

Abstract
Elevation of hypoxia‐inducible factor 1 protein has been shown to be protective in acute kidney injury and HIF1α enhancing drug therapies are currently in clinical trials for the treatment of anemia of chronic kidney disease. Despite its benefits, long‐term HIF1 elevation seems to be associated with additional effects in the kidneys such as tubulointerstitial fibrosis. To better understand the effects of prolonged HIF1 exposure, assessment of baseline and post‐therapy levels of HIF1α and other related biomarkers is essential. In this study, we assessed the effect of HIF1α enhancement using prolyl hydroxylase inhibitor (PHD‐I) DMOG, on a key profibrotic marker of kidney disease. In specific, we examined the change in expression of Collagen 4 subunit A2 in cultured urinary cells of CKD patients pre and post 24‐hour exposure to 1mM DMOG. Our results show that besides HIF1α enhancement, COL4A2 protein is suppressed in presence of DMOG. To determine if this effect is mediated by HIF1, we used HIF1α gene silencing in HEK293 cells and examined the effect of DMOG on protein and gene expression of COL4A2 post 24‐hour exposure. We showed that silencing HIF1α reverses and amplifies the expression of COL4A2 in HEK293 cells. Our data suggest that HIF1 directly regulates the expression of COL4A2 in kidney cells and that HIF1α enhancing therapy has suppressive effects on COL4A2 that may be clinically relevant and must be considered in determining the safety and efficacy of these drugs in the treatment of anemia.
Keywords: anemia, CKD, collagen IV, hypoxia inducible factor‐1, prolyl hydroxylse inhibitors, tubular fibrosis
In this report we have shown that PHD‐I treatment suppresses COL4A2 expression in CKD renal cells. The effect was shown to be transcriptionally regulated by HIF‐1 and silencing HIF‐1a gene reversed the suppression induced by PHD‐I in HEK293 cells.

Abbreviations
- CKD
chronic kidney disease
- COL4A2
collagen 4 alpha 2
- CTGF
connective tissue growth factor
- DMOG
dimethyloxalylglycine
- EGF
epidermal growth factor
- EMT
epithelial‐mesenchymal transition
- GBM
glomerular basement membrane
- KSF
keratinocyte serum free
- PAI‐1
plasminogen activator inhibitor 1
- PHD‐I
prolyl hydroxylase domain inhibitor
- TIMP1
tissue inhibitor of metalloproteinase 1
- UPC
urinary progenitor cell
1. INTRODUCTION
The use of agents to stimulate endogenous erythropoietin (EPO) production in anemic patients with chronic kidney disease (CKD) is a developing treatment approach. 1 If not properly treated, anemia increases cardiovascular risk, impairs quality of life, and increases the frequency of blood transfusions. 2 Current therapy with erythropoiesis‐stimulating agents (ESAs) or recombinant human erythropoietin increases hemoglobin (Hb) levels, decreasing the occurrence of blood transfusions. 1 Recent findings have highlighted the safety concerns associated with poor response to ESAs, primarily inflammation. Inflammation stimulates the hepatic secretion of hepcidin, a key regulator of iron metabolism, through the inflammatory cytokine IL‐6. 3 Elevated hepcidin levels lead to iron insufficiency which prevents the proper maturation of erythrocytes and leads to ESA resistance.3 One drug class under development stabilizes hypoxia‐inducible factor (HIF) through prolyl hydroxylase (PH) enzyme inhibition and is a potential alternative to ESAs when resistance occurs. Roxadustat, a prolyl hydroxylase inhibitor (PHD‐I), is currently undergoing phase III clinical trials and its use has shown a significant reduction in hepcidin while promoting the production of EPO. 4 , 5 , 6
Hypoxia‐inducible factor‐1 is a key factor in cellular response to hypoxia. It regulates gene expression for EPO, VEGF, and glycolytic enzymes to compensate for the reduced oxygen and nutrient supply in hypoxia. In normoxia, HIF1 alpha subunit is enzymatically hydroxylated by prolyl hydroxylase domain (PHD) enzymes, which enables the von Hippel–Lindau protein (pVHL) to bind, leading to ubiquitination and degradation of the protein. Under hypoxic conditions, HIF1α escapes degradation and binds to the aryl hydrocarbon receptor nuclear translocator (ARNT), and exerts its hypoxic response through binding to the cis‐consensus HIF1‐binding site, the hypoxia‐responsive element (HRE) to form an active HIF1 complex. 7 , 8 , 9 One agent, dimethyloxalylglycine (DMOG), inhibits both asparagine and proline hydroxylases, activating HIF1α gene expression. 2 Prolyl hydroxylase inhibitors such as DMOG reversely inhibit the catalytic action of PHD enzymes. This results from their binding to the ferrous‐iron‐containing active site of PHD, blocking the entry of the co‐substrate 2‐oxoglutarate, leading to HIF1 stabilization and its downstream activation of genes. 2 PHD‐I’s stimulate erythropoiesis with concentrations of endogenous EPO allowing for a lower exposure to EPO than that of which ESAs provide, which may reduce the associated adverse side effects.
