

Abstract
Although often regarded as a protean illness with myriad clinical and imaging manifestations, neurosarcoidosis typically presents as recognizable syndromes that can be approached in a rational, systematic fashion. Understanding of neurosarcoidosis has progressed significantly in recent years, including updated diagnostic criteria and advances in treatment. The diagnosis of neurosarcoidosis is established by the clinical syndrome, imaging and histopathological findings, and exclusion of other causes. Mounting evidence supports the use of tumor necrosis factor inhibitors as an important addition to the therapeutic armamentarium, along with glucocorticoids and steroid-sparing cytotoxic immunosuppressants. In this narrative review, we summarize recent advances in the diagnosis and treatment of neurosarcoidosis.
Sarcoidosis is an immune-mediated disorder characterized by granulomatous inflammation of affected organs.1 Compact, well-formed, coalescent non- or minimally necrotizing epithelioid granulomas with scattered lymphocytes are pathologic hallmarks of the disease.2 Common organs affected include the lungs (90%), skin (∼15%), eye (10–30%), liver (20–30%), and lymph nodes (10%–20%).1,3
Neurologic involvement of sarcoidosis (neurosarcoidosis, NS) can involve the CNS or peripheral nervous system (PNS) or both and can cause substantial morbidity. NS was historically reported to occur in 5%–10% of all patients with sarcoidosis, although this number could reflect sampling bias from pulmonary sarcoidosis-focused cohorts. Clinically occult NS is identified at autopsy in 15%–25% of patients, and a 2017 study identified NS in 234/290 (34%) of patients with systemic sarcoidosis.4 Neurologic manifestations are the presenting syndrome in 50%–70% of patients with NS, and a meta-analysis of 1,088 patients with NS found that only 31% had systemic disease at presentation, whereas 84% eventually developed systemic manifestations.5 Although sarcoidosis is classically considered a multisystem disease, including by American Thoracic Society sarcoidosis diagnostic criteria,6 approximately 10%–20% of patients with NS do not have identifiable systemic sarcoidosis (termed isolated NS).7-9 Over the last several years, there have been important advances in diagnosis and treatment for sarcoidosis and NS. In this review, we discuss the pathophysiology, diagnosis, and treatment of NS, emphasizing clinically relevant updates.
Epidemiology
Sarcoidosis occurs globally, with the highest incidence in populations of northern European and recent African descent. In the United States, sarcoidosis incidence ranges from 35 to 80/100,000 among African Americans to 3–10/100,000 among Caucasians; some studies report a slight female predominance.3,10 In southwest England and south Wales between 1990 and 2002, NS incidence was estimated to be 1/100,000.11 Neurologic involvement is one of several factors associated with increased mortality in sarcoidosis.3
Pathogenesis
Sarcoidosis is an inflammatory condition characterized by a heightened cell-mediated, granulomatous response to as-yet unidentified antigens.12 Although there is no consensus about whether sarcoidosis represents an autoimmune disease,13 evidence is accumulating that there may be a role for autoimmunity in some forms of sarcoidosis, such as the self-limiting form of Lofgren syndrome associated with individuals who carry the HLA-DRB1*03 allele.14,15 The current understanding of sarcoidosis pathogenesis is primarily derived from studies of pulmonary sarcoidosis. To what extent these observations translate to NS and whether the immunology of NS differs from sarcoidosis in other organ systems are areas of active investigation.9
Genetics
Complex genetic patterns of inheritance contribute to sarcoidosis risk, and nearly all associated susceptibility genes identified to date are important in immune function.16,17 The risk of sarcoidosis is 80 times higher for monozygotic twins, 7 times higher in dizygotic twins, and 2–5 times higher for nontwin siblings of a person with sarcoidosis, and there is variability of risk for sarcoidosis susceptibility by race and ethnicity.17 Organ system–specific studies are scarce and limited by small sample size; however, there appear to be haplotypes that increase the risk of specific sarcoidosis phenotypes, such as Lofgren syndrome.12 In contrast, there appears to be little to no concordance between affected siblings with sarcoidosis in phenotypic patterns or outcomes.18
Environmental Factors
Environmental antigens, both infectious and noninfectious, have been postulated to influence the development of sarcoidosis by directly inducing a granulomatous inflammatory response or by indirectly effecting epigenetic and immunologic changes associated with sarcoidosis.12 Similar to emerging models with other autoimmune diseases, it is likely that a combination of environmental exposures in genetically susceptible individuals contributes to risk of sarcoidosis. Nondegradable antigens can become a nidus for granuloma formation,19 and accumulation of serum amyloid A has also been observed as a possible biomarker of systemic sarcoidosis.19
Mechanisms of Inflammation and Granuloma Formation
The accumulation of granulomas that disrupt normal tissue microarchitecture and cause organ dysfunction is a central feature of sarcoidosis.20 Granulomas are composed of epithelioid macrophages and CD4+ T helper cells surrounded by a ring of fibroblasts with scattered B cells and CD8+ cytotoxic T cells at the periphery. Along with other inflammatory cytokines, activated macrophages avidly express tumor necrosis factor (TNF)-α, which, through stimulating naive CD4+ T cells, appears critical for the formation and maintenance of sarcoid granulomas.21 T helper cells produce IL-2 and interferon gamma, which increase immune cell proliferation and cytotoxicity, respectively. Th17-polarized effector T cells have been detected in sarcoidosis granulomas, blood, and bronchoalveolar lavage fluids from patients with pulmonary sarcoidosis, and the Th17 effector response (including the production of interferon-γ) may influence the course and severity of sarcoidosis.22,23 Whole-blood gene expression profiling from patients with sarcoidosis demonstrates shared gene pathways with inflammatory responses to active tuberculosis,24 which may indicate that there is harnessing of similar inflammatory pathways in sarcoidosis and TB and explain in part why there can be some overlap in clinical phenotype.25
Clinical Manifestations and Physical Examination
Approximately 75% of patients with systemic sarcoidosis who develop NS will do so within 2 years of sarcoidosis diagnosis.26 In clinical practice, NS is often the presenting clinical syndrome, and sarcoidosis is first considered and diagnosed as part of the neurologic evaluation. Although the spectrum of neurologic manifestations of sarcoidosis is broad, these can be systematically organized into recognizable patterns with more common phenotypes (Table 1). Patterns of referral and care delivery among subspecialists likely also influence patterns of disease manifestations reported in the literature and observed in clinical practice. For example, a high incidence of optic neuritis was observed in a series of patients from an MS clinic.27
Table 1.
