

Abstract
Objectives. To determine how deaths of infants with genetic diagnoses are described in national mortality statistics.
Methods. We present a retrospective cohort study of mortality data, obtained from the National Death Index (NDI), and clinical data for 517 infants born from 2011 to 2017 who died before 1 year of age in the United States.
Results. Although 115 of 517 deceased infants (22%) had a confirmed diagnosis of a genetic disorder, only 61 of 115 deaths (53%) were attributed to International Classification of Diseases, 10th Revision codes representing congenital anomalies or genetic disorders (Q00-Q99) as the underlying cause of death because of inconsistencies in death reporting. Infants with genetic diagnoses whose underlying causes of death were coded as Q00-Q99 were more likely to have chromosomal disorders than monogenic conditions (43/61 [70%] vs 18/61 [30%]; P < .001), which reflects the need for improved accounting for monogenic disorders in mortality statistics.
Conclusions. Genetic disorders, although a leading cause of infant mortality, are not accurately captured by vital statistics.
Public Health Implications. Expanded access to genetic testing and further clarity in death reporting are needed to describe properly the contribution of genetic disorders to infant mortality.
Genetic disorders and congenital anomalies are considered the leading cause of infant mortality in the United States, 1 , 2 based on data from the National Center for Health Statistics (NCHS) at the Centers for Disease Control and Prevention. 1 These data are generated from death certificates, on which a medical care provider specifies a series of events leading to death, starting from the most immediate cause (e.g., pulmonary hypoplasia) and ending with the underlying cause of death (e.g., congenital diaphragmatic hernia). These death certificates are translated by the NCHS into codes using the International Classification of Diseases, 10th Revision (ICD-10), and these codes are grouped to generate national mortality statistics. 3 A computer algorithm is used to code entries from a death certificate, assign the underlying cause of death, and produce multiple cause mortality data.
These data consistently demonstrate that the ICD-10 codes most frequently assigned as the underlying cause for infant deaths (approximately 20%) are in the Q00-Q99 category, which encompasses “congenital malformations, deformations and chromosomal abnormalities.” 1 This category includes well-defined syndromes such as Down syndrome (Q90.9) and congenital anomalies such as cystic kidney disease (Q61) and congenital cardiac and vascular malformations (Q20-Q28). Although these data are often interpreted to mean that genetic disorders are the leading cause of infant mortality, 4–6 many genetic disorders do not have their own ICD-10 codes, may not be included on the death certificate, or may not be reported and coded accurately because of errors at the provider level 7 or at the level of the NCHS. 8 The nature and scope of this misclassification and possible underestimation of the mortality burden of genetic disorders remain unknown, although a previous study identified that ICD-9 codes (the predecessor to ICD-10) as reflected in hospital discharge records were correct for only 18% of the live- or stillborn infants coded as having major anomalies because of both provider misdiagnosis and flaws in the coding system. 9 Therefore, the objective of this study was to analyze a diverse cohort of infant deaths spanning several years, identify deaths occurring in the setting of a confirmed genetic disorder, and subsequently review the ICD-10 codes used to report these deaths at the national level to evaluate their accuracy.
METHODS
We identified a cohort of 573 infants born over a 7-year period (2011–2017) who were registered to our hospital—a large, academic pediatric medical center in an urban area with a large referral network—and died before 1 year (365 days) of age, as indicated by a “deceased” status in our electronic medical record (EMR). These deaths may have occurred at our institution or elsewhere. We requested records for all infants from the National Death Index (NDI) per their protocol, 10 including the ICD-10 code representing the underlying cause of death, record, and entity-axis codes. A list of individuals meeting our criteria was sent to the NDI, and “matches” were returned to us. Each possible match retrieved from the NDI was evaluated and deemed to be a true match if the infant’s name, date of birth, date of death (when available), and state in which the death occurred (when available) were consistent with our EMR data. We also reviewed the available medical records of these infants, on whom we have previously published, 7 to identify the proportion with a laboratory-confirmed genetic disorder using our previously described criteria 11 as well as to extract their demographic data and phenotypic features. If a death occurred at our institution, the provider-completed death certificate worksheet was reviewed when available. Clinical data were abstracted from the EMR and entered into an electronic database (REDCap 12 ). Most infants were seen at our hospital in an inpatient or outpatient setting (527), although some EMR entries were limited to records review (28) or had no clinical information (18). We included all registered infants to maximize the size of this cohort and included infants who might have been enrolled in our research protocol, through which they had received a diagnosis of a genetic disorder.