CKD is primarily a hypoxic kidney pathology and HIF1α seems to play a key role in the defense of the kidney tubules to hypoxia. 10 , 11 , 12 In our previous work, we have shown HIF1α expression in CKD patients is increased 4‐fold compared to healthy individuals. 13 Although HIF1 activation would appear to have a cytoprotective effect on renal tissue, recent studies have demonstrated that constitutive HIF1 activation could accelerate fibrosis via upregulation of profibrogenic factors such as tissue inhibitor of metalloproteinase 1 (TIMP1), connective tissue growth factor (CTGF), and plasminogen activator inhibitor 1 (PAI‐1). 14 It is suggested that consistent noncontinuous low level HIF1 stimulation has the ability to improve erythropoiesis while limiting the adverse effects that arise from continuous HIF1 stimulation. 1
Renal fibrosis is caused by the disproportional formation and degradation of extracellular matrix proteins, including collagen. Type IV collagen is the main component of the glomerular basement membrane (GBM) and is composed of six α chains, α1(IV)–α6(IV). These chains are classified into two groups by their gene structure, α1, α3, and α5 are one set, and α2, α4, and α6 are another set. Most commonly, type IV collagen molecules contain α1 and α2 chains in a 2:1 ratio. In the GBM, however, this is replaced with a α3:α4:α5 chain composition. Collagen IV is more flexible compared to other collagen subtypes because of its lack of glycine in every third amino‐acid residue, which is quintessential for an otherwise tightened collagen helix. 15
Interestingly, dimethyloxalylglycine which activates HIF1 has been shown to reduce fibrosis in rat studies, as manifested by decreased type IV collagen and osteopontin expression. 16 On the contrary other studies have shown inhibition of type IV collagen assembly can lead to up‐regulation of transforming growth factor‐beta1, an inducer of type 2 epithelial‐mesenchymal transition (EMT). 17 Type 2 EMTs are found to be associated with fibrosis occurring in kidney, liver, lung, and intestine. 18 It appears that altered collagen IV levels are associated with several types of nephropathies such as Alport syndrome, diabetic nephropathy, IgA glomerulonephritis, and inherited nephropathies. 19 , 20 , 21 , 22 , 23 , 24 , 25 Studies have shown the involvement of various subunits in different types of nephropathies. A genome‐wide association study (GWAS) identified a common missense mutation in the alpha 3 chain that has been found to increase the risk of developing diabetic kidney disease. 25 The presence of IgA and IgG autoantibodies to the alpha 5 and 6 chains have been associated with glomerulonephritis. 23 Type IV collagen has been linked to the development of glomerulosclerosis in both experimental and human diabetic and nondiabetic renal disease. 19 , 20 Collagen IV alpha 2 mRNA levels have been shown to increase in isolated glomeruli undergoing experimental glomerulosclerosis prior to morphological alterations. 20 Additionally, glomerular collagen IV alpha 2 mRNA levels have been shown to significantly increase in the presence of diabetic nephropathy with lesions compared to diabetic nephropathy alone. 19
Amongst the various types of nephropathy, CKD with diabetic nephropathy is the most common, and changes in COL4A2 subunit seems to be most associated with CKD and diabetic nephropathy. Therefore, in this study, we examined the effect of PHD‐I on the expression of COL4A2 through HIF1 upregulation in cultured renal proximal tubular cells of patients with CKD as well as in HEK293 cell lines following DMOG exposure. We hypothesize that HIF1 upregulation alters the expression of COL4A2 subunit and this effect is more pronounced in CKD where baseline HIF1 levels are elevated.
2. MATERIALS AND METHODS
2.1. Subject recruitment
Upon approval from the Institutional Review Board of the Bernard J. Dunn School of Pharmacy, CKD patients (stage II to IV) were recruited into the study. Patients were recruited on predefined inclusion and exclusion criteria from a participating kidney and hypertension specialist clinic in Manassas, VA. CKD staging was defined based on the National Kidney Foundation's definitions of stage II‐V kidney disease. The study population included 18‐ to 90‐year‐old patients with existing renal failure with or without co‐existing cardiac dysfunction, who are being treated at the listed participating site, and who are identified by their authorized medical caregiver as a candidate for participation in the study. Exclusion criteria are renal transplantation, hemodialysis, immunocompromised, human immunodeficiency virus (HIV), hepatitis, amyloidosis, sarcoidosis, renal replacement therapy, autoimmune diseases (except diabetes type I and II), active infection, valvular heart diseases, diagnosed pulmonary hypertension, active illicit drug use, age <18 years, diabetic ketoacidosis, requiring bilevel positive airway pressure (BiPAP), poisoning, sickle cell anemia, active cancer, Fabry disease, and erythropoietin injection within the past 7 days. All of the above conditions may elevate/alter HIF1 levels independent of CKD.
2.2. Urine cell isolation
A total volume of 50 ml urine samples of CKD patients was collected. The samples were refrigerated or kept on ice and transported to the laboratory. In the laboratory, urine samples were centrifuged at 400 g at 4°C for 15 min. The supernatants were aspirated and pellets were resuspended in the wash buffer (1% 100 U/ml penicillin plus 100 μg/ml of streptomycin into phosphate‐buffered saline) and centrifuged at 400 g for 10 min. The pellets were transferred to either 24‐ or 6‐well plates. According to previously defined methods Urinary Progenitor Cell (UPC) media was used to promote primary cell growth in culture. 26 Media was added to the 24‐well plates and changed every 2–3 days. Cells were incubated over 2–3 weeks to ensure proper growth, after which they were passaged and transferred to larger plates.
2.3. Cell culture
2.3.1. Urinary progenitor cell medium
To prepare UPC medium, equal volumes of keratinocyte serum free (KSF) medium and progenitor cell (PC) Medium were mixed. The KSF Medium contained 5 ng/ml of epidermal growth factor (EGF), 50 ng/ml of bovine pituitary extract (BPE), 30 ng/ml of cholera toxin, and pen/strep solution (100 U/ml penicillin, 100 μg/ml streptomycin) that were added to the KSF medium. The PC Medium contained 4.24 g/500 ml Ham‐F12, 10% FBS, 0.4 μg/ml hydrocortisone, 0.1 nM cholera toxin, 5 ng/ml insulin, 1.8 × 10–4 M adenine, 5 μg/ml transferrin, 2 × 10–9 M triiodothyronine, 10 ng/ml EGF, and pen/strep solution that were added to the DMEM medium. 26
HEK 293 cells were cultured in DMEM with 10% fetal bovine serum and 1% pen/strep.