Select Diagnostic Considerations of Neurosarcoidosis by Phenotypic Manifestation

CNS Neurosarcoidosis
Meningitis
When NS affects the pachy- or leptomeninges, a subacute meningitis syndrome may develop that can persist to become chronic meningitis. Sarcoidosis leptomeningitis has a predilection for the base of the skull (basilar meningitis) and may extend to the spinal cord meninges. Leptomeningeal enhancement in sarcoidosis, often with a nodular component, may appear disproportionately worse on MRI than clinical symptoms might suggest. Leptomeningeal disease typically responds well to immunosuppressive treatment but may be complicated by cranial nerve dysfunction or seizures; communicating hydrocephalus is also characteristic, usually occurring secondary to chronic meningitis. Sarcoidosis less often causes pachymeningitis that must be distinguished from other causes of hypertrophic pachymeningitis.28
Cranial Neuropathy
Granulomatous infiltration of cranial nerve nuclei, fascicles, or nerves can produce cranial neuropathy, which is among the most commonly reported manifestations of NS. Multiple concurrent or serial cranial neuropathies should raise suspicion for NS. A subacute, progressive course is typical. Most patients (>80%) with cranial neuropathy from NS will have additional neurologic manifestations, and MRI often reveals gadolinium enhancement of the nearby leptomeninges.8 The reported frequency of individual cranial neuropathies varies form series to series, but the most commonly affected are the optic, facial, and vestibulocochlear nerves.8,26
NS can cause optic neuritis or perineuritis, which may involve the optic chiasm, or can produce compressive optic neuropathy from an infiltrating or mass-like lesion. Chiasmal involvement is often associated with sellar involvement given anatomic proximity.29 Optic neuritis is slightly more likely to be bilateral than unilateral in NS and visual recovery even with treatment can be poor.27 Facial nerve palsy that is recurrent, bilateral (simultaneous or sequential in 30%–40% in NS), or accompanied by other nearby cranial neuropathies should raise suspicion for NS as should leptomeningeal contrast enhancement and/or additional neurologic findings.8 Heerfordt syndrome (parotitis, facial nerve palsy, anterior uveitis, and low-grade fever) is a rare manifestation of facial nerve palsy in NS. Vestibulocochlear nerve involvement with vestibular dysfunction and/or hearing loss typically develops from leptomeningitis about the base of the brain. Other cranial neuropathies from NS occur less commonly.
Myelopathy
Sarcoidosis can affect the spinal cord via several mechanisms, including infiltration of the spinal cord parenchyma, leptomeninges, extradural space, or extraspinal tissues with compression of the spinal cord. Although historically reported as rare, contemporary studies have documented myelopathy in some 19%–26% of patients with NS, making it one of the more common neurologic manifestations.4,27 Characteristic imaging findings include nodular and linear leptomeningeal contrast enhancement associated with intraparenchymal T2 hyperintensity.30,31 Longitudinally extensive myelitis (LETM; ≥3 vertebral segments) from sarcoidosis is relatively common (75% of NS myelitis in 1 series)30 and must be distinguished from other causes of LETM. MRI clues that favor a diagnosis of sarcoidosis myelitis include a dorsal cord subpial pattern of gadolinium enhancement ≥2 spinal segments and persistence of enhancement for >2 months despite treatment.32 Central canal and dorsal subpial enhancement resembling a trident on axial MRI of the spinal cord also increases suspicion for NS diagnosis in patients with subacute myelitis.33 In a 2020 series, 4 main patterns of sarcoidosis myelitis were identified on MRI: longitudinally extensive myelitis (45%), short tumefactive myelitis (23%), meningitis/meningoradiculitis (23%), and anterior myelitis adjacent to disc degeneration (10%).34 Abnormal enhancement on MRI following the administration of gadolinium was observed in all but 1 patient in that series. As in the brain, mass-like spinal dural lesions can occur. Although cervical and thoracic cord pathologies are commonly discussed as usual sites of NS involvement in the literature, involvement of the conus medullaris and/or cauda equina also occurs.
Sellar Disease
Hypothalamic/pituitary involvement with consequent neuroendocrine dysfunction occurs in 10%–25% of cases. Endocrine dysfunction most often includes anterior hypopituitarism (LH/FSH 89%; TSH 67%; GH 50%; and ACTH 49%), hyperprolactinemia (49%), and diabetes insipidus (65%) and may be the presenting sign of NS in roughly half of patients with sellar disease.35 MRI findings include thickening and contrast enhancement of the pituitary gland or stalk sometimes with extension into the hypothalamus and often multifocal.36
Parenchymal Disease
Brain parenchymal NS may arise as a consequence of meningeal spread or vascular disease. Intraparenchymal mass-like lesions develop in roughly 15% of cases and can produce seizures and focal deficits. Characteristic MRI findings include contrast-enhancing lesions or T2 hyperintense and T1 isointense lesions that may or may not enhance (6%–37%). Cerebrovascular disease directly related to NS can also occur, including small, medium, or large vessel vasculitis or venulitis, vascular compression from granulomatous inflammation or mass lesion, or venous sinus thrombosis,37 but typical ischemic or hemorrhagic stroke in patients with known sarcoidosis can have many potential causes and may not be directly attributable to NS.
Encephalopathy
Subcortical encephalopathy including dementia may be seen in patients with NS. Nonspecific white matter T2/FLAIR hyperintense lesions without gadolinium enhancement are common and may be small and focal or larger, more diffuse lesions that resemble chronic vascular disease; their relationship to NS is uncertain as they are common even in the absence of NS, do not correlate with clinical disability in NS, nor do they abate with immunosuppressive treatment. In the absence of gadolinium enhancement, such lesions are unlikely to represent active NS and may sometimes reflect comorbid pathology, such as small vessel ischemic disease.
Neuropsychiatric Illness
Depression and other neuropsychiatric manifestations are nonspecific and may or may not be directly related to NS. Depression is reported in 60%–66% of patients with NS, and other neuropsychiatric manifestations including psychosis develop in up to 20%.26 Causes may include parenchymal NS, living with chronic illness, treatment complications (e.g., glucocorticoids), or other comorbidities.
