

Abstract
Glomerulopathy with Fibronectin Deposits (GFND) is a rare, autosomal dominant disease characterized by proteinuria, hematuria and progressive renal failure associated with glomerular deposition of fibronectin, frequently resulting in end-stage renal disease (ESRD). There is no established treatment for this condition beyond conservative measures such as blood pressure control and the use of angiotensin-converting enzyme (ACE) inhibitors. We present a case of GFND associated with progressive chronic kidney disease (CKD) and nephrotic range proteinuria showing a sustained response to prednisone treatment. A 27-year-old G2P2 Caucasian female presented with 3 g/day of proteinuria, serum creatinine (Cr) 0.7 mg/dL, inactive urinary sediment and normotension without medication. She was part of a large family with glomerular disease, including three members who died of cerebral hemorrhage or stroke in their thirties. The patient’s kidney biopsy showed mesangial deposition of fibronectin consistent with GFND. No interstitial fibrosis was seen. Genotyping revealed the Y973C FN1 gene mutation. Despite maximal tolerable ACE inhibition, proteinuria increased to 4–6 g/g Cr and serum Cr increased to 1.0 mg/dL. She was treated with prednisone 60 mg (~ 1 mg/Kg) daily for 2 mos and then tapered by ~ 0.2 mg/Kg every month for 6 mos of total therapy. Proteinuria decreased to ~ 1 g/g Cr for > 5 years and serum Cr stabilized in the 1.2 mg/dL range with treatment. No significant side effects were encountered. In conclusion, this protocol should be considered in GFND patients with nephrotic range proteinuria despite maximal angiotensin system inhibition who have relatively preserved renal function.
Keywords: Genetic kidney disease, Glomerular disease
Introduction
Glomerulopathy with Fibronectin Deposits (GFND) (OMIM 601894) or equivalently, Fibronectin Glomerulopathy, is a rare, autosomal dominant condition associated with proteinuria, microscopic hematuria, hypertension and progressive renal failure [1–4]. Glomerular deposition of fibronectin (FN) is seen on kidney biopsy. FN is a dimeric glycoprotein that exists in both cellular/extracellular matrix and circulating forms [5]. FN is involved in numerous important functional processes including cell adhesion, differentiation and development, migration and wound healing/thrombus formation [5, 6]. Antibody studies suggest that GFND results from the deposition of the circulating isoform [2] and GFND has been shown to recur after kidney transplant [2, 3, 7]. Limited studies have shown normal plasma levels of FN in GFND [1, 8, 9]. While age-related penetrance is quite variable, progression to ESRD is common and can occur as early as in the 2nd decade of life [2, 3, 10, 11].
Three missense mutations in the FN1 gene (locus 2q35) that codes for FN and underlies this condition were identified, all of which were in heparin-binding domains [10]. Additional FN mutations, including one in an integrin-binding domain, have also recently been reported [11]. Only 6 of the 15 affected pedigrees studied by Castelletti et al. had FN1 gene abnormalities, suggesting that GFND is likely a polygenetic syndrome. The pathogenesis of GFND remains unclear.
There is no established specific treatment for this condition beyond conservative measures such as blood pressure control and the use of ACE inhibitors. We present here a case of GFND associated with nephrotic range proteinuria in which treatment with a 6 month prednisone taper led to marked reduction in proteinuria and stabilization of renal function lasting at least 5 years.
Case report
Clinical history and initial laboratory data
A 27-year-old G2P2 Caucasian female was referred for proteinuria first noticed during her second pregnancy. Her past medical history was notable only for a seizure disorder that began following a motor vehicle accident 10 years ago, obesity and depression. Her first pregnancy was uncomplicated and she was unaware of any kidney issues until 16 weeks into her second pregnancy when she was found to have 3 + proteinuria. Renal function was normal, blood pressure was generally well controlled without medication and urine protein/creatinine ratio was ~ 1 g/g at 38 weeks. She had an uncomplicated cesarean section at 40 weeks and then returned 3 mos post-partum for further evaluation.