2.4. Immunocytochemistry
After 3–4th, primary cultured urine kidney cells were exposed to 1 mM PHD‐I (DMOG) for 24 hours. Following exposure, the cells were fixed with 1.5% formaldehyde. To stain for intracellular HIF1α, cells were permeabilized for 10 min with 0.01% Triton X‐100 in. Fixed cells were immunostained with 1% BSA/PBST to block nonspecific binding of the antibody. HIF1α and COL4A2 primary antibodies were added to the cells and they were incubated at 4°C overnight. The cells were then incubated with Rabbit IgG secondary antibody in 1% BSA/ PBST for 1 h at room temperature. The cells were washed with PBST and incubated with Horseradish peroxidase in 1% BSA for 30 min in the dark and then washed with PBS. To counterstain, the cells were incubated with DAPI (DNA stain) for 20 min until the change in color was seen. The cells were rinsed with tap water and viewed under a microscope for HIF1α and COL4A2 staining.
2.5. Flow cytometry
Flow cytometry experiments were performed to examine protein expression of HIF1a and COL4A2. The instrument used was Muse™ Cell Analyzer by Millipore Corporation, INC. Hayward, CA. To examine protein expression the cell count and viability assay and appropriate fluorescent‐tagged antibodies were used to detect fluorescent signals associated with the antibody of interest. Per‐protocol and to stabilize proteins the cells were fixed in 2% formaldehyde prior to antibody addition. Therefore, the measured signal was observed only in dead cells.
Primary urine isolated cells or HEK293 cells were treated for 24 h with 1 mM DMOG to measure HIF1α and collagen 4A2 levels. The cells were fixed in 2% formaldehyde. For HIF1α staining, cells were permeabilized with 0.01% Triton‐X in PBS. HIF1α or collagen 4A2 rabbit primary antibodies plus 0.5%–1% BSA (to prevent nonspecific binding) were added to the samples. Alexa Fluor 546 goat anti‐rabbit IgG was used as secondary conjugation. HIF1α and collagen 4A2 expressions were quantified as Mean Yellow Fluorescence (MYF) signal in the dead cell population in PHD‐I treated and non‐treated cells. Samples were measured in triplicates. Instrument system check was performed prior to each experiment to assure the accuracy of reading.
2.6. Real‐time reverse‐transcription polymerase chain reaction
RNA isolation was performed in a QIAcube system using the Qiagen RNeasy Plus Mini Kit. Total RNA of each sample was converted to cDNA using the high‐capacity reverse transcription kit. cDNA samples were amplified using Applied Biosystems 7300 PCR and the TaqMan gene expression assays protocol. Samples were loaded into 96‐well plates in triplicates. In order to determine gene stability, pairwise comparisons were made between genes using methods described by Vandesomple et al. in conjunction with qbase+ (geNorm) software produced by Biogazelle. 27 , 28 To take into account the effect of varying RNA quality and amount between samples, the previously described delta Ct method was utilized whereby each HIF1α and COL4A2 samples were normalized to their corresponding ACTB values and average of individual delta Ct values were used for comparison between groups. 27 , 28
2.7. RNAi cell transfection
In this experiment, short interfering RNA (SiRNA) for HIF1α interference was used to silence HIF1α. HEK 293 cells were plated in a six‐well plate and seeded to 70%–90% confluency. Lipofectamine 3000 was used to transfect cells with HIF1α SiRNA or negative control SiRNA. The transfected HEK293 cells were incubated for 48 h at 37°C then split and treated with DMOG 1mM on the third day. RT‐PCR and Flow cytometry experiments were then conducted to analyze HIF1α and COL4A2 expression change with and without exposure to DMOG.
2.8. Reagents and materials
The following materials were purchased from Sigma‐Aldrich (St. Louis, MO). Phosphate‐buffered saline with 10% bovine serum, human epidermal growth factor, human recombinant insulin, HEK293 human embryo kidney‐cell line, keratinocyte serum‐free growth medium, cholera toxin from Vibrio cholerae, adenine, apo‐transferrin human, 3,3′,5‐triiodo‐l‐thyronine, formaldehyde solution, triton X‐100, BSA (10X) in PBS.
The following items were purchased from Life Technologies (Carlsbad, CA): define keratinocyte‐SFM media, HIF 1α Taqman gene expression assay, ACTB Taqman gene expression assay, COL4A2 Taqman gene expression assay, AM16708 Silencer Pre‐Designed SIRNA, wild type SIRNA, HIF1α Recombinant Rabbit Monoclonal antibody, Alexa Fluor 546 goat anti−rabbit IgG, COL4A2 rabbit antibody.
High‐capacity reverse transcription kit was purchased from Applied Biosystems, Foster City, CA. The Lipofectamine 3000, RNase‐free was obtained from Invitrogen (Carlsbad, CA). Bovine Pituitary Extract (BPE) was obtained from Thermo Fisher Science (Carlsbad, CA). Prolyl‐Hydroxylase inhibitor 50 mg (DMOG) was purchased from Millipore Sigma (Rockville, MD).
2.9. Statistical analysis
Differences between groups were determined using Student's t‐test. Comparisons between groups were made either as paired or unpaired equal variance t‐test with one or two‐tailed distribution, and p < .05 was considered statistically significant. For RT‐PCR and Flow cytometry experiments in HEK293 cells, we used three to five experiments for statistical analysis. For the patient flow cytometry experiments sample size ranged from 7 to 13 patients. All samples were run and analyzed as triplicates in flow cytometry and RT‐PCR experiments.
2.10. Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 29 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 30
3. RESULTS
3.1. The effect of DMOG exposure on collagen 4A2 protein expression in CKD patient cells
A total of 13 patient samples with various stages of chronic kidney disease (II‐V) were included in our analysis from our participating kidney to hypertension clinic. Patients were consented to participate in the study and urine samples were collected and transferred to laboratories on the same day. Table 1 shows the basic characteristics of the patient population. The average eGFR of the patient was 33 ± 16 ml/min/1.73 m2. Patients were selected based on the inclusion/exclusion criteria and the other comorbidities are listed in Table 1. The incidence of diabetes mellitus (DM) in our patients was 69%. The most common comorbidity was essential hypertension (92%). Anemia was present in 39% of patients.
