Abstract
In the last decade immunotherapies such as immune checkpoint blockade (ICB) against the PD-1/PD-L1 system have revolutionised the treatment of numerous entities. To date, ovarian cancer has benefited very little from this success story. Possible causes include a rather low mutational burden compared to other tumour types, inadequate presentation of (neo-)antigens, and increased infiltration with immunosuppressive immune cells such as regulatory T cells and tumour-associated macrophages. In the clinical trials completed to date, the response rates to PD-1/PD-L1 checkpoint inhibitors have therefore been disappointingly low as well, although isolated long-term remissions have also been observed in ovarian cancer. The task now is to find suitable predictive biomarkers as well as to identify combination partners for ICB therapy that can increase the immunogenicity of ovarian cancer or overcome immunosuppressive resistance mechanisms. This paper provides an overview of the immune milieu in ovarian cancer, its impact on the effect of ICB, and summarises the clinical trial data available to date on ICB in ovarian cancer.
Key words: ovarian cancer, immune checkpoint blockade, PD-L1, tumour-infiltrating lymphocytes
Introduction
Although concepts on the interaction of tumours with the immune system were developed as early as the 19th century 1 , for much of the 20th century cancer research focused solely on the tumour cell itself. The success of this era was evident in chemotherapy, developed since the 1940s, and targeted therapy, developed since the 1990s. The surroundings of the tumour cell, the tumour microenvironment, and thus also the immune composition of a tumour, became the focus of research rather late 2 . Over the last forty years, however, the functional understanding of the interaction of the immune system with the tumour has expanded considerably, notably culminating in the breakthrough of immuno-oncology with the success of inhibitors against the immune checkpoint proteins CTLA-4 and PD-1/PD-L1 in malignant melanoma and other immunogenic tumours. In the long term, immuno-oncology promises to come closest to the ideal of personalised therapy. Interestingly enough, a substantial part of the effect of traditional chemotherapeutics and targeted substances also seems to arise from a stimulation of the tumour-suppressive immune response 3 . At present the landscape of oncological studies is dominated by immuno-oncological studies, especially those on immune checkpoint inhibition.
However, ovarian cancer has seen little of the success of immuno-oncology. Immunological treatment approaches such as the administration of inflammatory cytokines, adoptive T cell transfer, and vaccinations against common tumour antigens have only been partially successful and have not yet made it into routine clinical practice 4 . Most recently, the IMagyn050 trial, which was reported negative and tested the administration of the anti-PD-L1 antibody atezolizumab in addition to standard first-line therapy in advanced ovarian cancer, demonstrated that a “crushing victory” in immune checkpoint inhibition can no longer be expected 5 . It also remains to be seen whether the current trials in recurrent ovarian cancer will confirm a better effect of ICB mono-maintenance therapy, since the tumour tends to be immunologically “burnt out” in later lines of therapy. However, since in the phase I/II trials conducted to date long-term remissions have indeed been observed in some patients, there is still reason for optimism in ovarian cancer 7 , 8 .
Preclinically and clinically, therefore, three avenues are currently being explored: first, the mechanisms of immune escape in ovarian cancer need to be further elucidated, with an understanding of the immune milieu in ovarian cancer being absolutely essential. Second, this may also help in developing appropriate predictive biomarkers that can identify the (still small) population of patients actually benefiting from immune checkpoint inhibition. And third, ongoing trials currently recruiting are testing which combination of partners can be used to induce a substantial immune response against ovarian cancer, which may then be enhanced by ICB (e.g., PARP inhibitors).
This review therefore aims to provide an overview of the current understanding of the interaction of the immune system with epithelial ovarian cancer, which in turn is a prerequisite for understanding the effect, as well as the failure, of immunotherapies. The second part then provides an overview of the clinical trial results of immune checkpoint blockade to date.
The Immunogenicity of Ovarian Cancer: Mutational Burden, Tumour Antigens and Immune Infiltration
The ability of the immune system to fight tumour growth, and thus the success of immune checkpoint therapies, depends on several factors: intrinsic properties of the tumour itself such as mutational burden and (neo-)antigen presentation, immune cellular factors such as the extent and composition of the immune infiltrate within the tumour, and immune modulating qualities of the tumour microenvironment, e.g. the expression of immune checkpoint proteins. The time related dynamics of these factors in the course of the malignant disease further add to the complexity. The traditional concept is based on three succinct phases in which the immune system can initially fight the tumour effectively (elimination), but then weakly immunogenic tumour cell clones assert themselves under the selection pressure exerted in this way (equilibrium), and the tumour can finally escape completely from the control of the immune system (escape) 9 . These processes are also significantly affected by the systemic therapies.
In terms of the tumour mutational burden (TMB), ovarian cancer is in the lower midfield of all entities 10 , 11 . While malignant melanoma has mutation rates of about 14 – 47 mutations/Mb, most ovarian cancers exhibit only about 1 – 3.5 mutations/Mb 10 , 12 . Although some papers report higher mutation rates (20 – 40 mutations/Mb), only 1.3% of these mutations were even detected by autologous tumour-associated T cells 13 . TMB correlates with the number of neoantigens and the response to immune checkpoint blockade 14 . In addition, however, this also requires infiltration with appropriate immune effector cells. A recent paper studying these two factors (TMB, T cell signature) in numerous types of cancer revealed that (serous) ovarian cancer ranked almost at the bottom of the malignancies studied 15 . Interestingly enough, serous ovarian carcinomas with high T cell infiltration demonstrate a significantly lower clonal diversity and a lower neoantigen load, indicating that effective immuno-editing also takes place in serous ovarian carcinoma, i.e. effectively combating the majority of tumour cells, but also resulting in the selection of fewer, non-immunogenic tumour cell clones 16 . As TCGA analysis has shown, other mutations apart from p53 mutations are not very common in general 17 .
On the other hand, the low TMB is accompanied by a rather frequent expression of tumour antigens, especially cancer testis antigens (CTAs), against which a specific T cell response is often detected in vitro in ovarian cancer 18 . Nevertheless, antibody therapies against such antigens have failed in vivo, including against CA 125, which appears to be one of the most strongly presented immunogenic antigens in ovarian cancer 19 , 20 , 21 .
Another decisive factor determining tumour immunogenicity is immune infiltration. Tumour-infiltrating lymphocytes (TILs) as a prognostic marker in ovarian cancer were identified more than 30 years ago 22 , 23 . Further evidence of the immunogenicity of ovarian cancer comes from work developing molecular classifications of serous cancer based on genomic studies. Each of these trials was able to define an “immunoreactive subtype” based on the increased expression of genes, for example associated with IFN signalling, which demonstrated the best prognosis in all trials as well as in a meta-analysis 17 , 24 , 25 , 26 .
All in all, the immunogenicity of ovarian cancer therefore seems to be quite weak.
The Cellular Immune Milieu in Ovarian Cancer
Immune cells can both promote and inhibit tumour growth in ovarian cancer ( Fig. 1 ). The most important and best-studied immune cell populations in ovarian cancer are briefly presented below in terms of their effect on immune checkpoint therapy.
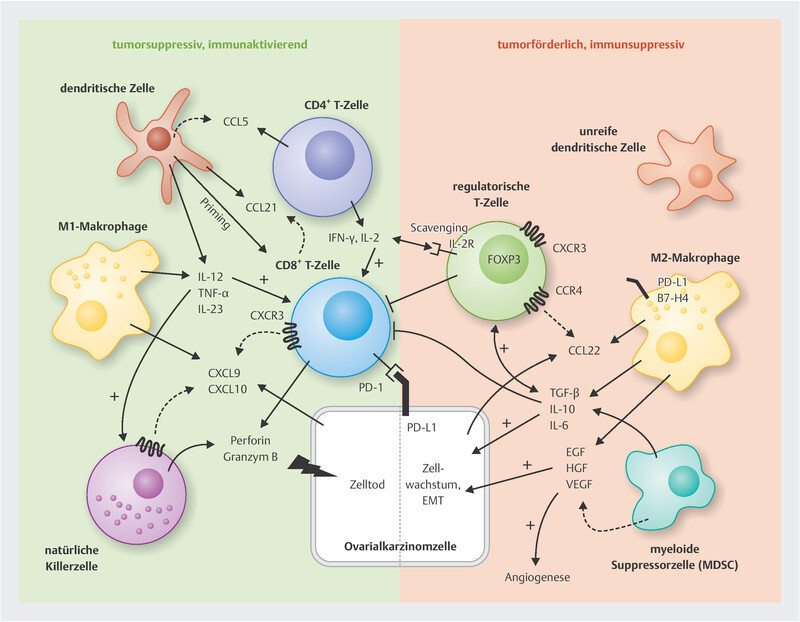
Fig. 1.
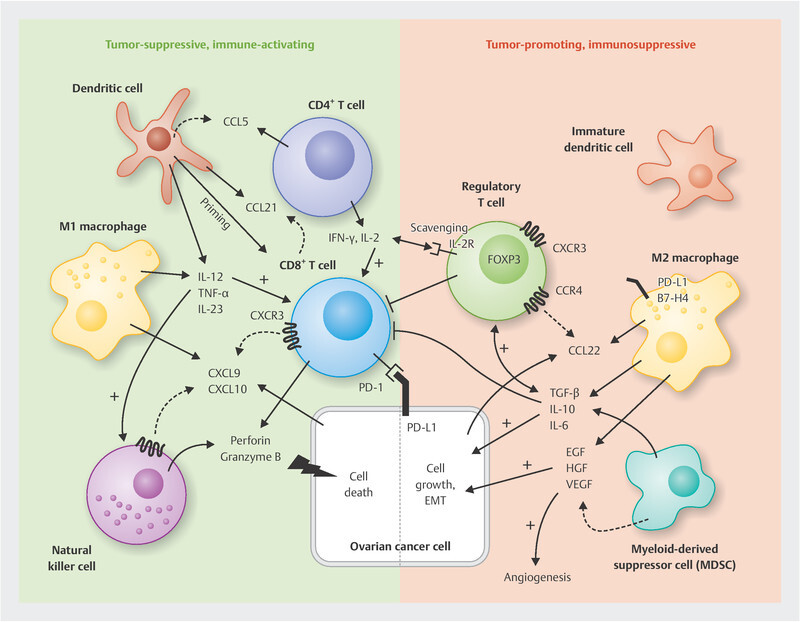
Cellular components of the immune milieu in ovarian cancer and selected important interaction mechanisms. CD8-positive cytotoxic T cells and natural killer cells are crucial mediators of the anti-tumour response in ovarian cancer. They are supported in different ways by dendritic cells, CD4-positive T helper cells and M1 macrophages. M2 macrophages and MDSCs, on the other hand, lead to immunosuppression and activation of regulatory T cells, e.g. through the release of IL-10 and TGF-β, which in turn can inhibit the function of other tumour-suppressive T cells. Details and other mechanisms in the main body of the text.
