

Abstract
The nuclear envelope compartmentalizes the eukaryotic genome, provides mechanical resistance, and regulates access to the chromatin. However, recent studies have identified several conditions where the nuclear membrane ruptures during interphase, breaking down this compartmentalization leading to DNA damage, chromothripsis, and kataegis. This review discusses three major circumstances that promote nuclear membrane rupture, nuclear deformation, chromatin bridges, and micronucleation, and how each of these nuclear catastrophes results in DNA damage. In addition, we highlight recent studies that demonstrate a single chromosome missegregation can initiate a cascade of events that lead to accumulating damage and even multiple rounds of chromothripsis.
Keywords: Chromothripsis, kataegis, nuclear membrane rupture, micronucleus, nuclear envelope, TREX1
1. Introduction
The nuclear envelope (NE) protects the eukaryotic genome by providing a physical barrier between the chromatin and cytosol. The major components of the NE are the nuclear membranes, which are contiguous with the ER and each other, nuclear pore complexes, which regulate large molecule transport, and the nuclear lamina (Figure 1). The nuclear lamina is a dense protein layer on the inner nuclear membrane comprised mainly of lamin filaments and NE transmembrane proteins. Most cells express four lamin proteins, lamin A, B1, B2, and C, which form independent meshworks [1,2]. The nuclear lamina has several critical functions. It regulates genome organization by binding heterochromatin, individual proteins function in major nuclear processes, including transcription, and DNA damage response [3], and together with the peripheral heterochromatin, it resists mechanical forces on the nuclear membrane [4]. Mutation or misexpression of nuclear lamina proteins disrupts nuclear lamina organization, are frequently observed in cancer cells, and cause a class of genetic diseases called laminopathies. Decreased mechanical resistance and cell signaling dysfunction were initially proposed to connect aberrant nuclear structure with disease pathologies [3]. However, recent studies have demonstrated that altered NE organization can also cause nuclear membrane rupture and repair, suggesting that loss of the barrier function could be a critical driver of DNA damage, genome evolution, and gene misexpression in disease.
Figure 1:
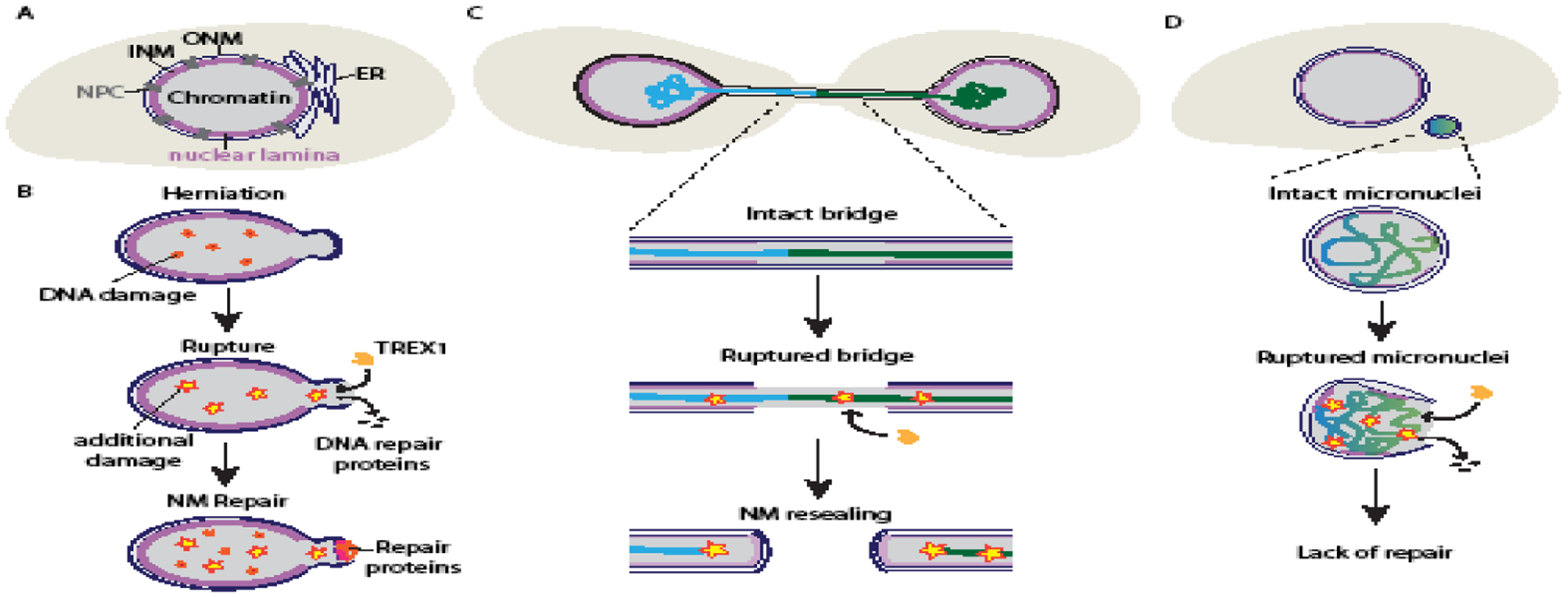
Mechanisms of nuclear membrane rupture and repair, and DNA damage.
(A) Major structures of the mammalian nucleus. The inner and outer nuclear membrane (INM and ONM) surround the chromatin and are contiguous with the ER and with each other at the nuclear pore complexes (NPCs). The nuclear lamina is a dense protein layer localized to the INM that includes transmembrane proteins and lamin filament meshworks. (B) Nuclear membrane rupture is often driven by mechanical deformation, which can disrupt the nuclear lamina and cause membrane blebs, chromatin herniation, and DNA damage. Membrane ruptures typically occur at nuclear lamina gaps and can result in DNA damage through loss of DNA damage repair proteins and infiltration of the TREX1 nuclease. Nuclear membrane (NM) ruptures are rapidly repaired by recruitment of chromatin binding proteins and the ESCRT-III membrane remodeling complex. (C) Chromatin bridges are formed from dicentric chromosomes are frequently depleted of nuclear lamina proteins and have elevated levels of nuclear membrane rupture. Membrane rupture permits TREX1 localization to the bridge, facilitating its breakage and potentially causing substantial DNA damage. (D) Micronuclei form around missegregated chromosomes and have a disorganized nuclear lamina and a high frequency of membrane rupture. Rupture causes DNA damage, loss of DNA damage repair factors, and localization of TREX1 to the exposed chromatin. Membrane repair is rarely observed in micronuclei.