TABLE 1.
Patient demographic information
| Gender, n (%) | |
| Male | 7 (54) |
| Female | 6 (46) |
| Age (yr) | 68 ± 13 |
| Race (%) | |
| Caucasian | 7 (54) |
| African American | 3 (23) |
| Asian | 3 (23) |
| GFR (ml/min/1.73m²) | 33 ± 16 |
| Comorbidities, n (%) | |
| Anemia | 5 (39) |
| Diabetes Mellitus | 9 (69) |
| Hypertension | 12 (92) |
| Hyperlipidemia | 4 (31) |
Abbreviation: GFR, glomerular filtration rate.
In order to examine the effect of PHD‐I treatment on kidney tubular cells we cultured urinary cells from collected urine samples of patients. For immunocytochemistry experiments, we used patient cells after 5‐weeks in culture and about the 3rd passage. Cells were exposed to 24 h of 1mM DMOG following which immunocytochemistry was performed to identify changes in HIF1α and COL4A2 expression. Figure 1A–D shows the result of immunostaining of COL4A2 and HIF1α in a patient urinary cell culture. In cells exposed to 1mM PHD‐I, COL4A2 decreased within 24 h in CKD patient cells. COL4A2 suppression was associated with increased HIF1α expression at 24 h.
FIGURE 1.
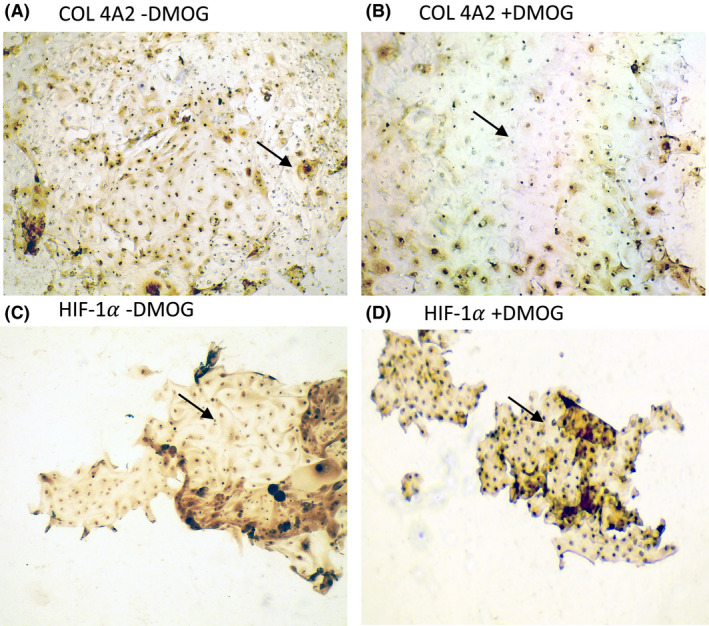
DMOG suppresses COL4A2 in cultured CKD urinary cells
To quantify the degree of suppression of COL4A2 subunit we performed flow cytometry experiments in isolated urinary cells. For these experiments, cells were cultured in six‐well plates after sample collection and 1 day post culture the cells were treated with 1mM PHD‐I or control. Cell culture duration was shortened to one day due to the necessity of high density of cells for flow cytometry experiments. Isolated urine samples have the highest number of fresh urinary cells within the first 24–48 h of collection. Flow cytometry experiments were performed after 24 h of exposure to 1 mM PHD‐I and results are shown in Figure 2. After 24 h of 1 mM PHD‐I treatment, there was a 32% reduction in the expression of collagen 4A2 and a 15% increase in protein expression of HIF1α (N = 7, p = .002; N = 13, p = .05, respectively).
FIGURE 2.
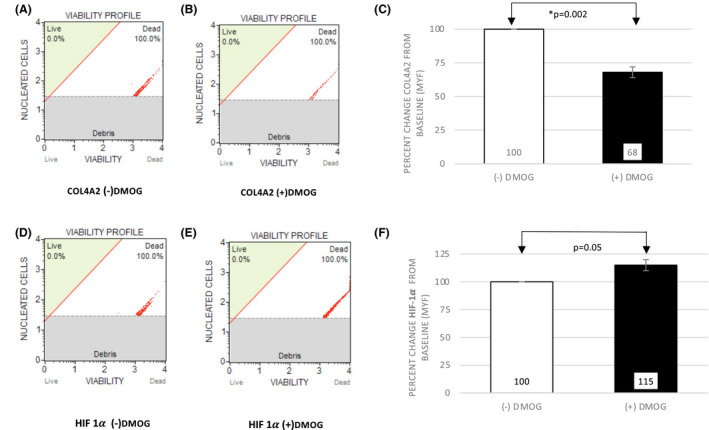
DMOG suppresses COL4A2 protein in CKD urinary cells analyzed by flow cytometry
3.2. The effect of HIF1α gene silencing on COL4A2 suppression by PHD‐I
In order to determine the role of HIF1 in the regulation of COL4A2 levels, we performed experiments in HEK 293 cells after silencing HIF1α gene. For these experiments, cells were transfected with HIF1α SiRNA ((+)SiRNA) or negative control SiRNA ((−)SiRNA) for 48hr and then incubated with or without 1mM PHD‐I for an additional 24 hours. After the treatment period, real‐time RT‐PCR and flow cytometry experiments were performed.