T cells (effector cells)
Collectively, T cells form the cornerstone of adaptive anti-tumour response and undoubtedly represent the most studied immune cell population in ovarian cancer. Ultimately, this includes various cell groups that can have both tumour-suppressive (T helper cells, cytotoxic T cells) and tumour-promoting (regulatory T cells, see below) qualities. They all have in common the surface marker CD3, which defines this cell type and which has been identified early on as a strong prognostic marker in ovarian cancer 23 . Since the aggregate of all CD3-positive T cells also contains FOXP3-positive regulatory T cells, subsequent studies established CD8, a characteristic receptor of cytotoxic T cells, as an even more robust prognostic marker 27 , 28 , 29 , 30 , 31 . The CD8/FOXP3 ratio, which takes into account the contrasting functions of cytotoxic T cells and regulatory T cells, is just as indicative. While most ovarian cancers contain immune cells in the tumour stroma, intratumoural T cells are found in only about half of all cases; they, in turn, have a stronger prognostic significance and therefore seem to be functionally more important 28 . CD4-positive T helper cells also correlate with better survival, especially the CD4 + CD25 + FOXP3 − cells, as CD25 is important for T cell expansion and activation 32 , 33 , 34 . Additional roles of CD4-positive T cells include CCL5-mediated recruitment of dendritic cells, which in turn prime CD8-positive cytotoxic T cells 35 .
Against the background of these observations, adequate infiltration of the tumour with T cells is not only functionally and prognostically significant, but also represents a basic prerequisite for the success of immunotherapies such as immune checkpoint blockade 36 , 37 . This recruitment of T cells is mediated by chemokines binding the corresponding chemokine receptors on the surface of the immune cells and chemotactically attracting them to the tumour. Such a function has been demonstrated in ovarian cancer especially for the CXCR3 ligands CXCL9 and CXCL10 38 , 39 , but also for CCL21/22 23 as well as CCL2, CCL4 and CCL5 40 .
The CXCR3 chemokine system plays a special role in solid tumours 41 , 42 . We demonstrated for the first time in humans that overexpression of the CXCR3 chemokines CXCL9 and CXCL10 is associated with increased infiltration of CD3-positive T cells and significantly improved survival in high-grade serous ovarian cancer (HGSOC) 38 . While we found the strongest expression in the tumour cells themselves, other groups localise these chemokines more in the macrophages and dendritic cells 39 . Our observations at the protein level were later confirmed by numerous other groups at the gene level as well: in particular, in the immunoreactive subtype of HGSOC, which has the best prognosis of all molecular subtypes, CXCR3 chemokines were the most upregulated genes 25 , 26 , 39 . CXCL9 was also the most upregulated gene in a comparison of regressed versus progressive metastases in a patient with high-grade serous ovarian cancer 43 . In a recent paper by the OTTA consortium, which identified a prognostic signature from 513 genes in HGSOC, once again CXCL9 was one of the five genes with the highest prognostic significance 44 . This work suggests a central role for the CXCR3 chemokine system in establishing or maintaining effective tumour-suppressive immune response. CXCR3 chemokines are also necessary for the success of PD-1/PD-L1 checkpoint inhibition and are involved in the immunomodulating activity of PARP inhibitors in ovarian cancer 39 , 45 , 46 . They can be induced not only by PARP inhibitors but also by cyclooxygenase inhibitors, which has led to the use of COX inhibitors as adjuvants in immune checkpoint inhibitor studies 32 , 38 , 47 .
Regulatory T cells (T regs )
Physiologically, regulatory T cells inhibit an exuberant immune response by suppressing the activities of T effector cells, B cells, macrophages, and NK cells 48 . Tumours can thus exploit T regs for immune escape. T regs are possibly commonly found in tumours because they suppress immune responses triggered primarily by self-antigens, and this correlates with the expression of tumour-associated antigens in the tumour 49 .
Immune response inhibition is achieved by the following mechanisms 48 , 49 : Secretion of immunosuppressive cytokines (IL-10, IL-35 and TGF-β); lysis of target cells by granzymes and perforins; interception of free IL-2 via the α-chain of the IL-2 receptor 50 so that it cannot activate CD8+ or NK cells; enzymatic cleavage of extracellular (proinflammatory) ATP into AMP and adenosine (via CD39 and CD73), which in turn suppresses the response of T, B and dendritic cells and macrophages via the A2A adenosine receptor; down-regulation of CD80 and CD86 on dendritic cells by binding to CTLA-4 on T regs .
The expression of the FOXP3 transcription factor characterises T regs ; on the one hand it is important for the differentiation towards T regs , on the other hand it is necessary for their suppressive function 51 , 52 , 53 . Its expression may therefore also be used to determine T reg infiltration in tumours 54 . Ovarian cancers appear to have a rather high number of T regs 55 , which may be one reason for the poor response to immune checkpoint therapies 54 , 56 . Accordingly, in most trials, increased FOXP3 + immune infiltration also correlates with a higher tumour stage and poorer prognosis 32 , 56 , 57 , 58 , 59 , 60 , 61 .
Different chemokine systems are thought to be responsible for the chemotactic recruitment of T regs into ovarian cancer. CCL28 is induced by hypoxia in preclinical ovarian cancer models, attracts CCR10-positive T regs and is associated with poor survival 62 . CCL22, which can be secreted by macrophages, recruits T regs via the CCR4 receptor 59 , 60 , 63 . However, T regs can also enter ovarian cancer via the CXCR3 receptor, which actually directs T effector cells 57 . CXCR3-positive T regs even seem to constitute the major part in ovarian cancer, correlate with the number of CXCR3-positive T effector cells, and inhibit them 64 . We were also able to show that in high-grade serous ovarian cancer the expression of CXCL9 and CXCL10 correlates not only with CD3-positive T cells overall, but also with FOXP3-positive regulatory T cells 38 . However, the strong protective effect of CXCR3 chemokines repeatedly noted in ovarian cancer shows that the net effect favours an enhanced, tumour-suppressive immune response. This is also confirmed by functional studies in syngeneic ovarian cancer mouse models 65 .
Basically, there are two approaches to prevent T reg -mediated immune escape, and thus possibly also to improve the effect of immune checkpoint inhibition: depletion of regulatory T cells or their reprogramming to IFN-γ producing, so-called Th1 T regs 49 , 66 , 67 , 68 . One possibility for selective T reg depletion is the administration of low-dose cyclophosphamide (< 250 mg/m 2 ) 69 , 70 , 71 . However, depletion does not appear to be very promising since the T regs quickly return and other T effector populations are usually suppressed as well 49 . However, reprogramming into an IFN-γ producing Th1 phenotype may succeed through CTLA-4 blockade, which could be a suitable combination partner for PD-1/PD-L1 inhibition in ovarian cancer 72 .
Tumour-associated macrophages (TAMs)
Macrophages are a heterogeneous group of myeloid cells and represent major type of immune cells in the ovarian cancer microenvironment, and may even exceed the number of tumor cells 73 . Tumour-associated macrophages (TAMs) either arise from immigration of blood-derived bone marrow monocytes or are derived from tissue-resident macrophages 74 . In analogy to the Th1/Th2 phenotypes of the T cell response, the tumour-suppressive M1 differentiation status is fundamentally distinguished from the tumour-promoting M2 activation status 75 . M1 macrophages may be activated by classical IFN-γ and produce proinflammatory cytokines such as IL-12, TNF-α and IL-23 and tumour-suppressive chemokines such as CXCL9 and CXCL10 39 , 76 . Redifferentiation into an M1 phenotype also appears to be part of the effect of chemotherapies, e.g., trabectidine or paclitaxel 77 , 78 . However, the bulk of TAMs are present in an M2 activation state, both in tumour samples from ovarian cancer patients and in preclinical models. This may be because the tumour cells themselves are able to polarise TAMs into an M2 phenotype 79 . CD163, CD204, CD206, and IL-10 are markers of an M2 phenotype 73 . Accordingly, increased levels of CD163 (or CD163-positive macrophages) in the ascites, blood and also in the tumour correlate with a higher tumour stage and a poorer prognosis 80 , 81 , 82 . On the other hand, several trials have found a correlation between an increased M1/M2 ratio and improved survival in ovarian cancer 82 .
M2 macrophages result in immunosuppression and increased tumour growth via various mechanisms. They secrete different growth factors such as VEGF, EGF, TGF-β, and HGF, thereby promoting angiogenesis, EMT, spheroid formation, and transcoelomic metastasis 83 , 84 , 85 . Secretion of these factors correlates with recurrence frequency, metastasis, chemoresistance, and poor survival in ovarian cancer 84 , 85 . Effective anti-tumour T cell response may also be inhibited in different ways by M2-TAMs: they induce the maturation of regulatory T cells by TGF-β and promote their recruitment into the tumour by CCL22 56 . T regs , on the other hand, promote the secretion of IL-6 and IL-10 by TAMs, leading to autocrine upregulation of the immune checkpoint B7-H4 in macrophages 60 . B7-H4 can block T cell function 60 , 86 , 87 . The expression of PD-L1 by macrophages can also contribute to immune escape.
Given this mode of action of M2-TAMs and their abundance in ovarian cancer, it is quite conceivable that they are an important resistance factor to PD-1/PD-L1-targeted checkpoint blockade 88 . Conversely, M1 macrophages can contribute to the success of anti-PD-1/PD-L1 therapy via the secretion of CXCL9 89 . Macrophage-depleting therapies and those allowing polarisation into an M1 phenotype could therefore be interesting combination partners in ICB 88 , 90 . Options for TAM suppression that already work in the preclinical setting and the resulting improvement in ICB include blockade of the colony-stimulating factor 1 receptor (CSF1R) and CCL2 inhibition 91 , 92 , 93 .
Other types of immune cells
Although other immune cells such as natural killer cells, dendritic cells, and myeloid-derived suppressor cells (MDSCs) certainly play an important role in ovarian cancer, their impact on the success or failure of immune checkpoint blockade is less obvious. Their role in ovarian cancer has recently been summarised elsewhere 94 . Preclinical studies also on ovarian cancer suggests that these cell types, rather than the tumour cells, may be crucial for PD-1/PD-L1-mediated T cell inactivation – a process that may not take place in the tumour at all, but in the draining lymph nodes 95 , 96 .
The PD-1/PD-L1 Immune Checkpoint in Ovarian Cancer
The expression of the immune checkpoint protein PD-L1 in the microenvironment of ovarian cancer provides the rationale for the use of appropriate inhibitory antibodies. In immunocompetent ovarian cancer mouse models, tumour growth was inhibited by inhibitors against PD-1 or PD-L1 97 , 98 .
PD-L1 expression was initially studied mostly in tumour cells. In particular, while previous reports showed a significant correlation of PD-L1 tumour cell expression with poor survival 28 , 97 , 99 , other groups demonstrated just the opposite, correlating PD-L1 with improved immune infiltration and prolonged survival 100 , 101 , 102 , 103 , 104 . This may be explained by a general increase in IFN-γ response in these tumours as IFN-γ on the one hand delivers an effective anti-tumour response, on the other hand it is a very potent inducer of PD-L1 expression in ovarian cancer cells 97 , 100 . This is in line with the observation that PD-L1 expression is particularly strong in the immunoreactive subtype of serous ovarian cancer, whose most highly expressed genes are induced primarily by IFN-γ 26 , 105 . The heterogeneity of the study populations, different staining techniques and methods of analysis may help explain these discrepant reports on the prognostic significance of PD-L1 in ovarian cancer.
There are also different claims regarding the main location of PD-L1 in the TME: while PD-L1 expression has traditionally been studied in tumour cells 28 , TAMs also appear to be a major site of PD-L1 location 102 . Functionally, PD-L1 on these myeloid cells may be significantly more important than tumour cell PD-L1 in suppressing T cell response. This is suggested by preclinical studies, also in ovarian cancer models 95 , 96 .