The patient has a strong family history of kidney disease, as shown in the pedigree (Fig. 1). The degree of renal dysfunction among affected family members is quite variable as has been reported in other pedigrees [2, 10, 11], suggesting that important additional genetic and/or environmental factors may be at play. Her father (III-9), age 50, has stable CKD II (Cr 1.1 mg/dL), low-level proteinuria (urine microalbumin to creatinine ratio (MA/Cr) 191 mg/g) and hypertension. Her 29 yr old brother (IV-8), found to have proteinuria on an employment screening, has CKD II (Cr 1.2 mg/dL), non-nephrotic range proteinuria (urine MA/Cr 1709 mg/g) and hypertension. Her sister (IV-12), age 25, appeared to be unaffected but eventually developed progressive CKD III and nephrotic range proteinuria in her thirties (see below). Meanwhile, a paternal first cousin (IV-3) started dialysis in his late teens and was eventually transplanted and a paternal aunt (III-2), age 42, recently presented with CKD III (Cr 1.9 mg/dL), 4 g/day of proteinuria and hypertension and subsequently underwent kidney biopsy (see below).
Fig. 1.

GFND Pedigree. Black shading, affected individuals; grey shading, status presently unknown. Arrow indicates the index case of this report. CVA, stroke; CH, cerebral hemorrhage; Bx, biopsy performed; Aut, autopsy performed; ESRD, end-stage renal disease
There is also a striking family history of early onset, fatal cerebrovascular events. The patient’s great grandmother (I-1) died of a “stroke” (details unknown) in her thirties and her grandmother (II-1) died of a cerebral hemorrhage at age 34. Her aunt (III-6) died of subarachnoid hemorrhage at age 33 and had “membranoproliferative kidney disease” on autopsy (GFND had yet to be defined at this time). The inheritance pattern suggests that all three possessed the FN1 mutation. Though not a typical association, clustering of cerebral hemorrhage within GFND families has been described [2]. The three patients reported by Strom et al. also had events in their late twenties and thirties. Stroke in this condition could simply be due to the development of malignant hypertension from CKD. However, the young age of these individuals raises the possibility that an additional FN-associated process may be at play. Indeed, the index patient’s aunt (III-6) reportedly had good blood pressure control on a single anti-hypertensive medication prior to her death, most likely from an intracranial aneurysm rupture. No extra-renal FN deposition has been reported in the few cases where autopsy was performed [2].
On physical exam, blood pressure was 116/61 mm Hg, wt was 212 lbs and she had no peripheral edema. Labs were noted for serum Cr 0.7 mg/dL, albumin 4.2 g/dL and urine protein/creatinine ratio of 4 g/g. She had no hematuria, pyuria or casts upon microscopic examination. She had 3 g of proteinuria on a 24 h collection. Antinuclear antibody screen was negative, rheumatoid factor titer was < 10 IU/ml and rapid plasma reagin was non-reactive. C3 was 191 mg/dL (normal 79–152), C4 was 37 mg/dL (normal 16–38) and hemoglobin A1c was 5.6%. Hepatitis B surface antigen, surface antibody and core antibody testing was negative as were antibody levels for hepatitis C and human immunodeficiency virus. Serum protein electrophoresis was normal. Urine protein electrophoresis had 196 mg/dL of protein and staining consistent with glomerular proteinuria; no paraprotein was seen. Renal ultrasound showed 12.9 and 11.7 cm non-echogenic kidneys. Given persistent nephrotic range proteinuria and family history, a kidney biopsy was performed.
Kidney biopsy
Light microscopy revealed enlarged glomeruli with marked lobular mesangial expansion and slight to moderate mesangial hypercellularity (Fig. 2a). Capillary loops were narrowed or obscured by the increased mesangial material. No interstitial inflammation or infiltrate was noted and arteries and arterioles appeared normal. Fibronectin immunostain showed intense, predominantly mesangial staining, not seen in an amyloid nephropathy negative control. Immunofluorescence (IF) (not shown) revealed low level (+/– to 1 +) staining for IgA, IgG, lambda and C3 in mesangium and segmentally in capillary walls. C3 additionally had 1–2 + arterial and tubular basement membrane staining. Absent or marginal (neg to +/–) glomerular staining for IgM, kappa, C1q, albumin and fibrin was observed. Low-level interstitial fibrin staining was seen. Electron microscopy (EM) revealed massive mesangial and subendothelial deposits of dense granular material (Fig. 2b) containing rare ~ 20 nm fibrillary/tubular deposits (latter not shown). Moderate to severe effacement of epithelial foot processes was seen.
