

Abstract
The past decade has seen an exponential increase in the identification of individual nucleobases that undergo base conversion and/or modification in transcriptomes. While the enzymes that catalyze these types of changes have been identified, the global interactome of these modifiers is still largely unknown. Furthermore, in some instances, redundancy among a family of enzymes leads to an inability to pinpoint the protein responsible for modifying a given transcript merely from high-throughput sequencing data. This chapter focuses on a method for global identification of transcripts recognized by an RNA modification/editing enzyme via capture of the RNAs that are bound in vivo, a method referred as RNA immunoprecipitation (RIP). We provide a guide of the major issues to consider when designing a RIP experiment, a detailed experimental protocol as well as troubleshooting advice. The RIP protocol presented here can be readily applied to any organism or cell line of interest as well as both RNA modification enzymes and RNA-binding proteins (RBPs) that regulate RNA modification levels. As mentioned at the end of the protocol, the RIP assay can be coupled to high-throughput sequencing to globally identify bound targets. For more quantitative investigations, such as how binding of an RNA modification enzyme/regulator to a given target changes during development/in specific tissues or assessing how the presence or absence of RNA modification affects transcript recognition by a particular RBP (irrespective of a role for the RBP in modulating modification levels); the RIP assay should be coupled to quantitative real-time PCR (qRT-PCR).
1. Introduction
For several decades, research has been geared towards understanding how somatic mutations and alterations in epigenetic marks affect gene expression in health and disease. However, programmed changes to RNA also occur and add an extra layer of complexity to gene expression regulation. These sequence-specific changes include insertion, deletion and/or base conversions (which as a group are historically referred to as RNA editing events (Gott & Emeson, 2000)) as well as chemical modification of nucleosides (Ontiveros, Stoute, & Liu, 2019). To date, over 170 RNA modifications have been documented (Boccaletto et al., 2018). Although the majority of these modifications have only been identified in abundant RNA species like transfer RNA (tRNA) and ribosomal RNA (rRNA), several modifications including inosine, N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C) and pseudouridine (Ψ) have been observed in messenger RNA (mRNA) and are collectively referred to as the epitranscriptome (Wiener & Schwartz, 2021; Zaccara, Ries, & Jaffrey, 2019). Ongoing efforts are focused on understanding the impact of RNA editing and modification on gene expression and have already revealed roles in regulating mRNA stability, decay, transport, translation, alternative splicing and polyadenylation (Erdmann, Mahapatra, Mukherjee, Yang, & Hundley, 2021; Zhao, Roundtree, & He, 2017). Given the critical role RNA modifications can play in regulating gene expression, it is not surprising that alterations in the epitranscriptome have been observed in a number of diseases, including cancers, cardiovascular and metabolic diseases as well as neurological disorders (Delaunay & Frye, 2019; Jain, Jantsch, & Licht, 2019; Jonkhout et al., 2017). Thus, gaining a systemic understanding of how individual modifications regulate mRNA expression and influence human disease biology is of the utmost importance.
To this end, the development of computational and experimental methods to identify RNA editing events and modifications in high-throughput mRNA sequencing data has been a major focus of the last decade. Base conversions that alter base-pairing and recognition by reverse transcription enzymes, such as Adenosine (A)-to-Inosine (I) editing, can be easily detected in RNA sequencing (RNA-seq) datasets (Ramaswami et al., 2013). However, many RNA modifications are not differentially recognized by reverse transcriptase and thus, methods to detect these changes rely on indirect detection by using chemical treatments that generate bulky adducts to block reverse transcriptase or promote nucleotide misincorporation (Accornero, Ross, & Alfonzo, 2020; Weichmann et al., 2020). The relatively low frequency of occurrence is another challenge to identifying targets of RNA editing and modifying enzymes via searching for changes present in RNA-seq datasets. Several methods to overcome this hurdle are being developed and center around the use of either a modification-specific antibody or proteins that recognize a specific modification to selectively enrich for edited and/or modified transcripts (Knutson & Heemstra, 2021). One limitation of these approaches is the availability of antibodies or proteins that specifically recognize modified nucleosides. Therefore, one strategy that researchers can employ to surmount the above-mentioned obstacles is to focus on RNA–protein interactions. As mRNA modifications are mediated by specific RNA-binding proteins (RBPs), capturing transcripts bound by RBPs can facilitate detection of modified transcripts (Anreiter, Mir, Simpson, Janga, & Soller, 2021). A growing school of thought also suggests that individual mRNA modifications have the potential to alter binding of other gene regulatory RBPs (Lewis, Pan, & Kalsotra, 2017). Furthermore, some enzymes, such as ADARs, bind to cellular RNAs and regulate gene expression independent of editing (Anantharaman et al., 2017). Thus, by capturing RNA–protein interactions, this approach would serve beneficial in detecting physiologically relevant targets that otherwise would not have been identified.
Biochemical approaches, including electromobility shift assays and nitrocellulose filter binding assays, can be used to examine interactions between RBPs and cognate RNA targets in vitro (Chen et al., 2000; Gallo, Keegan, Ring, & O’Connell, 2003; Kuttan & Bass, 2012; Rajendren et al., 2018). However, a major disadvantage with such biochemical approaches is that prior knowledge of the target RNA molecule is required. In addition, owing to the in vitro nature, information regarding the physiological composition and/or spatiotemporal regulation of ribonucleoprotein complexes cannot be obtained through such biochemical assays. RNA immunoprecipitation (RIP) is a powerful molecular approach that allows for identification of RNAs bound by an RBP in vivo. While several modifications of this technique have been used to identify targets of RBPs in vivo (Kaczynski, Hussain, & Farkas, 2019; Rajendren et al., 2021, 2018; Song et al., 2020; Sugimoto, Chakrabarti, Luscombe, & Ule, 2017; Sugimoto et al., 2015; Zarnegar et al., 2016), the basic workflow involves using a protein specific antibody to pull down an RBP of interest (along with bound targets) from cellular lysates. Following immunoprecipitation, bound RNA is extracted and is either coupled to quantitative real-time PCR (qRT-PCR) to quantitatively examine binding of specific targets or to high-throughput sequencing to get a global view of bound RNAs.
In general, RIP assays can be classified into two main categories based on whether the lysate is untreated (referred to as native) or treated with chemical crosslinkers (referred to as crosslinked) (see Fig. 1). Native RIPs are primarily used for immunoprecipitation of RBPs that are either highly expressed or have a strong affinity toward target RNA molecules (Cozzitorto et al., 2015; Keene, Komisarow, & Friedersdorf, 2006; Khalil et al., 2009; Zhao et al., 2010). However, since RNAs have been shown to associate with RBPs after cell lysis (Mili & Steitz, 2004), native RIPs might not be the technique of choice in identifying true RNA–RBP interactions. This is where use of chemical crosslinkers, such as formaldehyde or ultraviolet (UV) light, prove to be advantageous. In crosslinked RIPs, not only are RNA–protein interactions captured in real time; but additionally, identification of RNA targets that are either transient and/or to which the RBP has a weak affinity are improved. In addition, since crosslinking stabilizes RNA–protein interactions, the immunoprecipitated products can be subjected to stringent washing conditions that further decrease the chances of retrieving nonspecific interactions. It is noteworthy to mention that the choice of crosslinking agent being used in a given experiment can also influence retrieval of nonspecific interactors. For example, formaldehyde stabilizes protein–protein interactions in addition to crosslinking nucleic acid to protein molecules. Thus, the use of formaldehyde can increase detection of indirect interactions. Here, we describe a detailed protocol for immunoprecipitating an RBP of interest and include important considerations needed before starting the experiment as well as some troubleshooting advice.