Statistical analysis was performed using SPSS version 24 (IBM, Somers, NY), with continuous variables compared using a Mann-Whitney U test for nonparametric data and dichotomous variables compared using a 2-sided Fisher exact test (P < .05 considered significant).
RESULTS
Of the 573 deceased infants whom we identified in our EMRs, 517 (90%) were identified in the NDI. Infants not identified in the NDI (56/573) were more likely to have location of death unknown (27/56 [48%] vs 95/517 [18%]; P < .001), to have not been seen at our institution (23/56 [41%] vs 23/517 [4%]; P < .001), and to have had a younger median age at death (28 days [interquartile range (IQR) 1.5–100.5 days] vs 57 days [IQR 14–162 days]; P = .005) when compared with infants whom we successfully identified. Of the infants identified in the NDI, 115 of 517 (22%) had a genetic disorder confirmed by clinical or research laboratory testing (Table 1), and there was no significant difference in the proportion with a confirmed genetic disorder compared with infants not identified in the NDI (11/56 [20%]; P = .74). Of the 115 infants with a confirmed genetic disorder, 61 (53%) had an underlying cause of death attributed to ICD-10 codes Q00-Q99.
TABLE 1.
Genetic Diagnoses Identified in a Cohort of 517 Infant Deaths: United States, 2011–2018
| Syndrome (No.) | ICD-10 Code |
| Chromosomal (60) | |
| Down syndrome (18) | Q90 |
| Edward syndrome (6) | Q91-Q91.3 |
| Patau syndrome (5) | Q91.4-Q91.7 |
| Turner syndrome (2) | Q96 |
| Other trisomies (3) | Q92-Q92.1, Q92.8, Q92.9 Q99, Q99.8, Q99.9 |
| Jacobsen syndrome (3) | None |
| 22q11 deletion syndrome (8) | None |
| Large chromosomal rearrangements (6) or other deletion/duplication syndrome (9) | Q92, Q93, Q99 |
| Monogenic (55) | |
| Other (31) | 8/32 a |
| Spinal muscular atrophy (9) | G12.9 |
| CHARGE syndrome (3) | None |
| Autosomal recessive polycystic kidney disease (5) | Q61.1 |
| Smith-Lemli-Opitz syndrome (3) | None |
| Ornithine transcarbamylase deficiency (2) | E72.4 |
| Nonketotic hyperglycinemia (2) | E72.5 |
Note. ICD-10 = International Classification of Diseases, 10th Revision (Geneva, Switzerland: World Health Organization; 1992). Syndromes appearing more than once are tabulated; all others are grouped together.
a4/8 are Q00-Q99 codes.