Fig. 2.
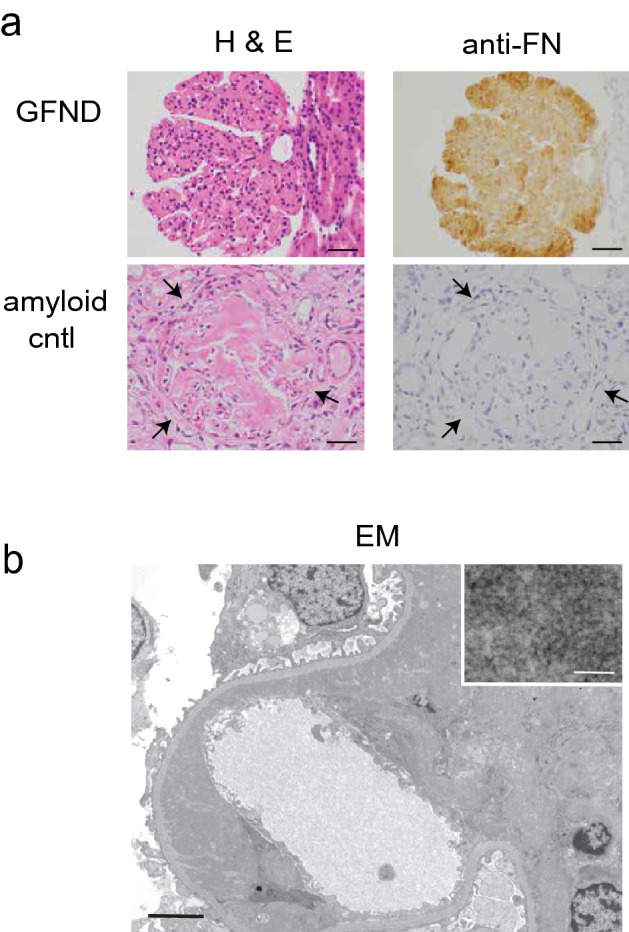
Kidney Biopsy. a Light microscopy. Hematoxylin and eosin staining for our GFND patient and an amyloid nephropathy control (left column). Scale markers 40 µm. Immunohistochemistry using anti-FN antibody reveals bright mesangial staining in our GFND patient but none in the amyloid nephropathy negative control (right column). High pH antigen retrieval solution (Dako) and EnVision FLEX Detection kit (Dako) were used. Primary antibody: mouse monoclonal anti-human FN (clone IST-4, 1:200; #F0916, Sigma-Aldrich). Scale markers 40 µm. b Electron microscopy shows marked mesangial and subendothelial deposition of dense granular material (original magnification × 4000; scale bar 4 µm) which is seen in greater detail in the representative inset (original magnification × 40,000; scale bar 100 nm)
Diagnosis
Kidney biopsy confirmed the diagnosis of GFND. Genotyping using published primers [10] revealed the Y973C missense mutation in FN1.
Clinical follow-up
The patient was started on lisinopril for persistent proteinuria which was titrated to 20–30 mg daily. Further increases were limited by hypotension. Despite this, her proteinuria increased to 4–6 g/g of Cr and serum Cr increased to 1.0 mg/dL (Fig. 3). The presence of foot process effacement and lack of significant interstitial fibrosis on her biopsy raised the possibility of steroid responsiveness. In addition, prednisone has shown some efficacy in patients with IgA nephropathy, another glomerular depositional disease, with persistent proteinuria despite maximal ACE inhibition [12, 13]. The patient was advised that steroids were an unproven therapy in GFND and the potential side effects were discussed. Given her worsening kidney function and proteinuria and the natural history of this disease, she opted to proceed. Based on a standard IgA protocol [14], she was started on prednisone 60 mg (~ 1 mg/Kg) daily for 2 mos and then tapered by ~ 0.2 mg/Kg every month for 6 mos of total therapy. Proteinuria decreased to below 1 g and remained ~ 1 g for more than 5 years (Fig. 3a). Serum Cr, which had been increasing, stabilized in the 1.2 mg/dL range (eGFR ~ 60 mL/min/1.73 m2) with treatment (Fig. 3b). No significant side effects were encountered.