Fig. 1.
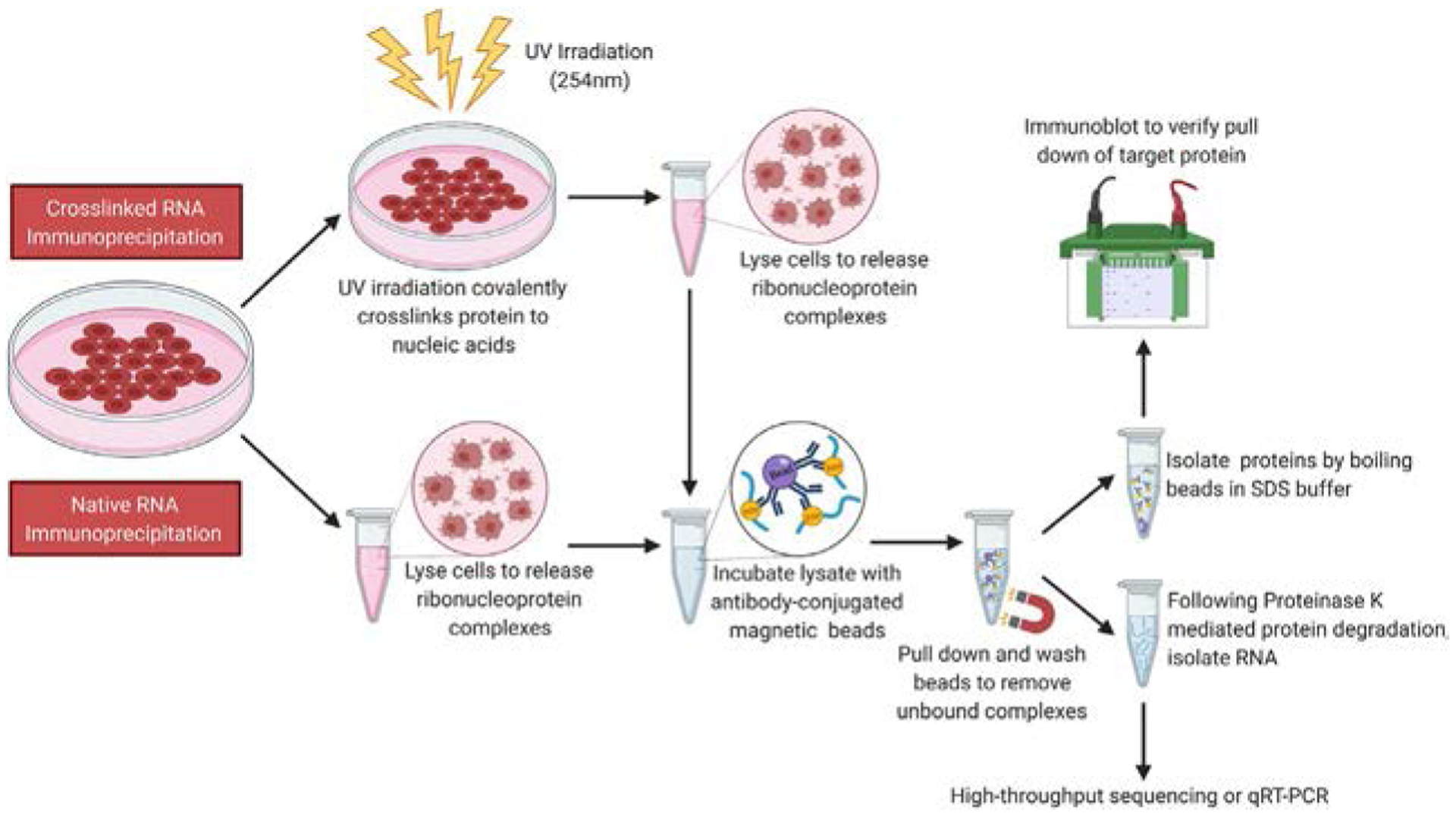
Schematic representation of a crosslinked and native RIP workflow. Cells (or other model organisms) can either be treated with a crosslinking agent that allows covalent crosslinking of proteins to bound RNA molecules (Crosslinked RIP) or not (Native RIP). Cells are lysed to release RBPs with bound RNAs, and the lysate is incubated with antibody-conjugated magnetic beads. Upon completion of incubation, beads are pulled down using a magnet and subjected to several rounds of stringent washing to remove nonspecific interactors. To examine immunoprecipitated protein, an SDS-containing buffer is added to the beads, and the mixture is boiled. The denatured protein lysate can then be resolved by SDS-PAGE, transferred to a nitrocellulose membrane and subjected to immunoblotting. To isolate immunoprecipitated protein bound RNA, the beads can be treated with Proteinase K. Released RNA can be extracted using standard phenol-chloroform based techniques and used for qRT-PCR or high-throughput sequencing.
2. Factors to consider when designing a RIP assay
The key to a successful RIP assay is to maximize the efficiency by which target RNA-protein complexes are pulled down while minimizing nonspecific interactions with the resin or beads used for immunoprecipitation. Here, we will mention several experimental aspects that should be considered when designing a RIP experiment.
• Quality of antibody
Efficient immunoprecipitation largely depends on the antibody employed. Antibodies are defined as monoclonal or polyclonal depending on the number of epitopes the antibody recognizes; one epitope for monoclonal and multiple epitopes for polyclonal. Due to the ability to recognize multiple epitopes, polyclonal antibodies are preferred for RBPs where the epitopes exposed in vivo are unknown. In addition, the ability of polyclonal antibodies to recognize multiple epitopes prevents the affinity of the antibody preparation from being significantly affected by minute changes in the antigen structure.
Moreover, to further enhance the RIP efficiency, polyclonal antibody preparations can be affinity purified. Briefly, when an animal is immunogenized using an antigen (against which the antibody is raised), the derived polyclonal serum not only comprises antibodies against the target antigen but also contain several other animal-specific antibodies. Thus, to enrich for antibodies specific to the protein of interest, antigen immobilized on a nitrocellulose membrane or resin can be used to affinity purify the polyclonal serum. This affinity purified antibody can then be used to perform RIPs. However, it should be noted that monoclonal antibodies have also been shown to successfully immunoprecipitate target proteins (Gilbert, Kristjuhan, Winkler, & Svejstrup, 2004; Rudel, Flatley, Weinmann, Kremmer, & Meister, 2008).