Nuclear membrane rupture has been observed in a variety of contexts, both in vitro and in vivo, and in model organisms from fission yeast to mouse tissue (Figure 1). Nuclear membrane rupture is transient, with many ruptures resealing within minutes [5–7]. Longer ruptures lasting several hours are survivable [8–10], but failure to restore nuclear compartmentalization eventually results in cell death [10]. In cells with a well-organized nuclear lamina, membrane rupture requires significant stress on the NE. In contrast, depletion of nuclear lamina proteins or expression of lamin mutants causes nuclear membrane rupture in a range of physiological settings, including muscle contraction and cell migration [11]. Another cause of nuclear membrane rupture is chromatin missegregation into either a chromatin bridge or a micronucleus (MN). Chromatin bridges form when dicentric chromosomes are pulled towards opposing spindle poles during mitosis. MN form when chromosomes or chromosome fragments missegregate during mitosis and assemble a separate NE. These structures have a high frequency of NE defects and increased membrane instability [12–16]. In contrast to the rapid repair of the nuclear membrane, MN membrane ruptures frequently fail to repair causing the chromatin to persist in the cytosol until the next mitosis [13].
Membrane integrity loss in either the nucleus or an MN causes DNA damage, activation of pro-inflammatory signaling, and invasive phenotypes. This review will focus specifically on DNA damage, but a broader discussion of the consequences of nuclear rupture can be found in [11]. Membrane rupture of MN or chromatin bridges can cause the DNA to shatter and become the substrate for chromothripsis and kataegis in subsequent cell divisions [14,16–18]. Chromothripsis occurs when part of the genome, a chromosome arm up to a few chromosomes, fragments and is religated in a single event [19,20]. In kataegis a cluster of hypermutations occur near chromosome rearrangement sites [21,22]. Chromothripsis has been identified in a broad range of cancer types, frequently as an early event that alters expression of driver genes [23]. Recent studies have identified multiple mechanisms of DNA damage from NE defects, some of which even occur independently of membrane rupture [16]. In addition, a single missegregation event can initiate multiple rounds of missegregation, damage, and chromothripsis [16,24]. Our understanding of membrane rupture dependent and independent mechanisms that cause DNA damage in nuclear abnormalities is rapidly evolving and this review will summarize recent findings in this area.
2. Deformation induced nuclear membrane rupture and DNA damage
2.1. Mechanisms of interphase nuclear membrane rupture and repair
A typical nuclear membrane rupture occurs at a gap in the nuclear lamina when mechanical stress causes this area of unsupported membrane to undergo blebbing and chromatin herniation, followed by membrane rupture (Figure 1). There is a strong correlation between nuclear lamina disorganization and nuclear membrane disruption. Nuclear membrane rupture is more frequent in laminopathy and cancer cells [6,7] and overexpression of nuclear lamina proteins is sufficient to inhibit rupture [7,14]. Recent work has demonstrated that substantial compressive or internal pressure is required for nuclear membrane rupture in cells where the nuclear periphery strongly resists mechanical force, likely due to the necessity of disrupting the nuclear lamina as well as the membrane [25–30]. However, when the nuclear lamina is disorganized, the forces generated by perinuclear actomyosin bundles or chromatin decompaction become sufficient to initiate membrane rupture [31,32]. Although a general model of force versus NE organization is sufficient to explain the majority of nucleus ruptures, examples exist where one or more of these features are missing, including lamina gaps [32] and membrane blebs [33]. In addition, other factors including membrane curvature [34], lipid identity [35], membrane recruitment [36,37], and chromatin softening [38], likely impact whether the membrane ruptures. Thus, further studies are needed to identify how these conditions connect to each other, and which ones are critical for maintaining nuclear membrane stability in vivo.
One of the most striking features of nucleus membrane rupture is its transience. Current models of membrane repair have focused on similarities between interphase membrane repair and nuclear membrane recruitment at the end of mitosis [39]. Many NE proteins involved in post-mitotic recruitment and resealing of the nuclear membrane also accumulate at rupture sites and are required for efficient membrane repair [8,9,26,27,40,41]. One of these is the ESCRT-III membrane remodeling complex, which is required to seal small holes in the post-mitotic nuclear membrane, but causes nuclear membrane rupture when misexpressed [42,43]. Depending on the cell model, depletion of ESCRT-III proteins either severely delays membrane repair [26,27], or has little to no effect [8,41]. These data suggest that different repair mechanisms are used in different situations, possibly correlating with membrane hole size [39]. More studies will be needed to understand membrane repair mechanism choice and identify whether interphase membrane repair is mediated by additional post-mitotic compartmentalization mechanisms [44,45].
2.2. Nuclear membrane rupture induced DNA damage
DNA double-stranded breaks (DSBs) are observed upon nuclear membrane rupture, but how damage occurs, and the relative contributions of membrane rupture and deformation are still being determined [26,27,46–48]. One mechanism for rupture-dependent DNA damage is based on the observation that several DSB repair proteins take longer than expected to recompartmentalize after nucleus rupture [34,48]. In this model, DNA damage occurs from standard sources, such as free radicals [28], but the DNA is repaired more slowly than in non-ruptured nuclei [48]. However, other analyses see evidence of DSBs occurring concurrently with membrane rupture during confined cell migration [26,27,47], suggesting that DNA damage can be an active process.
One mechanism of active DNA damage is mistargeting of cytoplasmic nucleases to chromatin during nuclear membrane rupture. When cancer cells are compressed, either mechanically or by high density growth conditions, nuclear membrane rupture increases and coincides with increased DSBs. These DSBs require TREX1 (three-prime repair exonuclease 1), an ER-associated nuclease that generates ssDNA by resection and normally degrades cytosolic DNA to suppress autoinflammation [49,50]. After nuclear membrane rupture, TREX1 accumulates on the exposed chromatin and co-localizes with DNA damage markers [49]. Interestingly, TREX1 does not localize to the nucleoplasm of ruptured nuclei, but to membrane inclusions at the nuclear periphery [49], similar to those observed after activation of ESCRT-III at the inner nuclear membrane [35,42,43,51]. These ESCRT-III foci drive accumulation of ER membranes, which are critical for TREX1 activity [52]. This suggests that the mechanism of nuclear membrane resealing could impact the extent of DNA damage.
Because most analysis of DNA damage after nuclear membrane rupture has been studied in highly confined cells, a persistent question has been how much of the damage is due to nuclear deformation. A recent paper addressed this question by observing the timing of DNA damage and nuclear membrane rupture during confined cell migration in a large group of transformed and normal cell lines. The authors identified several cell lines that acquired DSBs prior to or in the absence of rupture [47]. In these cell lines, DSBs only occurred in S/G2 phase and correlated with deformation induced replication stress. In contrast, nucleus rupture induced DSBs occurred in all the cell lines regardless of cell cycle stage [47]. These results strongly suggest that nucleus deformation and loss of compartmentalization have independent and additive effects on DNA damage.