PD-L1-positive tumours are most commonly found among serous carcinomas, significantly more so than in clear cell, mucinous and endometrioid cancers 102 , 104 . This is somewhat surprising, as clear cell cancers exhibit the best response to PD-1/PD-L1 checkpoint inhibition in current clinical trials, albeit with small case numbers (see below), and illustrates once again that PD-L1 expression per se is not a good predictive marker. BRCA1/2 mutation status did not result in a difference in PD-L1 expression 104 .
Effect of Cytotoxic Systemic Therapy, Surgery and BRCA Mutation Status on the Immune Milieu in Ovarian Cancer
Effect of chemotherapy
Even though the chemotherapeutic agents used in ovarian cancer were originally used purely for their direct cytotoxic properties, their effect also seems to be based, at least in part, on immunomodulatory properties 3 . Thus, the changes in the immune milieu occurring under chemotherapy offer additional opportunities for the correct timing of an immune checkpoint blockade.
Taxanes, for example, suppress regulatory T cells and MDSCs, enhance DC-mediated antigen presentation by upregulating MHC I, and activate NK and T cells as well as DCs through IL-12 and TNF-α secreted by macrophages 106 , 107 , 108 . Platinum salts can generate neoantigens through DNA damage and trigger an IFN type 1 response through activation of the STING signalling pathway, which is also associated with enhanced T cell infiltration, but on the other hand with the induction of PD-L1 109 . The suppression of regulatory T cells by low-dose cyclophosphamide has already been discussed (see above).
These preclinical findings have been confirmed in patients. The most reliable data in ovarian cancer come from studies of sequential tumour samples under neoadjuvant chemotherapy 110 . Neoadjuvant chemotherapy increases the number of CD4- and CD8-positive tumour-infiltrating T cells 111 , 112 , 113 , 114 , 115 , as well as B cells 111 and dendritic cells 114 . FOXP3-positive regulatory T cells, on the other hand, are downregulated 114 , which was a positive prognostic factor in two of the trials 112 , 113 . In addition, chemotherapy in ovarian cancer results in the induction of PD-1/PD-L1 in both tumour and immune cells 111 , 113 , 116 . This upregulation of checkpoint systems may also be the reason why TILs lose their prognostic value after neoadjuvant chemotherapy 110 .
The effect of surgery
The influence of surgery on the immune milieu in ovarian cancer is less well studied than that of chemotherapy. However, it has been shown that primary debulking leads to a reduction in regulatory T cells, a reduction in IL-10 and an improved function of CD8-positive T cells 117 . In addition, there is evidence that the prognostic benefit of chemokines or TIL subsets is particularly strong in patients without residual tumour after surgery 38 , 118 . Further studies are certainly necessary, especially with regard to lymphadenectomy and its effect on immunotherapies, since preclinical work, also in ovarian cancer, localise the functionally significant PD-1/PD-L1 interaction not in the tumour itself but rather in the lymph nodes 95 .
The effect of homologous recombination defects (e.g., BRCA mutations)
Patients with BRCA -mutated ovarian cancer have a significantly better prognosis than other patients, a fact mainly attributed to a better response to platinum-based chemotherapy. However, this could also have immunological reasons because BRCA -mutated tumours have a higher number of predicted neoantigens, more CD3- and CD8-positive lymphocytes, and stronger PD-L1 expression 27 , 119 . The reason for this could be the homologous recombination defect caused by the BRCA mutation, which ultimately results in STING activation and thus induction of an interferon response 120 .
Role of Immune Checkpoint Inhibition in the Treatment of Epithelial Ovarian Cancer
No immunotherapy in the strict sense has yet become clinical routine in ovarian cancer. Clinical testing of immune checkpoint inhibitors against the PD-1/PD-L1 system is the most advanced, with the first phase III trials now being completed. These therapies bring along completely novel challenges for the gynaecological oncologist in terms of adverse event management 121 . The most important study outcomes will be discussed below.
Monotherapy with immune checkpoint inhibitors
Due to the good outcomes of immune checkpoint inhibition in other cancers such as malignant melanoma and non-small cell lung cancer, these substances were initially tested as monotherapies in ovarian cancer as well. The rationale was the known expression of checkpoint proteins and their negative prognostic significance in ovarian cancer 28 , the strong prognostic impact of tumour-infiltrating T cells 23 , 122 , a TMB ranking at least in the middle of all entities 11 as well as numerous preclinical studies in animal models (see above). All this suggests an intrinsic immune response against ovarian cancer that can be unleashed by immune checkpoint inhibitors.
Table 1 summarises the important trials. The first trial tested the anti-PD-1 antibody nivolumab in 20 patients with histologically different ovarian cancers at two different doses (1 and 3 mg/kg) 123 . Despite a disappointing overall response rate (ORR) of 15%, four patients demonstrated a long-lasting response (> 12 months), two of whom achieved complete remission, including one patient with clear cell ovarian cancer. Interestingly enough, parts of one of the two carcinomas responding in the atezolizumab trial also featured clear cell histology 124 . Both patients in complete remission were dosed with 3 mg/kg. PD-L1 expression was not predictive of response, although the case numbers here were certainly too low.
Table 1 Comparison of the major clinical trials testing an immune checkpoint inhibitor as monotherapy in ovarian cancer. These were solely single-arm trials.
| Immune checkpoint inhibitor | Nivolumab | Pembrolizumab | Pembrolizumab | Avelumab | Atezolizumab |
|---|---|---|---|---|---|
| Abbreviations: CPS = combined positive score, CR = complete remission, DCR = disease control rate, ICB = immune checkpoint blockade, IHC = immunohistochemistry, IC = immune cells, CI = confidence interval, ORR = overall response rate, PD-1 = programmed cell death protein 1, PD-L1 = programmed cell death ligand 1, PFI = platinum-free interval, PFS = progression-free survival, Pt = platinum, TC = tumour cells | |||||
| Targeted structure | PD-1 | PD-1 | PD-1 | PD-L1 | PD-L1 |
| Trial | UMIN00000571 | NCT02054806 (KEYNOTE-028) | NCT02674061 (KEYNOTE-100) | NCT01772004 (JAVELIN Solid Tumour) | NCT01375842 |
| Dosing | 1 mg/kg q14 (n = 10) or 3 mg/kg q14 (n = 10) |
10 mg/kg q14 | 200 mg q21 | 10 mg/kg q14 | Dose ranging trial 9/12 patients: 15 mg/kg |
| Phase | Phase II | Phase Ib | Phase II | Phase Ib | Phase Ib |
| Design | Single-centre, open | Multicentre, open | Multicentre, open | Multicentre, open | Multicentre, open |
| No. of patients | 20 | 26 | 376 (A: 285, B: 91) | 125 | 12 |
| Important inclusion criteria | Pt-resistant (≤ 6 months) | PD-L1 positive | Cohort A: 3 – 12 months PFI since last Pt Cohort B: ≥ 3 months PFI since last Pt |
75% Pt-resistant (≤ 3 months) | |
| Histology | 75% serous 15% endometrioid 10% clear cell |
46% “adenocarcinoma” 46% high-grade serous 4% endometrioid 4% transitional cell carcinoma |
75% high-grade serous 7% endometrioid 6% low-grade serous 5% clear cell 7% unspecified |
74.4% serous 3.2% mucinous 2.4% endometrioid 1.6% clear cell 18.4% unspecified |
80% serous 10% endometrioid 10% mixed endometrioid/clear cell |
| Prior therapies | ≥ 2 lines (median 4) | 73% ≥ 3 lines (median 4) | Cohort A: 1 – 3 lines Cohort B: 4 – 6 lines |
65% ≥ 3 lines (median 3) | ≥ 2 lines (> 90%) |
| PD-L1 testing | TC-IHC (four-level) on primary surgical specimen | CPS ≥ 1% | CPS | TC-IHC a. o. | IC-IHC (≥ 5% of tumour) |
| ORR | 15% (3/20) | 12% (3/26) |
8.5% (32/376)
(A: 8.1%, B: 9.9%) |
9.6% (12/125) | 22% (2/9) |
| CR | 10% (2/20) | 4% (1/26) | 1.9% (7/376) | 0.8% (1/125) | 11% (1/9) |
| ORR (PD-L1 positive) | 12.5% (2/16) | 12% (3/26) | 13.8% (CPS ≥ 10) 8.0% (CPS ≥ 1) 5.0% (CPS < 1) |
No correlation with PD-L1 status | 25% (2/8) |
| ORR (PD-L1 negative) | 25% (1/4) | – | 0% (0/1) | ||
| DCR | 45% | 19% (5/26) | 37.2% (140/376) | 52% (65/125) | 22% (2/9) |
| DOR | 3.5 months | not met in the 3 responders (> 20.5 months) | 10.2 months A: 8.3 months B: 23.6 months |
10.4 months | not reported for entire cohort |
| PFS | 3.5 months (95% CI 1.7 – 3.9 months) | 1.9 months (95% CI 1.8 – 3.5 months) | 2.1 months (A and B) | 2.6 months (95% CI 1.4 – 2.8 months) | 2.9 months (95% CI 1.3 – 5.5 months) |
| References | Hamanishi et al. 123 | Varga et al. 168 | Matulonis et al. 125 , 169 | Disis et al. 127 | Liu et al. 124 |
The KEYNOTE-100 trial, with 376 patients the largest trial reported to date, tested the anti-PD-1 antibody pembrolizumab in a population with, in some cases heavily pretreated, mostly high-grade recurrent serous ovarian cancer 125 , 126 . Again, the overall response rate was at disappointing 8.5%, but with some long response intervals. In the subgroup analyses, the response did not correlate with platinum sensitivity or number of prior therapies. However, there was a dependence on PD-L1 expression as determined by the CPS score ( Table 1 ). Patients with a tumour CPS score of ≥ 10 were found to have a significantly better response (13.8%) than those with a score below 1 (5.0%). However, since the CPS thresholds (≥ 1, ≥ 10) were optimized based on a training collective (n = 100), these response rates may have overestimated the actual treatment effect 125 .
Interestingly enough, the clear cell subtype had the best numerical response in the KEYNOTE-100 trial as well (ORR 16%, n = 19). High-grade serous carcinomas responded in 8.5% (n = 283) of cases, and no response was observed in low-grade serous (n = 21) and endometrioid carcinomas (n = 28) 125 . Both patients with clear cell carcinoma in the JAVELIN solid tumour trial also exhibited a response to therapy 127 . Besides the genetic proximity to clear cell renal cell carcinomas 128 , 129 , one possible explanation is increased infiltration of these tumours with CD3-, CD8- and PD-1-positive immune cells as well as increased PD-L1 expression in the tumour cells, especially in tumours with microsatellite instability (MSI) 130 . Thus, MSI testing could be a possible criterion for stratification in clear cell ovarian cancer. The significance of immune checkpoint inhibition in clear cell ovarian cancer is currently being studied in the prospective PEACOCC trial (NCT03425565).
In summary, the monotherapy trials with inhibitors of the PD-1/PD-L1 immune checkpoint in ovarian cancer have shown a comparable response of around 10 – 15% in patients with mostly considerable prior treatment. However, the treatment effects observed in these patients, some of which were long-lasting, are encouraging. Another insight from these trials is the inadequate prediction of treatment success based solely on PD-L1 expression.