• Protein expression
Another determinant of efficient immunoprecipitation includes the relative abundance of the protein of interest. In general, immunoprecipitation of endogenous protein is considered ideal. Pulling down an exogenously expressed protein raises concerns as to whether the identified RNA targets are physiologically relevant or are a consequence of transgenic RBP expression promoting nonspecific interactions. However, depending on the type of protein or the model system being used, pulling down endogenous proteins may not be possible. In such cases, exogenously expressing the RBP to identify targets can be an important first step. An additional modification here can be to exogenously express the RBP fused to a small peptide (referred to as a “tag”) against which high-affinity, monoclonal antibodies are commercially available. In a recent study from our lab, switching from an affinity-purified polyclonal antibody of the RBP of interest (ADR-2) to a commercially available FLAG antibody improved the ability to identify physiological targets of the RBP, which was transgenically expressed in only one tissue of a multicellular organism (the nervous system of Caenorhabditis elegans) (Rajendren et al., 2021).
• Number of peptide tags
When epitope tags are employed, increasing the number of epitope tags fused to the protein of interest can enhance the efficiency of immunoprecipitation. In a previous study, using three tandem copies of the FLAG epitope instead of just one resulted in efficient immunoprecipitation of ADAR3 and bound RNAs (Oakes, Anderson, Cohen-Gadol, & Hundley, 2017).
• Nonspecific interactions
The ability of RBPs to associate with nonspecific RNAs after cell lysis, a phenomenon known as post-lysis reassortment (Mili & Steitz, 2004) makes identifying true RNA targets of any RBP critical and challenging. If protein lysates are derived from a multicellular organism, post-lysis reassortment can significantly affect signal-to-noise ratio as proteins expressed in one tissue can nonspecifically bind to RNAs expressed in other tissues thereby skewing our understanding of the biological function of the protein being studied. From personal experience, magnetic beads and agarose resin can nonspecifically bind RNA molecules. In fact, if possible, agarose resin should be avoided. To improve the signal-to-noise ratio and facilitate identification of physiological targets, bead volumes should be kept to a minimum. In addition, as described above, crosslinking before lysis can be employed. Several additional steps to reduce nonspecific binding are provided in the troubleshooting section at the end of this protocol.
• Use of controls
Successful identification of physiologically relevant targets needs to be validated using appropriate controls. When using a polyclonal antibody to perform RIP, immunoprecipitation using preimmune serum (derived from the same animal prior to immunogenization) can be performed as a negative control. The preimmune control helps to determine whether animal-derived antibodies and/or beads are contributing to nonspecific interactions with RNA (refer to the troubleshooting section at the end of this protocol for suggestions if nonspecific binding is observed). Binding of a known RNA target can serve as a positive control and be useful in optimizing the signal-to-noise ratio of the RIP assay. Ideally, lack of enrichment of a nonbinding RNA should also be monitored. Furthermore, when exogenously expressed proteins are employed, lysates lacking the overexpressed protein should be included as a negative control.
• Binding comparison of mutant RBPs and/or different genetic backgrounds
RIP can be useful in assessing how mutations affect the in vivo binding properties of a given RBP. When executing such an experiment, care needs to be taken that equal amounts of RBP (wildtype and mutant) are immunoprecipitated. Similar concerns occur regarding analysis of an RBP from different genetic backgrounds or at different times in development. If equal immunoprecipitation is not achieved, differences observed in enrichment of target RNAs cannot be attributed to the mutations in question but may rather result from differences in RBP immunoprecipitation efficiency. During a RIP assay, even if all reaction conditions, buffers and reagents are kept constant between different genetic backgrounds, equal amounts of RBP immunoprecipitation might not be achieved. Thus, before proceeding with reverse transcription, the immunoprecipitated protein should be subjected to immunoblotting, and the IP efficiencies compared. Depending on variations observed at the protein level, equivalent ratios of immunoprecipitated RNA need to be taken for further analysis.
3. Step-by-step method details
The protocol described below provides a general procedure for immunoprecipitation and may require optimization. The experiment should be performed in an RNase-free environment. All equipment and workspaces should be cleaned using water, then 70% ethanol and followed by RNase ZAP (Sigma, cat. no. R2020) and allowed to air dry. All solutions should be made with RNase-free chemicals and in glassware that has been baked overnight at 200°C. All tips used should contain filter barriers and be low retention; the latter of which also applies to microcentrifuge tubes and reduces loss of sample due to surface binding.
3.1. Materials and equipment
UV crosslinker (ex. Spectrolinker XL-1000 UV crosslinker)
Sonicator (ex. Misonix Ultrasonic liquid processors)
Mortar and pestle (ex. VWR, cat. no. 89038-146 and 89038-162)
Refrigerated benchtop microcentrifuge (ex. Eppendorf 5424)
Vortex mixer (ex. Fisher, cat. no. 02215365)
Benchtop centrifuge with swing bucket rotor (Eppendorf 5810R with A-4-62 rotor)
Spectrophotometer (ex. Nanodrop 2000c UV–Vis spectrophotometer)
Magnetic separation rack (ex. DynaMag™-2, Fisher, cat. no. 12321D)
1.5mL microcentrifuge tubes (ex. Thomas Scientific, cat. no. 1149X75)
Microcentrifuge Tube Rotator (ex. VWR, cat. no. 10136-084)
Thermomixer (ex. Benchmark Scientific, cat. no. H5000-HC)
15mL RNase-free tubes (ex. VWR, cat. no. 525-1068)
Heat block (ex. VWR, cat. no. 12621-104)
Water bath (ex. Fisher, cat. no. 15-462-10)
Real-Time PCR System (ex. Fisher QuantStudio 3)
96-well PCR plate (ex. Fisher, cat. no. AB-24400/W)
1. Cell lysate preparation
1.1. Reagents and Buffers
- Lysis buffer for mammalian cell culture
- 20mM Tris–HCl (pH 7.5) (Sigma, cat. no. T1503).
- 500mM NaCl (Fisher, cat. no. S271-1)
- 10mM EDTA (Acros Organics, cat. no. 118432500).
- 10% Glycerol (Sigma, cat. no. G5516).
- 0.1% NP-40 (Sigma, cat. no. 74385).
-
0.5% Triton-X 100 (Sigma, cat. no. T8787).Note: Detergents NP-40 and Triton-X 100 should be added to the buffer immediately before use.
- Lysis buffer for C. elegans
- 50mM HEPES (pH 7.4) (Sigma, cat. no. H3375).
- 70mM Potassium acetate (Sigma, cat. no. 236497).
- 5mM Magnesium acetate (Sigma, cat. no. M0631).
- 10% Glycerol (Sigma, cat. no. G5516).
- 0.05% NP-40 (Sigma, cat. no. 74385)
-
Protease inhibitor tablet (Roche, cat. no. 04693159001)
Note: Tablets should be dissolved in lysis buffer by rotating at 4°C immediately before use as protective activity is limited to 4h in solution.