3.3. Gene expression analysis in HIF1α
HIF1α is a key HIF1 subunit that leads to its stabilization and binding to DNA. Silencing HIF1α subunit thus inhibits the transcriptional activity of the protein. We, therefore, examined the effect of silencing HIF1α in HEK293 cells to determine if suppression of COL4A2 is transcriptionally mediated by HIF1. At baseline, HIF1α gene silencing suppressed the baseline mRNA expression of HIF1α by 78% (N = 4, p = .003) (Figure 3A). When treated with PHD‐I in control (−)SiRNA, HIF1α gene expression decreased by 61% (N = 4, p = .02) (Figure 3B). However, in silenced (+)SiRNA cells, HIF1α gene downregulated by 11% (N = 4, p = .26) in response to PHD‐I (Figure 3C). In comparison, there was an 85% difference in suppression of HIF1α gene in (‐)SiRNA compared to (+)SiRNA in presence of DMOG (N = 4, p = .002) (Figure 3D).
FIGURE 3.
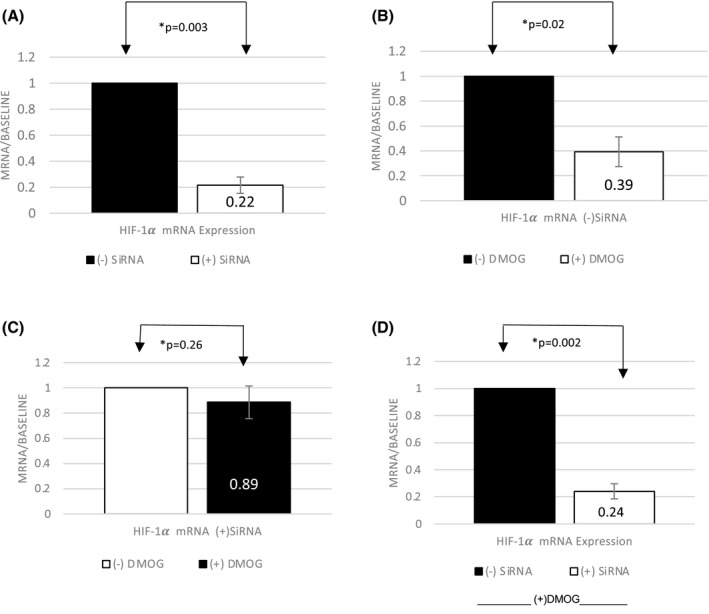
Effect of DMOG on HIF‐1α mRNA with or without gene silencing
3.4. Gene expression analysis in COL4A2
We also examined the effect of HIF1α gene silencing on COL4A2 gene expression. Since cultured primary cells are not as tolerant of transfection experiments and do not yield enough cells to perform either RT‐PCR or flow cytometry experiments post transfection we performed these experiments in HEK293 cells. At baseline, COL4A2 gene expression was not significantly different between HIF1α (+)SiRNA and (‐)SiRNA in the absence of PHD‐I (N = 4, p = .11) (Figure 4A). Treatment with PHD‐I however, reduced the baseline gene expression of COL4A2 by 36% compared to non‐treated cells in control HIF1α (‐)SiRNA (N = 4, p = .03) (Figure 4B). Furthermore, this suppression was significantly reversed and even increased 2‐fold when HIF1α was silenced via (+)SiRNA (N = 4, p = .04) (Figure 4C). In comparison, there was a 2.3‐fold increase in expression of COL4A2 in HIF1α (+)SiRNA compared to (−)SiRNA cells in presence of DMOG (N = 4, p = .03) (Figure 4D).
FIGURE 4.
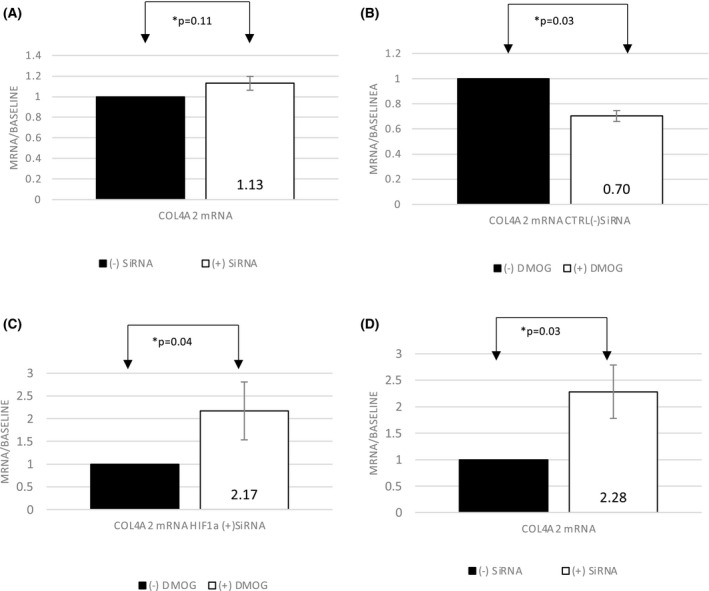
Effect of DMOG on COL4A2 mRNA with or without HIF‐1α gene silencing
3.5. Protein expression analysis in HIF1α and Collagen 4A2
To establish the phenotype associated with changes in COL4A2 gene expression in response to PHD‐I and HIF1α silencing, flow cytometry experiments were performed (Figures 5 and 6). In the negative control HIF1α ((−)SiRNA), PHD‐I resulted in an average of 10% decrease (Figure 5C) in COL4A2 protein expression and a 10% increase in the protein expression of HIF1α (N = 4, p = .028, and p = .01, respectively) (Figure 5C,F). In the HIF1α (+)SiRNA group, 24 hours of PHD‐I treatment, resulted in a 23% increase (N = 5, p = .018) (Figure 6C) in protein expression of COL4A2 which is consistent with the enhanced gene expression observed in RT‐PCR experiments. There was not a significant change in protein expression of HIF1α in the silenced cells treated with PHD‐I (N = 5, p = .07) (Figure 6F). In comparison to control, HIF1a gene silencing lead to a reversed suppressive effect of PHD‐I on COL4A2 by 33% (N = 4, p = .01) (Figure 6G). In all of our results, N represents the number of experiments performed to anlayze the data.