Combined treatment with chemotherapy and immune checkpoint inhibitors
The only combined treatments with immune checkpoint inhibitors in ovarian cancer studied at phase III level to date have been in combination with or as maintenance following chemotherapy ( Table 2 ). Against the backdrop of the immunomodulatory effect of many chemotherapeutics, this also makes sense, especially in first-line therapy as part of an “all-in” concept 3 . Clinical studies also confirm in ovarian cancer that platinum-based chemotherapy results in activation of an IFN response with increased immune infiltration of the tumour by CD4- and CD8-positive T cells, a reduction in the number of FOXP3-positive regulatory T cells, and an upregulation of PD-L1 114 , 131 , 132 . Since these effects may be greatest especially during chemotherapy, the addition of the immune checkpoint inhibitor should therefore take place early.
Table 2 Phase III trials on the combined treatment with immune checkpoint blockade and chemotherapy in advanced ovarian cancer.
| Trial | NCT02718417 (JAVELIN Ovarian 100) | NCT03038100 (IMagyn050) | NCT02580058 (JAVELIN Ovarian 200) |
|---|---|---|---|
| * did not meet predefined level of significance Abbreviations: ECOG PS = Eastern Co-operative Oncology Group Performance Status, ICB = immune checkpoint blockade, ICH = immunohistochemistry, HR = hazard ratio, OS = overall survival, PD-L1 = programmed cell death ligand 1, PLD = pegylated liposomal doxorubicin, PFS = progression-free survival | |||
| Immune checkpoint inhibitor used | Avelumab | Atezolizumab | Avelumab |
| Targeted ICB structure | PD-L1 | PD-L1 | PD-L1 |
| Arms | A) 6 × carboplatin/paclitaxel → avelumab for 24 months B) 6 × carboplatin/paclitaxel/avelumab → avelumab for 24 months C) 6 × carboplatin/paclitaxel |
A) 6 × carboplatin/paclitaxel/bevacizumab + placebo → maintenance with bevacizumab and placebo B) 6 × carboplatin/paclitaxel/bevacizumab + atezolizumab → maintenance with bevacizumab and atezolizumab |
A) Avelumab B) Avelumab + PLD C) PLD |
| Study design | Multicentre, randomised (1 : 1 : 1), open | Multicentre, randomised (1 : 1), double-blind | Multicentre, randomised (1 : 1 : 1), open |
| Primary endpoint(s) | PFS | PFS | PFS, OS |
| No. of patients (n) | 998 | 1301 | 566 |
| Population | First-line therapy FIGO III – IV, ECOG PS 0 – 1, after debulking or as neoadjuvant therapy (31.6% surgery without macroscopic residual tumour) |
First-line therapy FIGO III – IV, ECOG PS 0 – 2, after debulking (75%) or as neoadjuvant therapy/interval debulking (25%) (7.4% surgery without macroscopic residual tumour) |
Platinum-resistant/refractory recurrence ≤ 3 prior therapies no prior therapy in Pt-resistant recurrence |
| Histology | 76% high-grade serous 6.2% low-grade serous 5.5% clear cell 3.2% endometrioid 8.7% other |
76% high-grade serous 10% low-grade serous 12% high-grade non-serous 4% clear cell |
69% high-grade serous 4% low-grade serous 13% clear cell 3% endometrioid 10% other |
| Definition “PD-L1 positive” | ≥ 1% tumour cells positive and/or ≥ 5% immune cells positive (Ventana SP263 IHC assay) |
≥ 1% immune cells positive (Ventana SP142 IHC assay) |
≥ 1% tumour cells positive and/or ≥ 5% immune cells positive (Ventana SP263 IHC assay) |
| PD-L1 status | 48.8% PD-L1 positive 32.6% PD PD-L1 negative |
40% PD-L1 positive 60% PD PD-L1 negative |
57% PD-L1 positive |
| Median PFS | A) 16.8 months (HR 1.43 vs. C, p = 0.989) B) 18.1 months (HR 1.14 vs. C, p = 0.794) C) Median not reached |
A) 18.4 months B) 19.5 months (HR 0.92, p = 0.28) |
A) 1.9 months (HR 1.68 vs. C,> 0.999) B) 3.7 months (HR 0.78 vs. C, p = 0.030*) C) 3.5 months |
| Overall survival (OS) | No difference in OS to date | No difference in OS to date | A) 11.8 months (HR 1.14 vs. C, p = 0.8) B) 15.7 months (HR 0.89 vs. C, p = 0.21) C) 13.1 months |
| Median PFS (PD-L1 positive) | A) Median not met (HR 1.23 vs. C, p = 0.357) B) Median not met (HR 0.98 vs. C, p = 0.918) C) Median not met |
A) 18.5 months B) 20.8 months (HR 0.80, p = 0.038*) |
A) 1.9 months (HR 1.45 vs. C, p = 0.030) B) 3.7 months (HR 0.65 vs. C, p = 0.0149) C) 3.0 months |
| Median PFS (PD-L1 negative) | A) 16.8 months (HR 1.02 vs. C, p = 0.950) B) 13.9 months (HR 1.36 vs. C, p = 0.232) C) Median not met |
||
| References | Ledermann et al. 133 | Moore et al. 5 | Pujade-Lauraine et al. 135 |
However, this concept has not yet been confirmed in clinical trials and the addition of an immune checkpoint inhibitor has not resulted in any benefit.
The JAVELIN-Ovarian-100 trial (NCT02718417) studied the use of the anti-PD-L1 antibody avelumab either as maintenance treatment following or combined with first-line carboplatin/paclitaxel chemotherapy (n = 998). About 30% of patients had undergone surgery without residual tumour, and about 40% had received neoadjuvant chemotherapy. The trial was terminated prematurely because there was no benefit with regard to the primary endpoint PFS. Patients in the avelumab maintenance therapy arm had worse PFS than those in the control arm (HR 1.43; 95% CI 1.05 – 1.95; more deaths) 133 . Neither stratification by PD-L1 status, BRCA mutation nor CD8 expression was able to identify a subgroup benefiting from therapy 133 .
The IMagyn050 trial (NCT03038100) tested the addition of the anti-PD-L1 antibody atezolizumab to first-line carboplatin/paclitaxel/bevacizumab therapy. Atezolizumab was already initiated in parallel with chemotherapy and then continued in parallel with bevacizumab. Here, too, the primary endpoint (PFS) was not met 5 . Overall survival, albeit with very early data, also showed no improvement so far with the addition of the anti-PD-L1 antibody. As the only significant subgroup, patients whose tumours had more than 5% PD-L1-positive immune cells (about 20% of the study population) experienced a PFS benefit from atezolizumab (HR 0.64; 95% CI 0.43 – 0.96) 5 . Future biomarker trials on this population are expected.
The JAVELIN Ovarian 200 trial (NCT02580058, n = 566) examined the effect of combined treatment with avelumab and pegylated liposomal doxorubicin (PLD) in platinum-resistant/refractory recurrent ovarian cancer 135 . 48% of patients received second line of treatment, the remainder following two or three previous lines of treatment. Only in the PD-L1-positive subgroup did the combined treatment demonstrate some improvement over PLD monotherapy for both PFS and OS 135 .
Combined treatment with immune checkpoint and PARP inhibitors
The essential rationale for combined treatment with PD-1/PD-L1 and PARP inhibitors (PARPi) is the ability of PARP inhibitors to induce or at least enhance an anti-tumour immune response 137 , 138 ( Fig. 2 ). The original explanation was mainly the emergence of neoantigens or an increased tumour mutation burden due to the PARPi-induced reduced ability of DNA repair: especially in tumours that already have a homologous recombination (HR) defect, e.g., due to mutations in one of the BRCA genes, additional PARP inhibition leads to an increase in the already increased neoantigen burden 139 , 140 . Increased TMB, on the other hand, is a robust predictor of response to immune checkpoint inhibition 14 . However, the limited clinical data available to date demonstrate no significant correlation between BRCA1/2 mutation and improved response to immune checkpoint inhibition in ovarian cancer.
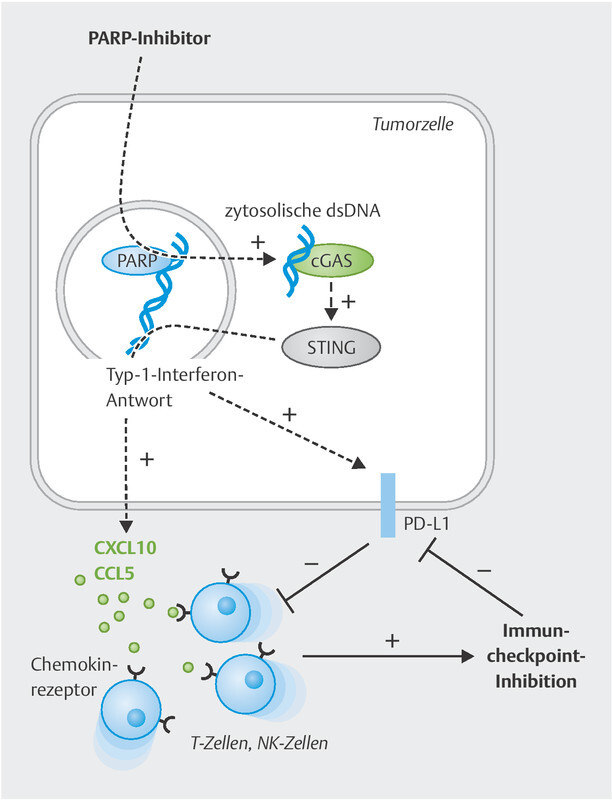
Fig. 2.
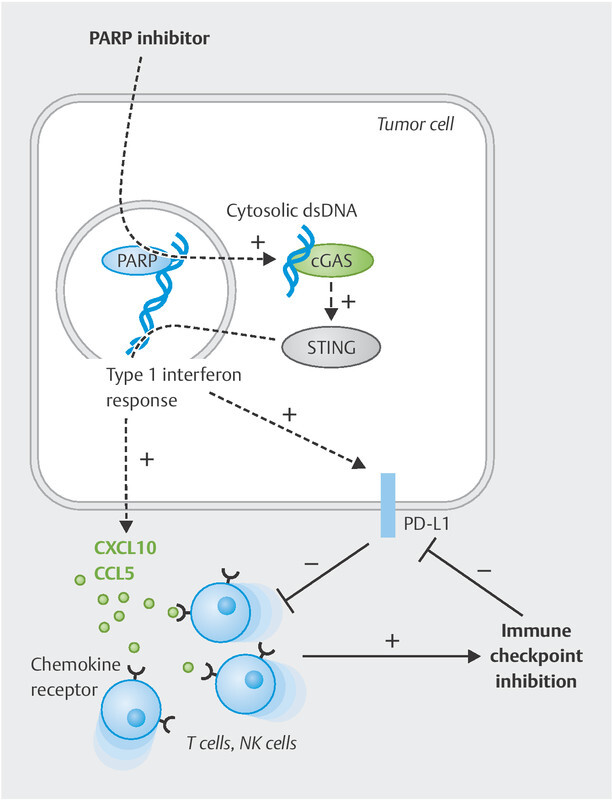
PARPi-mediated tumour cell effects allowing combined treatment with immune checkpoint blockade. PARP inhibition (similar to BRCA1/2 loss of function) leads to an accumulation of fragmented double-stranded DNA (dsDNA) in the cytosol of the tumour cell. This activates the cGAS/STING signalling pathway, which ultimately triggers a type I interferon response. This also releases downstream chemokines (CXCL10, CXCL11, CCL5), among others, which attract tumour-suppressive T and NK cells into the tumour. However, PD-L1 expression is also upregulated in the tumour microenvironment. Both immune cell recruitment and PD-L1 induction now permit effective immune checkpoint blockade.
