

Summary
Background:
Type 1 diabetes results from autoimmune-mediated destruction of beta cells. The tyrosine kinase inhibitor imatinib may impact relevant immunologic and metabolic pathways, and preclinical studies show that it reverses and prevents diabetes. Our goal was to evaluate the safety and efficacy of imatinib in preserving beta cell function in subjects with recent-onset type 1 diabetes.
Methods:
We conducted a phase 2, randomised, placebo-controlled, double blind clinical trial. Patients with recent-onset type 1 diabetes, aged 18-45 years, and with peak C-peptide ≥ 0.2 nmoles/L on mixed meal tolerance test (MMTT) were enrolled from 9 centres in the USA and Australia. Participants were randomly assigned 2:1 to receive either 400 mg imatinib or matching placebo for 26 weeks, respectively, using a computer-generated blocked randomisation scheme stratified by centre. The primary endpoint was the 2-hour area under the curve (AUC) C-peptide response to MMTT in the imatinib versus placebo group at 12 months, using an ANCOVA model adjusting for sex, baseline age, and baseline C-peptide, with further observation out to 24 months. Analyses were by intention to treat. This study is registered with ClinicalTrials.gov, number NCT01781975.
Findings:
Patients were screened and enrolled into the trial between February 12, 2014 and May 19, 2016. 45 patients were assigned to imatinib, 22 to placebo. The study met its primary endpoint: the adjusted mean difference between the imatinib and placebo-treated groups in 2-hour C-peptide AUC in response to an MMTT at 12 months was 0·0946 (90% CI: −0·00279, 0·191) (p=0·048, 1-tailed test). This effect was not sustained out to 24 months. Of the imatinib-treated subjects, 71% experienced a grade 2 or higher adverse event, compared to 59% of placebo-treated subjects. Per protocol guidelines, 38% of imatinib-treated subjects required a temporary modification in drug dosing and 13% permanently discontinued drug; 23% of the placebo group had temporary modifications in dosing.
Interpretation:
A 26-wk course of imatinib may preserve beta cell function at 12 months in adults with recent onset type 1 diabetes. Imatinib may offer a novel means to alter the course of type 1 diabetes, but requires careful monitoring for possible toxicities.
Funding:
Juvenile Research Diabetes Foundation
Introduction
Type 1 diabetes results from the autoimmune destruction of insulin-producing beta cells.1 Although exogenous insulin is widely available, affected individuals cannot consistently achieve euglycemia with current formulations and technologies, and as a result remain at risk for acute and long-term complications.2 Thus, preservation of remaining beta cell function, before or after diagnosis, offers the best means to control the disease process. We and others have conducted a series of clinical trials with various immunotherapies in order to preserve beta cell function in those with recent-onset type 1 diabetes. Despite our best efforts, we have not had robust success: a small number of larger placebo-controlled phase 2 studies have met their primary endpoint, but treated subjects must usually remain on exogenous insulin, and the effects wane over time as the therapies are withdrawn. 3–8
Therefore, investigators continue to search for novel therapies to interdict beta cell destruction, with particular attention to repurposing agents that are already approved for other indications, thereby accelerating their application to type 1 diabetes. In this quest, our attention has been drawn to imatinib mesylate (Gleevec), a first–in-class tyrosine kinase inhibitor that has had remarkable success as a therapy for chronic myelogenous leukemia (CML), 9 and that may also impact both immunologic and metabolic pathways. In preclinical studies, imatinib has been shown in the nonobese diabetes (NOD) mouse to both prevent diabetes and induce remission of new-onset diabetes without requiring continuous ongoing therapy.10, 11 Anti-CD3 monoclonal antibody and anti-thymocyte globulin have had similar effects in the NOD mouse, with promising effects when evaluated in clinical trials.3, 5 Further pre-clinical investigation suggests that imatinib may act at least in part via novel metabolic pathways, such as to counteract high endoplasmic reticulum (ER) stress in beta cells and reduce apoptosis, as well as to improve insulin sensitivity.10, 12–14
These preclinical observations have been extended to the clinical arena, where case reports and case series have shown positive effects of imatinib in patients with autoimmune conditions (such as rheumatoid arthritis15), and with type 1 as well as type 2 diabetes.16 To explore the role of imatinib in preservation of beta cell function in type 1 diabetes, we conducted a phase 2 trial with imatinib in patients with recent-onset disease, and report the efficacy and safety results herein for subjects followed out to 24 months.
Methods
Study design and patients
This phase 2, randomised, placebo-controlled, double-blinded clinical trial was conducted according to the Declaration of Helsinki and in accordance with good clinical practice guidelines, performed under an investigational new drug application (IND 117644) and was approved by independent institutional review boards at each participating center. All participants provided written informed consent. The study protocol is posted at https://www.protocols.io/view/collection-of-protocols-and-guidelines-for-safety-bvfqn3mw. An independent data and safety monitoring board conducted regular safety reviews.
Screening, enrollment, and subsequent study visits occurred at 9 clinical centres in the USA and Melbourne, Australia. Eligible participants were subjects aged 18-45 years at time of screening; < 100 days from diagnosis at the time of enrollment; positive for at least one diabetes-associated autoantibody (microinsulin autoantibodies (mIAA), tested only if duration of insulin therapy was <10 days; glutamate decarboxylase (GAD); islet-cell antigen-512 (ICA-512), zinc transporter 8 (ZnT8), or islet-cell autoantibodies (ICA)); peak stimulated C-peptide of > 0.2 nmoles/L during a mixed meal tolerance test (MMTT). Exclusion criteria included any serological or clinical evidence of infection; a positive PPD test; past infection with hepatitis B, C or HIV; significant past cardiac disease; anemia, leukopenia, thrombocytopenia, or neutropenia; liver or renal dysfunction; on-going use of diabetes medications other than insulin; vaccination with a live virus within 6 weeks before enrollment; prior treatment with imatinib or related tyrosine kinase inhibitor; inability to avoid medications that affect CYP3A4, or use of drugs that may have plasma concentrations altered by imatinib; malignancy, or any other condition that might compromise study participation or confound interpretation of the results. All participants had to consent to use reliable and effective forms of contraception, for females during the entire 2-year trial, and for males up to 3 months after the study drug dosing period.
Randomisation and masking
Eligible subjects were randomly assigned 2:1 to imatinib or placebo. The site-stratified randomisation scheme was computer generated at the data coordinating center using permuted-blocks of size 6. Site personnel randomised subjects via an interactive web-based randomisation system, which sent the treatment assignments directly to the unmasked site pharmacists.
Procedures
All investigators and study subjects were blinded to study group assignment. The drug treated group received imatinib mesylate as four 100 mg film-coated tablets daily for 26 weeks, and the placebo group received matching tablets (Novartis, East Hanover, NJ). Dose selection was based in part on extrapolation from preclinical studies, is the starting dose employed in oncology settings, and has been reported to have benefits for patients with other autoimmune diseases.9, 15, 17 Study drug administration was modified, as necessary, by clinical symptoms or laboratory abnormalities, based on algorithms derived from oncology settings. Briefly, for adverse events per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) V4.0 of grade 2 severity and considered likely related to imatinib, including gastrointestinal issues (vomiting, diarrhea), muscle cramping, edema, skin rash, or laboratory abnormalities (liver function changes, myelosuppression) the study drug/placebo dose was reduced by 50% until the issue resolved. Grade 3 events prompted discontinuation of study drug/placebo until the issue resolved, with re-challenge with study drug thereafter. Recurrent persisting grade 3AEs resulted in drug termination, and subject observation continued through the remainder of the trial. MMTTs were conducted at baseline, 3, 6, 12, 18, and 24 months (4-hr tests at baseline, 12 and 24 months, and 2-hr tests at other time points), with measurement of glucose, C-peptide, and insulin.
All participants received intensive diabetes management with the goal to achieve ADA-recommended HbA1C and glycemic targets. Subjects were encouraged to monitor blood glucose at least 4 times per day with a glucometer, and some elected to utilize continuous glucose monitoring. Per study protocol, a reportable hypoglycemic event was defined as those resulting in loss of consciousness, seizure, or requiring assistance of others due to altered state of consciousness, and a hyperglycemic event was one resulting in DKA. Insulin usage data was collected on all participants for the preceding 5 days before study visits.
Biochemical autoantibodies were assayed at the Barbara Davis Center (Aurora, CO) using radioimmunobinding assays, and ICA was measured at the University of Florida, as described previously.5 C-peptide, HbA1c, proinsulin, adiponectin, and serum chemistries were measured at the Northwest Lipid Research Laboratory (Seattle, WA). Beta cell death assay was conducted as described previously.18 All other routine laboratory measures were conducted locally.
Lymphocyte and myeloid cell subsets were evaluated from frozen peripheral blood mononuclear cells (PBMCs) isolated from whole blood and viably cryopreserved at the Immune Tolerance Network Core facility, as noted previously.5 Samples were assessed with multi-color flow cytometry (antibody panel configuration in Supplementary Table 1 using a LSRFortessa flow cytometer (BD Biosciences) with manual sequential gating performed in FlowJo version 9.9.6 (BD Biosciences).
The data was captured, managed, maintained and retrieved on an Oracle database management system. Clinical data was entered through web-based forms. Data checks were done centrally at regular intervals and questions or issues communicated back to the clinical sites for correction or clarification.