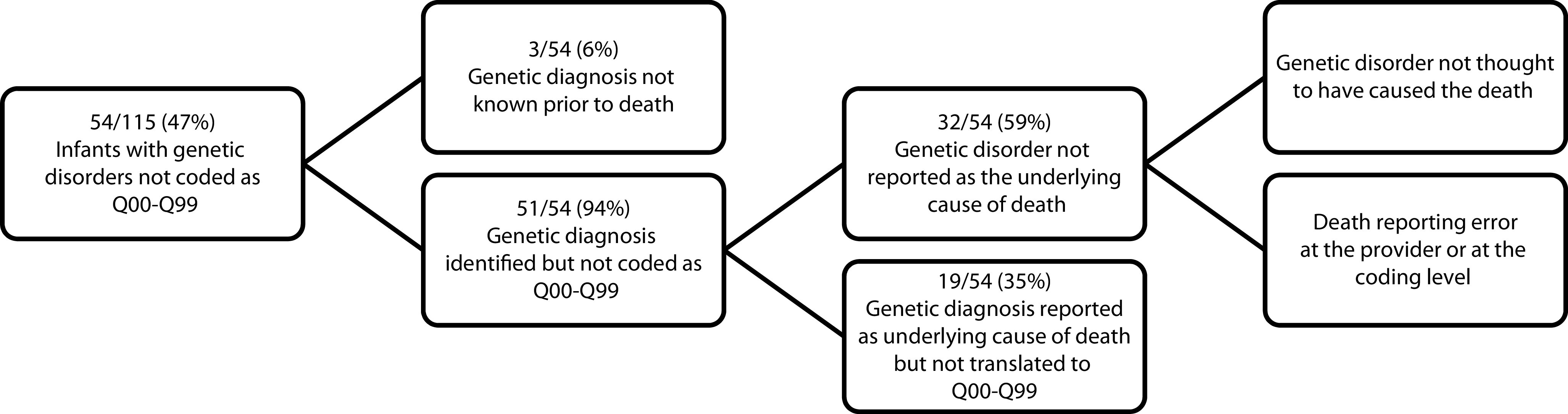
We then examined the 47% (54/115) of deceased infants with confirmed genetic disorders who were not coded as Q00-Q99 and identified three main reasons that a code in this category was not reported as the underlying cause of death: (1) the diagnosis was not known before death, (2) the diagnosis was known but not reported as the underlying cause of death, and (3) the diagnosis was known and reported as the underlying cause of death with an ICD-10 code that was not in the Q00-Q99 category (Figure 1).
FIGURE 1.
Possible Explanations for Infants With Genetic Disorders Not Being Coded as ICD-10 Q00-Q99: United States, 2011–2018
Note. ICD-10 = International Classification of Diseases, 10th Revision (Geneva, Switzerland: World Health Organization; 1992).
The diagnosis was not suspected or known before death in a small number of cases (3/54, 6%). In fact, although molecular diagnoses were identified postmortem in 13 of 54 infants (24%), a genetic disorder was suspected even before molecular confirmation in 10 (77%) of these 13 infants and was therefore included on the death certificate.
For 32 out of 54 infants (59%), the genetic disorder was known but not reflected in national mortality statistics as the underlying cause of death. This discrepancy might have occurred at the provider level. The genetic disorder may not have been noted as the underlying cause of death on the death certificate because it was not thought to have directly caused death or because provider error occurred when completing the death certificate. We were unable to confidently distinguish between purposeful and accidental omissions from EMR data. For example, a patient with Down syndrome may have died as a result of complications associated with a surgical procedure or from aspiration pneumonia, and one provider may have felt that those conditions represented death directly caused by Down syndrome whereas another provider did not. A third provider may not have been familiar with completion of death certificates and indicated only “respiratory failure” as the cause of death in either of those scenarios. The discrepancy may also have occurred at the national level, at which a provider may have included the diagnosis of a genetic disorder on the death certificate but chose an alternate condition as the underlying cause of death. Of the 32 infants with genetic disorders but underlying cause-of-death codes reflecting diagnoses that were not genetic (e.g., an infectious process), 23 (72%) had provider-completed death certificates in our EMR, and 15 of 23 certificates (65%) did include the genetic disorder, with 10 of 23 (43%) listing it as the underlying cause—indicating that the NCHS system then selected an alternative underlying cause of death during the coding process. The remaining 5 of 10 certificates listed the genetic disorder as a contributing but not underlying cause.