PNS Sarcoidosis
The spectrum of PNS sarcoidosis includes both large and small fiber polyneuropathies or polyradiculoneuropathy with pure motor, sensory, or mixed sensorimotor features, including a Guillain-Barre–like syndrome. Peripheral neuropathy is reported in some 15%–20% of patients, although the true incidence may be higher. Peripheral nerve vasculitis may produce mononeuritis multiplex with axonal features. Symmetric chronic sensorimotor neuropathy with axonal features on EMG is reported to be the most common noncranial nerve peripheral neuropathy. The association of neuropathy with sarcoidosis is best supported with biopsy confirmation, whereas causation is less clear with mild axonal polyneuropathies given the myriad potential causes. When biopsy is performed, both the muscle and nerve should be sampled when feasible as this allows examination of both tissues and likely improves the diagnostic yield, as there can be subclinical muscle involvement in up to 90% of nerve biopsies38 (and this can also help to distinguish sarcoidosis from leprosy, as the latter only involves peripheral nerves and not muscle).39
Small Fiber Neuropathy
Small fiber neuropathy is common in systemic sarcoidosis, although the pathogenesis is unclear.40 For this reason, current consensus diagnostic criteria for NS conceptualize small fiber neuropathy as a paraneurosarcoidosis manifestation that occurs in association with sarcoidosis but may not necessarily result from direct granulomatous infiltration.41 Other etiologies of small fiber neuropathy should also be considered before concluding that small fiber neuropathy in individuals with sarcoidosis is necessarily sarcoidosis related.42 A large 2017 study found that some 25% of patients with sarcoidosis with confirmed small fiber had an alternative or additional possible etiology.40 EMG with nerve conduction studies is generally normal in small fiber neuropathy unless there is also a concurrent large fiber neuropathy; quantitative sudomotor axon testing or skin biopsy with decreased nerve fiber layer density can provide objective evidence of small fiber nerve pathology, but such testing is not specific for sarcoidosis or other causes.42
Muscle Disease
A 2018 study of 48 patients with symptomatic muscle sarcoidosis identified 4 patterns based on clinical presentation, EMG, and pathology. These include nodular (27%); smoldering (29%); acute, subacute, or progressive myopathic (35%); and combined myopathic and neurogenic pattern (10%). The clinical course differed depending on phenotype—the nodular pattern was relapsing and remitting, whereas the myopathic pattern may have a progressive course.43
Diagnosis and Differential Diagnosis
Differential Diagnosis
The differential diagnosis of NS is summarized in Table 1 and is heavily influenced by the presenting neurologic syndrome. Sarcoidosis is fundamentally a pathologic diagnosis, and biopsy from affected tissue is strongly recommended to support a final diagnosis. Rigorous exclusion of mimics is particularly important, and even with pathologic confirmation of granulomatous inflammation consistent with sarcoidosis, there is also a histologic differential diagnosis to consider. Granulomatous infections and, rarely, granulomatous reactions associated with lymphoma can have histopathologic findings similar to those seen in sarcoidosis. A multisystem granulomatous syndrome phenotypically similar to sarcoidosis (and sometimes NS) has been observed to develop in 8%–22% of patients with common variable immunodeficiency, a heterogenous group of antibody deficiencies characterized by hypogammaglobulinemia and symptoms of immunodeficiency including infection and autoimmunity.44-46 Rigorous clinical-radiologic phenotyping and follow-up remain critical even with biopsy confirmation. Clear worsening or lack of a partial response to immunosuppressive treatment in patients with probable or possible NS warrants diagnostic reconsideration.
Neurosarcoidosis Diagnostic Criteria
Updated consensus NS diagnostic criteria were published in 2018 and categorize patients based on diagnostic certainty into definite, probable, and possible NS based on pathologic and clinical features, emphasizing clinical phenotype and biopsy confirmation (Table 2).41
Table 2.
Clinical Criteria for a Diagnosis of Neurosarcoidosis (2018 Neurosarcoidosis Consortium Consensus41)

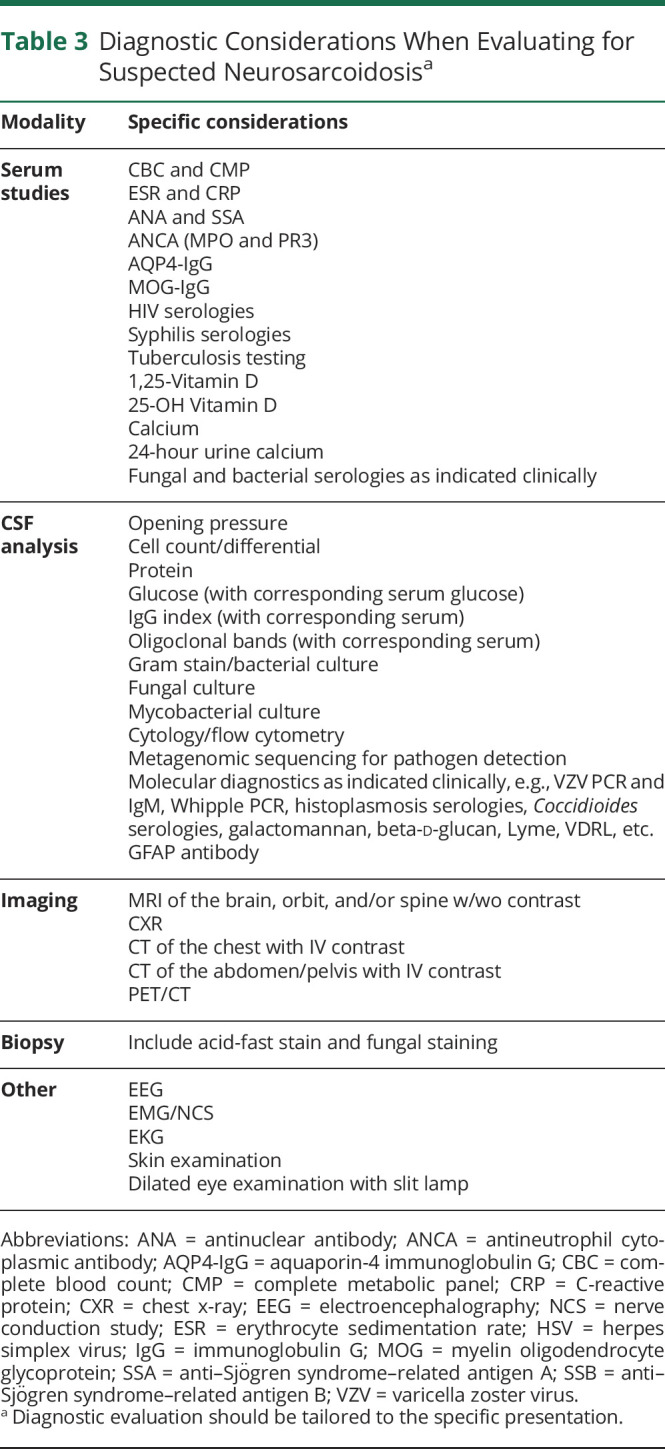
Serum Testing
Because neither sensitive nor specific serum tests for sarcoidosis diagnosis exist to date, the primary value of serum testing is to evaluate for alternative etiologies and for markers of other organ system manifestations of sarcoidosis (Table 3). Acute-phase reactants may be elevated but are non specific. Hypervitaminosis D and hypercalcemia are occasionally present and should prompt evaluation for hyperparathyroidism. Serum angiotensin-converting enzyme (ACE) levels are elevated in up to 60% of patients with pulmonary sarcoidosis, but are insensitive and nonspecific.47 A 2019 study identified serum soluble IL-2 receptor levels to be 88% sensitive and 85% specific for sarcoidosis compared with 62% and 76%, respectively, for ACE.48
Table 3.