Outcomes
The primary endpoint was the area under the stimulated C-peptide curve (AUC) mean over the first 2 hours of a 4-hour MMTT conducted at the month 12 visit in the imatinib versus placebo group, using an ANCOVA model adjusting for sex, baseline age, and baseline C-peptide. Pre-specified secondary outcomes included the 2-hour C-peptide AUC at 24 months; 4-hr C-peptide AUC at 12 and 24 months; C-peptide AUC over time to 24 months; exogenous insulin use at 12 and 24 months; major hypoglycemic events; HbA1C levels at 12 and 24 months; and frequency and severity of adverse events in the imatinib versus placebo groups. Pre-specified exploratory endpoints included: proportion of subjects who are exogenous insulin free at 12 and 24 months; proinsulin/C-peptide ratios; adiponectin concentrations; autoantibody titers and other immunologic measures; beta cell glucose sensitivity and insulin sensitivity (calculated from MMTT glucose, C-peptide, and insulin data, as noted previously).19, 20
Statistical analysis
The primary endpoint analysis was based on the pre-specified intention-to-treat (ITT) cohort defined as all randomised subjects with one-year (2-hour) C-peptide measured regardless of treatment adherence. The AUC mean was calculated applying the trapezoidal rule (base measured in minutes) to the six timed c-peptide values and dividing by 120 minutes. The AUC mean was transformed using the function: ln(YC–Pep +1) to provide better normal distributional behavior by the test statistic. The treatment group comparison was based on a Wald test using an ANCOVA model adjusting for sex, baseline age, and baseline C-peptide at the 0.05 alpha level (one-sided). The mean difference confidence interval was calculated using the bootstrap method. The predicted means for the confidence intervals (CI) for each treatment group were determined at the means of the other covariates. This same model was used for insulin use, HbA1c, serum adiponectin, ratio of fasting pro-insulin to C-peptide, and the autoantibody titer figures. The transformation applied to these endpoints varied to maintain approximately normal distributed residuals. For HbA1c a single outlier was removed because it violated the normal residual requirement of the model. For the proinsulin/C-peptide ratios, no transformation was adequate without removing outliers. When removing the 5 outliers (1 at 3 mos., 2 at 6 mos., and 2 at 12 mos.) not only did it normalize the residuals but there was evidence of a treatment effect at both 3 and 6 months. To assess the treatment effect without removing the outliers we took the residuals from the ANCOVA model after adjusting for the baseline value, age and sex and applied the 2-sample Wilcoxon test for treatment group difference. Only the primary endpoint C-peptide is reported as 1-sided with 90% confidence interval provided, all other significance levels are 2-sided with corresponding 95% confidence intervals. These analyses were conducted using TIBCO Spotfire S+ 8.2 software. Although an interim analysis had been planned, due the small sample size and limited data availability during the conduct of the trial, the DSMB did not feel that an interim analysis was necessary and thus was not performed.
Outcome measurements other than primary and secondary endpoints were all analyzed using the mixed model for repeated measures (MMRM). These outcomes include 2-hour plasma glucose AUC, 2-hour insulin secretion AUC, beta cell glucose sensitivity, weight, BMI, long and short acting insulin doses, fasting plasma glucose and insulin secretion, and insulin sensitivities. The same model was also applied to immune cell subsets. MMRM included study visits, treatment group, and interaction of treatment group by visits as fixed effects and baseline measurements as covariate. A compound symmetry variance–covariance structure was applied to model the within-patient random effects. P-values were then calculated to compare the differences of least square means between imatinib versus placebo groups at each study visit cross-sectionally. Multiple testing correction were made to the p-values across multiple visits. R software version 4.0.3 and nlme package were used to perform MMRM analyses. Not all analyses proposed in the study protocol are presented herein, due to space constraints, some analyses that remain ongoing, and change in priority of analyses over time.
Power and sample size
Using standard equations for the comparison of two means, a sample size of 40 imatinib-treated and 20 placebo treated subjects with C-peptide AUC mean measured would provide power of 85% to detect a 35% increase in the expected experimental treatment group mean (0.551 vs. 0.744, nano-grams/Liter scale; 0.439 vs. 0.556 on the transformed scale). This calculation is based on a two-sample t-test at the 0.05 level (one-sided) with 2:1 allocation. To address subjects missing the primary endpoint, the sample size goal was set at 66 subjects (44 + 22), allowing for as many as 10% of the subjects without a 1-year MMTT C-peptide AUC measurement.
This study is registered with ClinicalTrials.gov, NCT01781975.
Role of the funding source
The funder had no role in study design, data collection, data analysis, or data interpretation. The writing team had full access to all of the data and had final responsibility for submission of the manuscript for publication.
Results
Between February 12, 2014 and May 19, 2016, we screened 80 individuals, randomly allocating 45 to imatinib and 22 to the placebo group (figure 1). Demographic and baseline characteristics were comparable between the two groups, with the exception of somewhat higher weight and BMI in the imatinib group (table 1). Adherence with the study protocol was high, with 2 subjects in the imatinib group and 1 subject in the placebo group withdrawing prior to the primary endpoint assessment at 12 months. Adherence to study drug/placebo administration was high. With complete adherence of the 4 pills ingested daily for 26 weeks defined as 1.00, median pill consumption in the imatinib group was 0·883 (quartiles 0·757, 0·931; range 0·092-1.0) and 0·970 in the placebo group (quartiles: 0·857, 0·997; range 0·286-1·0). These calculations did not account for adjustments to drug dosing mandated per protocol for adverse events (see Safety).
Figure 1.
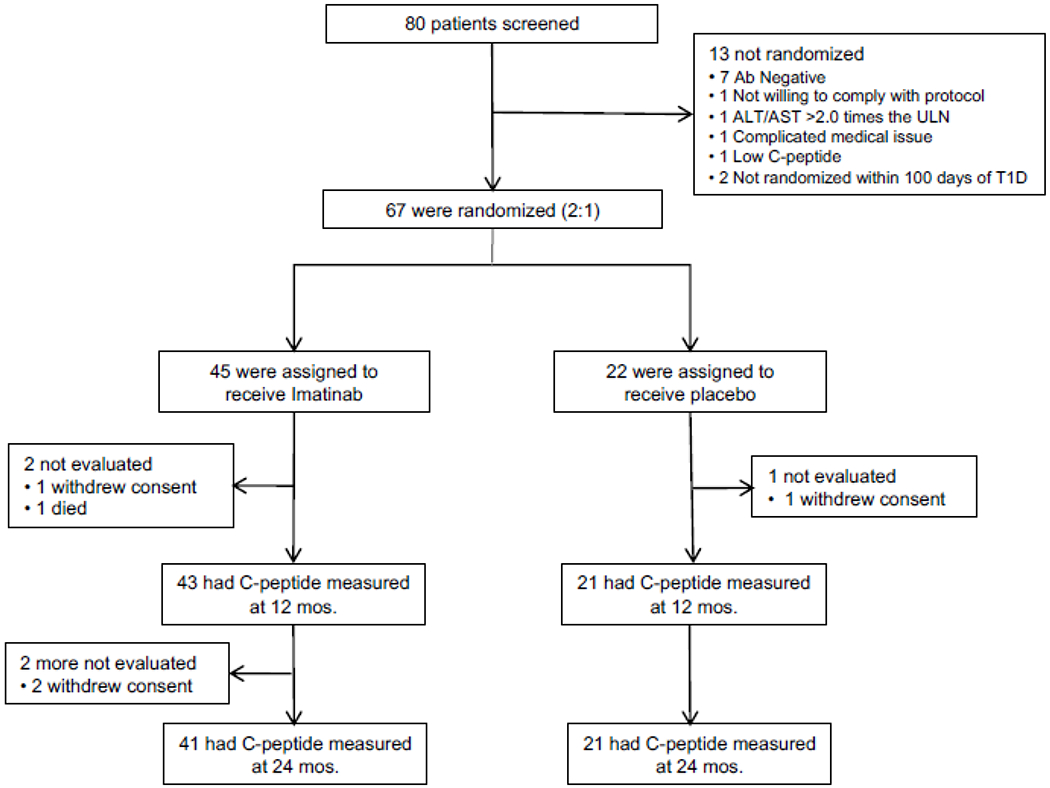
Consort Diagram
Table 1.