- Phosphate buffer saline (1× PBS, pH 7.4)
- 137mM NaCl (Fisher, cat. no. S271-1).
- 2.7mM KCl (Sigma, cat. no. 7300).
- 10mM Na2HPO4 (Sigma, cat. no. S3139).
- 1.8mM KH2PO4 (Sigma, cat. no. PX1565-1)
- 10 × M9 buffer
- 33.7mM Na2HPO4 (Sigma, cat. no. S3139).
- 22mM KH2PO4 (Sigma, cat. no. PX1565-1).
- 8.55mM NaCl (Fisher, cat. no. S271-1).
- 9.35mM NH4Cl (VWR, cat. no. BDH9208)
Bradford reagent (Sigma, cat. no. B6916)
1.2. Procedure
-
1.2.1.
Grow the cells/organism in the appropriate medium. The amount required will depend on the number of immunoprecipitations to be performed, the amount of protein extract used for each immunoprecipitation, and the expression level of your protein of interest
-
1.2.2.
Remove the growth media and wash the sample with balanced salt solution to remove any remaining growth media or other contaminants. For example, we use ice-cold 1× PBS for mammalian cell culture and 1× M9 buffer for C. elegans
-
1.2.3.
After washing, add a minimal amount of ice-cold salt solution to the sample. If working with an organism in liquid, spread the sample on a petri dish (containing agar) to form a thin layer
-
1.2.4.
After taking off the lid or any barrier between your samples and the light source, place your sample as close as possible to the lights within the UV crosslinker. Irradiate with energy (mJ/cm2) optimal for your sample. The value of energy used for irradiation varies depending on the sample. In our experience, 150mJ/cm2 for mammalian cell culture and 3000mJ/cm2 for C. elegans is ample irradiation energy
-
1.2.5.
Place the plates immediately on ice or at 4°C after irradiation
-
1.2.6.
Collect the irradiated samples in conical tubes and pellet by centrifugation at 4°C
-
1.2.7.
Remove supernatant and resuspend the sample in lysis buffer containing protease inhibitor by pipetting gently. At this stage, the lysate can be frozen in liquid nitrogen and stored at −80°C until proceeding to the next step. Store at least 50μL of lysis buffer (with detergents and protease inhibitor added) for step 1.2.11
-
1.2.8.
In addition to detergent, the cells are further lysed by physical methods such as sonication, mechanical disruption (ex. grinding with mortar and pestle) and freeze thaw cycles depending on sample type. As the physical methods can cause a rise in temperature, to avoid RBP denaturation, it is important to prechill equipment and keep the sample cold during lysis. For mammalian cells, cells are sonicated twice at 30% amplitude with a microtip for 20s. For C. elegans, the frozen pellets are ground using mortar and pestle on dry ice in a 4°C room
-
1.2.9.
Centrifuge the cell lysate at maximum speed in refrigerated (4°C) benchtop centrifuge for 10min to remove any cellular debris
-
1.2.10.
Transfer the supernatant to a new tube and place at 4°C. It is important to note that immunoprecipitation must be performed as soon as possible after lysis and that lysates cannot be stored for later use
-
1.2.11.
Determine the protein concentration of the lysate by Bradford assay
2. Preparation of magnetic beads for the RNA immunoprecipitation
2.1. Reagents and Buffers
Dynabeads M-280 Sheep anti-Rabbit IgG (Invitrogen, cat. no. 11203D) or Dynabeads M-280 Sheep anti-Mouse IgG (Invitrogen, cat. no. 11201D)
ANTI-FLAG M2 Magnetic Beads (Sigma, cat. no. M8823)
Antibody against your protein of interest
-
Antibody conjugation buffer—make fresh before use
0.1% (w/v) Bovine serum albumin (Fisher, cat. no. 9048-46-8).
2mM EDTA (Acros Organics, cat. no. 118432500).
1× PBS (refer to steps in list 1.1)
-
6× SDS buffer
37.5mM Tris (Sigma, cat. no. T1503).
12% SDS (Sigma, cat. no. L3771).
70% Glycerol (Sigma, cat. no. G5516).
0.06% Bromophenol blue (Sigma, cat. no. B0126).
12% β-mercaptoethanol (Sigma, cat. no. M3148).
2.2. Procedure
The antibody of interest can be covalently linked to agarose resin/magnetic beads or indirectly bound to beads coated with IgG. Agarose resin has a high capacity to bind but the sponge-like structure increases nonspecific binding (ThermoFisherScientific). Therefore, if using agarose resin, additional steps of blocking the beads, preclearing of lysates and extensive washing should be employed. Magnetic beads are nonporous and are recommended due to both low nonspecific binding and easy handling during washes. Magnetic beads with polyclonal α-rabbit or α-mouse IgG covalently bound to the bead surface can be noncovalently coupled with antibody that recognizes your target protein and used for immunoprecipitation. Alternatively, magnetic beads conjugated to monoclonal antibodies that recognize epitope, such as FLAG, can be used for immunoprecipitation of tagged proteins (ThermoFisherScientific, 2021).
Note: All of the following steps must be performed at 4°C.
-
2.2.1.
Resuspend the magnetic beads by pipetting, and aliquot the required amount of beads into a 1.5mL tube. The amount of beads used for each assay needs to be optimized to both ensure efficient immunoprecipitation of the RBP as well as minimal capture of nonspecific interactions. Typically, at least two immunoprecipitations for each cell type/condition are performed to allow one sample to be used to assess efficient protein immunoprecipitation by western blotting and the other sample for analysis of bound RNA bound by qRT-PCR or high-throughput RNA sequencing
-
2.2.2.
Place the beads on a magnetic rack for 1min and discard the supernatant
For magnetic beads covalently linked with antibody, skip to step 2.2.8.
-
2.2.3.
Resuspend the beads in 1mL antibody conjugation buffer and wash by rotating for 5min at 4°C
-
2.2.4.
Place the beads on a magnetic rack for 1min and remove buffer. Perform steps 2.2.3 and 2.2.4 for a total of three times
-
2.2.5.
Add the antibody recognizing your target protein of interest to the beads. If applicable, in a separate tube of beads, add the preimmune serum that serves as the negative control. Add antibody conjugation buffer to bring volume to 1mL
Note: For the magnetic beads listed above, the manufacturers recommend 0.4–4μg antibody/50μL of beads. The ratio of antibody to beads varies and should be optimized using the antibody of interest.
-
2.2.6.
Incubate the beads with antibody on a microcentrifuge rotator for at least 1h up and up to overnight at 4°C. The binding efficiency of antibody to beads may vary and can be optimized using different incubation times
-
2.2.7.
After incubation, place the beads on a magnetic rack for 1min and discard the unbound antibody. Optional: Take 100μL of the unbound antibody fraction to analyze by western blotting to determine the efficiency of antibody binding to beads for the corresponding incubation time
-
2.2.8.
Resuspend the beads in 1mL lysis buffer and wash by rotating for 5min at 4°C. This washing step removes any unbound antibody as well as equilibrates the antibody bound beads to the lysate buffer condition
-
2.2.9.