Confined cell migration causes nuclear membrane rupture and heritable changes in the genome [48], but the extent of genome rearrangements attributable to rupture is unclear. Sequencing of single cell clones after several rounds of constricted migration found a wide variation of copy number gains and losses compared to control cells [48]. However, these copy number changes were accompanied by other indications of genome instability, including MN, that could also be a source of structural variation. The tendency of nuclear lamina disruption or deformation to lead to additional defects in chromosome missegregation has been a challenge with other analyses of rupture-induced DNA damage as well [53]. In support of a model where a single burst of nuclear rupture likely has minimal effects on genome rearrangements, a single round of confined migration did not generate detectable variation between grown-out clones [48]. One question for future studies is whether increasing rupture duration would be sufficient to cause complex rearrangements, or whether additional changes to the underlying chromatin are necessary.
3. Chromatin bridge resolution and DNA damage
3.1. Dicentric chromosomes cause genome instability
Telomere shortening typically protects against tumor development, but aberrant recognition of telomere ends by DNA damage repair pathways causes telomere fusions that generate dicentric chromosomes and chromatids in a process called telomere crisis. Telomere crisis is a common occurrence in cancer and causes many pro-proliferative changes in genome structure, including amplification of cancer drivers by breakage-fusion-bridge (BFB) cycles [54]. Dicentric chromosomes can also form when DSBs on different chromosomes are ligated together and when neo-centromeres arise spontaneously [55]. Dicentric chromosomes/chromatids pulled towards opposing spindle poles during anaphase form a chromatin bridge that breaks during the next cell cycle [14,16]. Analysis of complex genome rearrangements after chromatin bridge breakage has been most heavily studied in telomere crisis models [14,16,56–58], but these mechanisms likely apply to all types of dicentric chromosomes.
3.2. Breaking chromatin bridges
New analyses of chromatin bridge breakage have shed light on how these events can lead to chromothripsis. Two recently proposed mechanisms for chromatin bridge breakage are nuclear membrane rupture followed by TREX1 exonuclease digestion [14,56,59], and mechanical stretching and breakage of bridges by cell migration and actomyosin forces [16,56] (Figure 2). In support of the nuclear membrane rupture model, described in Maciejowski et al., (2015), chromatin bridge breakage is frequently preceded by membrane rupture and TREX1 localization to the bridged chromatin. Loss of either membrane rupture or TREX1 inhibits ssDNA generation on the bridge, which significantly delays bridge resolution. These data suggest that nuclear membrane rupture is not required for bridge breakage, but substantially increases its efficiency. Currently, how the ssDNA breaks to resolve the bridge is unknown, as is the identity of the nickase that enables TREX1 resection in this nuclear compartment.
Figure 2:
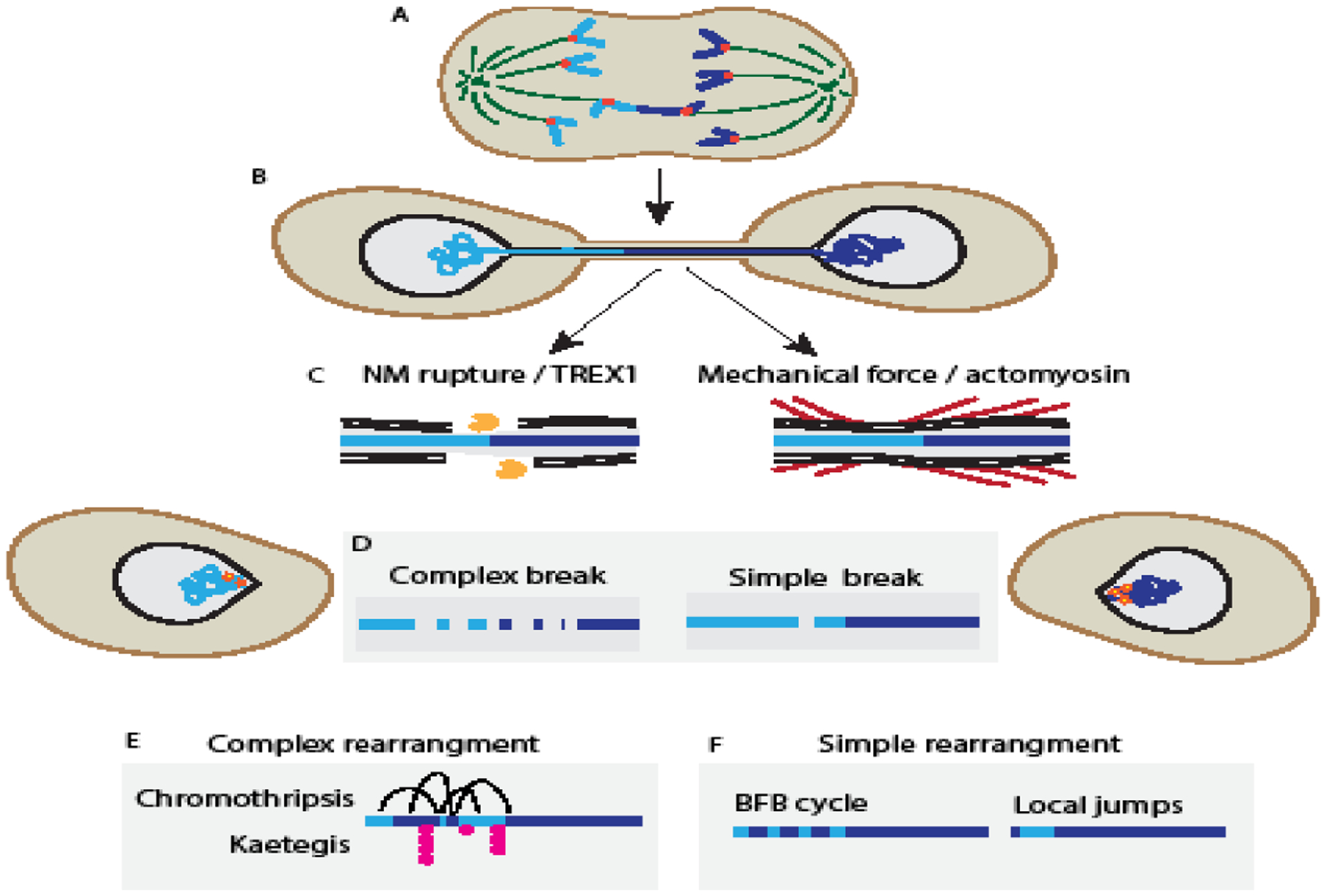
Chromatin bridge breakage causes complex and simple genome rearrangements
(A) Telomere crisis leads to telomere fusion and dicentric chromosomes, which form a (B) chromatin bridge when the two centromeres are pulled towards opposing spindle poles during mitosis. (C) Two models for interphase bridge resolution are left: nuclear membrane (NM) rupture causes bridge resection by the 3’-exonuclease TREX1, generating tracts of ssDNA, or right: cell migration causes bridge breakage through actomyosin-based forces (red). (D) Left: TREX1-dependent bridge breakage is associated with extensive DNA damage and chromatin fragmentation. Right: Whereas, mechanical breakage shows limited DNA damage, including simple breaks. (E) Chromosome fragmentation during bridge breakage results in chromothripsis from end-joining repair pathways and ssDNA exposure results in kataegis from APOBEC3B editing. (F) Limited DNA damage during bridge breakage causes highly local rearrangements, including local jumps and small insertions, and initiate breakage-fusion bridge (BFB) rearrangements.