Diagnostic Considerations When Evaluating for Suspected Neurosarcoidosisa
CSF Analysis
CSF analysis should be considered in the diagnostic evaluation of patients with CNS NS to evaluate for evidence of intrathecal inflammation and exclude alternative etiologies (Table 3), particularly when leptomeningitis is identified on MRI. Many patients with CNS sarcoidosis have abnormal CSF examinations, but no test is specific for NS. A mild to moderate pleocytosis (usually <100 cells/μL), with lymphocyte predominance and elevated protein, is typical; there may also be neutrophils present, particularly acutely; CSF eosinophils are rare. Isolated CSF protein elevation can be a marker of inflammation but is nonspecific, especially in the absence of MRI evidence of CNS sarcoidosis. NS is one of the few noninfectious etiologies that can cause hypoglycorrhachia, particularly in the context of leptomeningitis, but levels below 20 mg/dL should raise concern for an undiagnosed fungal, mycobacterial, or malignant etiology.49 Oligoclonal bands and elevated immunoglobulin G (IgG) index may be seen in ∼20%–40% of NS cases but are nonspecific.27,50 CSF ACE levels have poor sensitivity and specificity for NS.51 One series of patients with LETM from NMO, MS, or NS found that CSF hypoglycorrhachia and elevated CSF ACE levels were uncommon but exclusive to NS.32 One study including 43 patients with NS, 14 with multiple sclerosis, and 48 with other inflammatory disorders found that compared with MS and other inflammatory disorders, the CSF CD4/CD8 ratio and IL-6 concentration were elevated in neurosarcoidosis.52 CSF IL-6 concentration >50 pg/mL was also associated with progression or relapse of NS.52 Next-generation metagenomic deep sequencing of the CSF for pathogen detection may be helpful to evaluate for infection in addition to culture and more targeted molecular studies.53,54 CSF cytology and flow cytometry should be considered to evaluate for malignancy. CSF studies can also be helpful to monitor for NS disease activity over time, such as in response to treatment, particularly opening pressure, cell count, total protein, glucose, IgG index, and oligoclonal bands.
Neuroimaging
When evaluating a patient for suspected CNS NS, MRI with and without gadolinium is the most appropriate imaging modality (Figures 1 and 2). Gadolinium enhancement lacks specificity, but is a helpful marker of active CNS sarcoidosis, and failure to include contrast-enhanced sequences is a common diagnostic and monitoring pitfall for CNS sarcoidosis. In addition to MRI patterns illustrated in Figures 1 and 2, deep medullary vein engorgement and radial perivenular enhancement have also been described in NS.55 MRI has value as a biomarker for response to therapy and critically informs clinical decision making.
Figure 1. Brain MRI Manifestations of Neurosarcoidosis.

Typical patterns of NS observed on brain MRI including leptomeningeal and pachymeningeal disease, perivascular infiltration, MS-like lesions, which may represent coexistent disease, mass-like lesions, cranial nerve infiltration, and pituitary/meningeal disease. (A.a–A.b) Post-gadolinium T1 MRI from a patient with probable NS demonstrating leptomeningeal enhancement predominantly affecting the base of the brain (arrows). (B.a) T2/FLAIR MRI of definite NS demonstrates T2 hyperintensity in the occipital lobe (arrow) adjacent to an area of (B.b) dural thickening and enhancement (arrow) on post-gadolinium T1 MRI. (C) Post-gadolinium T1 MRI demonstrating multifocal perivascular meningeal enhancement in the subcortical white matter (arrows) and cortex (arrowhead) in definite NS. (D) Sagittal FLAIR MRI of the same patient from B, exhibited T2 hyperintense lesions in the cerebellum (not shown) as well as the periventricular and juxtacortical white matter (arrows). Atrophy associated with the occipital lobe T2 hyperintense lesion can be appreciated (arrowhead). This patient was diagnosed with multiple sclerosis and later developed pachymeningeal thickening biopsy proven to be sarcoidosis. (E.a) Post-gadolinium T1 MRI demonstrating a focal area of contrast enhancement in the left insula (arrow) with (E.b) parenchymal T2 hyperintensity on FLAIR in a patient with probable neurosarcoidosis. (F.a) Mass-like lesion in the right mesial temporal lobe with enhancement (arrow) on post-gadolinium T1 MRI with corresponding (F.b) swelling and T2 hyperintensity on FLAIR (arrow) in definite NS. Note that most mass-like lesions in NS have some meningeal component as seen in E and F. (G) Coronal post-gadolinium T1 MRI demonstrating left optic nerve enhancement (arrow) in a patient with probable NS who presented with left optic neuritis. (H) Axial post-gadolinium T1 MRI with contrast enhancement and swelling in Meckel cave about the left trigeminal nerve (arrow) in a patient with probable NS who developed left trigeminal neuropathy (pain and decreased sensation). (I) Axial post-gadolinium T1 MRI demonstrating contrast enhancement in both internal auditory meatuses (arrows) in a patient with probable NS and bilateral facial nerve palsy. (J) Sagittal post-gadolinium T1 MRI in a patient with definite NS who presented with hypopituitarism demonstrates abnormal contrast enhancement and swelling of the pituitary gland (arrowhead) and stalk (arrow).
Figure 2. Spinal Cord MRI Manifestations of Neurosarcoidosis.

Typical patterns of spinal cord involvement in NS including longitudinally extensive myelitis, short segment myelitis, perivascular and leptomeningeal lesions, mas-like dural and leptomeningeal lesions, and lumbosacral radiculitis. (A.a) Sagittal T2 MRI of the cervical spine demonstrating longitudinally extensive transverse myelitis (between arrowheads) in a patient with probable NS and (A.b) extensive contrast enhancement on post-gadolinium T1 MRI (arrow). (B.a) Sagittal T2 MRI of the thoracic spine demonstrating a short segment myelitis (arrow) in a patient with probable NS with (B.b) contrast enhancement on post-gadolinium T1 MRI (arrow). (C) Sagittal post-gadolinium T1 MRI of the cervical spine from the same patient in Figure 1C demonstrating multifocal areas of contrast enhancement (arrowheads) in an unusual pattern suggesting leptomeningeal and perivascular infiltration. (D.a) Sagittal T2 MRI of the cervical spine with longitudinally extensive T2 hyperintensity and cord deformity in a patient with definite NS. (D.b) Sagittal post-gadolinium T1 MRI from the same patient in (D.a) demonstrating multiple mass-like enhancing lesions surrounding and compressing the spinal cord (arrowheads) well as leptomeningeal enhancement about the medulla (arrow). (E) Sagittal post-gadolinium T1 MRI of the lumbar spine demonstrating nodular leptomeningeal enhancement about the cauda equina (arrowhead) and enhancement within the conus medullaris (arrow) in a patient with definite NS who presented with lumbosacral radiculomyelitis (Elsberg syndrome).