Place the beads on a magnetic rack for 1min and take off the buffer
-
2.2.10.
Perform steps 2.2.8 and 2.2.9 three times. The number of washing steps after the antibody conjugation depends on the strength of antibody-bead interaction and can be optimized to minimize the loss of antibody during wash
3. RNA-protein immunoprecipitation
3.1. Reagents and buffers
Protease inhibitor tablet (Roche, cat. no. 04693159001)
TRIzol reagent (Ambion, cat. no. 15596018)
-
Tris-buffered saline (1× TBS, pH 7.5)
16mM Tris (Sigma, cat. no. T1503).
110mM NaCl (Fisher, cat. no. S271-1)
-
High salt wash buffer for mammalian cell culture
20mM Tris–HCl (pH 7.5) (Sigma, cat. no. T1503).
500mM NaCl (Fisher, cat. no. S271-1).
10mM EDTA (Acros Organics, cat. no. 118432500).
10% Glycerol (Sigma, cat. no. G5516)
-
High salt wash buffer for C. elegans
16mM Tris (Sigma, cat. no. T1503).
500mM NaCl (Fisher, cat. no. S271-1)
6× SDS sample buffer (refer to steps in list 2.1)
Proteinase K, 800U/μL (New England Biolabs, cat. no. P8107S)
RNasin Plus RNase inhibitor (Promega, cat. no. N2618)
3.2. Procedure
-
3.2.1.
Dissolve one protease inhibitor tablet per 1mL of lysis buffer by rotating at 4°C
-
3.2.2.
Add the required volume of lysate to beads. Depending on the level of protein expression and efficiency of the immunoprecipitation, the amount of lysate can vary from 100μg to 5mg
-
3.2.3.
Add 100μL of lysis buffer from step 3.2.1. to each tube and bring total volume to 1mL using lysis buffer used in step 1.2.7
Note: The IgG Dynabeads capacity is 25 μg of target protein/ml of antibody bound beads. Based on the level of target protein in your sample, this step should be optimized.
-
3.2.4.
Incubate the antibody bound beads with the lysates rotating at 4°C for 1h
Note: Depending upon the immunoprecipitation efficiency the incubation time can be altered.
-
3.2.5.
Place 10% of the lysate used for each immunoprecipitation in a new tube labeled as “Input”. Add 6× SDS sample buffer and boil the sample at 100°C for 10 min. Store it in −20°C for western blot analysis to determine the efficiency of immunoprecipitation
-
3.2.6.
Take a portion of the lysate used for each immunoprecipitation and place in a new 15mL tube and add TRIzol reagent. The TRIzol volume should be 3× the sample volume. Vortex the lysate/TRIzol mixture for 1min and freeze in liquid nitrogen. Label the tube as “Input RNA” and store at −80°C until RNA isolation. The amount of each transcript detected in the target protein IP sample will be normalized by the transcript amount in the input RNA sample to control for differential gene expression between strains or conditions
-
3.2.7.
After incubating cell lysate with antibody-bound beads, place the tubes on a magnetic rack for 1min and discard the supernatant. Optional: place 100μL of the supernatant in a new tube labeled as “unbound lysate,” add 6× SDS sample buffer and boil at 100°C for 10min. Store at −20°C for western blot
-
3.2.8.
Resuspend the beads in 1mL of high salt wash buffer. Rotate the tubes for 5min at 4°C. Then, place the tubes on a magnetic rack and remove the supernatant. Repeat washing with high salt wash buffer for 3–5 times. The number of washes varies depending on the strength of antibody protein interaction and nonspecific interaction of other proteins to beads
-
3.2.9.
Spin down the tubes, place on the magnetic rack for 1min, and discard any remaining buffer
-
3.2.10.
Resuspend the beads in 100μL of 1× TBS and combine the beads from technical replicates of the immunoprecipitation into one 1.5mL tube. Mix the beads by pipetting gently and then redistribute 100μL of beads back into the same number of tubes. Note: This step minimizes the variation among IP samples used for different downstream analysis such as western blotting, RNA-seq, qRT-PCR
-
3.2.11.
Place the tubes on a magnetic rack and remove 1× TBS
-
3.2.12.
Add 30–100μL of 2× SDS sample buffer to the beads from step 3.2.11 for western blot analysis. Label the tube as “IP,” boil the sample at 100°C for 10min and store at −20°C. Keep at least one tube of each IP condition or sample for checking the IP efficiency
-
3.2.13.
Resuspend the sample that will be used for RNA analysis from step 10 in 100μL of 1× TBS
-
3.2.14.
Add 0.5μL Proteinase K and 1μL of RNasin Plus RNase inhibitor to each tube
-
3.2.15.
Incubate at 42°C for 15min in a thermomixer at 1200rpm
-
3.2.16.
After the incubation, spin down the samples and place them on a magnetic rack for 2min
-
3.2.17.
Collect the supernatant from the tubes of immunoprecipitation into 15mL tubes and add TRIzol (similar to step 3.2.6). Vortex for 1min, freeze in liquid nitrogen, and store at −80°C
-
3.2.19.
To check the immunoprecipitation efficiency for the RBP, perform immunoblotting using the appropriate primary and secondary antibody. Use the protein samples labeled “Input” and “IP” saved in −20°C from steps 3.2.5 and 3.2.12 Optional: The “unbound” lysate sample from step 3.2.7 can also be used. Boil the samples at 100°C and spin down. Place the tubes on a magnetic rack and load input, IP, and unbound lysate sample (optional) on a polyacrylamide gel of the percentage that provides the best resolution for your protein of interest
4. RNA extraction
4.1. Reagents
TRIzol reagent (Ambion, cat. no. 15596018)
Chloroform (Sigma, cat. no. C2432)
Isopropanol (Sigma, cat. no. 32727-2500)
Ethanol (KOPTEC, cat. no. 64-17-5)
RNase-free water
Glycoblue Coprecipitant (Invitrogen, cat. no. AM9515)
Turbo DNase (Invitrogen, cat. no. AM2239)
RNeasy Mini kit (Qiagen, cat. no. 74106)
RNeasy MinElute clean up kit (Qiagen, cat. no. 74204)
3M Sodium acetate (Sigma, cat. no. S7670)
RNasin Plus RNase inhibitor (Promega, cat. no. N2618)
4.2. Procedure
-
4.2.1.
Thaw the RNA samples stored at −80°C from step 3.2.6 and 3.2.17 in a 37°C water bath. Vortex for 1min
-
4.2.2.
Freeze the sample again in liquid nitrogen, then thaw at 37°C and vortex for 1min. Repeat this step twice
-
4.2.3.
Add chloroform to each tube and mix the samples by inverting approximately 15 times. The amount of chloroform added should be 0.2× the volume of the total sample
-
4.2.4.
Leave the tubes undisturbed for 3min at room temperature
-
4.2.5.
Centrifuge the sample at maximum speed (3220g) for 15min at 4°C to separate the phases
-
4.2.6.