Fig. 3.
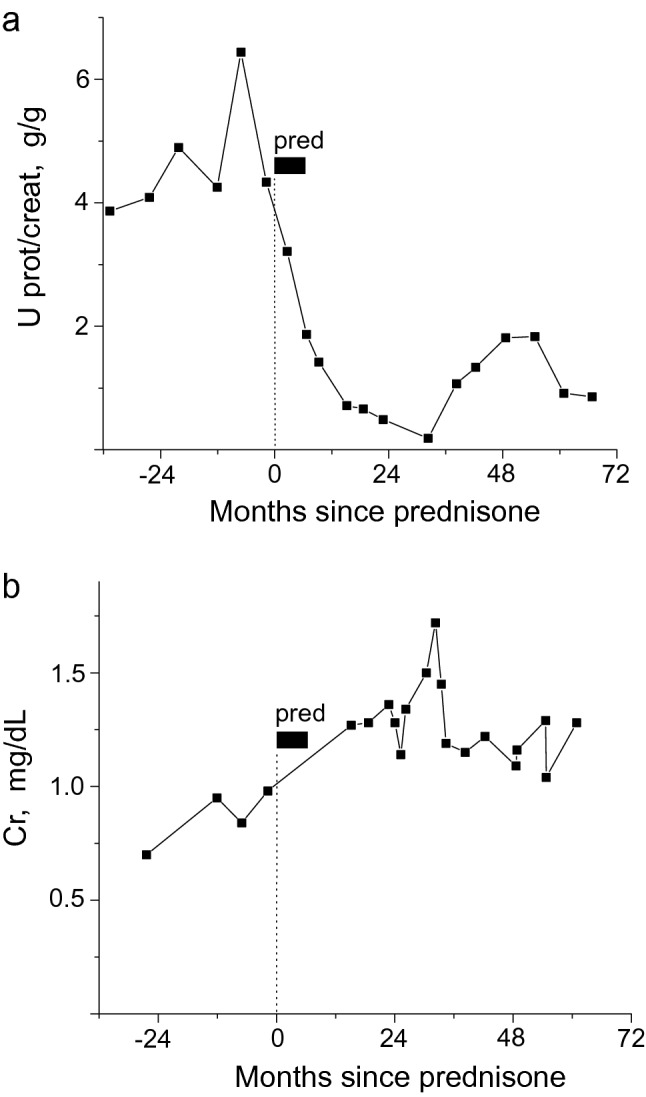
Prednisone reduced proteinuria a and stabilized serum creatinine b in our GFND patient. Prednisone was initiated at 60 mg daily and then tapered for 6 mos of total therapy (see text for details)
The patient’s paternal aunt (III-2) also underwent kidney biopsy around this time which showed GFND. She was treated with ACE inhibitors and initially, her nephrotic range proteinuria came down to the 2 g/g Cr range and her progressive CKD stabilized (serum Cr ~ 2.5 mg/dL). Unfortunately, her proteinuria eventually worsened, renal function declined and she in now on peritoneal dialysis and listed for transplant. Recently, the patient’s sister (IV-12) developed progressive CKD III, nephrotic range proteinuria and hypertension with kidney biopsy confirming GFND. We anticipate treating her with prednisone as well if proteinuria persists despite increased angiotensin receptor blocker (ARB) dosing.
Discussion
We show a significant and durable reduction in proteinuria and stabilization in serum creatinine in a young woman with GFND and nephrotic range proteinuria with relatively preserved renal function.