To investigate one possible explanation for why an infant with a genetic disorder might be coded with a “nongenetic” underlying cause of death, we evaluated for a difference in median age at death between infants with genetic disorders who were coded as Q00-Q99 versus infants not coded as Q00-Q99, and we did not find a significant difference (median 64 days, IQR 29–170 days vs median 122 days, IQR 30.75–178.5 days; P = .35). This finding suggested that older infants, who might have acquired medical issues secondary to or distinct from the genetic disorder, were not more likely to be coded with a cause of death outside of the Q00-Q99 category. Misreporting as a result of diagnoses being known, but not captured in the NDI as the underlying cause of death, also may have been attributed to problems at the coding level if the genetic disorder was included on the death certificate but not extracted by the NCHS system as the underlying cause. Of all infants with confirmed genetic disorders, 75 of 115 (65%) had a Q00-Q99 code listed in the entity or record axis data. Therefore, in 14 infants, a diagnosis in this category was on the death certificate but was not selected as the underlying cause of death by the NDI’s classification system.
For the remaining 19 of 54 infants (35%), the diagnosis of a genetic disorder was made either clinically or molecularly and was reported as the underlying cause of death but was not captured in the Q00-Q99 category. This was caused by the design of the ICD-10 classification system whereby genetic disorders without congenital anomalies are often reported in an organ system–based category instead. For example, spinal muscular atrophy (G12.9) and other genetic neuromuscular diseases are coded under diseases of the nervous system (G00-G98), and metabolic disorders (e.g., urea cycle disorders, E72.2) are coded under endocrine, nutritional, and metabolic diseases (E00-E88). Most monogenic disorders diagnosed in the infants in our cohort did not have a distinct ICD-10 code, and most of the codes that did exist were predominantly not in the Q00-Q99 category (Table 1).
Overall, we found that infants with diagnoses of genetic disorders that were coded as Q00-Q99 for the underlying cause of death were more likely to have chromosomal disorders (such as Down syndrome or other aneuploidy syndromes), whereas infants with diagnoses that were not coded as Q00-Q99 were more likely to have monogenic disorders (Table 2). Additionally, we found that of the 402 infants without a confirmed genetic disorder, 192 (48%) were categorized under the Q00-Q99 code for the underlying cause of death, reflecting deaths attributable to congenital anomalies that may or may not have an underlying Mendelian genetic etiology. The deaths of nearly half of the infants in our overall cohort (253/517, 49%) were attributed to a Q00-Q99 code, most often congenital anomalies of the heart or circulatory system (167/253, 66%); congenital diaphragmatic hernia, exomphalos, gastroschisis, and other malformations of the musculoskeletal system or integument (30/253, 12%); or chromosomal abnormalities (20/253, 8%). A minority of these infants with deaths caused by congenital anomalies had a confirmed molecular genetic disorder (61/253, 24%) that we ascertained from the records available, although a proportion may have had disorders that either were not identified because of lack of testing or were identified but not available to us in the EMR. Additional features of the infants with or without underlying cause of death codes in the Q00-Q99 category, such as sex and history of preterm birth, are presented in Table 2.
TABLE 2.
Comparison of Demographic and Clinical Features Between Infants With and Without an Underlying Cause of Death in the ICD-10 Q00-Q99 Category: United States, 2011–2018
| Infant Demographic | Underlying Cause-of-Death Code | P | ||
| Q00-Q99, No./Total No. (%) or Median (IQR) | Not Q00-Q99, No./Total No. (%) or Median (IQR) | Total, No./Total No. (%) or Median (IQR) | ||
| Sex | .42 | |||
| Female | 111/253 (44) | 106/264 (40) | 217/517 (42) | |
| Male | 142/253 (56) | 160/264 (60) | 302/517 (58) | |
| History of preterm delivery | < .001 | |||
| Yes | 86/239 (36) | 133/254 (52) | 219/517 (42) | |
| No | 153/239 (64) | 121/254 (48) | 274/517 (53) | |
| Unknown | 14/517 | 10/517 | 24/517 (5) | |
| Age at death, days | 57(15–163.5) | 56.5 (13–162) | .88 | 57 (14–162) |
| Genetic diagnosis | 61/253 (24) | 54/253 (21) | .34 | 115/517 (22) |
| Chromosomal | 43/61 (70) | 17/54 (31) | < .001 | 60/115 (52) |
| Monogenic | 18/61 (30) | 37/54 (69) | < .001 | 55/115 (48) |
Note. ICD-10 = International Classification of Diseases, 10th Revision (Geneva, Switzerland: World Health Organization; 1992); IQR = interquartile range. “Q00-Q99” refers to infant deaths classified with ICD-10 codes Q00-Q99 as the underlying cause of death, whereas “Not Q00-Q99” refers to infant deaths classified with other ICD-10 codes as the underlying cause of death.