Transfer the aqueous phase (top-most transparent layer) to siliconized microcentrifuge tubes. Be sure to collect as much of the aqueous layer as possible without disturbing the other layers. If the volume is more than 750μL, use multiple tubes
-
4.2.7.
Add an equal volume of isopropanol to each tube. To facilitate detection of pellets, add 1μL glycoblue to the immunoprecipitation samples
-
4.2.8.
Vortex and incubate at room temperature for 10min
-
4.2.9.
Centrifuge the tubes at maximum speed at 4°C for at least 30min. Discard the supernatant and add 1mL of freshly made 70% ethanol and gently mix a few times
-
4.2.10.
Centrifuge the tubes at maximum speed at 4°C for 5min. Quickly remove the supernatant and spin down the tubes again by a short spin and remove any remaining liquid by pipetting
-
4.2.11.
Leave the tubes open for 1min for RNA pellets to air dry
-
4.2.12.
Resuspend the pellet in 30–80μL RNase-free water by pipetting up and down several times. The RNA samples at this stage can be stored at −80°C
-
4.2.13.
Determine the concentration of input RNA samples using a nanodrop spectrophotometer. The RNA samples from RIP do not need to be quantified as the entire IP will be used for downstream analysis
-
4.2.14.
To remove any DNA contamination in the extracted RNA, treat the samples with Turbo DNase. Use 5μL of Turbo DNase per 100μg RNA of the input samples and 1μL Turbo DNase per IP samples
-
4.2.15.
Incubate RNA with Turbo DNase, 10× Turbo DNase buffer (10μL), RNasin (2μL), and RNase-free water (up to 100μL) at 37°C for 1h
-
4.2.16.
After incubation, purify the RNA samples using the Qiagen RNeasy kit according to the manufacturer’s instructions. Elute the RNA in 30–50μL of RNase-free water
Note: An alternative is to elute the RIP RNA samples in 10μL RNase-free water using the RNeasy MinElute clean up kit and skip to step 4.2.24.
-
4.2.17.
Concentrate the IP RNA sample by ethanol precipitation. Add three volumes of 100% ethanol and 1/10 volume of 3M NaOAc to the sample. Mix well. Add 1μL of glycoblue to the IP samples
-
4.2.18.
Incubate the tubes at −80°C for at least 30min to allow precipitation of RNA and centrifuge the tubes at maximum speed for at least 30min at 4°C
-
4.2.19.
Discard the supernatant and add 1mL of freshly made 70% ethanol and gently mix a few times
-
4.2.20.
Centrifuge the tubes at maximum speed at 4°C for 5min
-
4.2.21.
Quickly remove the supernatant and spin down the tubes again by a short spin and remove any remaining liquid by pipetting
-
4.2.22.
Leave tubes open for 1min for pellets to air dry
-
4.2.23.
Pipet up and down to resuspend pellets. Use a total of 30μL RNase-free water for input RNA samples and 10μL for IP RNA samples
-
4.2.24.
Determine the concentration of input RNA samples using a spectrophotometer. At this stage, RNA samples can be stored at −80°C
5. RT-qPCR analysis
5.1. Reagents
Superscript III Reverse transcriptase (Invitrogen, cat. no. 18080-044)
Random hexamer primer (Fisher, cat. no. S0142)
Oligo(dT) primer (Fisher, cat. no. S0132)
dNTPs (Promega, cat. nos. U120A, U121A, U122A, U123A)
RNasin Plus RNase inhibitor (Promega, cat. no. N2618)
KAPA SYBR FAST Universal (KAPA Biosystems, cat. no. KK4602)
Gene specific primers (5μM stock solution)
5.2. Procedure
-
5.2.1.
Perform reverse transcription of 2μg of each input RNA sample and all (10μL) the RNA immunoprecipitated from one entire IP using a thermostable reverse transcriptase such as Superscript II (200U/reaction). For abundant genes, use of random hexamers and/or oligo(dT) for priming is typically adequate. However, in some instances gene-specific primers (250nM) are needed to obtain ample product to detect by qRT-PCR. A control reaction lacking the reverse transcriptase is also recommended
-
5.2.2.
After reverse transcription, dilute the cDNA synthesized from the input and immunoprecipitated RNA by adding 20μL RNase-free water. For analysis of genes with low expression, use cDNA without dilution
-
5.2.3.
Perform qRT-PCR using gene specific primers and DNA polymerase optimal for qRT-PCR conditions, along with components to detect the amplification of DNA. We suggest the use of KAPA SYBR FAST qPCR master mix which has SYBR Green I fluorescent dye for detection and our instrument of choice is the QuantStudio 3. Perform the qPCR in triplicates for each sample for more accurate data
-
5.2.4.
The cDNA amplification of your gene of interest in each of the input and immunoprecipitation samples are calculated based on the Ct values of samples compared to that of a standard curve. The amount of binding in each condition is determined by dividing the cDNA value in the IP sample by the corresponding input sample (Fig. 2A, B). A gene is considered to be bound if significant enrichment is observed in the experimental IP sample compared to the negative control IP (Fig. 2A and C). Furthermore, the enrichment of a true target should be significantly higher than that for a random/nontarget RNA
Fig. 2.