An independent model of chromatin bridge breakage, proposed by Umbreit et al., (2020), found that it requires the forces generated by cell migration. Using similar models to Maciejowski et al., (2015), the authors observed that chromatin bridge breakage occurred independently of nuclear membrane rupture and that deleting TREX1 had no effect on the probability of bridge breakage. Instead, the authors observed that interphase bridge breakage was higher in migrating cell types and demonstrated that the actomyosin network is essential for this process. Because nuclear membrane rupture is also driven by actomyosin forces [11], a critical experiment was showing that increasing the number of rupturing nuclei did not increase the overall frequency of bridge breakage [16]. Together these data suggest that actomyosin, perhaps concentrated at focal adhesions [16], is sufficient to break chromatin bridges. What determines whether a chromatin bridge breaks by mechanical force alone or requires TREX1 is not understood but could correspond to the amount of chromatin in the bridge. For example, bridges containing multiple dicentric chromatids may require TREX1 digestion in addition to mechanical force to resolve them [56].
3.3. DNA damage from chromatin bridge formation and breakage
Sequence analysis of tumor samples and in vitro chromothripsis models identifies events where BFB is intertwined with chromothripsis and kataegis, consistent with telomere crisis being a significant mechanism of complex chromatin rearrangements [24,57,60]. The mechanism of bridge breakage may be a key determinant of the extent of rearrangement, as evident by the different genomic signatures in different studies of telomere crisis [14,16] (Figure 2). Cell populations sequenced several cell cycles after telomere crisis show a high frequency of chromosome arm-level chromothripsis and kataegis at breakpoints [14]. However, daughter cells sequenced right after bridge breakage found simple rearrangements and limited fragmentation at most break sites [16]. These simple patterns are also observed when TREX1 is knocked out [14], suggesting mechanical breakage leads to simple breaks and TREX1-associated breakage leads to fragmentation. TREX1 generates tracts of ssDNA, which can be hypermutated by APOBEC3B, a nuclear cystine deaminase. Interestingly hypermuation also increases the number of rearrangements at bridge break sites [14], suggesting that TREX1 initiates a cascade of DNA damage events. Critical questions for TREX1 dependent chromothripsis include how APOBEC3B increases DNA damage and how ssDNA generated by TREX1 is converted to dsDNA in rearranged chromosomes.
If a chromatin bridge avoids DNA damage during breakage, it can still be fragmented during the next mitosis or after missegregation into an MN. DNA replication is impaired in chromatin bridges both before and after breakage, and this under replicated DNA recruits damage response and replication markers in the next mitosis [16]. Single-cell sequencing of chromatin bridge-containing cells after this second mitosis showed an increased number of rearrangements at bridge breakpoints [16]. In addition, bridged chromatin has a high probability of being micronucleated in these “grand-daughter” cells and subject to additional DNA fragmentation events (see section 4) [16,24].
Analysis of rearrangement breakpoints in telomere crisis models of chromothripsis suggests that non-homologous end joining (NHEJ) and microhomology mediated end joining (MMEJ) are the predominant reassembly pathways [14,16,56]. Signatures of complex replication-associated DNA reassembly are also observed at rearrangement sites after chromatin bridge breakage [16,61], suggesting that multiple repair pathways can act at these broken DNA ends. The conditions favoring different DNA repair mechanisms are currently undefined, but could depend on cell cycle state and relative repair pathway activity in different cell types.
4. Micronuclei rupture and DNA damage
4.1. Introduction to micronuclei (MN)
The two major pathways of MN formation in human cells are whole chromosome missegregation, due to improper kinetochore-spindle attachments, and missegregation of chromatin fragments, due to DNA replication stress or unrepaired DNA damage. Most studies on MN membrane stability and DNA damage have focused on whole chromosome missegregation pathways, but both pathways are observed in vivo [62]. Although intact MN frequently appear structurally normal, recruitment or maintenance of nuclear pore complexes and nuclear lamina proteins can be severely compromised and major nuclear functions, including transcription and DNA replication, are reduced or delayed [13,15,17,63]. One complication in interpreting these analyses is that chromosome specific differences in heterochromatin amount and gene expression levels mean that individual MN will have a wide range of normal transcription levels and DNA replication timing. Thus, future studies comparing nuclear and micronuclear functioning for individual chromosomes will be critical to define how micronucleation impacts their function.
4.2. Mechanism of MN rupture
Similar to membrane rupture in the nucleus, MN membrane rupture occurs at gaps in the nuclear lamina and is inhibited by overexpressing lamin proteins [13]. MN have a high frequency of lamina gaps compared to the nucleus [13], and these are correlated with high membrane curvature and defective post-mitotic NE recruitment [15,28,64,65]. Lamina defects can appear hours before the MN ruptures [13], indicating that other factors trigger loss of MN integrity. In the nucleus, rupture coincides with increased force on the membrane, frequently from contractile actomyosin assemblies [11]. However, actomyosin inhibition has limited effects on MN rupture [15,31], suggesting that internal pressure from the chromatin or nucleoplasm may drive MN rupture rather than external mechanical force. A critical area of future research will be investigating how other factors that contribute to membrane stress, including lipid composition and chromatin compaction [32,35,64] impact membrane stability.
Unlike the nucleus, MN rarely repair their membrane after rupture [11,13]. Although NE proteins, including ESCRT-III, accumulate on ruptured MN (Figure 3), additional repair steps fail to occur [9,15,42,51]. One explanation for this is that over-recruitment of ESCRT-III in ruptured MN inhibits membrane repair [42]. Unlike the primary nucleus, where ESCRT-III binding is transient, most MN show persistent recruitment of this complex; the exception are the few MN that go on to repair their membrane [42,51]. However, knockdown of ESCRT-III only showed a modest increase in repaired MN [42], strongly suggesting that other factors inhibiting repair have yet to be uncovered.