Numerous recent papers have shown that PARP inhibitors and BRCA mutations not only induce neoantigens but also activate independent mechanisms improving the immune response to the tumour 46 , 141 , 142 , 143 , 144 , 145 ( Fig. 2 ). Incomplete DNA repair results in the accumulation of double-stranded DNA fragments in the cytosol of the tumour cell. This cytosolic dsDNA is recognised by the cyclic cGMP-AMP synthase (cGAS), which then produces cGAMP and thus activates the STING signalling pathway (“stimulator of interferon genes”). This STING activation is accompanied by phosphorylation of the transcription factors TBK1 and IRF3, which, together with activation of the NF-κB pathway, leads to secretion of the lymphocyte-recruiting chemokines CCL5 and CXCL10 145 . This cellular process, originally intended for viral infections, ensures increased recruitment of tumour-suppressive lymphocytes such as T cells or NK cells into the tumour microenvironment 46 , 141 , 142 , 143 , 144 , 146 . In preclinical models of ovarian cancer and TNBC, the PARPi effect even depended on this STING activation 46 , 143 . Studies of the tumour genome of BRCA -mutated vs. non-mutated tumours have revealed that this process is actually relevant in patients as well: CXCL10 was identified as one of the most upregulated genes 147 , 148 , 149 .
Moreover, cGAS/STING activation by PARP inhibitors induces PD-L1 expression in the tumour microenvironment 144 , 150 . This mechanism may also explain the increased PD-L1 expression observed in BRCA -mutated tumours 119 , 151 . Other mechanisms may also play a role here 152 , 153 . Since the chemokines induced by PARPi/STING (CCL5, CXCL10) are necessary for anti-PD-1/PD-L1 therapy to work 39 , 45 , this mechanism provides another strong rationale for combining PARP and immune checkpoint inhibitors 145 .
The therapeutic synergism was also confirmed preclinically in ovarian cancer mouse models both with and without BRCA mutation 46 , 142 , 154 . In addition, since both groups of substances have little overlapping profiles of side effects, combined treatment should also be tolerated clinically.
So far, three trials have studied the combination of PARP inhibitor and an anti-PD-1/PD-L1 antibody ( Table 3 ). In the MEDIOLA trial, the combination of olaparib and durvalumab (anti-PD-L1) was initially assessed in a population of 34 patients solely with BRCA germline mutations and recurrent platinum-sensitive ovarian cancer 155 . The overall response rate (ORR) was 72% and 22% achieved complete remission. Although difficult to answer in such a small population, ORR was not dependent on PD-L1 status or lymphocytic infiltration in the tumour. Whether the high response rate can actually be attributed to the synergistic action of olaparib and durvalumab must be examined in further trials because olaparib also achieved response rates of around 72% in the comparable population of the SOLO3 trial, with a rate of complete remissions comparable to the MEDIOLA trial 157 . In both trials, the response rate was better in earlier lines of therapy. The median PFS was 11.1 months in the MEDIOLA study and 14.3 months in the SOLO3 study. Even though by their very nature cross-trial comparisons are problematic, the question of synergism arises here, at least in BRCA -mutated recurrent ovarian cancer.
Table 3 Trials on combined treatment with immune checkpoint and PARP inhibitors.
| Trial | NCT02657889 (TOPACIO/KEYNOTE-162) | NCT02734004 (MEDIOLA) | NCT02734004 (MEDIOLA) | NCT02484404 |
|---|---|---|---|---|
| Abbreviations: CR = complete remission, DCR = disease control rate, HRD = homologous recombination deficit, ICB = immune checkpoint blockade, IHC = immunohistochemistry, IC = immune cells, CI = confidence interval, ORR = overall response rate, PD-1 = programmed cell death protein 1, PD-L1 = programmed cell death ligand 1, PFS = progression free survival, Pt = platinum, TC = tumour cells, WT = wild type | ||||
| Combined treatment | Niraparib + pembrolizumab | Olaparib + durvalumab | Olaparib + durvalumab + bevacizumab | Olaparib + durvalumab |
| Targeted ICB structure | PD-1 | PD-L1 | PD-L1 | PD-L1 |
| Dosing | Niraparib 200 mg qd Pembrolizumab 200 mg q21 |
Olaparib 300 mg bid after 4 weeks durvalumab 1500 mg q28 |
Olaparib 300 mg bid after 4 weeks durvalumab 1500 mg q28 + bevacizumab 10 mg/kg q14 |
Olaparib 300 mg bid Durvalumab 1500 mg q28 |
| Phase | Phase I/II (pooled) | Phase I/II | Phase II | Phase II |
| Study design | multicentre, open, single-arm | multicentre, open, single-arm | multicentre, open, single-arm | single-centre, open, single-arm |
| No. of patients | 62 (60 analysed) | 66 (64 analysed) | 31 | 35 |
| Population | Pt-resistant recurrence (response > 6 months to first-line Pt) 79% tBRCA-WT 18% tBRCA-mutated 35% HRD positive (Myriad) |
Pt-sensitive 50% gBRCA-WT 50% gBRCA-mutated |
Pt-sensitive 100% gBRCA-WT |
86% Pt-resistant (< 6 months) 77% BRCA-WT 23% BRCA-mutated 20% HRD positive (BROCA- HR) |
| Prior therapies | 1 – 5 (median 3) | gBRCA-mutated: 1 – 4 + lines (median 2) gBRCA-WT: 1 – 2 lines (median 1) |
1 – 2 lines (median 1) | 52% ≥ 4 lines (median 4) |
| Histology | not reported | 81% (26/32) serous 19% (6/32) non-serous not reported for gBRCA-WT |
not reported | 88% (31/35) high-grade serous 9% (3/35) endometrioid 3% (1/35) mucinous |
| PD-L1 status | 56% PD-L1 positive | |||
| ORR | 18% (11/60) | 53% (23/32) | 87% (27/31) | 14% (5/35) |
| CR | 5% (3/60) | 22% (7/32 gBRCA-mutated) | not reported | 0% (0/35) |
| ORR (BRCA mut ) | 18% (2/11) | 72% (23/32) | 37% (3/8) | |
| ORR (BRCA-WT) | 19% (9/47) | 34.4% (11/32) | 87% (27/31) | 7% (2/27) |
| ORR (PD-L1 positive) | 21% (7/33) | – | – | |
| ORR (PD-L1 negative) | 10% (2/21) | – | – | |
| ORR (HRD positive) | 14% (3/21) | – | – | |
| ORR (HRD negative) | 19% (6/32) | – | – | |
| DCR | 65% (39/60) | gBRCA-mutated: 66% gBRCA-WT: 28% |
77% (24/31) | 71% (25/35) |
| PFS | 3.4 months (95% CI 2.1 – 5.1 months) | 11.1 months (95% CI 8.2 – 15.9 months) 5.5 months (95% CI 3.6 – 7.5 months) |
14.7 months (95% CI 10.0 – 18.1 months) | 3.9 months (95% CI 2.0 – 7.25 months) |
| References | Konstantinopoulos et al. 8 | Drew et al. 155 , 156 | Drew et al. 156 | Lampert et al. 7 |
Much more promising, however, are the outcomes of this combination in the BRCA wild-type population of the MEDIOLA trial 156 . In the platinum-sensitive population with little prior treatment, the combined treatment showed response rates of 34%. The addition of the anti-VEGF antibody bevacizumab dramatically increased this to 87% with a PFS of 14.7 months 156 . Here, one may currently assume a synergistic effect of this triple combination, which will now be assessed in numerous phase III trials with different PARP and immune checkpoint inhibitors. The DUO-O trial (AGO-OVAR23/ENGOT-ov46/NCT03737643) is studying the role of durvalumab and olaparib as an add-on to first-line carboplatin/paclitaxel/bevacizumab therapy, mostly in the BRCA wild-type population. The FIRST trial (AGO-OVAR24/ENGOT-ov44/NCT03602859) is similarly examining the combination of dostarlimab and niraparib. KEYLYNK-001 (ENGOT-ov43, NCT03740165) is assessing the combination of pembrolizumab and olaparib as add-on to first-line chemotherapy and bevacizumab in patients without BRCA mutations. Other trials in the relapse setting include the ANITA trial (AGO-OVAR2.33, NCT03598270; atezolizumab and niraparib) and the AGO-OVAR2.29 trial (NCT03353831; atezolizumab and bevacizumab).
The TOPACIO trial studied a combination of pembrolizumab and niraparib in the platinum-resistant recurrent setting 8 . Cancers both with and without BRCA mutations were eligible. The overall response rate was 18% (5% complete remission). This did not depend on BRCA mutation status or HRD (homologous recombination defect as a marker of DNA repair that is also impaired outside BRCA 1/2). On the contrary, 5 of the 8 patients responding to therapy for more than 6 months had platinum-resistant/refractory BRCA -non-mutated cancer. Due to the single-arm design, only cross-trial comparisons can help assess the outcomes: The overall response rate of 19% in the platinum-resistant BRCA wild-type population is significantly higher than the figures from trials with PARPi as monotherapy reporting a response rate of 0 – 5% 159 , 160 . It also exceeds the response rates of nearly 10% of pembrolizumab as monotherapy in the KEYNOTE-100 trial 125 . On the other hand, in the BRCA -mutated population of the TOPACIO trial (response rate 18%) there was no improvement compared to other trials with PARPi as monotherapy reporting response rates of 0 – 14% (platinum-refractory) or 25 – 30% (platinum-resistant), such as the ARIEL2 trial 161 , 162 .
Since the traditional predictive biomarkers such as PD-L1 expression, HRD and BRCA status did not work in the TOPACIO trial, an elaborate biomarker project was conducted to search for other predictive markers within the trial population 131 . It was shown that a recently published HRD-associated mutation signature 163 and interferon signalling in the CD8-positive immune cell compartment were predictive of treatment response. These markers identified all patients responding to this treatment 131 . However, as this is a singular “training population”, these findings need to be validated in other populations receiving similar therapies.
Another trial (NCT02484404) investigated the combination of olaparib and durvalumab from the MEDIOLA trial, this time in a mixed, but mainly platinum-resistant (86%, < 6 months), mainly BRCA -non-mutated population of 35 patients with significant prior treatment 7 . The overall response rate was 14% (0% complete remission). There was no dependence on PD-L1 status, but a significantly better numerical response rate in the BRCA -mutated group (3/8 patients vs. 2/27 patients, Table 3 ). It is interesting, however, that despite a heavily pretreated study population, long-lasting remissions of up to 2 years were also seen in platinum-resistant, BRCA -non-mutated tumours 7 .
The latter trial also conducted an extensive biomarker study by obtaining tumour and serum samples from 20 patients before starting therapy and after 15 days 7 . Both the preclinically postulated increase in tumour mutational burden and the induction of interferon response were studied. Altogether, all samples showed a low mutational burden of < 5 somatic mutations/Mb, irrespective of the BRCA mutation status. PARP inhibitor therapy did not increase this TMB, although here the rather short period between the sample collections (15 days) may also have been too short to detect any effects. Tumour mutation burden was not predictive of response to immune checkpoint blockade. The study also confirmed the preclinically proven induction of an IFN response with the upregulation of IFN-γ, the CXCR3 chemokines CXCL9 and CXCL10, CCL5, the induction of PD-L1, and the increased accumulation of tumour-infiltrating lymphocytes. IFN-γ serum level elevation was predictive of treatment response, as was the immunoreactive HGSOC subtype 24 .