Baseline Demographic and Laboratory Characteristics of Participants at Entry
| Imatinib N=45 | Placebo N=22 | |
|---|---|---|
|
| ||
| Age- yr | ||
| Median | 26.6 | 23.4 |
| 1st and 3rd quartiles | 22.4 – 32.4 | 21.6 – 29.9 |
| Range | 19 – 45.7 | 18.8 – 40.6 |
|
| ||
| Male sex- no. of patients (%) | 27 (60.0) | 10 (45.5) |
|
| ||
| Race - no. of patients (%) | ||
| White | 44 (97.8) | 21 (95.5) |
|
| ||
| Ethnicity - no. of patients (%) | ||
| Non-Hispanic | 43 (95.6) | 19 (86.4) |
|
| ||
| Autoantibodies Positive - no. of patients (%) | ||
| GAD65H | 39 (86.7) | 20 (90.9) |
| IA2H | 28 (62.2) | 15 (68.2) |
| mIAA | 23 (51.1) | 13 (59.1) |
| ICA* | 24 (54.5) | 14 (63.6) |
| ZNT8 | 22 (48.9) | 12 (54.5) |
|
| ||
| No. of Autoantibodies Positive - no. of patients (%) | ||
| 1 | 9 (20.0) | 4 (18.2) |
| 2 | 7 (15.6) | 4 (18.2) |
| 3 | 12 (26.7) | 1 (4.5) |
| 4 | 8 (17.8) | 6 (27.3) |
| 5 | 9 (20.0) | 7 (31.8) |
|
| ||
| No. of days from diagnosis to first infusion - Median | 82 | 83.5 |
| 1st and 3rd quartiles | 70 - 91 | 73.3 - 95.8 |
|
| ||
| Weight (kg) - Median | 73.4 | 65.3 |
| 1st and 3rd quartiles | 66.6 - 80.5 | 59.6 - 79.5 |
|
| ||
| Body Mass Index** (kg/m2) - Median | 24.1 | 22.3 |
| 1st and 3rd quartiles | 22.1 - 26 | 20.1 - 25.1 |
|
| ||
| AUC Median | 0.736 | 0.679 |
| 1st and 3rd quartiles | 0.544 - 1 | 0.614 - 0.877 |
|
| ||
| Glycated hemoglobin at baseline (HbA1c -%) - median | 7.4 | 7.05 |
| 1st and 3rd quartiles | 6.6 - 8.1 | 5.9 - 8.8 |
|
| ||
| Total daily insulin dose at baseline* (U/kg) - median | 0.222 | 0.242 |
| 1st and 3rd quartiles | 0.087 – 0.376 | 0.0645 – 0.374 |
Missing data: 1 patient missing ICA status
Note: BMI was greater for the Imatinib Group than the Placebo Group (this stemmed from the difference in weight distribution)
Subjects were followed over 24 months, with active treatment for the first 26 weeks. The pre-specified primary outcome was a measure of endogenous insulin secretion, the 2-hr C-peptide AUC in response to a 4-hr MMTT at 12 months in the imatinib versus placebo groups, using an ANCOVA model adjusting for sex, baseline age, and baseline C-peptide. The graph in figure 2 summarises the predicted treatment group means of the C-peptide AUC mean over time from baseline to 24 months (see also supplementary table 2). The 12-mo estimates are 0·583 (90% CI: 0·529, 0·639) and 0·489 (90% CI: 0·417, 0·564) nmoles/L for the imatinib and placebo groups, respectively. The adjusted mean difference between study groups is 0.0946 (90% CI: −0·00279, 0·191), and constitutes a 19% treatment effect (difference in the adjusted 12-mo means divided by the placebo group mean). The 12-mo group means of the AUC C-peptide means were compared within the ANCOVA model at 12 months and were significant, with the Wald test from the model of 1·66 (p = 0·048, 1-tail test). This effect subsequently waned at the later assessments out to 24 months. The 4-hr AUC mean C-peptide data was consistent with the 2-hr analyses: model adjusted mean was 0.581 nmoles/L [95%CI: 0.518, 0.646] for the imatinib group and 0·479 nmoles/L [95% CI: 0·395, 0·568] for the placebo group, with p=0–03 (1-sided). We also evaluated whether the placebo group had the expected decline in C-peptide over time, and based on a model using TrialNet data, the 12-month AUC mean of C-peptide for the placebo group declined as expected.21
Figure 2:
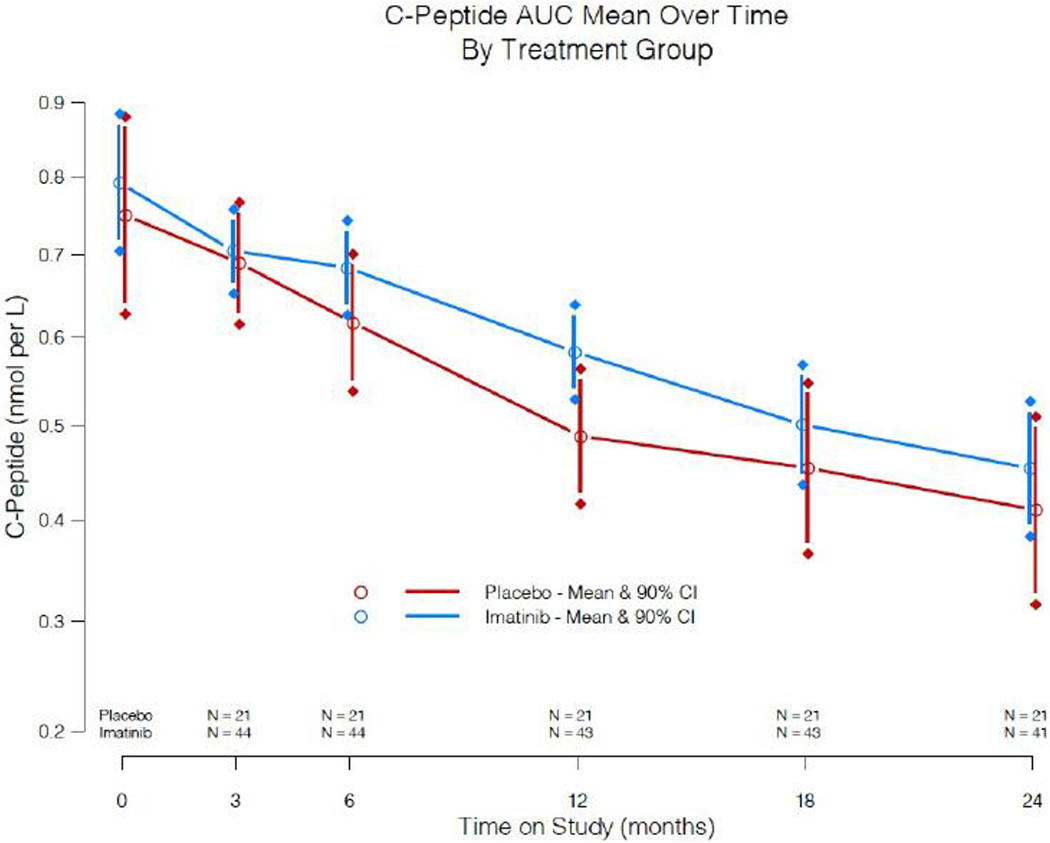
2-hr C-peptide AUC mean levels and 90% CI obtained from 4-hr MMTTs performed over the 24 month study in imatinib-treated versus placebo groups. The significance level at 12 months is p = 0·048 (one-sided); all other comparisons are not significant. In all figures, the imatinib-treated group is shown in blue, and the placebo-treated group in red.
In secondary analysis, exogenous insulin use was comparable at baseline between the two groups, but trended lower in the imatinib treatment group as compared to placebo at the initial assessment at 3 months and persisted through the 6-mo treatment period (at 6 months, mean difference −0·137 units/kg, [95% CI: −0·260, −0·0458]), with no statistically significant difference thereafter (Figure 3A, supplementary table 2). Short-acting insulin use made up the majority of the group difference (supplementary Table 2). Body weight and BMI did not have a statistically significant change over the 24-month study period in either group. For glycemic control during the study, both groups were treated to the same HbA1C target goal of ≤ 7%. HbA1c initially declined below baseline in both groups, but trended lower in the imatinib than placebo group during the active treatment phase, with the greatest difference in groups at 3 months (mean difference −0·422%, [95% CI: −0·772, −0·0676] (Figure 3B, supplementary table 2).
Figure 3:
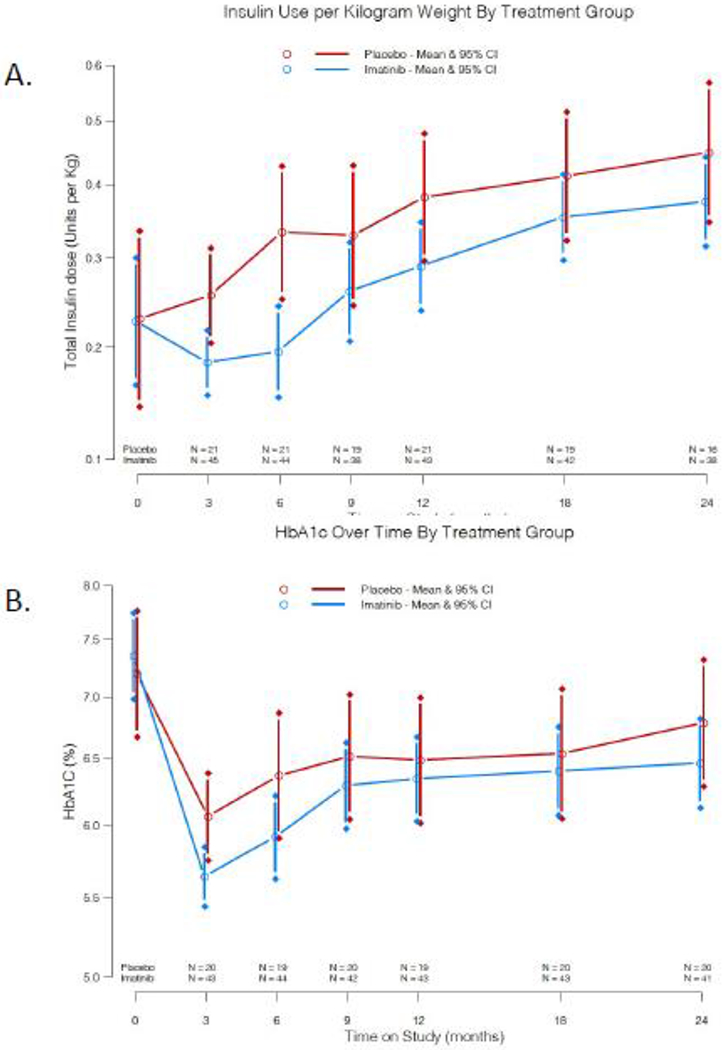
A) Exogenous insulin use per kilogram body weight means and 95% CI over the 24 month study according to treatment group. B) Mean HbA1c levels and 95% CI over the 24 months according to treatment group.