FIGURE 5.
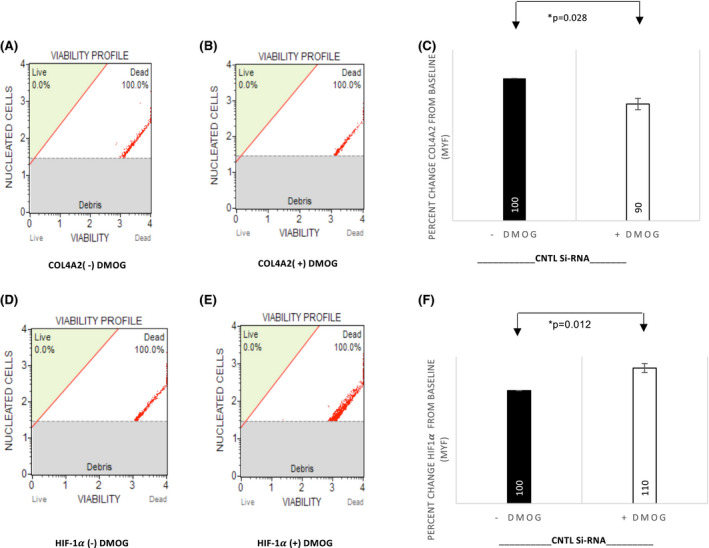
DMOG suppresses COL4A2 protein levels in control HEK293 cells
FIGURE 6.
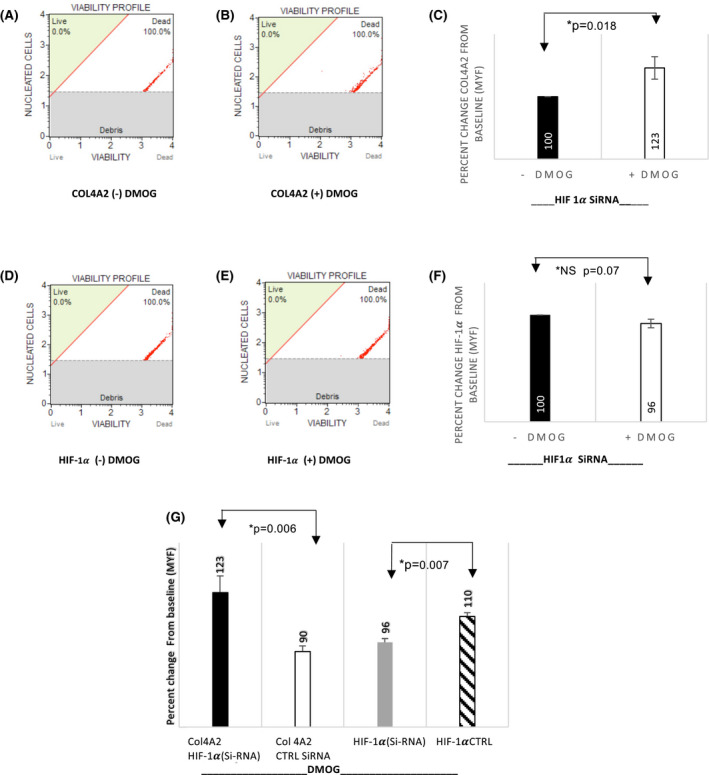
HIF‐1α gene silencing reverses the suppression of COL4A2 protein in HEK293 cells
4. DISCUSSION
In this report, we investigated the gene expression and protein yield of COL4A2, in presence of DMOG, a PHD inhibitor. We found that COL4A2 protein levels decrease in the presence of DMOG in CKD patient urinary cells (Figure 2). We observed a similar effect but to a lesser extent in HEK293 cell lines (10% vs 32%) (Figure 5). It must be noted that HEK293 cells were chosen as an alternate non‐disease cell model and were not intended to resemble kidney cells. The lower response of HEK293 cells to DMOG can be attributed to the fact that patient cells have a higher baseline HIF1α expression compared to normal. 13 Additionally, HIF1 levels are higher at baseline in hypoxic conditions, such as CKD, compared to the normoxic conditions in which HEK293 experiments were conducted. Lastly, the CKD cells are a model of kidney disease that may have a differential response to COL4A2 suppression compared to non‐disease HEK293 cells. Nevertheless, the degree of COL4A2 suppression was correlated with the increase in HIF1α levels in both CKD and HEK293 cells. A similar pattern of COL4A2 suppression was also seen in HEK293 cells similar to CKD patient cells. Therefore, it is safe to assume that HEK293 cells are a suitable secondary model to explore the transcriptional regulation of COL4A2 by HIF1.
We further explored the effect of gene silencing to determine if HIF1 is responsible for changes in COL4A2 levels. Our SiRNA experiments yielded up to 79% suppression of HIF1α which validates the efficiency of the method used (Figure 3D). Our results confirmed that HIF1α silencing eliminates the suppression of COL4A2 both in gene and protein levels. What is most noteworthy is that COL4A2 protein levels measured through flow cytometry experiments show an increase of 23% in presence of PHD‐I in the knockout HIF1α cells while a decrease of 10% in the wild type HIF1α (−)SiRNA) cells (Figure 6G), which verifies that HIF1 activity is directly involved in COL4A2 gene and protein expression. This supports our hypothesis that HIF1 directly impacts the expression of COL4A2. These findings are novel, to our knowledge, there are no reports to date that investigate the COL4A2 phenotype in relation to HIF1 enhancement both in vitro, as well as in patient cells with existing CKD. We also show, that the regulation of COL4A2 by PHD‐I is transcriptionally regulated via HIF1 and that silencing HIF1α reverses the suppression induced by PHD‐I. We noted an increase in gene expression of COL4A2 (2‐fold) in the absence of HIF1α in response to DMOG. This finding suggests that DMOG exposure can increase the expression of COL4A2 in the absence of HIF1. This effect was seen to a lesser degree (23% increase) in protein expression of COL4A2 in HIF1α silenced HEK293 cells post DMOG exposure. Since the increase in gene expression of COL4A2 was more robust than its protein levels, it suggests that loss of HIF1α may act directly or through other genes that enhance COL4A2 response to DMOG. Additional experiments are needed to explore the exact mechanism of this effect. As to the direct mechanism of HIF1 suppression of COL4A2 gene, there is evidence that HIF1 downregulates the promoter of collagen I through the SP3 transcription factor in chondrocytes. 31 Similar binding sites are present in the collagen 4A1,2 common promoter region. It is possible that HIF1 suppression of collagen 4A2 is via a similar mechanism.