Figure 3:
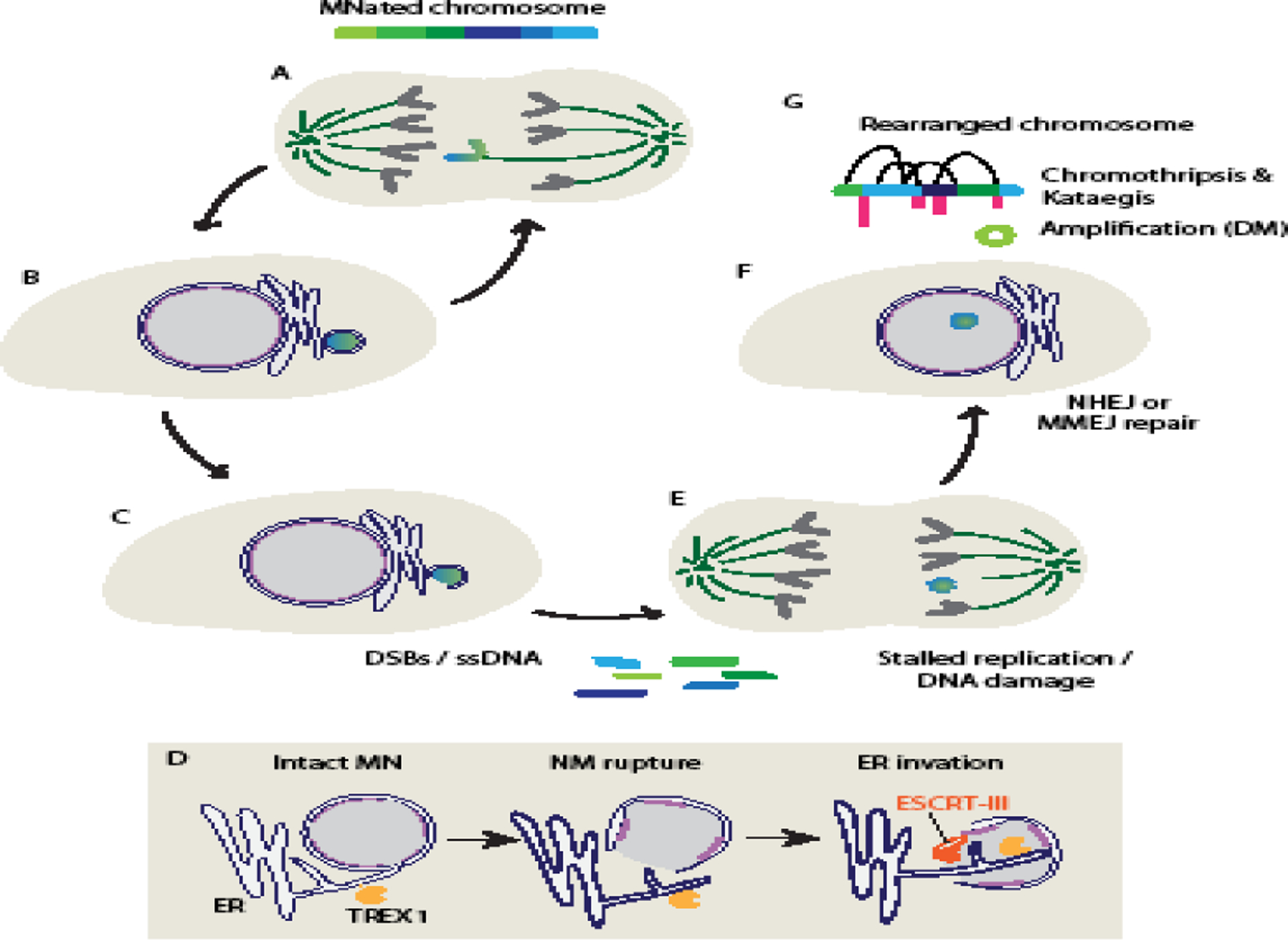
Micronucleation causes complex genome rearrangements
(A - B) Micronuclei (MN) form when missegregated chromosomes recruit an independent nuclear membrane at the end of mitosis. This chromatin is prone to continued missegregation and frequently forms an MN in the next cell cycle as well. (C) Interphase MN rupture causes both double-stranded DNA breaks (DSBs), likely from replication fork collapse, and ssDNA generation that lead to chromosome fragmentation. (D) Upon nuclear membrane (NM) rupture in MN, overactivation of the ESCRT-III membrane remodeling complex drives ER invasion and activation of the TREX1 exonuclease on MN DNA, leading to ssDNA accumulation and DSBs. (E) Many MN fail to finish DNA replication prior to mitosis and acquire additional DNA damage upon nuclear envelope breakdown. (F) When the fragmented chromatin is surrounded again by nucleoplasm, the fragments are reassembled by the end-joining DNA damage repair pathways, NHEJ and MMEJ, with some contributions from additional replication-based mechanisms. (G) The result of these events is a high frequency of chromothripsis, sometimes accompanied by kateagis. In addition, unassembled fragments can circularize to become double minutes (DMs) and undergo repeated rounds of amplification. Abbreviations: NHEJ, non-homologous end-joining, MMEJ: microhomology-mediated end joining.
4.3. NE defects and membrane rupture result in MN DNA damage
Membrane rupture precedes formation of DSBs and ssDNA in MN, which undergo chromosome fragmentation, chromothripsis, and kataegis at a high frequency [13,17,18,52] (Figure 3). However, the molecular mechanisms of DNA damage in MN, and whether membrane rupture is required, are still being elucidated. MN that rupture during DNA replication show a massive accumulation of the DSB marker gH2AX [12,13,17], consistent with DNA fragmentation after replication fork collapse. DNA replication in MN is frequently delayed or reduced compared to the nucleus, such that intact MN are often still replicating when the cell enters mitosis [12,16]. Similar to chromatin bridges, this under replicated DNA shows evidence of mitotic replication and DNA damage, which leads to a slight increase in chromothripsis frequency in the next cell cycle [16]. These data suggest that even if membrane rupture is inhibited, micronucleation is sufficient to cause chromosome rearrangements.