Looking at the trial outcomes available so far, it appears that the combination of PARP and immune checkpoint inhibitors demonstrates the preclinically assumed synergistic effect, especially in the BRCA wild-type population. It may be further improved by the addition of an anti-angiogenic agent 156 , 158 , 164 .
Immune checkpoint inhibitors combined with other immunotherapies
Approaches in immunotherapy that induce a specific immune response by the adaptive immune system, such as adoptive T cell transfer or CAR T cell therapy, have already shown some efficacy in ovarian cancer 165 , 166 . Since in these therapies anergy of the immune effector cells involved may also be induced by immune checkpoints such as the PD-1/PD-L1 system, ICB could also be a promising combination partner for improving therapeutic success 167 .
Conclusions
Despite success in other malignancies, the clinical response rates of immune checkpoint blockade in ovarian cancer have so far been disappointing and limited to only a few patients. The combination of immune checkpoint and PARP inhibition would seem to make sense and shows a possible benefit, especially in the BRCA wild-type population, presumably because it is precisely in this population that the immunostimulatory activity of PARP inhibitors comes into play. All in all, the few trial data available so far suggest that the response is better, in early lines of therapy. In the future, immune checkpoint blockade could also be useful in other immunotherapy approaches, such as adoptive T cell transfer and CAR T cell therapy, where the induced adaptive immune response can be further disinhibited by the ICB.
Further research is now warranted to search for appropriate biomarkers to identify subpopulations that will benefit, and on the other hand, to identify other combination partners that can induce an immune response against ovarian cancer. The phase III trials currently underway, in particular on the triple combination of ICB, PARPi and anti-angiogenic therapy, will hopefully secure ICB a role in the treatment of advanced ovarian cancer.
Acknowledgements
The author would like to thank the Deutsche Forschungsgemeinschaft (BR4733/1-3, BR4733/2-1, BR4733/3-1, BR4733/4-1) and the Wilhelm Sander Foundation (2019.085.1) for their generous support.
Danksagung
Der Autor dankt der Deutschen Forschungsgemeinschaft (BR4733/1-3, BR4733/2-1, BR4733/3-1, BR4733/4-1) sowie der Wilhelm Sander-Stiftung (2019.085.1) für die großzügige Unterstützung.
Footnotes
Conflict of Interest/Interessenkonflikt Lecturing, travel fees and/or consulting activities for AstraZeneca, Clovis, GSK, Pfizer, PharmaMar, Roche, Tesaro, and Teva./Vortragstätigkeit, Reisehonorare oder Beratertätigkeit von AstraZeneca, Clovis, GSK, Pfizer, PharmaMar, Roche, Tesaro und Teva.
References/Literatur
- 1.Tyzzer E E. Tumor Immunity. Am J Cancer Res. 1916;1:125–156. [Google Scholar]
- 2.Maman S, Witz I P. A history of exploring cancer in context. Nat Rev Cancer. 2018;18:359–376. doi: 10.1038/s41568-018-0006-7. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Buque A, Kepp O. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28:690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Marth C, Wieser V, Tsibulak I. Immunotherapy in ovarian cancer: fake news or the real deal? Int J Gynecol Cancer. 2019;29:201–211. doi: 10.1136/ijgc-2018-000011. [DOI] [PubMed] [Google Scholar]
- 5.Moore K N, Bookman M, Sehouli J. Primary results from IMagyn050/GOG 3015/ENGOT-OV39, a double-blind placebo (pbo)-controlled randomised phase III trial of bevacizumab (bev)-containing therapy +/- atezolizumab (atezo) for newly diagnosed stage III/IV ovarian cancer (OC) Ann Oncol. 2020;31:S1161–S1162. doi: 10.1016/j.annonc.2020.08.2261. [DOI] [Google Scholar]
- 6.Moore K. Primary results from IMagyn050/GOG 3015/ENGOT-OV39, a double-blind placebo-controlled randomised phase 3 trial of bevacizumab-containing therapy ± atezolizumab for newly diagnosed stage III/IV ovarian cancer. ESMO 2020. J Clin Oncol. 2021;39:1842–1855. doi: 10.1200/JCO.21.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lampert E J, Zimmer A, Padget M. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: a Proof-of-Concept Phase II Study. Clin Cancer Res. 2020;26:4268–4279. doi: 10.1158/1078-0432.CCR-20-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstantinopoulos P A, Waggoner S, Vidal G A. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination With Pembrolizumab in Patients With Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.2019.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunn G P, Old L J, Schreiber R D. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 10.Alexandrov L B, Nik-Zainal S, Wedge D C. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schumacher T N, Schreiber R D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 12.Chalmers Z R, Connelly C F, Fabrizio D. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wick D A, Webb J R, Nielsen J S. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clin Cancer Res. 2014;20:1125–1134. doi: 10.1158/1078-0432.CCR-13-2147. [DOI] [PubMed] [Google Scholar]
- 14.Yarchoan M, Johnson B A, 3rd, Lutz E R. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:569. doi: 10.1038/nrc.2017.74. [DOI] [PubMed] [Google Scholar]
- 15.Cristescu R, Mogg R, Ayers M. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018 doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang A W, McPherson A, Milne K. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell. 2018;173:1755–1.769E25. doi: 10.1016/j.cell.2018.03.073. [DOI] [PubMed] [Google Scholar]
- 17.The Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang R F, Wang H Y. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017;27:11–37. doi: 10.1038/cr.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuster H, Peper J K, Bosmuller H C. The immunopeptidomic landscape of ovarian carcinomas. Proc Natl Acad Sci U S A. 2017;114:E9942–E9951. doi: 10.1073/pnas.1707658114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Want M Y, Lugade A A, Battaglia S. Nature of tumour rejection antigens in ovarian cancer. Immunology. 2018;155:202–210. doi: 10.1111/imm.12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez G M, Galpin K JC, McCloskey C W. The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers (Basel) 2018;10:242. doi: 10.3390/cancers10080242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma D, Gu M J. Immune effect of tumor-infiltrating lymphocytes and its relation to the survival rate of patients with ovarian malignancies. J Tongji Med Univ. 1991;11:235–239. doi: 10.1007/BF02888158. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Conejo-Garcia J R, Katsaros D. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 24.Konecny G E, Wang C, Hamidi H. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst. 2014 doi: 10.1093/jnci/dju249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tothill R W, Tinker A V, George J. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–5208. doi: 10.1158/1078-0432.CCR-08-0196. [DOI] [PubMed] [Google Scholar]
- 26.Wang C, Armasu S M, Kalli K R. Pooled Clustering of High-Grade Serous Ovarian Cancer Gene Expression Leads to Novel Consensus Subtypes Associated with Survival and Surgical Outcomes. Clin Cancer Res. 2017;23:4077–4085. doi: 10.1158/1078-0432.CCR-17-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke B, Tinker A V, Lee C H. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22:393–402. doi: 10.1038/modpathol.2008.191. [DOI] [PubMed] [Google Scholar]
- 28.Hamanishi J, Mandai M, Iwasaki M. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104:3360–3365. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knutson K L, Maurer M J, Preston C C. Regulatory T cells, inherited variation, and clinical outcome in epithelial ovarian cancer. Cancer Immunol Immunother. 2015;64:1495–1504. doi: 10.1007/s00262-015-1753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montfort A, Owen S, Piskorz A M. Combining measures of immune infiltration shows additive effect on survival prediction in high-grade serous ovarian carcinoma. Br J Cancer. 2020;122:1803–1810. doi: 10.1038/s41416-020-0822-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato E, Olson S H, Ahn J. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamanishi J, Mandai M, Abiko K. The comprehensive assessment of local immune status of ovarian cancer by the clustering of multiple immune factors. Clin Immunol. 2011;141:338–347. doi: 10.1016/j.clim.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Tsiatas M L, Gyftaki R, Liacos C. Study of T lymphocytes infiltrating peritoneal metastases in advanced ovarian cancer: associations with vascular endothelial growth factor levels and prognosis in patients receiving platinum-based chemotherapy. Int J Gynecol Cancer. 2009;19:1329–1334. doi: 10.1111/IGC.0b013e3181b7a40e. [DOI] [PubMed] [Google Scholar]
- 34.deLeeuw R J, Kroeger D R, Kost S E. CD25 identifies a subset of CD4(+)FoxP3(−) TIL that are exhausted yet prognostically favorable in human ovarian cancer. Cancer Immunol Res. 2015;3:245–253. doi: 10.1158/2326-6066.CIR-14-0146. [DOI] [PubMed] [Google Scholar]
- 35.Nesbeth Y C, Martinez D G, Toraya S. CD4+ T cells elicit host immune responses to MHC class II-negative ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J Immunol. 2010;184:5654–5662. doi: 10.4049/jimmunol.0903247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abastado J P. The next challenge in cancer immunotherapy: controlling T-cell traffic to the tumor. Cancer Res. 2012;72:2159–2161. doi: 10.1158/0008-5472.CAN-11-3538. [DOI] [PubMed] [Google Scholar]
- 37.Chen D S, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 38.Bronger H, Singer J, Windmuller C. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115:553–563. doi: 10.1038/bjc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dangaj D, Bruand M, Grimm A J. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell. 2019;35:885–9.0E12. doi: 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zsiros E, Duttagupta P, Dangaj D. The Ovarian Cancer Chemokine Landscape Is Conducive to Homing of Vaccine-Primed and CD3/CD28-Costimulated T Cells Prepared for Adoptive Therapy. Clin Cancer Res. 2015;21:2840–2850. doi: 10.1158/1078-0432.CCR-14-2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melero I, Rouzaut A, Motz G T. T-cell and NK-cell infiltration into solid tumors: a key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014;4:522–526. doi: 10.1158/2159-8290.CD-13-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagarsheth N, Wicha M S, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017 doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jimenez-Sanchez A, Memon D, Pourpe S. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell. 2017;170:927–9.38E22. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Millstein J, Budden T, Goode E L. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann Oncol. 2020 doi: 10.1016/j.annonc.2020.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chow M T, Ozga A J, Servis R L. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity. 2019;50:1498–1.512E8. doi: 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen J, Zhao W, Ju Z. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019;79:311–319. doi: 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M, Matsumura N, Mandai M. Classification using hierarchical clustering of tumor-infiltrating immune cells identifies poor prognostic ovarian cancers with high levels of COX expression. Mod Pathol. 2009;22:373–384. doi: 10.1038/modpathol.2008.187. [DOI] [PubMed] [Google Scholar]