DISCUSSION
Our data demonstrated that genetic disorders, although a leading cause of infant mortality, are not accurately captured by national mortality statistics, as reflected in the 54 of 117 infants with genetic disorders whose underlying cause of death was not reported in the Q00-Q99 category that is commonly used to represent genetic conditions. This inaccuracy is a particular issue for monogenic conditions, many of which either do not have a unique ICD-10 code or have one but are reported within an organ system–based category. It reflects the design of the ICD-10 coding system, which groups infants who have common chromosomal abnormalities with infants who have structural malformations in the same Q00-Q99 category. Our data also showed that many Mendelian disorders either do not cause birth defects or are not properly translated from the death report description to an ICD-10 code reflecting the genetic disorder. Indeed, few Mendelian genetic conditions have a distinct ICD-10 code, and it would be difficult for any system to correctly classify the many eponymous disorders causing congenital anomalies into the Q00-Q99 category. Thus, a certain proportion of diagnoses within each ICD-10 organ system category actually represents genetic disorders. Categories that seem particularly enriched for Mendelian genetic disorders are diseases of the nervous system (G00-G98) and endocrine, nutritional, and metabolic diseases (E00-E88). Conversely, even within the Q00-Q99 category, not all conditions are genetic because certain congenital anomalies may arise as a result of teratogenic exposure (e.g., diabetic embryopathy or fetal hydantoin syndrome) or deformations or disruptions (e.g., amniotic band sequence). The scope of the total contribution of Mendelian genetic disorders, combined across all categories, to infant mortality is therefore unclear with current reporting techniques.
Further insight into this scope is important because the identification of a Mendelian genetic disorder may suggest interventions at the individual or broader public health level. Multiple previous studies have demonstrated that genetic syndromes, whether monogenic or chromosomal, underlie a substantial proportion of stillbirths 13 and infant deaths. 7 , 11 , 14 , 15 A recent publication of a large cohort of infants diagnosed by exome sequencing demonstrated that infants with diagnoses of genetic disorders were more likely to die in the first 120 days of life than infants without, and nearly half of all infant deaths in that study occurred in the setting of a confirmed molecular disorder. 6 We also previously demonstrated that genetic disorders affect a substantial proportion of infants admitted to our neonatal intensive care unit who die in early childhood 11 and suggested inaccuracies in death reporting in this population. 7 Death certificates for neonates and infants are known to be highly inaccurate, with previous studies revealing discordance between information reported on the death certificate compared with medical records 16 or autopsy results. 17
This study provided further support for the inaccuracy of infant mortality data and expanded on our previous work by analyzing the consequences of delayed diagnoses and inaccurate completion of the death certificate, 7 , 11 as reflected in the high proportion of infants with genetic disorders that were not reported as such after death. Our genotype and phenotype data permitted an evaluation of why this occurs and we found that, most often, the explanation related to either inaccurate death reporting, which may be modifiable, 18 , 19 or shortfalls of the current coding system, which are more difficult to modify. 9 , 20 For similar reasons, other conditions are also underrepresented or misrepresented in mortality statistics. For example, epilepsy has been shown to be underreported on death certificates and was identified as an underlying or contributing cause for only 7% of individuals known to have epilepsy in a large clinical cohort. 21
The contribution of prematurity to infant mortality has also been shown to be underreported, 20 which also may occur either at the provider level or at the coding level. 8 , 22 , 23 The prematurity experience is particularly informative because it has shown that attributing deaths to organ system–based diagnoses suggests that strategies to reduce infant deaths ideally would be developed to treat these failing organs rather than prevent the root cause, such as preterm birth or low birth weight 20 , 24 or—as we would argue—monogenic Mendelian disorders. Indeed, preterm birth was previously suggested as the leading cause of infant mortality in the United States if all prematurity-related causes were combined, 20 and a modified classification scheme has been proposed (Dollfus et al., 24 updated in 2015 by Nakamura et al. 25 ) to understand infant mortality, taking this proposed scheme into account. It is unclear how the contribution of prematurity, top-ranked by the Dollfus classification system, would compare with deaths caused by genetic disorders if all genetic disorders were appropriately characterized in a cohesive group because these deaths face a similar categorization challenge when distributed by organ system. Further updates to the Dollfus classification scheme might take this consideration into account, particularly because the genetic basis for many congenital disorders is understood or will be understood because of advances in genomic technology.