Systemic Evaluation
Part of the early diagnostic strategy to support a diagnosis of NS is to search for evidence of systemic sarcoidosis, with a goal of establishing histopathologic confirmation of the underlying illness. This includes a comprehensive physical examination. A dilated eye examination can also identify evidence of ocular sarcoidosis that may support the diagnosis. Roughly half of patients with CNS-predominant sarcoidosis have an abnormal chest x-ray at the time of neurologic presentation.11,27 Chest, abdominal, and pelvic CT with IV contrast can be valuable for identifying sarcoidosis when chest x-ray is normal and clinical suspicion for sarcoidosis is high. When structural imaging does not reveal a biopsy target, combined fluorodeoxyglucose PET (FDG-PET)/CT can reveal metabolically active lymph nodes or otherwise occult lesions that may appear normal on CT.56 Combined whole-body FDG-PET/CT scan documented abnormalities consistent with sarcoidosis in 15/19 (78%) patients in 1 series5 and 22% of patients with suspected sarcoidosis in another.57 FDG-PET may also show metabolically active lesions in the brain or spinal cord, but these are almost always better seen on MRI.
Biopsy
Diagnostic confidence of NS is greatest when there is biopsy confirmation of sarcoidosis in the nervous system (what is termed definite neurosarcoidosis in the 2018 consensus diagnostic criteria); however, in some cases, the neuroanatomic localization may preclude biopsy due to concerns about morbidity or opportunity to secure a diagnosis by less invasive means. In such cases, it may be preferable to establish the diagnosis of systemic sarcoidosis in the context of a neurologic syndrome consistent with NS (probable neurosarcoidosis). When there is evidence of concurrent pulmonary sarcoidosis, a tissue sample can usually be acquired by endobronchial ultrasound-guided transbronchial biopsy of mediastinal or hilar lymphadenopathy with a high diagnostic yield; mediastinoscopy may be required in some cases depending on the affected anatomy.58 Comprehensive skin and eye examination should be pursued as either may reveal systemic evidence of sarcoidosis via relatively noninvasive biopsy. There should be caution when relying on skin biopsy as the only pathologic foundation to support the sarcoidosis diagnosis given the broad differential diagnosis of cutaneous granulomas and granulomatous dermatitis, as well as caution relying solely on superficial lymph node biopsy. In cases in which body imaging is unrevealing, biopsy of a neurologic target may be necessary to exclude malignancy or other etiologies.
Although sarcoidosis granulomas are generally non-necrotizing and any necrotizing granulomas or poorly formed granulomas should raise caution about sarcoidosis as the final diagnosis, in bona fide sarcoidosis, there can sometimes be rare areas of necrosis on biopsy, particularly in larger samples.59 Sarcoidosis granulomas are not histologically distinct from other granulomatous conditions, and special staining for acid-fast bacilli, fungi, and cultures is important for excluding infectious etiologies.2
Treatment
The goal of disease-modifying NS treatment is to reduce or prevent organ system damage from harmful effects of granulomatous inflammation (Table 4). In some circumstances, such as mild or transient disease, immunosuppression may not be necessary, but for most patients with CNS sarcoidosis and for many with large-fiber peripheral nervous system NS involvement, immunosuppression is indicated early to minimize neurologic injury and disability. Treatments focused on symptom management and rehabilitation are also important in the comprehensive care of patients with NS. When there is multisystem involvement, an interdisciplinary approach can be helpful to coordinate what is often complex care and decision making. No randomized trials exist yet to guide the treatment of NS; therefore, treatment is based on expert opinion and observations from case series and single reports.
Table 4.
Medications Commonly Used in the Treatment of Neurosarcoidosis

Treatment in NS should be individualized, weighing risks to benefit, with appropriate counseling to guide shared decision making about disease-modifying treatment, while also incorporating treatment considerations for sarcoidosis involving other organ systems as applicable. In general, glucocorticoids are standard first-line treatment. For more severely affected patients, consideration of initial IV or PO bioequivalent pulse dosing (e.g., 1 g IV methylprednisolone for 3–5 days) followed by a few month oral glucocorticoid taper (e.g., prednisone 1 mg/kg/d and tapering lower) would be typical, adjusting speed and taper duration based on severity, risk, tolerability, clinical, and imaging response. For patients with less severe disease, a moderate, shorter course of glucocorticoids may be sufficient with close monitoring. Given concerns about safety and toxicity associated with glucocorticoid exposure, early steroid-sparing therapy with methotrexate or azathioprine should be considered (typically reserving mycophenolate mofetil given data about comparative lesser efficacy in retrospective studies), although these can take several months to achieve clinical efficacy. In severer cases, when there is an incomplete response to glucocorticoids, or when there is particular concern about glucocorticoid toxicity risk, earlier use of TNF inhibitors is increasingly considered.
Acute Treatment
Glucocorticoids are widely considered to be first-line therapy for NS and work rapidly in most patients.7,26,27 Patients with severe presentations may require pulse-dose IV methylprednisolone, 1 g daily for 3–5 days followed by a prolonged oral glucocorticoid taper. For less severe presentations, prednisone 0.5–1 mg/kg/d or bioequivalent doses in other glucocorticoid formulations may be effective. For patients with mild or moderate presentations, monotherapy with prednisone may be sufficient and when the clinical and imaging response is adequate, prednisone can be tapered gradually over several months. Care should be taken to consider and evaluate for recurrence or worsening when tapering glucocorticoids in NS.
A meta-analysis from 2016 noted a favorable outcome with glucocorticoids in 71% of patients with NS.5 However, a significant portion of patients with NS will be refractory to glucocorticoids or will experience recurrence of disease activity when attempting to taper to more tolerable or safer doses.7,27 Glucocorticoid doses required to achieve and maintain remission can also sometimes be prohibitive due to steroid toxicity.60 Therefore, steroid-sparing agents should generally be given to patients with NS with moderate to severe presentations or in patients who are unable to achieve treatment goals balanced against risks with oral steroids.
Steroid-Sparing Agents
A number of steroid-sparing agents have been used in the treatment of NS, including azathioprine, methotrexate, mycophenolate mofetil, hydroxychloroquine, cyclophosphamide, and TNF inhibitors. Choosing among these options is based on clinical judgment, comorbidities, and the available literature, including observations from treatment of systemic sarcoidosis. A 2016 retrospective study of 40 patients with NS suggested greater efficacy of methotrexate over mycophenolate.e1 Another retrospective study of 234 patients with NS in 2017 noted lower risk for relapse among patients treated with methotrexate, cyclophosphamide, or infliximab and greater relapse risk with mycophenolate.4 Agents like azathioprine, methotrexate, and mycophenolate mofetil can take several months to achieve full immunosuppressive effects clinically, during which time it may be helpful to continue oral glucocorticoids. Close clinical and radiologic follow-up is important when tapering glucocorticoids given frequent recurrence of disease activity on steroid withdrawal.