- 48.Plitas G, Rudensky A Y. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res. 2016;4:721–725. doi: 10.1158/2326-6066.CIR-16-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li C, Jiang P, Wei S. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19:116. doi: 10.1186/s12943-020-01234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakaguchi S, Sakaguchi N, Asano M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 51.Fontenot J D, Gavin M A, Rudensky A Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 52.Gavin M A, Rasmussen J P, Fontenot J D. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 53.Williams L M, Rudensky A Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 54.Sehouli J, Loddenkemper C, Cornu T. Epigenetic quantification of tumor-infiltrating T-lymphocytes. Epigenetics. 2011;6:236–246. doi: 10.4161/epi.6.2.13755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woo E Y, Chu C S, Goletz T J. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- 56.Curiel T J, Coukos G, Zou L. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 57.Barnett J C, Bean S M, Whitaker R S. Ovarian cancer tumor infiltrating T-regulatory (T(reg)) cells are associated with a metastatic phenotype. Gynecol Oncol. 2010;116:556–562. doi: 10.1016/j.ygyno.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 58.Wolf D, Wolf A M, Rumpold H. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005;11:8326–8331. doi: 10.1158/1078-0432.CCR-05-1244. [DOI] [PubMed] [Google Scholar]
- 59.Fialova A, Partlova S, Sojka L. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer. 2013;132:1070–1079. doi: 10.1002/ijc.27759. [DOI] [PubMed] [Google Scholar]
- 60.Kryczek I, Wei S, Zhu G. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67:8900–8905. doi: 10.1158/0008-5472.CAN-07-1866. [DOI] [PubMed] [Google Scholar]
- 61.Curiel T J, Cheng P, Mottram P. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004;64:5535–5538. doi: 10.1158/0008-5472.CAN-04-1272. [DOI] [PubMed] [Google Scholar]
- 62.Facciabene A, Peng X, Hagemann I S. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 63.Wertel I, Surowka J, Polak G. Macrophage-derived chemokine CCL22 and regulatory T cells in ovarian cancer patients. Tumour Biol. 2015;36:4811–4817. doi: 10.1007/s13277-015-3133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Redjimi N, Raffin C, Raimbaud I. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res. 2012;72:4351–4360. doi: 10.1158/0008-5472.CAN-12-0579. [DOI] [PubMed] [Google Scholar]
- 65.Au K K, Peterson N, Truesdell P. CXCL10 alters the tumour immune microenvironment and disease progression in a syngeneic murine model of high-grade serous ovarian cancer. Gynecol Oncol. 2017;145:436–445. doi: 10.1016/j.ygyno.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Di Pilato M, Kim E Y, Cadilha B L. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. 2019;570:112–116. doi: 10.1038/s41586-019-1215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Overacre-Delgoffe A E, Chikina M, Dadey R E. Interferon-gamma Drives Treg Fragility to Promote Anti-tumor Immunity. Cell. 2017;169:1130–1.141E14. doi: 10.1016/j.cell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung K, Kim J A, Kim Y J. A Neuropilin-1 Antagonist Exerts Antitumor Immunity by Inhibiting the Suppressive Function of Intratumoral Regulatory T Cells. Cancer Immunol Res. 2020;8:46–56. doi: 10.1158/2326-6066.CIR-19-0143. [DOI] [PubMed] [Google Scholar]
- 69.Kandalaft L E, Chiang C L, Tanyi J. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J Transl Med. 2013;11:149. doi: 10.1186/1479-5876-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh M, Loftus T, Webb E. Minireview: Regulatory T Cells and Ovarian Cancer. Immunol Invest. 2016;45:712–720. doi: 10.1080/08820139.2016.1186689. [DOI] [PubMed] [Google Scholar]
- 71.de Boo L W, Vulink A JE, Bos M. Metronomic cyclophosphamide-induced long-term remission after recurrent high-grade serous ovarian cancer: A case study. Mol Clin Oncol. 2017;7:1130–1134. doi: 10.3892/mco.2017.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang R, Cai Z, Zhang Y. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res. 2006;66:6807–6815. doi: 10.1158/0008-5472.CAN-05-3755. [DOI] [PubMed] [Google Scholar]
- 73.Colvin E K. Tumor-associated macrophages contribute to tumor progression in ovarian cancer. Front Oncol. 2014;4:137. doi: 10.3389/fonc.2014.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Finkernagel F, Reinartz S, Lieber S. The transcriptional signature of human ovarian carcinoma macrophages is associated with extracellular matrix reorganization. Oncotarget. 2016;7:75339–75352. doi: 10.18632/oncotarget.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang M, McKay D, Pollard J W. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018;78:5492–5503. doi: 10.1158/0008-5472.CAN-18-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duluc D, Corvaisier M, Blanchard S. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125:367–373. doi: 10.1002/ijc.24401. [DOI] [PubMed] [Google Scholar]
- 77.Germano G, Frapolli R, Belgiovine C. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–262. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 78.Wanderley C W, Colon D F, Luiz J PM. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018;78:5891–5900. doi: 10.1158/0008-5472.CAN-17-3480. [DOI] [PubMed] [Google Scholar]
- 79.Hagemann T, Wilson J, Burke F. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006;176:5023–5032. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- 80.Reinartz S, Schumann T, Finkernagel F. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer. 2014;134:32–42. doi: 10.1002/ijc.28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.No J H, Moon J M, Kim K. Prognostic significance of serum soluble CD163 level in patients with epithelial ovarian cancer. Gynecol Obstet Invest. 2013;75:263–267. doi: 10.1159/000349892. [DOI] [PubMed] [Google Scholar]
- 82.Yuan X, Zhang J, Li D. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol Oncol. 2017;147:181–187. doi: 10.1016/j.ygyno.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 83.Yin M, Li X, Tan S. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. 2016;126:4157–4173. doi: 10.1172/JCI87252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aras S, Zaidi M R. TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer. 2017;117:1583–1591. doi: 10.1038/bjc.2017.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Yao H, Song G. Regulation of epithelial-mesenchymal transition by tumor-associated macrophages in cancer. Am J Transl Res. 2015;7:1699–1711. [PMC free article] [PubMed] [Google Scholar]
- 86.Kryczek I, Zou L, Rodriguez P. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sica G L, Choi I H, Zhu G. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 88.Cassetta L, Kitamura T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front Cell Dev Biol. 2018;6:38. doi: 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qu Y, Wen J, Thomas G. Baseline Frequency of Inflammatory Cxcl9-Expressing Tumor-Associated Macrophages Predicts Response to Avelumab Treatment. Cell Rep. 2020;32:107873. doi: 10.1016/j.celrep.2020.107873. [DOI] [PubMed] [Google Scholar]
- 90.Worzfeld T, Pogge von Strandmann E, Huber M. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol. 2017;7:24. doi: 10.3389/fonc.2017.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mantovani A, Marchesi F, Malesci A. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu Y, Knolhoff B L, Meyer M A. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moughon D L, He H, Schokrpur S. Macrophage Blockade Using CSF1R Inhibitors Reverses the Vascular Leakage Underlying Malignant Ascites in Late-Stage Epithelial Ovarian Cancer. Cancer Res. 2015;75:4742–4752. doi: 10.1158/0008-5472.CAN-14-3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baci D, Bosi A, Gallazzi M. The Ovarian Cancer Tumor Immune Microenvironment (TIME) as Target for Therapy: A Focus on Innate Immunity Cells as Therapeutic Effectors. Int J Mol Sci. 2020;21:3125. doi: 10.3390/ijms21093125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin H, Wei S, Hurt E M. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128:805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang H, Liang Y, Anders R A. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest. 2018;128:580–588. doi: 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abiko K, Mandai M, Hamanishi J. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin Cancer Res. 2013;19:1363–1374. doi: 10.1158/1078-0432.CCR-12-2199. [DOI] [PubMed] [Google Scholar]
- 98.Krempski J, Karyampudi L, Behrens M D. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186:6905–6913. doi: 10.4049/jimmunol.1100274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xue C, Xu Y, Ye W. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol Oncol. 2020;157:222–233. doi: 10.1016/j.ygyno.2019.12.012. [DOI] [PubMed] [Google Scholar]
- 100.Abiko K, Matsumura N, Hamanishi J. IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112:1501–1509. doi: 10.1038/bjc.2015.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Darb-Esfahani S, Kunze C A, Kulbe H. Prognostic impact of programmed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor-infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget. 2016;7:1486–1499. doi: 10.18632/oncotarget.6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Webb J R, Milne K, Kroeger D R. PD-L1 expression is associated with tumor-infiltrating T cells and favorable prognosis in high-grade serous ovarian cancer. Gynecol Oncol. 2016;141:293–302. doi: 10.1016/j.ygyno.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 103.Hogdall E, Hogdall C, Vo T. Impact of PD-L1 and T-cell inflamed gene expression profile on survival in advanced ovarian cancer. Int J Gynecol Cancer. 2020;30:1034–1042. doi: 10.1136/ijgc-2019-001109. [DOI] [PubMed] [Google Scholar]
- 104.Chen H, Molberg K, Strickland A L. PD-L1 Expression and CD8+ Tumor-infiltrating Lymphocytes in Different Types of Tubo-ovarian Carcinoma and Their Prognostic Value in High-grade Serous Carcinoma. Am J Surg Pathol. 2020;44:1050–1060. doi: 10.1097/PAS.0000000000001503. [DOI] [PubMed] [Google Scholar]
- 105.Chin C D, Fares C M, Campos M. Association of PD-L1 expression by immunohistochemistry and gene microarray with molecular subtypes of ovarian tumors. Mod Pathol. 2020;33:2001–2010. doi: 10.1038/s41379-020-0567-3. [DOI] [PubMed] [Google Scholar]
- 106.Peng J, Hamanishi J, Matsumura N. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015;75:5034–5045. doi: 10.1158/0008-5472.CAN-14-3098. [DOI] [PubMed] [Google Scholar]
- 107.Kodumudi K N, Woan K, Gilvary D L. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 2010;16:4583–4594. doi: 10.1158/1078-0432.CCR-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang L, Dermawan K, Jin M. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol. 2008;129:219–229. doi: 10.1016/j.clim.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 109.Mouw K W, Konstantinopoulos P A. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. 2018;118:933–935. doi: 10.1038/s41416-018-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Khairallah A S, Genestie C, Auguste A. Impact of neoadjuvant chemotherapy on the immune microenvironment in advanced epithelial ovarian cancer: Prognostic and therapeutic implications. Int J Cancer. 2018;143:8–15. doi: 10.1002/ijc.31200. [DOI] [PubMed] [Google Scholar]
- 111.Lo C S, Sanii S, Kroeger D R. Neoadjuvant Chemotherapy of Ovarian Cancer Results in Three Patterns of Tumor-Infiltrating Lymphocyte Response with Distinct Implications for Immunotherapy. Clin Cancer Res. 2017;23:925–934. doi: 10.1158/1078-0432.CCR-16-1433. [DOI] [PubMed] [Google Scholar]
- 112.Pölcher M, Braun M, Friedrichs N. Foxp3(+) cell infiltration and granzyme B(+)/Foxp3(+) cell ratio are associated with outcome in neoadjuvant chemotherapy-treated ovarian carcinoma. Cancer Immunol Immunother. 2010;59:909–919. doi: 10.1007/s00262-010-0817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bohm S, Montfort A, Pearce O M. Neoadjuvant Chemotherapy Modulates the Immune Microenvironment in Metastases of Tubo-Ovarian High-Grade Serous Carcinoma. Clin Cancer Res. 2016;22:3025–3036. doi: 10.1158/1078-0432.CCR-15-2657. [DOI] [PubMed] [Google Scholar]