Our results and those of others 9 , 26 also reveal the limitations of using ICD-9 or -10 data to study the prevalence of either genetic disorders or congenital anomalies related to mortality or other outcomes. For rare diseases, however, this remains one of the only options, with enrollment in registries, surveillance programs, and natural history studies representing another approach. 9 , 26 The use of billing or cause-of-death codes in a large, electronic data set, as has been done in other studies, 27 compared with the use of data acquired through programs such as the National Birth Defects Surveillance Study 28 illustrates a trade-off between strength in numbers (from population databases) and increased accuracy in genotyping and phenotyping (from surveillance programs with active case-finding and confirmation), and the method that we used for the current study is less feasible at scale. Nevertheless, improvements to the ICD coding system are needed to more accurately track outcomes related to these conditions, and other methods to acquire these data should be explored. The lack of ability to distinguish between genetic and teratogenic causes of birth defects was described many years ago with relation to ICD-9, which highlighted the need for dedicated codes for particular genetic syndromes. 9 Although the number of codes related to congenital anomalies increased with ICD-10, the ability to distinguish genetic from teratogenic causes of congenital anomalies was not addressed. 9 The introduction of ICD-11 represents another opportunity to incorporate genomic knowledge into public health databases.
Limitations
Limitations to this study included the nature of our pediatric hospital, which is not a birth hospital; thus, our population is enriched for older infants with congenital anomalies or other subspecialty-based concerns who have survived long enough to be transferred. This likely also impacted our ability to identify deceased infants in the NDI because some deaths may have occurred outside the United States if an infant was transferred home for palliative care or if an infant’s name was changed from the name assigned at the birth hospital without our medical record system being updated. Additionally, we relied on the information available to us via our research study or the EMR to determine the proportion of infants with genetic diagnoses, so additional infants may have had diagnoses of which we were not aware. This was also a single institution study, and although our institution is a large, academic hospital that receives many national and international referrals for specialty care, this cohort primarily represented infants from our catchment area. However, although the etiologic landscape of infant deaths in other regions may differ, particularly related to use of pregnancy-terminating procedures, 29 the underlying reporting and coding issues should be similar. Finally, relying on “deceased” status in our EMR may not have captured all deaths, particularly infants who died outside of our institution, because we may not have been notified of all deaths.
Public Health Implications
Current understanding of the impact of inaccurate death reporting on mortality statistics is incomplete, 30 and further efforts should be made to address this knowledge deficit. It would require not only improved reporting of deaths and possible further updating of the Dollfus classification scheme to include all Mendelian genetic disorders in one category but also expanded access to genetic testing to identify the mortality burden of rare genetic conditions. These results have important implications. Because the decision of whether to offer treatment to a critically ill infant, continue life-sustaining measures, or continue a pregnancy in the case of a confirmed genetic disorder may hinge on the chances of survival—for example, extracorporeal membrane oxygenation is not routinely offered for infants with “lethal” genetic conditions such as Trisomy 13 or 18 31 —accurate mortality statistics are necessary to guide these decisions. Additionally, public health resources, particularly funding to drive innovative research, are often allocated using published mortality rates to address conditions with the highest public health impact. Accurate classification of infant deaths has been recognized as a critical component of prevention efforts. 22 Thus, attempts to mitigate the leading cause of infant mortality depend on producing higher quality data to augment our understanding of these diseases and their mortality burden.