TNF Inhibitors
TNF-alpha has been targeted in the treatment of sarcoidosise2 and NS.9 The best-studied TNF-alpha antagonist in NS is infliximab, a chimeric monoclonal antibody to TNF-alpha, which appears capable of inhibiting the formation of granulomas in sarcoidosis and inducing apoptosis via complement-dependent and antibody-dependent cytotoxicity.e3-e5 Case reports and small series have suggested efficacy of infliximab for NS.9,e6,e7 In 2017, a multi-institutional study reported benefit with infliximab for CNS sarcoidosis.9 A favorable clinical response was seen in 77% (29% of whom achieved complete remission) and a favorable MRI response in 82% (complete remission in 44%). Favorable outcomes with infliximab were reported in other series, including 87% with clinical stability or improvement in a series from Utahe8 and clinical stability or improvement in 96% (remission in 21%, improvement in 50%, and stability in 25%) in a series from the Netherlands.e9
Treatment is generally targeted to remission and often continued for a few to several years in patients with a history of severe disease. When discontinuing a TNF inhibitor in NS, it is important to monitor the patient clinically and by MRI for recurrence, which can occur as early as 3–6 months after stopping. In our multi-institutional study, disease recurred in 56% of patients who discontinued therapy after achieving remission, typically in the same neuroanatomic location.9 Similar recurrence rates on infliximab discontinuation in NS were reported in a French series,e10 whereas 31% of patients relapsed on tapering or discontinuing infliximab in a cohort from the Netherlands.e9 Other TNF antagonists, such as adalimumab, may also be effective in neurosarcoidosis.e11
Adverse effects of TNF-alpha inhibition include leukopenia, elevated liver enzymes, infusion-related reactions, infection (including reactivation of herpes zoster and latent tuberculosis as well as new infection with fungi such as histoplasmosis),e12 hypersensitivity reactions, malignancy, CNS inflammatory demyelination,e13,e14 and rarely progressive multifocal leukoencephalopathy. There are also rare reports of paradoxical granulomatous reactions associated with TNF antagonists, particularly etanercept.e15 Immunogenicity from neutralizing antibodies to infliximab occurs in a minority of patients and is associated with infusion reactions and reduced efficacy related to increased clearance or infliximab neutralization.e16,e17 It is common in NS to continue a lower-dose cytotoxic immunosuppressant such as methotrexate or azathioprine concurrently with infliximab to attenuate the formation of infliximab antibodies in addition to potential synergistic immunosuppressive benefits, although there are also emerging data that suggest favorable outcomes with TNF inhibitor monotherapy in NS.e18
B Cell–Targeted Therapy
Rituximab has been reported to have some efficacy in systemic sarcoidosise19 and in probable NS.e20 However, the role of anti-CD20 B cell–depleting therapy in neurosarcoidosis and sarcoidosis more broadly is unclear,e21,e22 and there may exist a publication bias. Two patients included in a 2017 multicenter study progressed despite rituximab but responded to subsequent infliximab,9 and rituximab was observed to be one of the least effective treatments in a 2020 series from Utah.e8
Emerging Immunosuppressive Approaches
Tofacitinib (a JAK inhibitor) has been observed to be favorable for treatment refractory cutaneous sarcoidosis, with molecular analysis of skin lesions pre- and post-treatment suggesting a role for JAK-STAT signaling,e23 and this approach has also led to improvement in multiorgan system sarcoidosis.e24 There are also emerging reports of benefit in multiorgan system sarcoidosis with baricitinib.e25 Clinical improvement with tocilizumab (an anti–IL-6 therapy) has also been observed in treatment refractory sarcoidosis with pulmonary, sinus, and cutaneous involvement.e26 Whether such strategies will prove to have a role in neurosarcoidosis remains to be determined.
Monitoring Response to Treatment
The goal of immunosuppression in NS is to minimize the risk of neurologic injury from granulomatous inflammation. In many patients, the goal may be complete remission of the neuroinflammatory response. In others, suppressing the inflammatory response, even if not achieving complete remission, may be the appropriate balance of therapeutic risk. Monitoring of treatment response includes history, physical examination, and typically MRI with and without contrast, which may be able to detect worsening or recurrent disease before new or recurrent clinical symptoms develop. In a patient with abnormal MRI findings attributable to NS who is doing well clinically, repeat imaging 2–4 months after treatment initiation is one reasonable approach. MRI monitoring frequency can then be gradually spaced out over time based on treatment response. When there are no abnormal findings on MRI, changes on MRI of uncertain etiology, or when there is incongruence between clinical symptoms and MRI findings (particularly when the CSF examination demonstrated intrathecal inflammation previously in the disease course), repeat CSF examination can be helpful clinically to monitor disease activity and confirm remission. Clinical response may lag behind MRI response—if there was underlying injury from the inflammatory process, neurologic impairment may not necessarily improve, but it should not worsen. Althogh the disease may remit and some patients can discontinue therapy, there is risk of recurrence with treatment discontinuation. When there is apparent worsening of NS, it is also important to avoid premature diagnostic closure, and clinicians should consider infectious complications (such as VZV infection or fungal infection) in the differential diagnosis even in patients with known NS.e12
Outcomes
Clinical outcome varies substantially among patients with neurosarcoidosis depending on the severity, extent, and neuroanatomic localization of the underlying disease. For example, patients with facial nerve palsy related to NS have historically been reported to regain facial movements in 80% of cases.e27 In contemporary studies, complete neurologic recovery was noted in only 29–33% of patients in patients with severe, treatment refractory NS that ultimately led to treatment with TNF inhibitors.e10,e28 A 2021 study of NS optic neuropathy found that only 24% of patients had a visual acuity less than 20/200 in the affected eye at last follow-up.e29 A 2020 study of sarcoid myelopathy found that 52% of patients had a modified Rankin Scale score of 3 or greater (indicating moderate level of disability), and about 30% used a cane or walker or more for ambulatory support on follow-up 1 year later.34
Conclusions
Recent advances in the diagnosis and treatment of NS include updated diagnostic criteria and mounting evidence supporting a role of TNF-alpha inhibition for aggressive and/or refractory cases. Randomized clinical trials are needed to determine optimal treatment strategies.