It must be noted that DMOG has a potency range of 0.1–1 mM. Although in in vivo experiments DMOG has been used at the lower concentrations, dose response studies in in vitro experiments have shown HIF1 stabilization without toxicity in the 1mM concentration of DMOG. 32 Additionally, this concentration corresponds to similar HIF1 stabilization as Roxadustat 10 µM concentration. 33 We therefore, elected to perform our in vitro experiments using the 1 mM concentration of DMOG.
Among other key profibrotic factors such as Matrix Metalloproteinases (MMPs), type IV collagen is a key structural component of the GBM and its increased expression is correlated with renal fibrosis and diabetic nephropathy. Other collagen types (I, III, and XVIII) are also involved in the development of interstitial fibrosis. 34 Our findings showed a significant reduction (32%) in the expression of COL4A2 in response to PHD‐I, through the upregulation of HIF1 in CKD cells. Although recent evidence has shown that HIF1 enhancement can increase fibrosis through the upregulation of profibrogenic factors such as TIMP1, CTGF, and PAI‐1, based on our findings this does not appear to be the case in terms of type IV collagen. 34
In terms of clinical application of our study, the use of PHD‐I’s is a developing treatment approach in anemia of kidney disease. Phase III clinical trials of Roxadustat, a PHD‐I, have found it to be safe and effective in promoting the production of EPO in CKD patients. 5 , 6 ESA resistance can occur due to a common side effect of treatment, inflammation, PHD‐I’s are a viable alternative when this occurs. Our findings suggest that PHD‐I use may also play a role in slowing down the progression of renal fibrosis, thus having added benefits in CKD.
There are several limitations of the current research. Our study was primarily modeled in CKD patient urine as a source of kidney tubular cells. It is important to examine the effect in an animal model of kidney disease to further establish the HIF1 mediated suppression of COL4A2. Another potential limitation of our study is the lack of data in healthy kidney cells. In our preliminary experiments, we examined the effect in healthy subject urinary cells and did not find consistent suppression of COL4A2 by DMOG (data not shown). Given that our aim was to study the effect of PHD‐I in CKD patients, we elected to focus our investigation in disease urinary cell model. We did, however, examine HEK‐293 cells as non‐disease cell model to explore the gene transcription changes in response to HIF‐1 enhancement. It would be informative to establish similar results in healthy subject urinary cells in future studies. Furthermore, for the reasons described above we chose to study COL4A2 subunit in this study. It is important to further explore the role of other type IV collagen subunits in similar models to better understand the contribution of collagen IV expression to kidney fibrosis. Collagens in kidney tubules and glomeruli play major roles in the structural integrity of the kidney. Along these lines, the role of other collagen types (I, III, and XVIII) in response to HIF1 and PHD‐I also needs to be investigated. Another potential limitation of our study is the timing of exposure to DMOG to establish long‐term effects. In our study, we examined exposures of 4, 8, 12, 24, and 36 h and noted 8–24 h of DMOG exposure to produce steady‐state effects on HIF1 stabilization, after which the effect begins to wear off (data not shown). Given that in cell culture gene transcription occurs in as early as 4 h, we considered 24 h to be relatively long term. However, to better examine long‐term effects, future studies conducted in animal models would strongly support our findings.
CONFLICT OF INTEREST
Author do not claim any conflict of interest.
AUTHOR CONTRIBUTIONS
Wrote the manuscript: Movafagh, Kamal, Handy, Salaheldin, and Moore. Participated in Research Design: Movafagh, and Sanaei Ardekani. Conducted Experiments: Movafagh, Kamal, Handy, Salaheldin, Alam, and Moore. Performed Data Analysis: Sanaei Ardekani, Movafagh, Kamal, Handy, Alam, and Moore.
ACKNOWLEDGMENTS
None.
Sanaei‐Ardekani M, Kamal S, Handy W, et al. Suppression of collagen IV alpha‐2 subunit by prolyl hydroxylase domain inhibition via hypoxia‐inducible factor‐1 in chronic kidney disease. Pharmacol Res Perspect. 2021;9:e00872. doi: 10.1002/prp2.872
Shyreen Kamal, Whitney Handy, Sidrah Alam, and Aya Salaheldin contributed equally to the manuscript.
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this published article.