There are only a couple of limited reports of the use of immunosuppressive therapy in GFND. Prednisolone with mizoribine (for 1 year) was used in an atypical, sporadic case of fibronectin glomerulopathy [15] and a “multi-drug regimen” (details not provided) was used in a GFND patient that led to stable creatinine and ~ 2 g/day of proteinuria after 20 mos [9].
This is, therefore, one of the first published reports of effective treatment with immunosuppressive therapy and to our knowledge (based on negative PubMed searches) the first report of effective prednisone monotherapy for this condition which often progresses to ESRD. This is, of course, a single case report, so additional experience with steroids in GFND is needed before any more robust conclusions can be drawn. Since this is a rare disease, randomized controlled trials may not be feasible but tracking experience via a registry may be helpful. An advantage of this treatment regimen is that it is of relatively short duration. Nevertheless, exacerbation of diabetic control, increased risk of infections, gastrointestinal complications and bone resorption are of concern and appropriate Pneumocystis pneumonia and gastrointestinal prophylaxis should be addressed.
While marked glomerular FN deposition characteristic of GFND was the predominant finding on kidney biopsy, low level IF staining for IgA was also noted, raising the possibility of superimposed IgA nephropathy which could be steroid responsive. However, a clinically significant component of IgA nephropathy is less likely given the relatively weak IF signal, absence of demonstrable immune deposits on EM corresponding to the IF findings (though admittedly, these could be obscured by FN) and absence of microhematuria.
This kindred harbors the Y973C mutation, the most common FN1 mutation in the series reported by Castelletti et al. [10]. It is located within a heparin-binding domain (Hep-III) and these domains are believed to regulate FN assembly into organized fibrils [16]. The mechanism by which this mutation leads to glomerular deposition of FN is unknown. The mutation adds an additional cysteine to this region which may result in disulfide bond formation, leading to altered protein folding and function [10].
There are multiple potential explanations for the benefit of steroids suggested by this case report. One possibility is that deposition of fibronectin may lead to a steroid-responsive inflammatory reaction mediated by proliferating mesangial cells as has been observed with IgA nephropathy [12]. Alternatively, steroids may alter the expression of proteins that affect the glomerular filtration barrier as has been seen in models of minimal change disease [17]. Better knowledge of the pathogenesis of GFND may lead to more targeted therapies with fewer systemic side effects. Finally, while GFND is rare, FN deposition is seen in several common conditions such as diabetic nephropathy, lupus nephritis and IgA nephropathy [18–20] so that understanding GFND may provide insight into disease pathogenesis in these more prevalent conditions.
In conclusion, a steroid trial should be considered in a GFND patient with nephrotic range proteinuria, despite maximal ACE or ARB treatment, in the absence of considerable interstitial fibrosis or advanced CKD.
Acknowledgements
The authors would like to thank family members for their helpfulness in providing clinical history and family contact information necessary to put together the pedigree (Fig. 1). We would also like to thank Sierra Kovar and Karen Vanderbilt (Pathology and Laboratory Medicine, University of Rochester) as well as Chris Larsen (Arkana Laboratories, Little Rock, AR) for technical assistance with the fibronectin immunohistochemistry.
Declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Ethical standards
The present report was performed in accordance with the ethical standards of the institutional research committee and with the Helsinki declaration.