Glossary
- ACE
angiotensin-converting enzyme
- FDG
fluorodeoxyglucose
- IgG
immunoglobulin G
- LETM
longitudinally extensive myelitis
- PNS
peripheral nervous system
- TNF
tumor necrosis factor
Appendix. Authors

Contributor Information
Siddharama Pawate, Email: siddharama.pawate@vanderbilt.edu.
Laura L. Koth, Email: laura.koth@ucsf.edu.
Tracey A. Cho, Email: tracey-cho@uiowa.edu.
Jeffrey M. Gelfand, Email: jeffrey.gelfand@ucsf.edu.
Study Funding
No targeted funding reported.
Disclosure
M.J. Bradshaw reports personal compensation for medical legal counseling. S. Pawate, L.L. Koth, and T.A. Cho report no disclosures. J.M. Gelfand reports scientific consulting fees from Biogen, research support to UCSF from Genentech, and personal compensation for medical legal consulting. Go to Neurology.org/NN for full disclosures.
References
- 1.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155-1167. [DOI] [PubMed] [Google Scholar]
- 2.Rossi G, Cavazza A, Colby TV. Pathology of sarcoidosis. Clin Rev Allergy Immunol. 2015;49(1):36-44. [DOI] [PubMed] [Google Scholar]
- 3.Ungprasert P, Carmona EM, Utz JP, Ryu JH, Crowson CS, Matteson EL. Epidemiology of sarcoidosis 1946-2013: a population-based study. Mayo Clin Proc. 2016;91(2):183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joubert B, Chapelon-Abric C, Biard L, et al. Association of prognostic factors and immunosuppressive treatment with long-term outcomes in neurosarcoidosis. JAMA Neurol. 2017;74(11):1336-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fritz D, van de Beek D, Brouwer MC. Clinical features, treatment and outcome in neurosarcoidosis: systematic review and meta-analysis. BMC Neurol. 2016;16(1):220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160(2):736-755. [DOI] [PubMed] [Google Scholar]
- 7.Scott TF, Yandora K, Valeri A, Chieffe C, Schramke C. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol. 2007;64(5):691-696. [DOI] [PubMed] [Google Scholar]
- 8.Carlson ML, White JR Jr., Espahbodi M, et al. Cranial base manifestations of neurosarcoidosis: a review of 305 patients. Otol Neurotol. 2015;36(1):156-166. [DOI] [PubMed] [Google Scholar]
- 9.Gelfand JM, Bradshaw MJ, Stern BJ, et al. Infliximab for the treatment of CNS sarcoidosis: a multi-institutional series. Neurology. 2017;89(20):2092-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc. 2016;13(8):1244-1252. [DOI] [PubMed] [Google Scholar]
- 11.Joseph FG, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009;80(3):297-304. [DOI] [PubMed] [Google Scholar]
- 12.Moller DR, Rybicki BA, Hamzeh NY, et al. Genetic, immunologic, and environmental basis of sarcoidosis. Ann Am Thorac Soc. 2017;14(suppl 6):S429–S436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today. 1993;14(9):426-430. [DOI] [PubMed] [Google Scholar]
- 14.Berlin M, Fogdell-Hahn A, Olerup O, Eklund A, Grunewald J. HLA-DR predicts the prognosis in Scandinavian patients with pulmonary sarcoidosis. Am J Respir Crit Care Med. 1997;156(5):1601-1605. [DOI] [PubMed] [Google Scholar]
- 15.Grunewald J, Eklund A. Löfgren's syndrome: human leukocyte antigen strongly influences the disease course. Am J Respir Crit Care Med. 2009;179(4):307-312. [DOI] [PubMed] [Google Scholar]
- 16.van Moorsel CH, Christiani DC. Genetic susceptibility to sarcoidosis, a chronic inflammatory disorder. Am J Respir Crit Care Med. 2012;186(9):816-818. [DOI] [PubMed] [Google Scholar]
- 17.Fingerlin TE, Hamzeh N, Maier LA. Genetics of sarcoidosis. Clin Chest Med. 2015;36(4):569-584. [DOI] [PubMed] [Google Scholar]
- 18.Judson MA, Hirst K, Iyengar SK, et al. Comparison of sarcoidosis phenotypes among affected African-American siblings. Chest. 2006;130(3):855-862. [DOI] [PubMed] [Google Scholar]
- 19.Chen ES, Moller DR. Etiologies of sarcoidosis. Clin Rev Allergy Immunol. 2015;49(1):6-18. [DOI] [PubMed] [Google Scholar]
- 20.Iannuzzi MC, Fontana JR. Sarcoidosis: clinical presentation, immunopathogenesis, and therapeutics. JAMA. 2011;305(4):391-399. [DOI] [PubMed] [Google Scholar]
- 21.Baughman RP, Strohofer SA, Buchsbaum J, Lower EE. Release of tumor necrosis factor by alveolar macrophages of patients with sarcoidosis. J Lab Clin Med. 1990;115(1):36-42. [PubMed] [Google Scholar]
- 22.Ramstein J, Broos CE, Simpson LJ, et al. IFN-gamma-Producing T-helper 17.1 cells are increased in sarcoidosis and are more prevalent than T-helper type 1 cells. Am J Respir Crit Care Med. 2016;193(11):1281-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broos CE, Koth LL, van Nimwegen M, et al. Increased T-helper 17.1 cells in sarcoidosis mediastinal lymph nodes. Eur Respir J. 2018;51(3). [DOI] [PubMed] [Google Scholar]
- 24.Berry MP, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koth LL, Solberg OD, Peng JC, Bhakta NR, Nguyen CP, Woodruff PG. Sarcoidosis blood transcriptome reflects lung inflammation and overlaps with tuberculosis. Am J Respir Crit Care Med. 2011;184(10):1153-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krumholz A, Stern BJ. Neurologic manifestations of sarcoidosis. Handb Clin Neurol. 2014;119:305-333. [DOI] [PubMed] [Google Scholar]
- 27.Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM. 2009;102(7):449-460. [DOI] [PubMed] [Google Scholar]
- 28.Mekinian A, Maisonobe L, Boukari L, et al. Characteristics, outcome and treatments with cranial pachymeningitis: a multicenter French retrospective study of 60 patients. Medicine (Baltimore). 2018;97(30):e11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koczman JJ, Rouleau J, Gaunt M, Kardon RH, Wall M, Lee AG. Neuro-ophthalmic sarcoidosis: the University of Iowa experience. Semin Ophthalmol. 2008;23(3):157-168. [DOI] [PubMed] [Google Scholar]
- 30.Durel CA, Marignier R, Maucort-Boulch D, et al. Clinical features and prognostic factors of spinal cord sarcoidosis: a multicenter observational study of 20 biopsy-proven patients. J Neurol. 2016;263(5):981-990. [DOI] [PubMed] [Google Scholar]