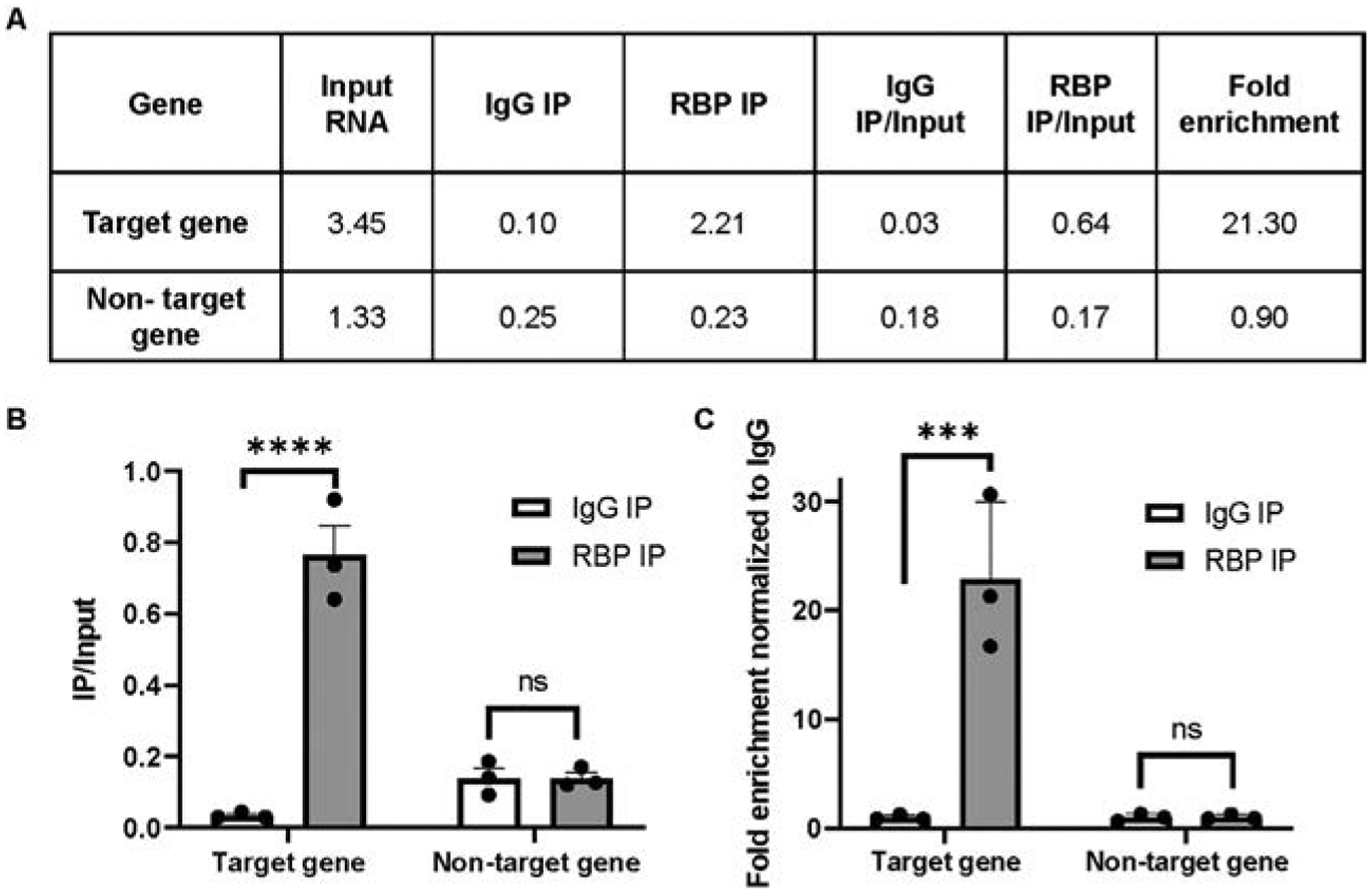
Representative RIP qRT-PCR results. (A) The table lists the amount of cDNA amplification detected by qRT-PCR in RNA samples for Input lysate as well the IgG and RBP immunoprecipitations. For each gene under study, the amount of RNA detected in the IP samples is normalized by the amount of corresponding gene in the input. (B) Bar graph represents the cDNA present in the RIPs divided by cDNA present in the input lysates for each gene. (C) The bar graph represents the fold enrichment of cDNA present in the RBP RIP divided by cDNA present in the input lysates normalized to the same ratio in the IgG control RIP. The mean of replicates is plotted with error bars representing standard error of the mean. Statistical significance was determined by two-way ANOVA, where **** represents P<0.0001.
6. RIP-Seq
Global views of the transcripts interacting with an RBP can be determined by performing high-throughput sequencing on the input and immunoprecipitated RNA samples. RNA libraries can be generated using commercially available kits. It is important to note that for modifications that occur in highly repetitive regions, such as A-to-I editing, maintaining the strand information is critical to successfully mapping the sequence to the correct genomic location as well assigning the correct type of RNA modification (Oakes, Vadlamani, & Hundley, 2017). Therefore, commercial kits that maintain this information, such as the KAPA Strand-Specific RNA Library Kit are preferred. Before library preparation, RNA from RIP assays can be subjected to poly(A) selection and/or ribosomal RNA depletion depending upon whether bound targets are expected to contain poly(A) tails. If looking for small RNAs bound to an RBP, size selection of RNAs will also be necessary.
RNA sequencing reads can be processed and aligned to the reference genome using STAR (Dobin et al., 2013). The RNA reads are analyzed using DESeq2 (Love, Huber, & Anders, 2014) or EdgeR (Robinson, McCarthy, & Smyth, 2010) to estimate the abundance of reads mapping to specific transcripts in input and immunoprecipitated samples. The number of reads of each transcript in the RIP sample is normalized by the number of reads of that transcript in the corresponding input RNA sample to account for any gene expression variation. The normalized read count of a given transcript immunoprecipitated by the protein of interest is then compared with the normalized read count in the RIP negative control.
4. Troubleshooting
| Issue | Probable cause | Solution |
|---|---|---|
| Low RNA yield | RNase contamination | Ensure all instruments, workspaces and reagents are RNase-free. While working with RNA, routine cleanup with RNase ZAP is beneficial |
| IP of nonspecific and/or degraded proteins | Post-cell lysis | Increase stringency of washes |
| Protein degradation during sonication | Ensure samples do not overheat during sonication. Keep samples on ice at all times and increase duration of intervals between consequent sonication cycles | |
| Low IP efficiency | Immunoprecipitated protein is too low | Combine immunoprecipitated proteins from multiple technical replicates |
| Antibody unable to pull down protein of interest | Insert single (or tandem repeats) of an epitope tag to the protein of interest. Commercially available antibodies can then be used to immunoprecipitate target protein with higher efficiency | |
| Low signal-to-noise ratio | Too much lysate | Decrease amount of lysate used. A common misconception is that increasing the amount of input lysate increases IP efficiency. However, increasing the lysate not only increases the target protein concentration but also the pool of nonspecific RNAs |
| RNA is nonspecifically binding to beads and/or target protein | Preclear protein lysates by incubating with antibody-free beads prior to incubation with antibody-coated beads | |
| Coat beads with a layer of nonspecific protein like Bovine Serum Albumin (BSA) prior to immunoprecipitation | ||
| Increase stringency of washing by replacing mild buffers with high salt containing buffers (greater than or equal to 0.5M NaCl) |
5. Summary
The RIP workflow described above is versatile and can easily be modified to suit any model organism or study being performed. Similar to chromatin immunoprecipitation (ChIP), a widely used technique to study epigenetic modifications, RIP has proven to be an indispensable tool in the field of RNA biology. For several years now, RIPs have facilitated specific as well as systemic identification of RNA molecules bound by a target protein (Kaczynski et al., 2019; Oakes, Anderson, et al., 2017; Rajendren et al., 2021; Song et al., 2020; Sugimoto et al., 2015). By comparing RIP-seq and RNA-seq profiles in presence or absence of a given RNA modification, researchers now have the potential to connect the recognition of modified transcripts by RBPs to the impact of these factors on gene expression.
Acknowledgment
We thank Alfa Dhakal, Emily A. Erdmann, Ananya Mahapatra and Boyoon Yang for helpful comments regarding this protocol. Work in the laboratory of H.A.H. is supported by the National Science Foundation (Award Number 1917050) and the National Institutes of Health (R01GM130759). R.K. is supported by the Wright fellowship (IUSM-Bloomington). Fig. 1 was created with Biorender.com.