In further exploratory analysis of data from the MMTTs, fasting plasma glucose concentrations and fasting insulin secretion rates were similar in the two groups at baseline and did not differ significantly thereafter (Supplementary Table 3). By contrast, the MMTT-stimulated responses are distinctly different between the imatinib and placebo groups during the treatment period (figure 4, supplementary figure 1). This difference was most notable for the 2-hour glucose concentration, which was lower in the imatinib group at 3 months (mean difference −2·74 mmol/L [95% CI: −1·17, −4·07] and 6 months (mean difference of −4·1 mmol/L [95% CI: −2·3, −5·62] compared to the placebo group (figure 4A). Despite the lower glucose excursion, 2-hr insulin secretion AUC during the MMTTs was comparable between the imatinib and placebo treatment groups (figure 4B). Consequently, the insulin secretion/plasma glucose dose-response curves fell flatter in the placebo than the imatinib group (figure 5A). Beta cell glucose sensitivity, which is the slope of the insulin secretion/plasma glucose dose-response function,19 increased above baseline while on imatinib for 3 months and stabilized out to 6 months, but declined thereafter off of study drug; by contrast, the measure steadily declined in the placebo group over time, with a difference between the groups at 6 months (mean difference of 9·4 pmol/min/ m2/mmol/L [95% CI: 2·23, 21·5] (figure 5B). In line with the notion that lower glucose concentrations stimulated greater insulin release during imatinib treatment, insulin secretion rates calculated at matched glycemia (7 mmol/L) were higher in the imatinib than placebo group at 6 months (mean difference of 23·2 pmol/min/ m2 [95% CI: 4·3, 53·8]) (supplementary table 3). Based on plasma glucose and insulin levels measured during the MMTTs, insulin sensitivity was estimated to be similar in the two groups at baseline, and remained stable in the imatinib group while on active therapy but declined steadily in the placebo group (mean difference at 6 months of 1·2 mL/min/kg [95% CI: 0·6, 1·8]; these differences resolved off of active therapy at 12 months (supplementary table 3).
Figure 4:
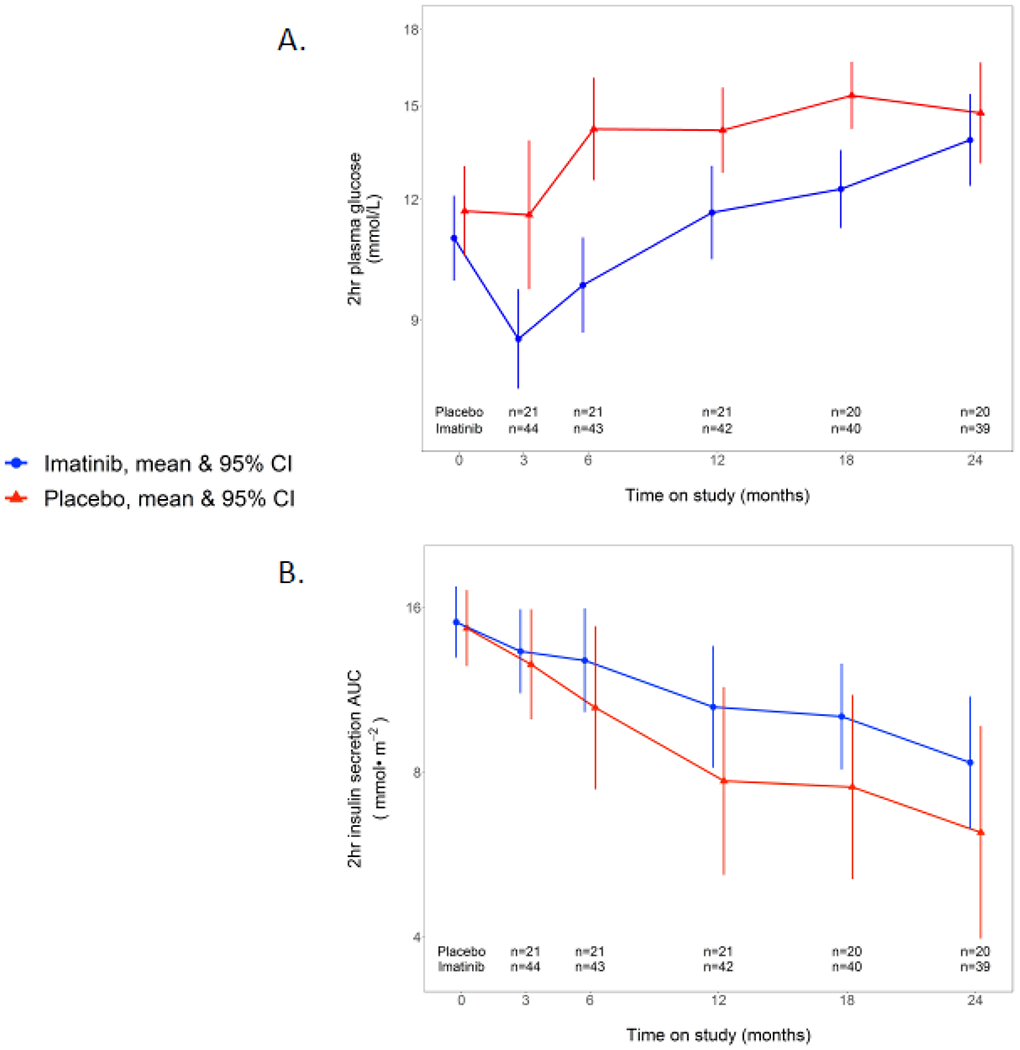
MMTT responses over time: a) 2-hr plasma glucose and b) 2-hr serum insulin secretion AUC in response to MMTT over time in imatinib versus placebo group. Plots are means and 95% CI.
Figure 5:
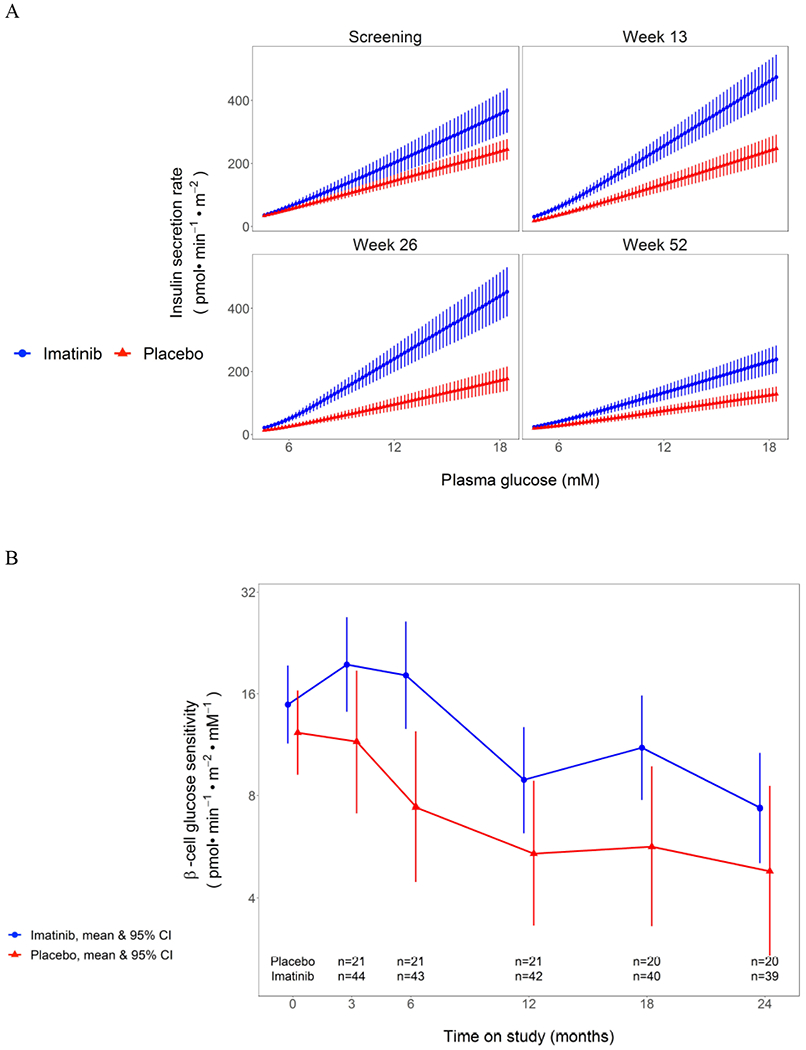
A) Dose-response curve of insulin secretion rates versus plasma glucose levels during the serial MMTTs in imatinib versus placebo groups. Data are mean with SEM. The mean slope of the dose response is beta-cell glucose sensitivity, presented in B) as change in beta cell glucose sensitivity over time in imatinib versus placebo study groups. Plots are means with 95% CI.
Additional exploratory analyses were conducted to investigate possible mechanisms of imatinib action on metabolism in treated subjects compared to placebo. With the effects noted on beta cell glucose sensitivity, and possible direct impact of imatinib on beta cell function and survival from animal models,10, 12 we evaluated changes in pro-insulin/C-peptide ratios over time. Fasting proinsulin to C-peptide ratios were stable in the drug-treated group while on active treatment, but trended steadily upward in the placebo group over time; due to outliers when calculating the ratio, we conducted a non-parametric test on the treatment group difference and noted a statistically significant difference at both the 3-mo (p =0·04) and 6-mo assessments (p = 0·03)(figure 6A). To determine if imatinib therapy had an impact on beta cell apoptosis, we evaluated change in beta cell death over time, utilizing a PCR based assay for the cell-free preproinsulin gene (INS) (supplementary figure 2).18 Low level values were noted at baseline, and we observed no significant change over time in unmethylated, methylated, nor in the ratio of unmethylated to methylated insulin DNA, comparing drug-treated to placebo groups. Finally, with the impact of imatinib on insulin sensitivity, we measured serum adiponectin concentrations. Adiponectin increased in the imatinib treated group during the 6 months while on therapy (mean difference 5·75 μg/mL at 6 months, [95% CI: 2·89, 8·59]), and by 12 months dropped back down to baseline levels indistinguishable from the placebo group (figure 6B).
