, 57, 2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hsu JM, Tathireddy P, Rieth L, Normann AR, Solzbacher F, Thin Solid Films 2007, 516, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Deku F, Cohen Y, Joshi-Imre A, Kanneganti A, Gardner TJ, Cogan SF, J. Neural Eng 2018, 15, 016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Beygi M, Bentley JT, Frewin CL, Kuliasha CA, Takshi A, Bernardin EK, a Via FL, Saddow SE, Micromachines 2019, 10, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Önneby C, Pantano CG, J. Vac. Sci. Technol., A 1997, 15, 1597. [Google Scholar]
- [32].Robertson J, Mater. Sci. Eng., B 2002, 37, 129. [Google Scholar]
- [33].Viana GA, Motta EF, Da Costa MEHM, Freire FL, Marques FC, Diamond Relat. Mater 2010, 19, 756. [Google Scholar]
- [34].Mutsukura N, Miyatani K, Diamond Relat. Mater 1995, 4, 342. [Google Scholar]
- [35].Caschera D, Cossari P, Federici F, Kaciulis S, Mezzi A, Padeletti G, Trucchi DM, Thin Solid Films 2011, 519, 4087. [Google Scholar]
- [36].Capote G, Bonetti LF, Santos LV, Trava-Airoldi VJ, Corat EJ, Thin Solid Films 2008, 516, 4011. [Google Scholar]
- [37].Ţucureanu V, Matei A, Avram AM, Crit. Rev. Anal. Chem 2016, 46, 502. [DOI] [PubMed] [Google Scholar]
- [38].Lazar G, Zellama K, Vascan I, Stamate M, Lazar I, Rusu I, J. Optoelectron. Adv. Mater 2005, 7, 647. [Google Scholar]
- [39].Brown HR, Yang ACM, Russell TP, Volksen W, Kramer EJ, Polymer 1988, 29, 1807. [Google Scholar]
- [40].Miwa T, Tawata R, Numata S, Polymer 1993, 34, 621. [Google Scholar]
- [41].Chan CM, Ko TM, Hiraoka H, Surf. Sci. Rep 1996, 24, 1. [Google Scholar]
- [42].Liston EM, Martinu L, Wertheimer MR, J. Adhes. Sci. Technol 1993, 7, 1091. [Google Scholar]
- [43].Kim SH, Na SW, Lee NE, Nam YW, Kim YH, Surf. Coat. Technol 2005, 200, 2072. [Google Scholar]
- [44].Lacombe R, Adhesion Measurement Methods, Taylor and Francis, Boca Raton, FL: 2005. [Google Scholar]
- [45].Ordonez JS, Rudmann L, Cvancara P, Bentler C, Stieglitz T, Proc. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. EMBS 2015, 2015, 1045. [DOI] [PubMed] [Google Scholar]
- [46].Inagaki N, Tasaka S, Hibi K, J. Adhes. Sci. Technol 1994, 8, 395. [Google Scholar]
- [47].Freilich SC, J. Vac. Sci. Technol., A 1986, 4, 1039. [Google Scholar]
- [48].Pappas DL, Cuomo JJ, Sachdev KG, J. Vac. Sci. Technol., A 1991, 9, 2704. [Google Scholar]
- [49].Buchwalter LP, J. Adhes. Sci. Technol 1990, 4, 697. [Google Scholar]
- [50].Madou MJ, Fundamentals of Microfabrication and Nanotechnology, CRC Press, Boca Raton, FL: 2011. [Google Scholar]
- [51].Georgiev DG, Baird RJ, Newaz G, Auner G, Witte R, Herfurth H, Appl. Surf. Sci 2004, 236, 71. [Google Scholar]
- [52].Girardeaux C, Druet E, Demoncy P, Delamar M, Electron Spectrosc J. Relat. Phenom 1995, 74, 57. [Google Scholar]
- [53].Nakamura Y, Suzuki Y, Watanabe Y, Thin Solid Films 1996, 290, 367. [Google Scholar]
- [54].Flitsch R, Shih D, J. Vac. Sci. Technol., A 1990, 8, 2376. [Google Scholar]
- [55].Bin Lee S, Kim YK, Plasma Process. Polym 2009, 6, 5525. [Google Scholar]
- [56].Chou NJ, Tang CH, J. Vac. Sci. Technol., A 1984, 2, 751. [Google Scholar]
- [57].Jordan JL, Kovac CA, Morar JF, Pollak RA, Phys. Rev. B 1987, 36, 1369. [DOI] [PubMed] [Google Scholar]
- [58].Haight R, White RC, Silverman BD, Ho PS, J. Vac. Sci. Technol., A 1988, 6, 2188. [Google Scholar]
- [59].Nandi M, Sen A, Chem. Mater 1989, 1, 291. [Google Scholar]
- [60].US Food Drug Administration, 2019, p. 1.