Informed consent
Written consent was obtained from the patient discussed and documentation is available for review upon request.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mazzucco G, Maran E, Rollino C, Monga G. Glomerulonephritis with organized deposits: a mesangiopathic, not immune complex-mediated disease? A pathologic study of two cases in the same family. Hum Pathol. 1992;23(1):63–68. doi: 10.1016/0046-8177(92)90013-S. [DOI] [PubMed] [Google Scholar]
- 2.Strom EH, et al. Glomerulopathy associated with predominant fibronectin deposits: a newly recognized hereditary disease. Kidney Int. 1995;48(1):163–170. doi: 10.1038/ki.1995.280. [DOI] [PubMed] [Google Scholar]
- 3.Gemperle O, Neuweiler J, Reutter FW, Hildebrandt F, Krapf R. Familial glomerulopathy with giant fibrillar (fibronectin-positive) deposits: 15-year follow-up in a large kindred. Am J Kidney Dis. 1996;28(5):668–675. doi: 10.1016/S0272-6386(96)90247-4. [DOI] [PubMed] [Google Scholar]
- 4.Assmann KJ, Koene RA, Wetzels JF. Familial glomerulonephritis characterized by massive deposits of fibronectin. Am J Kidney Dis. 1995;25(5):781–791. doi: 10.1016/0272-6386(95)90555-3. [DOI] [PubMed] [Google Scholar]
- 5.Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115(Pt 20):3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- 6.Carsons SE. Fibronectin in health and disease. Boca Raton: CRC Press Inc; 1989. [Google Scholar]
- 7.Otsuka Y, et al. A recurrent fibronectin glomerulopathy in a renal transplant patient: a case report. Clin Transplant. 2012;26(Suppl 24):58–63. doi: 10.1111/j.1399-0012.2012.01644.x. [DOI] [PubMed] [Google Scholar]
- 8.Niimi K, Tsuru N, Uesugi N, Takebayashi S. Fibronectin glomerulopathy with nephrotic syndrome in a 3-year-old male. Pediatr Nephrol. 2002;17(5):363–366. doi: 10.1007/s00467-002-0833-2. [DOI] [PubMed] [Google Scholar]
- 9.Brcic I, Brcic L, Kuzmanic D, Coric M, Coric M. Fibronectin glomerulopathy in a 34-year-old man: a case report. Ultrastruct Pathol. 2010;34(4):240–242. doi: 10.3109/01913121003783209. [DOI] [PubMed] [Google Scholar]
- 10.Castelletti F, et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc Natl Acad Sci U S A. 2008;105(7):2538–2543. doi: 10.1073/pnas.0707730105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtsubo H, et al. Identification of mutations in FN1 leading to glomerulopathy with fibronectin deposits. Pediatr Nephrol. 2016;31(9):1459–1467. doi: 10.1007/s00467-016-3368-7. [DOI] [PubMed] [Google Scholar]
- 12.Coppo R. Corticosteroids in IgA nephropathy: lessons from recent studies. J Am Soc Nephrol. 2017;28(1):25–33. doi: 10.1681/ASN.2016060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group: Clinical Practice Guidelines for Glomerulonephritis. Immunoglobulin A nephropathy. Kidney Int 2, 139–274, 2012.
- 14.Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized controlled clinical trial of corticosteroids plus ACE-inhibitors with long-term follow-up in proteinuric IgA nephropathy. Nephrol Dial Transplant. 2009;24(12):3694–3701. doi: 10.1093/ndt/gfp356. [DOI] [PubMed] [Google Scholar]
- 15.Yoshino M, et al. Clinicopathological analysis of glomerulopathy with fibronectin deposits (GFND): a case of sporadic, elderly-onset GFND with codeposition of IgA, C1q, and fibrinogen. Intern Med. 2013;52(15):1715–1720. doi: 10.2169/internalmedicine.52.0046. [DOI] [PubMed] [Google Scholar]
- 16.Maqueda A, Moyano JV, Hernandez Del Cerro M, Peters DM, Garcia-Pardo A. The heparin III-binding domain of fibronectin (III4-5 repeats) binds to fibronectin and inhibits fibronectin matrix assembly. Matrix Biol. 2007;26(8):642–651. doi: 10.1016/j.matbio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Chugh SS, Clement LC, Mace C. New insights into human minimal change disease: lessons from animal models. Am J Kidney Dis. 2012;59(2):284–292. doi: 10.1053/j.ajkd.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klemis V, et al. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017;91(6):1374–1385. doi: 10.1016/j.kint.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Mattii L, et al. Kidney expression of RhoA, TGF-beta1, and fibronectin in human IgA nephropathy. Nephron Exp Nephrol. 2005;101(1):e16–23. doi: 10.1159/000086035. [DOI] [PubMed] [Google Scholar]
- 20.Assad L, Schwartz MM, Virtanen I, Gould VE. Immunolocalization of tenascin and cellular fibronectins in diverse glomerulopathies. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;63(5):307–316. doi: 10.1007/BF02899277. [DOI] [PubMed] [Google Scholar]