ACKNOWLEDGMENTS
M. H. Wojcik received support from the National Institutes of Health, National Institute of Child Health and Human Development (NIH/NICHD; K23 HD102589).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
HUMAN PARTICIPANT PROTECTION
This study was approved by the institutional review board of Boston Children’s Hospital as a retrospective study with a waiver of informed consent because of the nature of the study. Infants diagnosed via research testing were enrolled in a separate institutional review board–approved study at Boston Children’s Hospital.
References
- 1.Xu JQMS, Kochanek KD, Arias E. NCHS Data Brief. 2018;355:2020. [PubMed] [Google Scholar]
- 2.Murphy SL, Mathews TJ, Martin JA, Minkovitz CS, Strobino DM. Annual summary of vital statistics: 2013-2014. Pediatrics. 2017;139(6):e20163239. doi: 10.1542/peds.2016-3239. [DOI] [PubMed] [Google Scholar]
- 3.Moriyama IM, Loy RM, Robb-Smith AHT. Classifying diseases for primary mortality tabulations and problems of joint causes of death. In: Rosenberg HM, Hoyert DL, editors. History of the Statistical Classification of Diseases and Causes of Death. Hyattsville, MD: National Center for Health Statistics; 2011. pp. 23–35. [Google Scholar]
- 4.Kingsmore SF, Cakici JA, Clark MM, et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. 2019;105(4):719–733. doi: 10.1016/j.ajhg.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira CR. The burden of rare diseases. Am J Med Genet A. 2019;179(6):885–892. doi: 10.1002/ajmg.a.61124. [DOI] [PubMed] [Google Scholar]
- 6.Meng L, Pammi M, Saronwala A, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171(12):e173438. doi: 10.1001/jamapediatrics.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wojcik MH, Schwartz TS, Thiele KE, et al. Infant mortality: the contribution of genetic disorders. J Perinatol. 2019;39(12):1611–1619. doi: 10.1038/s41372-019-0451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carver JD, McDermott RJ, Jacobson HN, et al . Infant mortality statistics do not adequately reflect the impact of short gestation. Pediatrics. 1993;92:229–232. [PubMed] [Google Scholar]
- 9.Holmes LB, Westgate MN. Using ICD-9 codes to establish prevalence of malformations in newborn infants. Birth Defects Res A Clin Mol Teratol. 2012;94(4):208–214. doi: 10.1002/bdra.23001. [DOI] [PubMed] [Google Scholar]
- 10.National Center for Health Statistics. National Death Index User’s Guide. Hyattsville: MD; 2013. [Google Scholar]
- 11.Wojcik MH, Schwartz TS, Yamin I, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities. Genet Med. 2018;20(11):1396–1404. doi: 10.1038/gim.2018.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris PA, Taylor R, Thielke R, Payne J,, Gonzalez N, Conde JG. Research electronic data capture (REDCap): a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamseldin HE, Kurdi W, Almusafri F, et al. Molecular autopsy in maternal-fetal medicine. Genet Med. 2018;20(4):420–427. doi: 10.1038/gim.2017.111. [DOI] [PubMed] [Google Scholar]
- 14.Hudome SM, Kirby RS, Senner JW, Cunniff C. Contribution of genetic disorders to neonatal mortality in a regional intensive care setting. Am J Perinatol. 1994;11(2):100–103. doi: 10.1055/s-2007-994565. [DOI] [PubMed] [Google Scholar]
- 15.Kingsmore SF, Henderson A, Owen MJ, et al. Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. NPJ Genom Med. 2020;5(1):49. doi: 10.1038/s41525-020-00155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole SK. Accuracy of death certificates in neonatal deaths. Community Med. 1989;11(1):1–8. doi: 10.1093/oxfordjournals.pubmed.a042440. [DOI] [PubMed] [Google Scholar]
- 17.Seske LM, Muglia LJ, Hall ES, Bove KE, Greenberg JM. Infant mortality, cause of death, and vital records reporting in Ohio, United States. Matern Child Health J. 2017;21(4):727–733. doi: 10.1007/s10995-016-2159-x. [DOI] [PubMed] [Google Scholar]