- [61].Barceloux DG, J. Toxicol., Clin. Toxicol 1999, 37, 173. [DOI] [PubMed] [Google Scholar]
- [62].Vomero M, Castagnola E, Ciarpella F, Maggiolini E, Goshi N, Zucchini E, Carli S, Fadiga L, Kassegne S, Ricci D, Sci. Rep 2017, 7, 40332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vomero M, Castagnola E, Ordonez JS, Carli S, Zucchini E, Maggiolini E, Gueli C, Goshi N, Ciarpella F, Cea C, Fadiga L, Ricci D, Kassegne S, Stieglitz T, Adv. Biosyst 2018, 2, 1. [Google Scholar]
- [64].Nimbalkar S, Fuhrer E, Silva P, Nguyen T, Sereno M, Kassegne S, Korvink J, Microsystems Nanoeng. 2019, 5, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Eliaz N, Materials 2019, 12, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stieglitz T, Beutel H, Keller R, Schuettler M, Meyer JU, Proc. Seventh Int. Conf. Microelectron. Neural, Fuzzy Bio-Inspired Syst 1999, 112. [Google Scholar]
- [67].Stieglitz T, Beutel H, Meyer J-U, Sens. Actuators, A 1997, 60, 240. [Google Scholar]
- [68].Kelly SK, Shire DB, Chen J, Gingerich MD, Cogan SF, Drohan WA, Ellersick W, Krishnan A, Behan S, Wyatt JL, Rizzo JF, Electron. Proc. 2013 IEEE Int. Conf. Multimed. Expo Work. ICMEW 2013 2013, 1. [Google Scholar]
- [69].Cogan SF, Ehrlich J, Plante TD, Smirnov A, Shire DB, Gingerich M, Rizzo JF, J. Biomed. Mater. Res., Part B 2009, 89, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tooker A, Tolosa V, Shah KG, Sheth H, Felix S, Delima T, Pannu S, Proc. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. EMBS 2012, 5999. [DOI] [PubMed] [Google Scholar]
- [71].Chung T, Wang JQ, Wang J, Cao B, Li Y, Pang SW, J. Neural Eng 2015, 12, 056018. [DOI] [PubMed] [Google Scholar]
- [72].Kato YX, Furukawa S, Samejima K, Hironaka N, Kashino M, Front. Neuroeng 2012, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Martinez WE, Gregori G, Mates T, Thin Solid Films 2010, 518, 2585. [Google Scholar]
- [74].Poate JM, Gold Bull. 1981, 14, 2. [Google Scholar]
- [75].Faber EJ, Wolters RAM, Schmitz J, Appl. Phys. Lett 2011, 98, 082102. [Google Scholar]
- [76].Pan JT, Blech IA, Thin Solid Films 1984, 113, 129. [Google Scholar]
- [77].Ohring M, Engineering Materials Science, Academic, San Diego, CA: 1995. [Google Scholar]
- [78].Zhang W, Brongersma SH, Richard O, Brijs B, Palmans R, Froyen L, Maex K, Microelectron. Eng 2004, 76, 146. [Google Scholar]
- [79].Fan P, Yi K, a Shao JD, Fan ZX, J. Appl. Phys 2004, 95, 2527. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.