Figure 6:
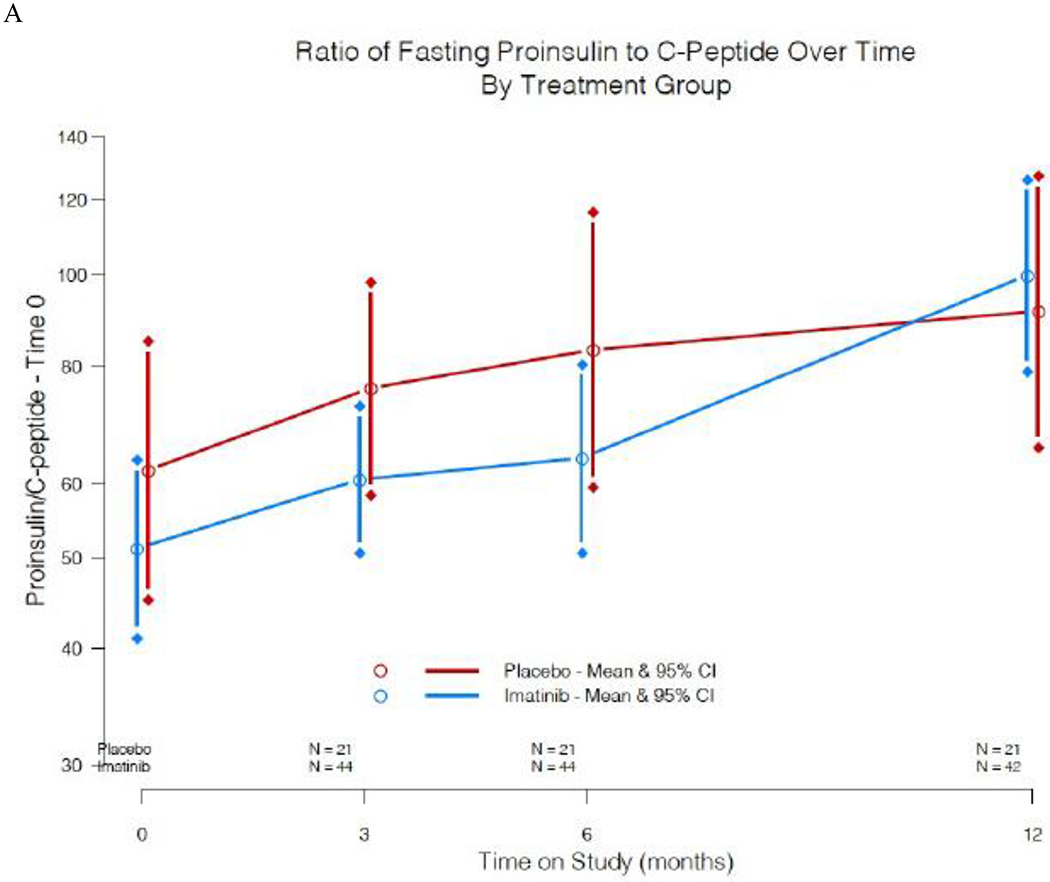
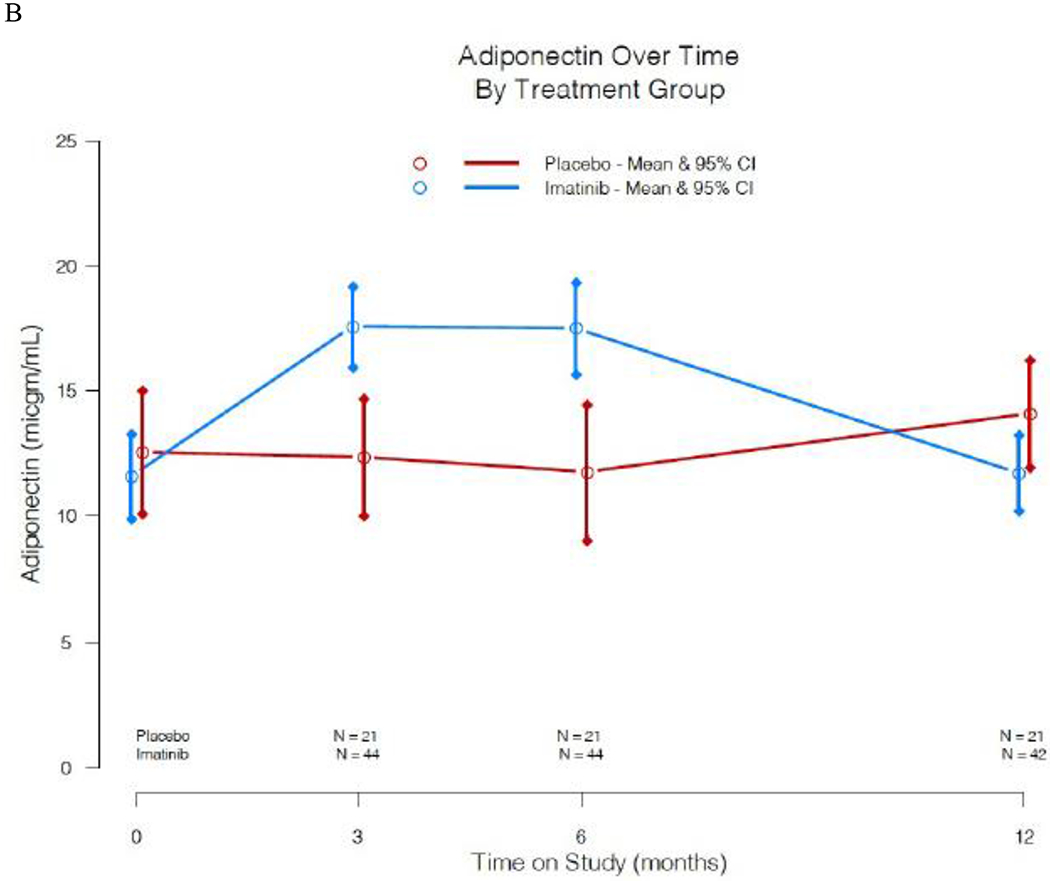
Additional assessments of imatinib effects on beta cell function and metabolism. A) Pro-insulin/C-peptide ratios over time in imatinib versus placebo study groups. Plots are means and 95% CI. Due to outliers when calculating the ratio, we conducted a Wilcoxon 2-sample test on the treatment group difference and noted a statistically significant difference at both 3 months (p =0·04) and 6 months (p =0·03). B) Serum adiponectin concentration over time in imatinib versus placebo study groups. Plots are means and 95% CI.
As imatinib has been reported to affect various immune cell types, we performed comprehensive immune phenotyping over the course of the study. No statistically significant changes in B cell function via autoantibody titers were noted over time in drug treated versus placebo subjects (supplementary figure 3). Immune phenotyping by flow cytometry indicated no significant differences in B cell, T cell, and myeloid cells between the drug treated and placebo groups (supplementary figure 4). In further analyses, we noted a transient reduction in frequencies of CD141+ DC (cDC1) and CD95+ IgD-CD27+ memory B cells with therapy as compared to baseline and the placebo group (supplementary figure 5), suggesting an impact on rare and metabolically active cells. In evaluating non-inflammatory immune cell subsets in the upper versus lower quartile of C-peptide AUC responders amongst the drug treated group, we did not observe any differences during the course of the study (supplementary figure 6). Additional exploratory immunologic analyses are ongoing.
Adverse events (AEs) of grade 2 severity or higher were collected during the study, and 71% of drug-treated and 59% of placebo treated subjects reported at least one such AE (table 2). The distribution of AEs across the grading levels were similar between the two groups, with most rated as mild to moderate in severity. However, when evaluating the AEs deemed likely attributed to study drug, there was a significantly higher proportion of subjects in the imatinib group who had AEs, with the bulk again in the mild to moderate level. Of the most common AEs, the notable differences between study groups occurred with: 1) gastrointestinal issues, 13% in the drug-treated subjects vs 0% in the placebo group, primarily nausea; and 2) additional laboratory investigations, with 22% in the imatinib group vs 9% in placebo treated subjects primarily per protocol assessments to evaluate and track abnormal complete blood cell counts and liver function tests. Infections were reported in 27% of the imatinib treated group versus 18% of the placebo group, with 26 and 9 events, respectively. No opportunistic infections were noted, and all were confined to grade 2 severity, with the exception of one subject with skin infection in the imatinib group (see below). In terms of AEs related to diabetes management, one subject in each group experienced severe hypoglycemia, and 1 subject in the placebo group had a single episode of diabetic ketoacidosis.
Table 2A:
Safety Experience of Trial Participants: Adverse Events Severity by Treatment Group
| Severity (Grade) | Treatment Group | |
|---|---|---|
| Imatinib No. of subjects (%) | Placebo No. of subjects (%) | |
| 0/1 | 13 (28.9) | 9 (40.9) |
| 2 | 21 (46.7) | 10 (45.5) |
| 3 | 7 (15.6) | 2 (9.1) |
| 4 | 3 (6.7) | 1 (4.5) |
| 5 | 1 (2.2) | 0 (0.0) |
| Total | 45 (100.0) | 22 (100.0) |
In evaluation of the higher grade AEs, a single grade 5 AE occurred in the drug-treated group, and was deemed unrelated to study drug. The grade 3 and 4 AEs that occurred in the drug-treated subjects related primarily to anticipated issues with shifts in liver function and blood cell counts. These events all resolved uneventfully but required adjustments to study drug administration, as specified in the protocol. Of the 45 subjects in the drug-treated group, 20 had no modification in study drug administration, 17 had temporary modifications to dosing (7 for neutropenia, 4 for rashes, 2 for abnormal liver function tests, and 1 each for gastrointestinal issues, thrombocytopenia, cramps, and mental health concerns) , and 6 permanently discontinued (4 for liver function abnormalities; 1 for pre-existing cardiac arrhythmia noted after randomisation, and 1 for persisting allergic skin rash). By contrast, 15 of 22 placebo subjects had no drug modification, and 5 had transient adjustments (3 for thrombocytopenia, 1 for neutropenia, 1 for diabetic ketoacidosis). In each group, although there were no indications to alter drug administration, 1 subject temporarily discontinued study drug/placebo and 1 subject in each group elected to permanently discontinue study drug.