- 31.Soni N, Bathla G, Pillenahalli Maheshwarappa R. Imaging findings in spinal sarcoidosis: a report of 18 cases and review of the current literature. Neuroradiol J. 2019;32(1):17-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flanagan EP, Kaufmann TJ, Krecke KN, et al. Discriminating long myelitis of neuromyelitis optica from sarcoidosis. Ann Neurol. 2016;79(3):437-447. [DOI] [PubMed] [Google Scholar]
- 33.Zalewski NL, Krecke KN, Weinshenker BG, et al. Central canal enhancement and the trident sign in spinal cord sarcoidosis. Neurology. 2016;87(7):743-744. [DOI] [PubMed] [Google Scholar]
- 34.Murphy OC, Salazar-Camelo A, Jimenez JA, et al. Clinical and MRI phenotypes of sarcoidosis-associated myelopathy. Neurol Neuroimmunol Neuroinflamm. 2020;7(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anthony J, Esper GJ, Ioachimescu A. Hypothalamic-pituitary sarcoidosis with vision loss and hypopituitarism: case series and literature review. Pituitary. 2016;19(1):19-29. [DOI] [PubMed] [Google Scholar]
- 36.Langrand C, Bihan H, Raverot G, et al. Hypothalamo-pituitary sarcoidosis: a multicenter study of 24 patients. QJM. 2012;105(10):981-995. [DOI] [PubMed] [Google Scholar]
- 37.Bathla G, Watal P, Gupta S, Nagpal P, Mohan S, Moritani T. Cerebrovascular manifestations of neurosarcoidosis: an underrecognized aspect of the imaging spectrum. AJNR Am J Neuroradiol. 2018;39(7):1194-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kupersmith MJ, Alban T, Zeiffer B, Lefton D. Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. Brain. 2002;125(pt 4):812-822. [DOI] [PubMed] [Google Scholar]
- 39.Vital A, Vital C. Clinical Neuropathology practice guide 3-2014: combined nerve and muscle biopsy in the diagnostic workup of neuropathy—the Bordeaux experience. Clin Neuropathol. 2014;33(3):172-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med. 2017;126:135-138. [DOI] [PubMed] [Google Scholar]
- 41.Stern BJ, Royal W III, Gelfand JM, et al. Definition and consensus diagnostic criteria for neurosarcoidosis: from the neurosarcoidosis Consortium Consensus Group. JAMA Neurol. 2018;75(12):1546-1553. [DOI] [PubMed] [Google Scholar]
- 42.Heij L, Dahan A, Hoitsma E. Sarcoidosis and pain caused by small-fiber neuropathy. Pain Res Treat. 2012;2012:256024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen Aubart F, Abbara S, Maisonobe T, et al. Symptomatic muscular sarcoidosis: lessons from a nationwide multicenter study. Neurol Neuroimmunol Neuroinflamm. 2018;5(3):e452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abbott JK, Gelfand EW. Common variable immunodeficiency: diagnosis, management, and treatment. Immunol Allergy Clin North Am. 2015;35(4):637-658. [DOI] [PubMed] [Google Scholar]
- 45.Najem CE, Springer J, Prayson R, et al. Intra cranial granulomatous disease in common variable immunodeficiency: case series and review of the literature. Semin Arthritis Rheum 2018;47(6):890-896. [DOI] [PubMed] [Google Scholar]
- 46.Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and detection of sarcoidosis. An official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26-e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ungprasert P, Carmona EM, Crowson CS, Matteson EL. Diagnostic utility of angiotensin-converting enzyme in sarcoidosis: a population-based study. Lung. 2016;194(1):91-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eurelings LEM, Miedema JR, Dalm VASH, et al. Sensitivity and specificity of serum soluble interleukin-2 receptor for diagnosing sarcoidosis in a population of patients suspected of sarcoidosis. PLoS One. 2019;14(10):e0223897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baud MO, Vitt JR, Robbins NM, et al. Pleocytosis is not fully responsible for low CSF glucose in meningitis. Neurol Neuroimmunol Neuroinflamm. 2018;5(1):e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leonhard SE, Fritz D, Eftimov F, van der Kooi AJ, van de Beek D, Brouwer MC. Neurosarcoidosis in a tertiary referral center: a cross-sectional cohort study. Medicine (Baltimore). 2016;95(14):e3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bridel C, Courvoisier DS, Vuilleumier N, Lalive PH. Cerebrospinal fluid angiotensin-converting enzyme for diagnosis of neurosarcoidosis. J Neuroimmunol. 2015;285:1-3. [DOI] [PubMed] [Google Scholar]
- 52.Chazal T, Costopoulos M, Maillart E, et al. The cerebrospinal fluid CD4/CD8 ratio and interleukin-6 and -10 levels in neurosarcoidosis: a multicenter, pragmatic, comparative study. Eur J Neurol. 2019;26(10):1274-1280. [DOI] [PubMed] [Google Scholar]
- 53.Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med. 2014;370(25):2408-2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salzberg SL, Breitwieser FP, Kumar A, et al. Next-generation sequencing in neuropathologic diagnosis of infections of the nervous system. Neurol Neuroimmunol Neuroinflamm. 2016;3(4):e251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zamora C, Hung SC, Tomingas C, Atkinson C, Castillo M. Engorgement of deep medullary veins in neurosarcoidosis: a common-yet-underrecognized cerebrovascular finding on SWI. AJNR Am J Neuroradiol. 2018;39(11):2045-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Treglia G, Annunziata S, Sobic-Saranovic D, Bertagna F, Caldarella C, Giovanella L. The role of 18F-FDG-PET and PET/CT in patients with sarcoidosis: an updated evidence-based review. Acad Radiol. 2014;21(5):675-684. [DOI] [PubMed] [Google Scholar]
- 57.Fritz D, van de Beek D, Brouwer MC, Booij J. Whole-body 18F-FDG PET-CT in the diagnosis of neurosarcoidosis. Mayo Clin Proc. 2020;95(5):1082-1084. [DOI] [PubMed] [Google Scholar]
- 58.Nakajima T, Yasufuku K, Fujiwara T, Yoshino I. Recent advances in endobronchial ultrasound-guided transbronchial needle aspiration. Respir Investig. 2016;54(4):230-236. [DOI] [PubMed] [Google Scholar]
- 59.Kleinschmidt-DeMasters BKTT, Rodriguez F. Neurosarcoidosis. Diagnostic Pathology: Neuropathology: Elsevier; 2016:703-707. [Google Scholar]
- 60.Miloslavsky EM, Naden RP, Bijlsma JW, et al. Development of a Glucocorticoid Toxicity Index (GTI) using multicriteria decision analysis. Ann Rheum Dis. 2017;76(3):543-546. [DOI] [PubMed] [Google Scholar]
- Data available from Dryad (Additional References, References 1-29): inks.lww.com/NXI/A604