- 114.Yaniz E, Genestie C, Klein C. Impact of chemotherapy alone or in combination with an anti-angiogenic on the immune tumor microenvironment (TME) of ovarian cancer: Data from the randomized CHIVA trial (a GINECO –GINEGEPS study) J Clin Oncol. 2020;38 (no. 15_suppl):6011. doi: 10.1200/JCO.2020.38.15_suppl.6011. [DOI] [Google Scholar]
- 115.Wu X, Feng Q M, Wang Y. The immunologic aspects in advanced ovarian cancer patients treated with paclitaxel and carboplatin chemotherapy. Cancer Immunol Immunother. 2010;59:279–291. doi: 10.1007/s00262-009-0749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mesnage S JL, Auguste A, Genestie C. Neoadjuvant chemotherapy (NACT) increases immune infiltration and programmed death-ligand 1 (PD-L1) expression in epithelial ovarian cancer (EOC) Ann Oncol. 2017;28:651–657. doi: 10.1093/annonc/mdw625. [DOI] [PubMed] [Google Scholar]
- 117.Napoletano C, Bellati F, Landi R. Ovarian cancer cytoreduction induces changes in T cell population subsets reducing immunosuppression. J Cell Mol Med. 2010;14:2748–2759. doi: 10.1111/j.1582-4934.2009.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bosmuller H C, Wagner P, Peper J K. Combined Immunoscore of CD103 and CD3 Identifies Long-Term Survivors in High-Grade Serous Ovarian Cancer. Int J Gynecol Cancer. 2016 doi: 10.1097/IGC.0000000000000672. [DOI] [PubMed] [Google Scholar]
- 119.Strickland K C, Howitt B E, Shukla S A. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–13598. doi: 10.18632/oncotarget.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cardenas H, Jiang G, Thomes Pepin J. Interferon-gamma signaling is associated with BRCA1 loss-of-function mutations in high grade serous ovarian cancer. NPJ Precis Oncol. 2019;3:32. doi: 10.1038/s41698-019-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Khoja L, Day D, Wei-Wu Chen T. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol. 2017;28:2377–2385. doi: 10.1093/annonc/mdx286. [DOI] [PubMed] [Google Scholar]
- 122.Hwang W T, Adams S F, Tahirovic E. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124:192–198. doi: 10.1016/j.ygyno.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hamanishi J, Mandai M, Ikeda T. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015;33:4015–4022. doi: 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 124.Liu J F, Gordon M, Veneris J. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol Oncol. 2019;154:314–322. doi: 10.1016/j.ygyno.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 125.Matulonis U A, Shapira-Frommer R, Santin A D. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: results from the phase II KEYNOTE-100 study. Ann Oncol. 2019;30:1080–1087. doi: 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 126.Matulonis U, Shapira R, Santin A. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Final results from the Phase 2 KEYNOTE-100 study. ASCO 2020. J Clin Oncol. 2020;38 (15_Suppl):6005. doi: 10.1093/annonc/mdz135. [DOI] [PubMed] [Google Scholar]
- 127.Disis M L, Taylor M H, Kelly K. Efficacy and Safety of Avelumab for Patients With Recurrent or Refractory Ovarian Cancer: Phase 1b Results From the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019;5:393–401. doi: 10.1001/jamaoncol.2018.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Motzer R J, Escudier B, McDermott D F. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zorn K K, Bonome T, Gangi L. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin Cancer Res. 2005;11:6422–6430. doi: 10.1158/1078-0432.CCR-05-0508. [DOI] [PubMed] [Google Scholar]
- 130.Howitt B E, Strickland K C, Sholl L M. Clear cell ovarian cancers with microsatellite instability: A unique subset of ovarian cancers with increased tumor-infiltrating lymphocytes and PD-1/PD-L1 expression. Oncoimmunology. 2017;6:e1277308. doi: 10.1080/2162402X.2016.1277308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Färkkila A, Gulhan D C, Casado J. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun. 2020;11:1459. doi: 10.1038/s41467-020-15315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Coleman S, Clayton A, Mason M D. Recovery of CD8+ T-cell function during systemic chemotherapy in advanced ovarian cancer. Cancer Res. 2005;65:7000–7006. doi: 10.1158/0008-5472.CAN-04-3792. [DOI] [PubMed] [Google Scholar]
- 133.Ledermann J A, Colombo N, Oza A M.Avelumab in combination with and/or following chemotherapy vs. chemotherapy alone in patients with previously untreated epithelial ovarian cancer: Results from the phase 3 javelin ovarian 100 trial Gynecol Oncol 202015913–14. 10.1016/j.ygyno.2020.06.02532771275 [DOI] [Google Scholar]
- 134.Ledermann J A.Avelumab in combination with and/or following chemotherapy vs. chemotherapy alone in patients with previously untreated epithelial ovarian cancer: Results from the phase 3 trial SGO-Meeting 2020 Gynecol Oncol 20201590113–14. 10.1016/j.ygyno.2020.06.02532771275 [DOI] [Google Scholar]
- 135.Pujade-Lauraine E, Fujiwara K, Ledermann J A. Avelumab alone or in combination with pegylated liposomal doxorubicin versus pegylated liposomal doxorubicin alone in platinum-resistant or refractory epithelial ovarian cancer: Primary and biomarker analysis of the phase III JAVELIN Ovarian 200 trial. Gynecol Oncol. 2019;154:21–22. doi: 10.1016/j.ygyno.2019.04.053. [DOI] [Google Scholar]
- 136.Pujade-Lauraine E. Avelumab alone or in combination with pegylated liposomal doxorubicin (PLD) vs. PLD alone in platinum-resistant or refractory epithelial ovarian cancer: primary and biomarker analysis of the phase 3 trial. SO Meeting 2019. Gynecol Oncol. 2019;154 01:21–22. doi: 10.1016/j.ygyno.2019.04.053. [DOI] [Google Scholar]
- 137.Stewart R A, Pilie P G, Yap T A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018;78:6717–6725. doi: 10.1158/0008-5472.CAN-18-2652. [DOI] [PubMed] [Google Scholar]
- 138.Peyraud F, Italiano A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers (Basel) 2020;12:1502. doi: 10.3390/cancers12061502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Germano G, Lamba S, Rospo G. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552:116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 140.Mouw K W, Goldberg M S, Konstantinopoulos P A. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017;7:675–693. doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chabanon R M, Muirhead G, Krastev D B. PARP inhibition enhances tumor cell-intrinsic immunity in ERCC1-deficient non-small cell lung cancer. J Clin Invest. 2019;129:1211–1228. doi: 10.1172/JCI123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ding L, Kim H J, Wang Q. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018;25:2972–2.98E8. doi: 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pantelidou C, Sonzogni O, De Oliveria Taveira M. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019;9:722–737. doi: 10.1158/2159-8290.CD-18-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sen T, Rodriguez B L, Chen L. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019;9:646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee E K, Konstantinopoulos P A. PARP inhibition and immune modulation: scientific rationale and perspectives for the treatment of gynecologic cancers. Ther Adv Med Oncol. 2020;12:1.758835920944116E15. doi: 10.1177/1758835920944116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zitvogel L, Galluzzi L, Kepp O. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 147.Mulligan A M, Raitman I, Feeley L. Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the Ontario Familial Breast Cancer Registry. Clin Cancer Res. 2013;19:336–346. doi: 10.1158/1078-0432.CCR-11-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mulligan J M, Hill L A, Deharo S. Identification and validation of an anthracycline/cyclophosphamide-based chemotherapy response assay in breast cancer. J Natl Cancer Inst. 2014;106:djt335. doi: 10.1093/jnci/djt335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Parkes E E, Walker S M, Taggart L E. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst. 2017 doi: 10.1093/jnci/djw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gao Y, Yang J, Cai Y. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int J Cancer. 2018;143:931–943. doi: 10.1002/ijc.31357. [DOI] [PubMed] [Google Scholar]
- 151.Gottlieb C E, Mills A M, Cross J V. Tumor-associated macrophage expression of PD-L1 in implants of high grade serous ovarian carcinoma: A comparison of matched primary and metastatic tumors. Gynecol Oncol. 2017;144:607–612. doi: 10.1016/j.ygyno.2016.12.021. [DOI] [PubMed] [Google Scholar]
- 152.Jiao S, Xia W, Yamaguchi H. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017;23:3711–3720. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sato H, Niimi A, Yasuhara T. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017;8:1751. doi: 10.1038/s41467-017-01883-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang Z, Sun K, Xiao Y. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep. 2019;9:1853. doi: 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Drew Y, Kaufman B, Banerjee S.Phase II study of olaparib plus durvalumab (MEDIOLA): Updated results in germline BRCA-mutated platinum-sensitive relapsed (PSR) ovarian cancer (OC) Ann Oncol 201930051190PD31198952 [Google Scholar]
- 156.Drew Y, Penson R T, OʼMalley D M. Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC) Ann Oncol. 2020;31:S615–S616. doi: 10.1016/j.annonc.2020.08.953. [DOI] [Google Scholar]
- 157.Penson R T, Valencia R V, Cibula D. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J Clin Oncol. 2020;38:1164–1174. doi: 10.1200/JCO.19.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Drew Y. Phase II study of olaparib plus durvalumab and bevacizumab (MEDIOLA): initial results in patients with non-germline BRCA-mutated platinum sensitive relapsed ovarian cancer. ESMO 2020. Ann Oncol. 2020;31 04:S615–S616. doi: 10.1016/j.annonc.2020.08.953. [DOI] [Google Scholar]
- 159.Gelmon K A, Tischkowitz M, Mackay H. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–861. doi: 10.1016/S1470-2045(11)70214-5. [DOI] [PubMed] [Google Scholar]
- 160.Sandhu S K, Schelman W R, Wilding G. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–892. doi: 10.1016/S1470-2045(13)70240-7. [DOI] [PubMed] [Google Scholar]
- 161.Domchek S M, Aghajanian C, Shapira-Frommer R. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140:199–203. doi: 10.1016/j.ygyno.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Oza A M, Tinker A V, Oaknin A. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol. 2017;147:267–275. doi: 10.1016/j.ygyno.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 163.Gulhan D C, Lee J J, Melloni G EM. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51:912–919. doi: 10.1038/s41588-019-0390-2. [DOI] [PubMed] [Google Scholar]
- 164.Liu J F, Herold C, Gray K P. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019 doi: 10.1001/jamaoncol.2019.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pedersen M, Westergaard M CW, Milne K. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: a pilot study. Oncoimmunology. 2018;7:e1502905. doi: 10.1080/2162402X.2018.1502905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yan W, Hu H, Tang B. Advances Of Chimeric Antigen Receptor T Cell Therapy In Ovarian Cancer. Onco Targets Ther. 2019;12:8015–8022. doi: 10.2147/OTT.S203550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kverneland A H, Pedersen M, Westergaard M CW. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget. 2020;11:2092–2105. doi: 10.18632/oncotarget.27604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Varga A, Piha-Paul S, Ott P A. Pembrolizumab in patients with programmed death ligand 1-positive advanced ovarian cancer: Analysis of KEYNOTE-028. Gynecol Oncol. 2019;152:243–250. doi: 10.1016/j.ygyno.2018.11.017. [DOI] [PubMed] [Google Scholar]
- 169.Matulonis U A, Shapira R, Santin A. Final results from the KEYNOTE-100 trial of pembrolizumab in patients with advanced recurrent ovarian cancer. J Clin Oncol. 2020;38 (no. 15_suppl):6005. doi: 10.1200/JCO.2020.38.15_suppl.6005. [DOI] [Google Scholar]