Seven subjects experienced a total of 11 serious adverse events (SAEs), 4 subjects (8 events) in the treatment group and 3 subjects (3 events) in the placebo group (supplementary table 4). Two participants in the imatinib-treated group each had 3 SAEs. Two of these events were considered possibly related to study drug (skin infections in a single subject at a 2 month interval), and the remainder were considered to be unrelated to study drug.
Discussion
Given the challenges in managing type 1 diabetes, clinical trialists have sought agents to safely and effectively block autoimmune mediated destruction of beta cells, often utilizing immunotherapies targeting T cells. Transient effects have been observed with a handful of such agents,3–8 prompting the search for new approaches. Missing from our current armamentarium has been a therapy that also impacts metabolism, and improves beta cell health. Herein, we report the first new onset type 1 diabetes clinical trial conducted with a tyrosine kinase inhibitor, utilizing the first-in-class drug member imatinib. Although initially developed for use in CML, preclinical studies and clinical observations suggest that imatinib may offer a novel means to treat type 1 diabetes, targeting both immunologic and metabolic pathways.10, 11, 15–17, 22 In designing this trial, we sought to evaluate response while on therapy, and to determine if there was a persisting effect off of therapy, as was observed in prior NOD mouse studies after 10 weeks of imatinib exposure.11 The trial achieved the pre-specified primary outcome, with adult participants treated for 26 weeks exhibiting greater C-peptide AUC in response to MMTT at 12 months, compared to the placebo subjects. However, the effect was not sustained during a second year of observation off of therapy. As compared to the placebo group, imatinib-treated subjects had lower exogenous insulin needs and trended towards lower HbA1C while on therapy, but these effects waned in the ensuing months off of therapy. These metabolic effects may stem from imatinib’s impact on improved beta cell function and peripheral insulin sensitivity.
Imatinib may act via a variety of different mechanisms to alter the course of type 1 diabetes. Imatinib was first developed as a specific inhibitor of the Abl kinases to target the Bcr-Abl fusion protein in CML. However, imatinib may also have broader clinical utility, as it also inhibits other tyrosine kinases, including platelet-derived growth factor receptor (PDGFR), c-kit (CD117), macrophage colony stimulating factor receptor (c-fms), Abl-related gene, and Lck.9, 15, 17, 22 In evaluation of the potential metabolic effects of imatinib, preclinical studies report effects on both the beta cell and insulin sensitivity. Direct effects of imatinib on beta cell function include increased glucose-stimulated insulin secretion, and enhanced beta cell survival in the face of various stressors, including high fat diet, cyclophosphamide, streptozotocin, and autoimmunity.10, 11, 23 Han et al reported lower ER stress and increased beta cell mass in db/db mice treated with imatinib.14 Morita et al expanded on these observations in the NOD mouse, noting that in the face of ER stress, imatinib antagonises the interaction between ABL and the ER transmembrane kinase/endoribonuclease (RNase) IRE1α, thereby blunting IRE1α hyperactivity, reducing beta cell apoptosis, and reversing diabetes.12
Several of our trial observations suggest that imatinib improved beta cell function during the course of this trial. First, beta cell glucose sensitivity improved during imatinib treatment, but diminished after it was withdrawn (figure 5). This analysis has typically been used to evaluate beta cell function in type 2 diabetes, but has also been used previously to evaluate an at-risk type 1 diabetes population, and we have now applied this methodology to evaluation post diagnosis.24 Rather than simply utilizing insulin secretion in response to a MMTT, beta cell glucose sensitivity evaluates the ability of the beta cell to respond with insulin secretion to a given glucose level, and may thus provide a better overall measure of beta cell function. If the impact of imatinib had been mediated solely via a change in insulin resistance, rather than also on beta cell function, then one would expect less insulin secreted for a given glucose level rather than our observation of greater insulin secretion, and thus the findings support an improvement in beta cell function. Second, the pro-insulin to C-peptide ratio remains lower during imatinib therapy (figure 6A), which has been linked with lower ER stress and improved beta cell function.25, 26 A third line of evidence in support of imatinib’s impact on beta cell health is its effect on adiponectin concentrations (figure 6B): although an increase in adiponectin is often associated with improved insulin sensitivity, prior studies suggest that adiponectin may decrease beta cell apoptosis and improve function through direct actions mediated by adiponectin receptors on beta cells.27 One last assay of interest in assessing the impact of imatinib on beta cell health was the beta cell death assay, utilizing PCR amplification of the INS gene from sera. From our clinical samples, we did not see a difference in the imatinib versus placebo groups over time. However, we note that the values measured were in the lower range of the assay, and we may therefore have lacked the sensitivity to detect a difference in beta cell death.
Imatinib may also improve insulin sensitivity. In high fat-fed mice, imatinib blocks PPARγ phosphorylation, which in turn improves insulin sensitivity and promotes browning of adipose tissue.28 Imatinib has also been shown to improve insulin sensitivity of rats fed a high fat diet, and induces remission of diabetes in db/db mice.13, 14 Several case reports and case series note that patients with type 2 diabetes have experienced significant improvement or disease resolution on imatinib.16 As noted previously29 and in this study, imatinib therapy increases serum adiponectin levels, which has been associated with improvement in insulin sensitivity. No clinical studies to date have formally assessed changes in insulin sensitivity with imatinib; in our study, we did note a dramatic decrease in exogenous insulin needs while on therapy. We utilized glucose and insulin secretion data from MMTTs to estimate insulin sensitivity using a validated method,20 and found a significant difference between imatinib and placebo-treated groups at 6 months. Future formal testing with hyperglycaemic-euglycemic clamp studies will help clarify the impact of imatinib on insulin sensitivity.
Our interest in evaluating imatinib in this clinical trial also stemmed from possible immunologic effects, and its potential to block further autoimmune destruction of beta cells, as suggested by preclinical studies and clinical reports and small case series in various autoimmune conditions.15, 17, 22 In the past NOD mouse studies with imatinib, no significant changes were noted in the various immunologic mechanistic assays, including T cell effector function and trafficking to the islets, insulitis scores, CD4+/CD8+ ratios in spleen and pancreatic lymph nodes, and regulatory T cell function.11 Similarly, we found no immunologic readout from peripheral blood samples that clearly delineated drug-treated subjects from placebo, with no significant changes in autoantibody levels over time, nor changes in immune cell subsets by flow cytometry. These latter assessments are limited to immunophenotyping, and there may be effects on immunologic function. Furthermore, our assessments were limited to sampling from the peripheral circulation, and thus we cannot determine if immunologic changes may have occurred in imatinib-treated subjects within the pancreatic lymph node or islets.
In planning this study, we were mindful of potential safety issues associated with imatinib, but anticipated that it may be better tolerated than in oncology settings, as we were working with a younger otherwise healthy population. Indeed, in general, imatinib was well tolerated, and if subjects did develop AEs, they tended to occur early in the course of study drug administration, to be milder than that described in the oncology literature,30 and usually resolved in the ensuing days and weeks with ongoing therapy. We adopted a more conservative algorithm for surveillance and modification of drug dosing for use in our T1D study, enabling investigators to detect potential drug toxicities early in the treatment course. Approximately one third of imatinib-treated subj ects required a temporary modification in dosing, and 13% had to permanently discontinue imatinib. Thus, subjects treated with imatinib must be carefully monitored, and this algorithm can be used to guide drug administration in future studies.
Limitations of this study include the fact that it was modest in size; was confined only to adults and included almost exclusively Caucasians; and was associated with possible safety issues, with the more frequent dose adjustments and side effect profile for those on imatinib possibly weakening the blinding of study group assignment.. Furthermore, the study evaluated the impact of only a 6 month treatment period. Nonetheless, in looking across results from successful phase 2 new onset type 1 diabetes trials, the 19% effect size with imatinib noted at 12 months compares favorably with several other agents, including rituximab, alefacept, and abatacept.31 Thus, further evaluation of the use of imatinib in type 1 diabetes may be warranted. Although imatinib is approved for use in children with CML down to age 3, we were limited in this study to evaluate only adults, in order to clarify potential safety issues and document a prospect of benefit for this population. The rate of beta cell loss occurs more slowly in adults compared to children,32 and there may be some fundamental differences in the disease process that associates with age of type 1 diabetes presentation. As suggested with other agents, including CTLA4-Ig, anti-CD20, and anti-CD3 monoclonal antibodies3, 4, 7, this therapy may prove to be more efficacious in children with new onset type 1 diabetes than adults. The ideal dose and duration of imatinib therapy requires further evaluation. We utilized a standard starting dose employed in oncology, but benefits in type 1 diabetes may occur at lower doses, and with reduced risk for adverse effects. Unlike in the NOD mouse, the metabolic benefits realized by treated subjects were not sustained as imatinib was withdrawn, and thus continuous or chronic intermittent therapy may be required. An additional question is whether imatinib will offer an additive or synergistic response when used in combination with a drug that works by an alternate mechanism, such as via T cell immunomodulation with teplizumab or ATG. Imatinib may also prove even more efficacious if used earlier in the course of disease, such as at stage 2 when beta cell function is declining but prior to frank hyperglycemia.33 Finally, the list of approved tyrosine kinase inhibitors with varied specificities continues to grow steadily, and as we learn more about the most critical pathways to target in type 1 diabetes, there may be a better drug identified in this class to consider for treatment of type 1 diabetes.
In summary, this phase 2 study showed that 26 weeks of treatment with imatinib slowed decline of beta cell function out to 12 months, and may have novel effects on metabolism, with improved beta cell function and insulin sensitivity. These initial observations suggest future study considerations with imatinib in type 1 diabetes, provided treated subjects are closely monitored for possible toxicities.
Supplementary Material
Supplementary Figure 1: Time-course of plasma glucose (A) and C-peptide concentrations (B) during the MMTT at screening and week 26 in the imatinib and placebo groups. Plots are mean with 95% CI.