Reference
- Accornero F, Ross RL, & Alfonzo JD (2020). From canonical to modified nucleotides: Balancing translation and metabolism. Critical Reviews in Biochemistry and Molecular Biology, 55(6), 525–540. 10.1080/10409238.2020.1818685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman A, Tripathi V, Khan A, Yoon JH, Singh DK, Gholamalamdari O, et al. (2017). ADAR2 regulates RNA stability by modifying access of decay-promoting RNA-binding proteins. Nucleic Acids Research, 45(7), 4189–4201. 10.1093/nar/gkw1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anreiter I, Mir Q, Simpson JT, Janga SC, & Soller M (2021). New twists in detecting mRNA modification dynamics. Trends in Biotechnology, 39(1), 72–89. 10.1016/j.tibtech.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, et al. (2018). MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Research, 46(D1), D303–D307. 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, & Nishikura K (2000). A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA, 6(5), 755–767. 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzitorto JA, Jimbo M, Chand S, Blanco F, Lal S, Gilbert M, et al. (2015). Studying RNA-binding protein interactions with target mRNAs in eukaryotic cells: Native ribonucleoprotein immunoprecipitation (RIP) assays. Methods in Molecular Biology, 1262, 239–246. 10.1007/978-1-4939-2253-6_14. [DOI] [PubMed] [Google Scholar]
- Delaunay S, & Frye M (2019). RNA modifications regulating cell fate in cancer. Nature Cell Biology, 21(5), 552–559. 10.1038/s41556-019-0319-0. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann EA, Mahapatra A, Mukherjee P, Yang B, & Hundley HA (2021). To protect and modify double-stranded RNA—The critical roles of ADARs in development, immunity and oncogenesis. Critical Reviews in Biochemistry and Molecular Biology, 56(1), 54–87. 10.1080/10409238.2020.1856768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Keegan LP, Ring GM, & O’Connell MA (2003). An ADAR that edits transcripts encoding ion channel subunits functions as a dimer. The EMBO Journal, 22(13), 3421–3430. 10.1093/emboj/cdg327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Kristjuhan A, Winkler GS, & Svejstrup JQ (2004). Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Molecular Cell, 14(4), 457–464. 10.1016/s1097-2765(04)00239-4. [DOI] [PubMed] [Google Scholar]
- Gott JM, & Emeson RB (2000). Functions and mechanisms of RNA editing. Annual Review of Genetics, 34, 499–531. 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- Jain M, Jantsch MF, & Licht K (2019). The Editor’s I on disease development. Trends in Genetics, 35(12), 903–913. 10.1016/j.tig.2019.09.004. [DOI] [PubMed] [Google Scholar]
- Jonkhout N, Tran J, Smith MA, Schonrock N, Mattick JS, & Novoa EM (2017). The RNA modification landscape in human disease. RNA, 23(12), 1754–1769. 10.1261/rna.063503.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczynski T, Hussain A, & Farkas M (2019). Quick-irCLIP: Interrogating protein-RNA interactions using a rapid and simple cross-linking and immunoprecipitation technique. MethodsX, 6, 1292–1304. 10.1016/j.mex.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, & Friedersdorf MB (2006). RIP-chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nature Protocols, 1(1), 302–307. 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proceedings of the National Academy of Sciences of the United States of America, 106(28), 11667–11672. 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SD, & Heemstra JM (2021). Protein-based molecular recognition tools for detecting and profiling RNA modifications. Current Opinion in Structural Biology, 69, 1–10. 10.1016/j.sbi.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttan A, & Bass BL (2012). Mechanistic insights into editing-site specificity of ADARs. Proceedings of the National Academy of Sciences of the United States of America, 109(48), E3295–E3304. 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CJ, Pan T, & Kalsotra A (2017). RNA modifications and structures cooperate to guide RNA-protein interactions. Nature Reviews. Molecular Cell Biology, 18(3), 202–210. 10.1038/nrm.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, & Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, & Steitz JA (2004). Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses. RNA, 10(11), 1692–1694. 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes E, Anderson A, Cohen-Gadol A, & Hundley HA (2017). Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. The Journal of Biological Chemistry, 292(10), 4326–4335. 10.1074/jbc.M117.779868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes E, Vadlamani P, & Hundley HA (2017). Methods for the detection of adenosine-to-inosine editing events in cellular RNA. Methods in Molecular Biology, 1648, 103–127. 10.1007/978-1-4939-7204-3_9. [DOI] [PubMed] [Google Scholar]
- Ontiveros RJ, Stoute J, & Liu KF (2019). The chemical diversity of RNA modifications. The Biochemical Journal, 476(8), 1227–1245. 10.1042/BCJ20180445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendren S, Dhakal A, Vadlamani P, Townsend J, Deffit SN, & Hundley HA (2021). Profiling neural editomes reveals a molecular mechanism to regulate RNA editing during development. Genome Research, 31(1), 27–39. 10.1101/gr.267575.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendren S, Manning AC, Al-Awadi H, Yamada K, Takagi Y, & Hundley HA (2018). A protein-protein interaction underlies the molecular basis for substrate recognition by an adenosine-to-inosine RNA-editing enzyme. Nucleic Acids Research, 46(18), 9647–9659. 10.1093/nar/gky800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O’Connell MA, et al. (2013). Identifying RNA editing sites using RNA sequencing data alone. Nature Methods, 10(2), 128–132. 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, & Smyth GK (2010). edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel S, Flatley A, Weinmann L, Kremmer E, & Meister G (2008). A multifunctional human Argonaute2-specific monoclonal antibody. RNA, 14(6), 1244–1253. 10.1261/rna.973808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Yang W, Fu Q, Wu L, Zhao X, Zhang Y, et al. (2020). irCLASH reveals RNA substrates recognized by human ADARs. Nature Structural & Molecular Biology, 27(4), 351–362. 10.1038/s41594-020-0398-4. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Chakrabarti AM, Luscombe NM, & Ule J (2017). Using hiCLIP to identify RNA duplexes that interact with a specific RNA-binding protein. Nature Protocols, 12(3), 611–637. 10.1038/nprot.2016.188. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D’Ambrogio A, et al. (2015). hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature, 519(7544), 491–494. 10.1038/nature14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ThermoFisherScientific. 2021. Retrieved from https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fbrochures%2Freproducible-immunoprecipitation-brochure.pdf&title=RHluYWJlYWRzIGltbXVub3ByZWNpcGl0YXRpb24gYnJvY2h1cmU=.
- Weichmann F, Hett R, Schepers A, Ito-Kureha T, Flatley A, Slama K, et al. (2020)Validation strategies for antibodies targeting modified ribonucleotides. RNA, 26(10), 1489–1506. 10.1261/rna.076026.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener D, & Schwartz S (2021). The epitranscriptome beyond m(6)A. Nature Reviews. Genetics, 22(2), 119–131. 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, & Jaffrey SR (2019). Reading, writing and erasing mRNA methylation. Nature Reviews. Molecular Cell Biology, 20(10), 608–624. 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, & Khavari PA (2016). irCLIP platform for efficient characterization of protein-RNA interactions. Nature Methods, 13(6), 489–492. 10.1038/nmeth.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Molecular Cell, 40(6), 939–953. 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, & He C (2017). Post-transcriptional gene regulation by mRNA modifications. Nature Reviews. Molecular Cell Biology, 18(1), 31–42. 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]