REFERENCES
- 1. Gupta N, Wish JB. Hypoxia‐inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis off J Natl Kidney Found. 2017;69:815‐826. [DOI] [PubMed] [Google Scholar]
- 2. Del Vecchio L, Locatelli F. Investigational hypoxia‐inducible factor prolyl hydroxylase inhibitors (HIF‐PHI) for the treatment of anemia associated with chronic kidney disease. Expert Opin Investig Drugs. 2018;27:613‐621. [DOI] [PubMed] [Google Scholar]
- 3. Yan Z, Xu G. A novel choice to correct inflammation‐induced anemia in CKD: oral hypoxia‐inducible factor prolyl hydroxylase inhibitor roxadustat. Front Med. 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charytan C, Manllo‐Karim R, Martin ER, et al. A randomized trial of roxadustat in anemia of kidney failure: SIERRAS study. Kidney Int Rep. 2021;6(7):1829‐1839, Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen N, Hao C, Liu B‐C, et al. Roxadustat treatment for anemia in patients undergoing long‐term dialysis. N Engl J Med. 2019;381(11):1011‐1022, Massachusetts Medical Society. [DOI] [PubMed] [Google Scholar]
- 6. Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381(11):1001‐1010, Massachusetts Medical Society. [DOI] [PubMed] [Google Scholar]
- 7. Ayrapetov MK, Xu C, Sun Y, et al. Activation of Hif1α by the prolylhydroxylase inhibitor dimethyoxalyglycine decreases radiosensitivity. PLoS ONE. 2011;6(10):e26064, Public Library of Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kimura K, Iwano M, Higgins DF, et al. Stable expression of HIF‐1α in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol ‐ Ren Physiol. 2008;295:F1023‐F1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tanaka T, Miyata T, Inagi R, Fujita T, Nangaku M. Hypoxia in renal disease with proteinuria and/or glomerular hypertension. Am J Pathol. 2004;165:1979‐1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosenberger C, Heyman SN, Rosen S, et al. Up‐regulation of HIF in experimental acute renal failure: evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005;67:531‐542. [DOI] [PubMed] [Google Scholar]
- 11. Tanaka S, Tanaka T, Nangaku M. Hypoxia as a key player in the AKI‐to‐CKD transition. Am J Physiol ‐ Ren Physiol. 2014;307:F1187‐F1195. [DOI] [PubMed] [Google Scholar]
- 12. Tanaka T, Nangaku M. Drug discovery for overcoming chronic kidney disease (CKD): prolyl‐hydroxylase inhibitors to activate hypoxia‐inducible factor (HIF) as a novel therapeutic approach in CKD. J Pharmacol Sci. 2009;109:24‐31. [DOI] [PubMed] [Google Scholar]
- 13. Movafagh S, Raj D, Sanaei‐Ardekani M, et al. Hypoxia inducible factor 1: a urinary biomarker of kidney disease. Clin Transl Sci. 2017;10:201‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF‐1 stimulation of epithelial‐to‐mesenchymal transition. J Clin Invest. 2007;117:3810‐3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abreu‐Velez AM, Howard MS. Collagen IV in normal skin and in pathological processes. North Am J Med Sci. 2012;4:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song YR, You SJ, Lee Y‐M, et al. Activation of hypoxia‐inducible factor attenuates renal injury in rat remnant kidney. Nephrol Dial Transplant. 2010;25:77‐85. [DOI] [PubMed] [Google Scholar]
- 17. Zeisberg M, Bonner G, Maeshima Y, et al. Renal fibrosis: collagen composition and assembly regulates epithelial‐mesenchymal transdifferentiation. Am J Pathol. 2001;159:1313‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119:1420‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adler SG, Feld S, Striker L, et al. Glomerular type IV collagen in patients with diabetic nephropathy with and without additional glomerular disease. Kidney Int. 2000;57:2084‐2092. [DOI] [PubMed] [Google Scholar]
- 20. Bergijk EC, Van Alderwegen IE, Baelde HJ, et al. Differential expression of collagen IV isoforms in experimental glomerulosclerosis. J Pathol. 1998;184:307‐315. [DOI] [PubMed] [Google Scholar]
- 21. Cosgrove D, Liu S. Collagen IV diseases: a focus on the glomerular basement membrane in Alport syndrome. Matrix Biol J Int Soc Matrix Biol. 2017;57–58:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Esposito C, Striker LJ, Patel A, et al. Molecular analysis of glomerular diseases in renal biopsies: initial results of a collaborative international study. The International Study Group for Molecular Study of Kidney Biopsies. Proc Assoc Am Physicians. 1996;108:209‐217. [PubMed] [Google Scholar]
- 23. Ghohestani RF, Rotunda SL, Hudson B, et al. Crescentic glomerulonephritis and subepidermal blisters with autoantibodies to alpha5 and alpha6 chains of type IV collagen. Lab Investig J Tech Methods Pathol. 2003;83:605‐611. [DOI] [PubMed] [Google Scholar]
- 24. Matthaiou A, Poulli T, Deltas C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review. Clin Kidney J. 2020;13:1025‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salem RM, Todd JN, Sandholm N, et al.; SUMMIT Consortium, DCCT/EDIC Research Group, GENIE Consortium . Genome‐wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J Am Soc Nephrol. 2019;30:2000‐2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afzal MZ, Strande JL. Generation of induced pluripotent stem cells from muscular dystrophy patients: efficient integration‐free reprogramming of urine derived cells. J VIs Exp JoVE. 2015;52032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101‐1108. [DOI] [PubMed] [Google Scholar]
- 28. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:research0034.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: introduction and other protein targets. Br J Pharmacol. 2019;176:S1‐S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duval E, Bouyoucef M, Leclercq S, Baugé C, Boumédiene K. Hypoxia inducible factor 1 alpha down‐regulates type i collagen through Sp3 transcription factor in human chondrocytes. IUBMB Life. 2016;68:756‐763. [DOI] [PubMed] [Google Scholar]
- 32. Rahman SU, Lee M‐S, Baek J‐H, Ryoo H‐M, Woo KM. The prolyl hydroxylase inhibitor dimethyloxalylglycine enhances dentin sialophoshoprotein expression through VEGF‐induced runx2 stabilization. PLoS ONE. 2014;9:e112078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhute VJ, Harte J, Houghton JW, Maxwell PH. Mannose binding lectin is hydroxylated by collagen prolyl‐4‐hydroxylase and inhibited by some PHD inhibitors. Kidney360. 2020;1(6):447‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bülow RD, Boor P. Extracellular matrix in kidney fibrosis: more than just a scaffold. J Histochem Cytochem. 2019;67(9):643‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.