The ssDNA binding complex, RPA, is also enriched on ruptured MN, in a cell cycle independent manner [12,15,42,52]. RPA co-localizes with ESCRT-III foci on ruptured MN and its recruitment is dependent on TREX1 nuclease activity [42,52]. Because TREX1 activity is dependent on ER membrane-binding, a current model of ssDNA generation on ruptured MN is that unrestrained ESCRT-III activity causes accumulation of ER membrane, containing TREX1 and its nickase, APE-1, on the exposed MN chromatin [52]. Knockout of TREX1 also decreases DSBs in ruptured MN [52], but how the ssDNA is broken is still unknown. One suggestion, based on observations of chromatin compaction in ruptured MN, is that torsional stress drives ssDNA conversion to DSBs [13,42]. Alternatively, ssDNA could persist until mitosis and break due to mitotic chromatin condensation [66].
4.4. Mechanisms of DNA reassembly after micronucleation
After mitosis, chromatin fragments from ruptured MN are reincorporated into either the nucleus or an MN, which allows DNA repair factors to bind to the fragments initiating chromothripsis [12,13,17,66]. Analysis of chromothriptic breakpoints from several systems suggest that the majority of MN fragments are reassembled by NHEJ or MMEJ, similar to chromatin bridges [17,18,65]. In support of NHEJ being a critical reassembly pathway, loss of NHEJ activity significantly decreases the rearrangement frequency of micronucleated chromosomes and increases the number of DNA fragments visible in subsequent mitoses [18,24,66]. A small proportion of breakpoints have small insertions consistent with a replication-dependent repair pathway, but the identity of this pathway is unknown [17,18,65]. In addition, some breakpoints show evidence of kataegis [18], consistent with ssDNA being a DSB intermediate in some conditions. Examples of chromothripsis events with all of these reassembly signatures have been identified in human cancers [19,23,58,67–69]. A question for future studies will be whether there are biases in DNA damage reassembly mechanisms depending on the specific cancer type or the specific micronucleated chromosomes.
5. Cascading catastrophes accelerate genome evolution
One emerging property of nuclear membrane rupture associated DNA damage is that complex rearrangements often arise through multiple rounds of aberrant events. For example, chromatin trapped in bridges frequently becomes micronucleated in the next cell cycle [16,24,70], likely due to defective centromere assembly [14,16,59]. While some DNA damage occurs when the bridge initially breaks, even greater damage can occur when the chromatin becomes micronucleated, leading to an increased frequency and complexity of rearrangements [16]. These fragmentation events can also occur before or after canonical BFB cycles and appear interwoven with BFB in chromothripsis examples from sequenced cancers [16,24,60].
Micronucleation can also initiate a cascade of mutational events. Similar to chromatin bridges, centromere assembly is often defective in MN and correlates with frequent re-missegregation into an MN or chromatin bridge [65,71,72]. Not only do these second-generation MN and bridges have the same risk of fragmentation as the original, DNA damage repair is likely inefficient in these compartments [12,16,28], possibly leading to substantial fragment loss or increased error-prone replication-associated repair. In addition, chromosome fragmentation can lead to additional rounds of rearrangement via the production of extra-chromosomal DNA. Unincorporated DNA fragments after chromothripsis can circularize into objects called double minutes and become highly amplified [17,24]. Multiple double minutes can then aggregate and segregate as MN and undergo an additional round of fragmentation [24]. Evidence of multiple rounds of chromothripsis on the same chromatin has been identified in whole genome analyses of patient tumors [23,68], consistent with these cascading events occurring in vivo and facilitating cancer evolution.
6. Conclusions and future directions
The combination of rigorous cell biology experiments defining new NE dynamics and advanced single-cell and whole genome sequencing analysis methods has redefined the possibilities for studying complex genome evolution in cancer. Although many questions remain about the molecular details, recent studies linking DNA damage and reassembly to loss of nuclear membrane integrity, nuclear deformation, and nuclear assembly defects, have identified several high-frequency mechanisms of chromothripsis. These studies have shown that chromothripsis is a critical early event in many cancers that can affect driver genes, improve aneuploidy tolerance, and drive resistance to chemotherapeutics [23,24,65]. Understanding the mechanisms of chromothripsis may also be critical to understanding anti-cancer immunity. Recent work strongly suggests that the type and extent of DNA damage after nuclear membrane rupture could impact immune signaling and invasiveness. Large-scale chromosome rearrangements trigger targeting of the affected cell by the immune system [73], potentially enhanced by pro-inflammatory signaling from ruptured MN [74,75]. However, recent work also demonstrates that TREX1-dependent DNA damage actually limits innate immune pathway signaling [52], suggesting that the timing and degree of nuclear membrane alterations may determine the early immunogenic consequences of DNA fragmentation. In addition, nuclear membrane rupture is linked to upregulation of pro-invasion genes through non-canonical innate immune signaling and DNA damage, which may have similar dependencies [49,76]. Going forward, the challenge will be to identify which mechanisms of nuclear membrane rupture, DNA damage, and reassembly are active in vivo and what conditions favor cascading rearrangements. New studies examining the effects of altering membrane rupture frequency in animal models of early cancer development will be critical to link changes in nuclear membrane stability to disease.