Supplementary Figure 2: Circulating unmethylated and methylated preproinsulin (INS) DNA, and ratio of unmethylated to total INS DNA, in imatinib and placebo study groups over time. Plots represent median and inter-quartile range. No statistically significant differences were noted between groups.
Supplementary Figure 3: Autoantibody titers over time in imatinib versus placebo group, for a) mIAA , b) IA2, c) GAD, and d) ZnT8 autoantibodies. No transformation was found to normalize any of the four autoantibody titers, and therefore the confidence intervals are distorted.
Supplementary Figure 4: Immune cell subsets in imatinib versus placebo groups over time, as assessed by flow cytometry. A) B cells, B) dendritic cells, C) monocyte, D) CD4+ Tregs, E) CD4+ non-Tregs, and F) CD8+ T cell populations over time in the drug treated and placebo groups. No statistically significant differences were noted from baseline.
Supplementary Figure 5: Highly metabolically active rare cell subsets in imatinib versus placebo groups over time: a) cDC1 of dendritic cells (CDC 141+ of CD14−, CD16-HLA-DR+), and b) CD95 of IgD-CD27+ switched memory B cells. Differences between the imatinib and placebo groups were noted at the end of the 6-mo treatment period (p=0·0059 and 0·0034, respectively, corrected for baseline), and then resolved off of therapy by 12 months.
Supplementary Figure 6: Potentially protective immune cell subsets in the upper versus lower quartile of C-peptide AUC responders in the imatinib treated group over time, shown in purple and orange respectively, for A) HLA DR on DCs, B) partially exhausted CD8+ T cells, and C) CD4+Tregs. Although differences are noted between the upper and lower quartile at 6 and 12 months (5A, 5B), there is no significant change longitudinally from baseline to 6 or 12 months for either of these groups; no differences were noted at baseline or over time in FOXP3+ CD4 Treg (5C).
Supplementary Table 1: Flow cytometry antibody panels.
Supplementary Table 2: Efficacy Table of primary and secondary endpoints of group mean and mean difference estimates (including 90% or 95% confidence intervals)
Supplementary Table 3: Change in metabolic parameters over time in drug-treated versus placebo groups, median (interquartile range) displayed.
Supplementary Table 4: Serious adverse events by category and treatment assignment, with number of total events in a given category, number (percentage) of subjects in each treatment group, and possible attribution to study drug.
Table 2B:
Number of Events and Number of Subject for each Adverse Effect Type by Treatment Group
| Adverse Effect Category | Imatinib N= 45 | Placebo N = 22 | ||
|---|---|---|---|---|
| No. of events | No. of subjects (%) | No. of events | No. of subjects (%) | |
| Infections and infestations | 26 | 12 (26.7) | 9 | 4 (18.2) |
| Eye disorders | 2 | 2 (4.4) | 0 | 0 (0.0) |
| Musculoskeletal and connective tissue disorders | 4 | 3 (6.7) | 1 | 1 (4.5) |
| Nervous system disorders | 4 | 4 (8.9) | 0 | 0 (0.0) |
| Gastrointestinal disorders | 13 | 6 (13.3) | 0 | 0 (0.0) |
| Metabolism and nutrition disorders | 11 | 5 (11.1) | 2 | 2 (9.1) |
| Reproductive system and breast disorders | 1 | 1 (2.2) | 0 | 0 (0.0) |
| Psychiatric disorders | 9 | 5 (11.1) | 0 | 0 (0.0) |
| Respiratory, thoracic and mediastinal disorders | 6 | 6 (13.3) | 1 | 1 (4.5) |
| Skin and subcutaneous tissue disorders | 6 | 5 (11.1) | 3 | 2 (9.1) |
| Ear and labyrinth disorders | 1 | 1 (2.2) | 0 | 0 (0.0) |
| Cardiac disorders | 6 | 4 (8.9) | 0 | 0 (0.0) |
| General disorders/Administration site conditions | 2 | 2 (4.4) | 0 | 0 (0.0) |
| Laboratory Investigations | 18 | 10 (22.2) | 4 | 2 (9.1) |
| Pregnancy, puerperium and perinatal conditions | 0 | 0 (0.0) | 1 | 1 (4.5) |
| Injury, poisoning and procedural complications | 4 | 2 (4.4) | 0 | 0 (0.0) |
| Immune system disorders | 0 | 0 (0.0) | 1 | 1 (4.5) |
| Hepatobiliary disorders | 5 | 3 (6.7) | 2 | 2 (9.1) |
| Surgical and medical procedures | 0 | 0 (0.0) | 2 | 2 (9.1) |
| Blood and lymphatic system disorders | 7 | 4 (8.9) | 0 | 0 (0.0) |
| Endocrine disorders | 45 | 4 (8.9) | 2 | 1 (4.5) |
| Renal and urinary disorders | 2 | 1 (2.2) | 0 | 0 (0.0) |
| Total Events | 172 | --- | 28 | --- |
Research in Context:
Evidence before this study:
A series of clinical trials have been conducted in new onset type 1 diabetes in an attempt to preserve beta cell function, primarily using immunotherapies and often targeting T cells; some of these efforts have had modest initial success, but lack robust durable effects. In looking for new approaches, we searched the PubMed database for articles published in any language up to December 1, 2020 with the search terms imatinib, tyrosine kinase inhibitor (TKI), diabetes, and autoimmunity. We found publications suggesting that imatinib may target both immunologic and metabolic pathways, and offer a novel way to treat type 1 diabetes. Several preclinical studies in rodent models suggest that imatinib has effects on beta cell function and insulin sensitivity. Furthermore, case reports and case series suggest benefits of imatinib in treating some autoimmune diseases, as well as salutary effects on type 1 as well as on type 2 diabetes.
Added value of this study:
This clinical trial is the first phase 2 study to be conducted in recent onset type 1 diabetes with a tyrosine kinase inhibitor, utilizing the first-in-class member imatinib. This study showed that 26-wk treatment with imatinib slowed decline of beta cell function out to 12 months. Secondary and exploratory analyses did not reveal any overt impact of this treatment on immune responses, but did point to a series of unique effects on metabolism, with improved beta cell function and insulin sensitivity. Treated subjects must be monitored closely for possible side effects and toxicities that may require modification to imatinib dosing.
Implications of all the available evidence:
Imatinib may offer unique benefits to patients with type 1 diabetes, and may provide a novel means to target beta cell health and insulin sensitivity, but close monitoring for potential safety issues is necessary. Possible future studies with imatinib in type 1 diabetes include exploring lower doses and longer duration of therapy; extending related studies to children and adolescents; use in an at-risk population to delay or prevent progression to disease; and use in combination with drugs that work by complimentary mechanisms, such as one of the previously successful and more traditional immunotherapies.
Acknowledgements
The trial was sponsored by the Juvenile Diabetes Research Foundation. The sample collection and storage for mechanistic assays reported in this publication were also supported by the Immune Tolerance Network (ITN), which is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health under Award Number UM1AI109565. We thank Peter Sayre, MD, PhD, and Elisavet Serti, PhD, from the ITN for their contributions as study medical monitor and in oversight of mechanistic studies, respectively, and Sheila Scheiding from the HIP Core at Benaroya Research Institute for her assistance with mechanistic study analyses.
The project was supported in part by NIH/NCRR Clinical & Translational Awards grants UL1TR000004 (UCSF), UL1TR002537 (Iowa), 1UL1TR001108 (Indiana), UL1TR001878 (Children’s Hospital of Philadelphia), and P30DK036836 (Joslin Diabetes Research Center, NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Novartis Pharmaceuticals Corporation (Cambridge, Massachusetts, USA) supplied study drug and matching placebo, and gave input regarding dosage and safety, but had no direct involvement with study design, conduct, or management; data collection, analysis or interpretation; or manuscript preparation. There are no agreements concerning confidentiality of the data between the sponsor and the authors or the institutions named in the credit lines.
Conflicts of Interest:
SG has served on advisory boards for Avotres, Provention Bio, and Tolerion, and participated in clinical trials with Caladrius, Intrexon, Janssen, Provention Bio, and Tolerion. JG has consulted for Vertex Pharmaceuticals, Inc, and Regeneron Pharmaceuticals, Inc, and participated in clinical trials for Avotres, Caladrius, Janssen, and Tolerion. PG has served on advisory boards for Caladrius, Bristol Myers Squibb, and Lilly, received grant support from Caladrius, Novo Nordisk and Pfizer, and is co-founder and chief medica officer for ImmunoMolecular Therapeutics, Inc. RM has a patent application for DNA methylation in inflammatory disease. SW reports serving on advisory boards for Boehringer Ingelheim Pharmaceuticals, Inc. and Medtronic, and a DSMB for the National Institutes of Health. All other authors report no potential conflicts of interest.
Footnotes
Data Sharing
Data collected for the study and presented herein will be made available to others. Data will be organized in a data dictionary, and participant data will be de-identified. Related study documents, including the study protocol, informed consent forms, and statistical analysis plan, will also be available. Data requests should be sent to S.Gitelman via email, and he will partner with the coordinating center at the University of South Florida (J. Krischer) to supply the requested information via disk.
Contributor Information
Stephen E Gitelman, University of California San Francisco, San Francisco, CA, USA.
Brian N Bundy, University of South Florida, Tampa, Florida, USA.
Ele Ferrannini, CNR Institute of Clinical Physiology, Pisa, Italy.
Noha Lim, Immune Tolerance Network, Bethesda, MD, USA.
J Lori Blanchfield, Benaroya Research Institute, Seattle, WA, USA.
Linda A DiMeglio, Indiana University, Indianapolis, Indianapolis, IN, USA.
Eric I Felner, Emory University, Atlanta, GA, USA.
Jason L Gaglia, Joslin Diabetes Center, Boston, MA, USA.
Peter A Gottlieb, Barbara Davis Center, University of Colorado, Aurora, CO, USA.