- 18.Madsen A, Thihalolipavan S, Maduro G, et al . An intervention to improve cause-of-death reporting in New York City hospitals. Prev Chronic Dis. 2012;9(120071):2009–2010. doi: 10.5888/pcd9.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ong P, Gambatese M, Begier E, Zimmerman R, Soto A, Madsen A. Effect of cause-of-death training on agreement between hospital discharge diagnoses and cause of death reported, inpatient hospital deaths. Prev Chronic Dis. 2015; 2008;12:140299. doi: 10.5888/pcd12.140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callaghan WM, MacDorman MF, Rasmussen SA, Qin C, Lackritz EM. The contribution of preterm birth to infant mortality rates in the United States. Pediatrics. 2006;118(4):1566–1573. doi: 10.1542/peds.2006-0860. [DOI] [PubMed] [Google Scholar]
- 21.Bell GS, Gaitatzis A, Johnson AL, Sander JW. Predictive value of death certification in the case ascertainment of epilepsy. J Neurol Neurosurg Psychiatry. 2004;75(12):1756–1758. doi: 10.1136/jnnp.2003.029918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirby RS. Classifying infant deaths with a focus on prevention strategies. Public Health Rep. 2015;130(6):570–572. doi: 10.1177/003335491513000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sowards KA. What is the leading cause of infant mortality? A note on the interpretation of official statistics. Am J Public Health. 1999;89(11):1752–1754. doi: 10.2105/AJPH.89.11.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dollfus C, Patetta M, Siegel E, Cross AW. Infant mortality: a practical approach to the analysis of the leading causes of death and risk factors. Pediatrics. 1990;86:176–183. [PubMed] [Google Scholar]
- 25.Nakamura AM, Dove MS, Minnal A, Damesyn M, Curtis MP. Infant mortality: development of a proposed update to the Dollfus classification of infant deaths. Public Health Rep. 2015;130(6):632–642. doi: 10.1177/003335491513000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyle B, Addor MC, Arriola L, et al. Estimating global burden of disease due to congenital anomaly: an analysis of European data. Arch Dis Child Fetal Neonatal Ed. 2018;103(1):F22–F28. doi: 10.1136/archdischild-2016-311845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzaludo N, Belmont JW, Gainullin VG, Taft RJ. Estimating the burden and economic impact of pediatric genetic disease [published correction appears. in Genet Med. 2019;21(9):2161]. Genet Med. 2019;21(8):1781–1789. doi: 10.1038/s41436-018-0398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reefhuis J, Gilboa SM, Anderka M, et al. The National Birth Defects Prevention Study: a review of the methods. Birth Defects Res A Clin Mol Teratol. 2015;103(8):656–669. doi: 10.1002/bdra.23384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bairoliya N, Fink G. Causes of death and infant mortality rates among full-term births in the United States between 2010 and 2012: an observational study. PLoS Med. 2018;15(3):e1002531. doi: 10.1371/journal.pmed.1002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGivern L, Shulman L, Carney JK, Shapiro S, Bundock E. Death certification errors and the effect on mortality statistics. Public Health Rep. 2017;132(6):669–675. doi: 10.1177/0033354917736514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cairo SB, Arbuthnot M, Boomer LA, et al. Controversies in extracorporeal membrane oxygenation (ECMO) utilization and congenital diaphragmatic hernia (CDH) repair using a Delphi approach: from the American Pediatric Surgical Association Critical Care Committee (APSA-CCC) Pediatr Surg Int. 2018;34(11):1163–1169. doi: 10.1007/s00383-018-4337-y. [DOI] [PubMed] [Google Scholar]