Acknowledgements
We would like to thank members of Dr. Susan Parkhurst’s lab and the Hatch lab for critical reading of this manuscript. E.M.H was supported by the National Institutes of Health (R35GM124766-02); and the Rita Allen Foundation Scholars Program. A.E.M. was supported by the National Cancer Institute of the National Institutes of Health training grant (5T32CA009657-29)
Abbreviations:
- APOBEC3B
apolipoprotein B mRNA-editing enzyme
- ESCRT-III
endosomal sorting complexes required for transport III
- TREX1
three-primer repair exonuclease I
- gH2AX
gamma histone H2AX
- RPA
replication protein A
- APE-1
apurinic/apyrimidinic endonuclease 1
- INM
inner nuclear membrane
- NPC
nuclear pore complex
- ONM
outer nuclear membrane
- NE
nuclear envelope
- MN
micronucleus
- BFB
breakage-fusion-bridge
- NHEJ
non-homologous end-joining
- MMEJ
microhomology mediated end-joining
- DM
double minute chromosomes
- DSB
DNA double strand break
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Shimi T, Kittisopikul M, Tran J, Goldman AE, Adam SA, Zheng Y, et al. Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol Biol Cell 2015;26:4075–86. 10.1091/mbc.e15-07-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K, et al. The molecular architecture of lamins in somatic cells. Nature 2017;543:261. 10.1038/nature21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wong X, Stewart CL. The Laminopathies and the Insights They Provide into the Structural and Functional Organization of the Nucleus. Annu Rev Genom Hum G 2020;21:263–88. 10.1146/annurev-genom-121219-083616. [DOI] [PubMed] [Google Scholar]
- [4].Stephens AD, Banigan EJ, Marko JF. Chromatin’s physical properties shape the nucleus and its functions. Curr Opin Cell Biol 2019;58:76–84. 10.1016/j.ceb.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Noronha CMC de, Sherman MP, Lin HW, Cavrois MV, Moir RD, Goldman RD, et al. Dynamic Disruptions in Nuclear Envelope Architecture and Integrity Induced by HIV-1 Vpr. Science 2001;294:1105–8. 10.1126/science.1063957. [DOI] [PubMed] [Google Scholar]
- [6].Vos WHD, Houben F, Kamps M, Malhas A, Verheyen F, Cox J, et al. Repetitive disruptions of the nuclear envelope invoke temporary loss of cellular compartmentalization in laminopathies. Hum Mol Genet 2011;20:4175–86. 10.1093/hmg/ddr344. [DOI] [PubMed] [Google Scholar]
- [7].Vargas JD, Hatch EM, Anderson DJ, Hetzer MW. Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 2012;3:88–100. 10.4161/nucl.18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Robijns J, Molenberghs F, Sieprath T, Corne TDJ, Verschuuren M, Vos WHD. In silico synchronization reveals regulators of nuclear ruptures in lamin A/C deficient model cells. Sci Rep-Uk 2016;6:srep30325. 10.1038/srep30325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Young AM, Gunn AL, Hatch EM. BAF facilitates interphase nuclear membrane repair through recruitment of nuclear transmembrane proteins. Mol Biol Cell 2020;31:1551–60. 10.1091/mbc.e20-01-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chen NY, Yang Y, Weston TA, Belling JN, Heizer P, Tu Y, et al. An absence of lamin B1 in migrating neurons causes nuclear membrane ruptures and cell death. Proc National Acad Sci 2019;116:25870–9. 10.1073/pnas.1917225116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Maciejowski J, Hatch EM. Nuclear Membrane Rupture and Its Consequences. Annu Rev Cell Dev Bi 2020;36:1–30. 10.1146/annurev-cellbio-020520-120627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012;482:53. 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 2013;154:47–60. 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015;163:1641–54. 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu S, Kwon M, Mannino M, Yang N, Renda F, Khodjakov A, et al. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018;561:551–5. 10.1038/s41586-018-0534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Umbreit NT, Zhang C-Z, Lynch LD, Blaine LJ, Cheng AM, Tourdot R, et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020;368:eaba0712. 10.1126/science.aba0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature 2015;522:179. 10.1038/nature14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ly P, Brunner SF, Shoshani O, Kim DH, Lan W, Pyntikova T, et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet 2019;51:705–15. 10.1038/s41588-019-0360-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011;144:27–40. 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Korbel JO, Campbell PJ. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013;152:1226–36. 10.1016/j.cell.2013.02.023. [DOI] [PubMed] [Google Scholar]
- [21].Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 2014;14:786–800. 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Nik-Zainal S, Alexandrov LB, Wedge DC, Loo PV, Greenman CD, Raine K, et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012;149:979–93. 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD, et al. Pan-cancer analysis of whole genomes. Nature 2020;578:82–93. 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2020:1–5. 10.1038/s41586-020-03064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mistriotis P, Wisniewski EO, Bera K, Keys J, Li Y, Tuntithavornwat S, et al. Confinement hinders motility by inducing RhoA-mediated nuclear influx, volume expansion, and blebbingConfinement perturbs nuclear flux homeostasis. J Cell Biology 2019;218:4093–111. 10.1083/jcb.201902057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Denais CM, Gilbert RM, Isermann P, McGregor AL, Lindert M te, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science 2016;352:353–8. 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Raab M, Gentili M, Belly H de, Thiam H-R, Vargas P, Jimenez AJ, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016;352:359–62. 10.1126/science.aad7611. [DOI] [PubMed] [Google Scholar]
- [28].Xia Y, Pfeifer CR, Zhu K, Irianto J, Liu D, Pannell K, et al. Rescue of DNA damage after constricted migration reveals a mechano-regulated threshold for cell cycleDamage checkpoint in 3D migration. J Cell Biology 2019;218:2545–63. 10.1083/jcb.201811100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thiam H-R, Vargas P, Carpi N, Crespo CL, Raab M, Terriac E, et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat Commun 2016;7:10997. 10.1038/ncomms10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Deviri D, Pfeifer CR, Dooling LJ, Ivanovska IL, Discher DE, Safran SA. Scaling laws indicate distinct nucleation mechanisms of holes in the nuclear lamina. Nature Physics 2019;50:1. 10.1038/s41567-019-0506-8. [DOI] [Google Scholar]
- [31].Hatch EM, Hetzer MW. Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol 2016;215:jcb.201603053. 10.1083/jcb.201603053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, et al. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell 2018;29:220–33. 10.1091/mbc.e17-06-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen NY, Kim P, Weston TA, Edillo L, Tu Y, Fong LG, et al. Fibroblasts lacking nuclear lamins do not have nuclear blebs or protrusions but nevertheless have frequent nuclear membrane ruptures. Proc National Acad Sci 2018;115:201812622. 10.1073/pnas.1812622115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xia Y, Ivanovska IL, Zhu K, Smith L, Irianto J, Pfeifer CR, et al. Nuclear rupture at sites of high curvature compromises retention of DNA repair factorsHigh curvature correlates with nuclear rupture. J Cell Biology 2018;217:3796–808. 10.1083/jcb.201711161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thaller DJ, Tong D, Marklew CJ, Ader NR, Mannino PJ, Borah S, et al. Direct binding of ESCRT protein Chm7 to phosphatidic acid–rich membranes at nuclear envelope herniations. J Cell Biol 2021;220. 10.1083/jcb.202004222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lomakin AJ, Cattin CJ, Cuvelier D, Alraies Z, Molina M, Nader GPF, et al. The nucleus acts as a ruler tailoring cell responses to spatial constraints. Science 2020;370:eaba2894. 10.1126/science.aba2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Venturini V, Pezzano F, Castro FC, Häkkinen H-M, Jiménez-Delgado S, Colomer-Rosell M, et al. The nucleus measures shape changes for cellular proprioception to control dynamic cell behavior. Science 2020;370:eaba2644. 10.1126/science.aba2644. [DOI] [PubMed] [Google Scholar]