S Alice Long, Benaroya Research Institute, Seattle, WA, USA.
Andrea Mari, CNR Institute of Neurosciences, Padua, Italy.
Raghavendra G Mirmira, Indiana University, Indianapolis, Indianapolis, IN, USA.
Philip Raskin, University of Texas Southwestern, Dallas, Texas, USA.
Srinath Sanda, University of California San Francisco, San Francisco, CA, USA.
Eva Tsalikian, University of Iowa, Iowa City, IA, USA.
John M Wentworth, Walter and Eliza Hall Institute and Royal Melbourne Hospital, Melbourne, Australia.
Steven M Willi, Children’s Hospital of Philadelphia and University of Pennsylvania, Philadelphia, PA, USA.
Jeffrey P Krischer, University of South Florida, Tampa, Florida, USA.
Jeffrey A Bluestone, University of California San Francisco, San Francisco, CA, USA.
References
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. 2014;383(9911):69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foster NC, Beck RW, Miller KM, Clements MA, Rickels MR, DiMeglio LA, et al. State of Type 1 Diabetes Management and Outcomes from the T1D Exchange in 2016-2018. Diabetes Technol Ther. 2019;21(2):66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes. 2013;62(11):3766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361(22):2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haller MJ, Long SA, Blanchfield JL, Schatz DA, Skyler JS, Krischer JP, et al. Low-Dose Anti-Thymocyte Globulin Preserves C-Peptide, Reduces HbA1c, and Increases Regulatory to Conventional T-Cell Ratios in New-Onset Type 1 Diabetes: Two-Year Clinical Trial Data. Diabetes. 2019;68(6):1267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest. 2015;125(8):3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet. 2011;378(9789):412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quattrin T, Haller MJ, Steck AK, Felner EI, Li Y, Xia Y, et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N Engl J Med. 2020;383(21):2007–17. [DOI] [PubMed] [Google Scholar]
- 9.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–53. [DOI] [PubMed] [Google Scholar]
- 10.Hagerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J. 2007;21(2):618–28. [DOI] [PubMed] [Google Scholar]
- 11.Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, et al. Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A. 2008;105(48):18895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1alpha Signaling Spares ER-Stressed Pancreatic beta Cells to Reverse Autoimmune Diabetes. Cell Metab. 2017;25(4):883–97 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagerkvist R, Jansson L, Welsh N. Imatinib mesylate improves insulin sensitivity and glucose disposal rates in rats fed a high-fat diet. Clin Sci (Lond). 2008;114(1):65–71. [DOI] [PubMed] [Google Scholar]
- 14.Han MS, Chung KW, Cheon HG, Rhee SD, Yoon CH, Lee MK, et al. Imatinib mesylate reduces endoplasmic reticulum stress and induces remission of diabetes in db/db mice. Diabetes. 2009;58(2):329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Aura Swanson C, Paniagua RT, Lindstrom TM, Robinson WH. Tyrosine kinases as targets for the treatment of rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(6):317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fountas A, Diamantopoulos LN, Tsatsoulis A. Tyrosine Kinase Inhibitors and Diabetes: A Novel Treatment Paradigm? Trends Endocrinol Metab. 2015;26(11):643–56. [DOI] [PubMed] [Google Scholar]
- 17.Azizi G, Mirshafiey A. Imatinib mesylate: an innovation in treatment of autoimmune diseases. Recent Pat Inflamm Allergy Drug Discov. 2013;7(3):259–67. [DOI] [PubMed] [Google Scholar]
- 18.Fisher MM, Watkins RA, Blum J, Evans-Molina C, Chalasani N, DiMeglio LA, et al. Elevations in Circulating Methylated and Unmethylated Preproinsulin DNA in New-Onset Type 1 Diabetes. Diabetes. 2015;64(11):3867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mari A, Tura A, Gastaldelli A, Ferrannini E. Assessing insulin secretion by modeling in multiple-meal tests: role of potentiation. Diabetes. 2002;51 Suppl 1:S221–6. [DOI] [PubMed] [Google Scholar]
- 20.Stumvoll M, Mitrakou A, Pimenta W, Jenssen T, Yki-Jarvinen H, Van Haeften T, et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care. 2000;23(3):295–301. [DOI] [PubMed] [Google Scholar]
- 21.Bundy B, Krischer JP. A quantitative measure of treatment response in recent-onset type 1 diabetes. Endocrinology, Diabetes and Metabolism. 2020;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zitvogel L, Rusakiewicz S, Routy B, Ayyoub M, Kroemer G. Immunological off-target effects of imatinib. Nat Rev Clin Oncol. 2016;13(7):431–46. [DOI] [PubMed] [Google Scholar]
- 23.Duggan BM, Cavallari JF, Foley KP, Barra NG, Schertzer JD. RIPK2 Dictates Insulin Responses to Tyrosine Kinase Inhibitors in Obese Male Mice. Endocrinology. 2020;161(8). [DOI] [PubMed] [Google Scholar]
- 24.Ferrannini E, Mari A, Nofrate V, Sosenko JM, Skyler JS, Group DPTS. Progression to diabetes in relatives of type 1 diabetic patients: mechanisms and mode of onset. Diabetes. 2010;59(3):679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snorgaard O, Hartling SG, Binder C. Proinsulin and C-peptide at onset and during 12 months cyclosporin treatment of type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1990;33(1):36–42. [DOI] [PubMed] [Google Scholar]
- 26.Sims EK, Evans-Molina C, Tersey SA, Eizirik DL, Mirmira RG. Biomarkers of islet beta cell stress and death in type 1 diabetes. Diabetologia. 2018;61(11):2259–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao C, Sifuentes A, Holland WL. Regulation of glucose and lipid homeostasis by adiponectin: effects on hepatocytes, pancreatic beta cells and adipocytes. Best Pract Res Clin Endocrinol Metab. 2014;28(1):43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi SS, Kim ES, Jung JE, Marciano DP, Jo A, Koo JY, et al. PPARgamma Antagonist Gleevec Improves Insulin Sensitivity and Promotes the Browning of White Adipose Tissue. Diabetes. 2016;65(4):829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fitter S, Vandyke K, Schultz CG, White D, Hughes TP, Zannettino AC. Plasma adiponectin levels are markedly elevated in imatinib-treated chronic myeloid leukemia (CML) patients: a mechanism for improved insulin sensitivity in type 2 diabetic CML patients? J Clin Endocrinol Metab. 2010;95(8):3763–7. [DOI] [PubMed] [Google Scholar]
- 30.Deininger MW, O’Brien SG, Ford JM, Druker BJ. Practical management of patients with chronic myeloid leukemia receiving imatinib. J Clin Oncol. 2003;21(8):1637–47. [DOI] [PubMed] [Google Scholar]
- 31.Jacobsen LM, Bundy BN, Greco MN, Schatz DA, Atkinson MA, Brusko TM, et al. Comparing Beta Cell Preservation Across Clinical Trials in Recent-Onset Type 1 Diabetes. Diabetes Technol Ther. 2020;22(12):948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenbaum CJ, Beam CA, Boulware D, Gitelman SE, Gottlieb PA, Herold KC, et al. Fall in C-peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes. 2012;61(8):2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogun MM, Bundy BN, Goland RS, Greenbaum CJ. C-Peptide Levels in Subjects Followed Longitudinally Before and After Type 1 Diabetes Diagnosis in TrialNet. Diabetes Care. 2020;43(8):1836–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Time-course of plasma glucose (A) and C-peptide concentrations (B) during the MMTT at screening and week 26 in the imatinib and placebo groups. Plots are mean with 95% CI.
Supplementary Figure 2: Circulating unmethylated and methylated preproinsulin (INS) DNA, and ratio of unmethylated to total INS DNA, in imatinib and placebo study groups over time. Plots represent median and inter-quartile range. No statistically significant differences were noted between groups.
Supplementary Figure 3: Autoantibody titers over time in imatinib versus placebo group, for a) mIAA , b) IA2, c) GAD, and d) ZnT8 autoantibodies. No transformation was found to normalize any of the four autoantibody titers, and therefore the confidence intervals are distorted.
Supplementary Figure 4: Immune cell subsets in imatinib versus placebo groups over time, as assessed by flow cytometry. A) B cells, B) dendritic cells, C) monocyte, D) CD4+ Tregs, E) CD4+ non-Tregs, and F) CD8+ T cell populations over time in the drug treated and placebo groups. No statistically significant differences were noted from baseline.
Supplementary Figure 5: Highly metabolically active rare cell subsets in imatinib versus placebo groups over time: a) cDC1 of dendritic cells (CDC 141+ of CD14−, CD16-HLA-DR+), and b) CD95 of IgD-CD27+ switched memory B cells. Differences between the imatinib and placebo groups were noted at the end of the 6-mo treatment period (p=0·0059 and 0·0034, respectively, corrected for baseline), and then resolved off of therapy by 12 months.
Supplementary Figure 6: Potentially protective immune cell subsets in the upper versus lower quartile of C-peptide AUC responders in the imatinib treated group over time, shown in purple and orange respectively, for A) HLA DR on DCs, B) partially exhausted CD8+ T cells, and C) CD4+Tregs. Although differences are noted between the upper and lower quartile at 6 and 12 months (5A, 5B), there is no significant change longitudinally from baseline to 6 or 12 months for either of these groups; no differences were noted at baseline or over time in FOXP3+ CD4 Treg (5C).
Supplementary Table 1: Flow cytometry antibody panels.
Supplementary Table 2: Efficacy Table of primary and secondary endpoints of group mean and mean difference estimates (including 90% or 95% confidence intervals)
Supplementary Table 3: Change in metabolic parameters over time in drug-treated versus placebo groups, median (interquartile range) displayed.
Supplementary Table 4: Serious adverse events by category and treatment assignment, with number of total events in a given category, number (percentage) of subjects in each treatment group, and possible attribution to study drug.