- [38].Nava MM, Miroshnikova YA, Biggs LC, Whitefield DB, Metge F, Boucas J, et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020;181:800–817.e22. 10.1016/j.cell.2020.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lusk CP, Ader NR. CHMPions of repair: Emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr Opin Cell Biol 2020;64:25–33. 10.1016/j.ceb.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Penfield L, Wysolmerski B, Mauro M, Farhadifar R, Martinez MA, Biggs R, et al. Dynein-pulling forces counteract lamin-mediated nuclear stability during nuclear envelope repair. Mol Biol Cell 2018;29:mbc.E17-06-0374. 10.1091/mbc.e17-06-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Halfmann CT, Sears RM, Katiyar A, Busselman BW, Aman LK, Zhang Q, et al. Repair of nuclear ruptures requires barrier-to-autointegration factor. J Cell Biology 2019;218:jcb.201901116. 10.1083/jcb.201901116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vietri M, Schultz SW, Bellanger A, Jones CM, Petersen LI, Raiborg C, et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat Cell Biol 2020;22:856–67. 10.1038/s41556-020-0537-5. [DOI] [PubMed] [Google Scholar]
- [43].Thaller DJ, Allegretti M, Borah S, Ronchi P, Beck M, Lusk CP. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. Elife 2019;8:e45284. 10.7554/elife.45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Samwer M, Schneider MWG, Hoefler R, Schmalhorst PS, Jude JG, Zuber J, et al. DNA Cross-Bridging Shapes a Single Nucleus from a Set of Mitotic Chromosomes. Cell 2017;170:956–972.e23. 10.1016/j.cell.2017.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Penfield L, Shankar R, Szentgyörgyi E, Laffitte A, Mauro MS, Audhya A, et al. Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J Cell Biol 2020;219. 10.1083/jcb.201908179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Earle AJ, Kirby TJ, Fedorchak GR, Isermann P, Patel J, Iruvanti S, et al. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat Mater 2020;19:464–73. 10.1038/s41563-019-0563-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Shah P, Hobson CM, Cheng S, Colville MJ, Paszek MJ, Superfine R, et al. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Curr Biol 2020. 10.1016/j.cub.2020.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, et al. DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Curr Biol 2017;27:210–23. 10.1016/j.cub.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nader GPF, Agüera-Gonzalez S, Routet F, Gratia M, Maurin M, Cancila V, et al. Compromised nuclear envelope integrity drives tumor cell invasion. Biorxiv 2020:2020.05.22.110122. 10.1101/2020.05.22.110122. [DOI] [PubMed] [Google Scholar]
- [50].Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008;134:587–98. 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Willan J, Cleasby AJ, Flores-Rodriguez N, Stefani F, Rinaldo C, Pisciottani A, et al. ESCRT-III is necessary for the integrity of the nuclear envelope in micronuclei but is aberrant at ruptured micronuclear envelopes generating damage. Oncogenesis 2019;8:29. 10.1038/s41389-019-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mohr L, Toufektchan E, Chu K, Maciejowski J. ER-Directed TREX1 Limits cGAS Recognition of Micronuclei. Ssrn Electron J 2020. 10.2139/ssrn.3606800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Karoutas A, Szymanski W, Rausch T, Guhathakurta S, Rog-Zielinska EA, Peyronnet R, et al. The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat Cell Biol 2019;21:1248–60. 10.1038/s41556-019-0397-z. [DOI] [PubMed] [Google Scholar]
- [54].Maciejowski J, Lange T de . Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Bio 2017;18:175–86. 10.1038/nrm.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Barra V, Fachinetti D. The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat Commun 2018;9:4340. 10.1038/s41467-018-06545-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Maciejowski J, Chatzipli A, Dananberg A, Chu K, Toufektchan E, Klimczak LJ, et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet 2020;52:884–90. 10.1038/s41588-020-0667-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mardin BR, Drainas AP, Waszak SM, Weischenfeldt J, Isokane M, Stütz AM, et al. A cell‐based model system links chromothripsis with hyperploidy. Mol Syst Biol 2015;11:828–828. 10.15252/msb.20156505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Garsed DW, Marshall OJ, Corbin VDA, Hsu A, Di Stefano L, Schröder J, et al. The Architecture and Evolution of Cancer Neochromosomes. Cancer Cell 2014;26:653–67. 10.1016/j.ccell.2014.09.010. [DOI] [PubMed] [Google Scholar]
- [59].Fouquerel E, Barnes RP, Uttam S, Watkins SC, Bruchez MP, Opresko PL. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol Cell 2019;75:117–130.e6. 10.1016/j.molcel.2019.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014;508:98–102. 10.1038/nature13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cleal K, Jones RE, Grimstead JW, Hendrickson EA, Baird DM. Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin. Genome Res 2019;29:737–49. 10.1101/gr.240705.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Guo X, Ni J, Liang Z, Xue J, Fenech MF, Wang X. The Molecular Origins and Pathophysiological Consequences of Micronuclei: New Insights into an Age-old Problem. Mutat Res Rev Mutat Res 2018;779:1–35. 10.1016/j.mrrev.2018.11.001. [DOI] [PubMed] [Google Scholar]
- [63].Afonso O, Matos I, Pereira AJ, Aguiar P, Lampson MA, Maiato H. Feedback control of chromosome separation by a midzone Aurora B gradient. Science 2014;345:332–6. 10.1126/science.1251121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sepaniac LA, Martin W, Dionne LA, Stearns TM, Reinholdt LG, Stumpff J. Micronuclei arising due to loss of KIF18A form stable micronuclear envelopes and do not promote tumorigenesis. Biorxiv 2020:2020.11.23.394924. 10.1101/2020.11.23.394924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kneissig M, Keuper K, Pagter MS de, Roosmalen MJ van, Martin J, Otto H, et al. Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. Elife 2019;8:e50292. 10.7554/elife.50292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 2016;19:68–75. 10.1038/ncb3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 2020;52:331–41. 10.1038/s41588-019-0576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020;578:112–21. 10.1038/s41586-019-1913-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Voronina N, Wong JKL, Hübschmann D, Hlevnjak M, Uhrig S, Heilig CE, et al. The landscape of chromothripsis across adult cancer types. Nat Commun 2020;11:2320. 10.1038/s41467-020-16134-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hoffelder DR, Luo L, Burke NA, Watkins SC, Gollin SM, Saunders WS. Resolution of anaphase bridges in cancer cells. Chromosoma 2004;112:389–97. 10.1007/s00412-004-0284-6. [DOI] [PubMed] [Google Scholar]
- [71].Soto M, García-Santisteban I, Krenning L, Medema RH, Raaijmakers JA. Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 2018;131:jcs.214742. 10.1242/jcs.214742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].He B, Gnawali N, Hinman AW, Mattingly AJ, Osimani A, Cimini D. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 2019;10:2660–74. 10.18632/oncotarget.26853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018;174:1347–60. 10.1016/j.cell.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466. 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548:461. 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553:467. 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]