

Abstract
The synthesis of π‐extended porphyrins containing anthracenyl moieties still represents an important challenge. Here, we report on the synthesis of a series of unsubstituted naphthyl‐, pyrenyl‐ and anthracenyl‐fused zinc porphyrin derivatives. To this aim, meso‐substitued porphyrins are synthesized and the fusion of the PAHs (Polycyclic Aromatic Hydrocarbon) on the β‐positions are performed through thermally induced dehydro‐aromatization. The fused zinc‐porphyrin derivatives are fully characterized and their optical absorption and photoluminescence properties are reported. We also demonstrate that zinc can be removed from the porphyrin core, giving rise to pure C, H, N materials. This work constitutes the first step towards the synthesis of the fully‐fused tetra‐anthracenylporphyrin.
Keywords: anthracene, fused porphyrin, NIR dyes, polycyclic aromatic hydrocarbons, thermal cyclodehydrogenation
A series of π‐extented porphyrins are synthesized by thermal cyclodehydrogenation of porphyrins functionalized with unsubstituted PAHs. The fused porphyrin derivatives can be demetalated to give rise to the unsubstituted and metal‐free porphyrin dyes.

1. Introduction
The formation of π‐extended porphyrins have been extensively reported for the last fifteen years. π‐extended porphyrins can be seen as a porphyrinoid with an extended skeleton that is able to participate to the delocalization of the electrons and thus exhibits structures, electronic and optical properties significantly different from those of the parent porphyrins.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ] The conjugation of polycyclic aromatic hydrocarbons (PAHs) substituents, attached in the meso positions on the β‐positions of the pyrrolic rings, is a popular method to extend porphyrins. Recent works demonstrated that anthracene or anthracene‐like moieties could be fused on the porphyrin core, leading to a large shift of the absorption properties toward the near infrared (NIR).[ 7 , 8 , 9 , 12 ]
The general term of “fusion” describes the set of reactions able to connect molecular fragments to the porphyrin core by at least two bonds that prevent the free rotation between subunits, thus flattening the molecular assemblies and affording conjugated systems. The fusion of PAHs on porphyrins is generally achieved by cyclodehydrogenation using oxidants such as iron(III) chloride or 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ) in the presence of a Lewis acid and is referred to as Scholl reaction. In most cases, the reaction requires the presence of Ni(II) inside the porphyrin core and substituents on the PAH moieties.[ 3 , 8 , 9 , 12 , 13 ] This requirement was attributed to the high oxidation potential of non‐activated PAHs and of porphyrin. [10] Nevertheless, in 2012, the group of Thompson reported the thermally induced dehydro‐aromatization of PAHs (naphthalene, pyrene and coronene) [10] and later 2,3‐dihydro‐1H‐phenalene [14] on the porphyrin core. In this case, neither the presence of electron‐donating groups nor the presence of a nickel cation in the porphyrin were required for the reaction. An alternative approach based on surface activation has also been explored. In particular, it was demonstrated that the phenyl rings of tetraphenylporphyrin derivatives can be fused on the β‐position[ 15 , 16 , 17 , 18 , 19 ] and recently, the formation of open‐shell derivatives[ 20 , 21 ] and oligomers was achieved.[ 22 , 23 ]
Aiming at synthesizing the so far unattainable fused tetraanthracenylporphyrin, [24] we started investigating the fusion of anthracene moieties on the porphyrin core. Because of the symmetry of anthracene, the fusion of a 9‐anthracenyl moiety (attached in meso position) would lead to the formation of two new C−C bonds to the porphyrin core. Anthracene exhibits an oxidation potential very similar to the one of pyrene (0.886 V and 0.893 V vs. Ferrocenium/Ferrocene couple, respectively, according to Davis and Fry, Supporting Information of ref. [25]) that makes it a non‐activated PAH which cannot be oxidized in solution, using FeCl3 or DDQ, in the presence of a Lewis acid (Scholl reaction). Thus, we turned to thermal activation to perform the fusion reaction.
In this work, we report on the formation of three new and unsubstituted fused PAH‐porphyrin derivatives (i. e. naphthalene, pyrene and anthracene – Figure 1). [26] The use of mono‐functionalized porphyrins permits to better understand the reaction and the formation of by‐products; it also permits to obtain single derivatives rather than mixtures of isomers as observed, for example, for bis‐ or tetra‐substituted compounds. Finally, the fused porphyrin/anthracenyl derivative constitutes the first step towards the fused tetra‐anthracenylporphyrin.
Figure 1.

Structures of the fused PAH‐porphyrin derivatives Fused‐1, Fused‐2 and Fused‐3.
2. Results and Discussion
The synthesis of the fused porphyrins is depicted in Scheme 1. The mono‐substituted naphthyl‐, pyrenyl‐ and anthracenyl‐porphyrins 1–3 were synthesized in two steps from the commercially available free‐base 5‐bromo‐10,15,20‐triphenylporphyrin 4. First, Suzuki‐Miyaura couplings between porphyrin 4 and 1‐naphthaleneboronic acid, 1‐pyreneboronic acid or 9‐anthraceneboronic acid in the presence of Pd2(dba)3 and 2‐dicyclohexylphosphino‐2′,6′‐dimethoxybiphenyl (SPhos) gave porphyrins 5, 6 and 7, respectively. These porphyrins were then metalated using Zn(OAc)2. Finally, the fused porphyrins Fused‐1, Fused‐2 and Fused‐3 were obtained by thermal treatment of the parent porphyrins (1, 2, 3) in a tubular quartz oven in nitrogen atmosphere; the reaction conditions are summarized in Table 1.
Scheme 1.

i) Pd2(dba)3, SPhos, K3PO4, toluene/water, 85 °C, 48 h, 98 % (1), 96 % (2), 52 % (3); ii) Zn(OAc)2, CH2Cl2, MeOH, 40 °C, (1) 2 h, 93 %, (2) 2 h, 87 %, (3) overnight, 87 %; iii) 495 °C, 180 s; iv) 495 °C, 120 s; v) 495 °C, 165 s.
Table 1.
Temperature and time conditions for the thermal oxidation of porphyrin 1–3.
|
Porphyrin |
Temperature [°C] |
Reaction time [s] |
|---|---|---|
|
Fused‐1 |
495 |
180 |
|
Fused‐2 |
495 |
120 |
|
Fused‐3 |
495 |
165 |
The investigation of redox behavior of the Zn‐porphyrin species 1, 2 and 3 bearing polyaromatic hydrocarbon moieties has been carried out in a strictly dried dichloromethane solution of tetrabutylammonium hexafluorophosphate (TBAH), devoting particular attention to the oxidation processes. Under the experimental conditions used, the oxidized species are rather stable due to the low nucleophilicity of the electrolyte system.
The cyclic voltammetric curves of the three porphyrins 1–3 are reported in Figure 2. For all compounds, the observed redox processes studied lie within the positive potential window between about +0.5 V and +2.4 V and all occur in the positive potentials region. The species show three voltammetric waves for 1 and four processes for 2 and 3, respectively. Compounds 2 and 3 show the first three peaks as reversible, voltammetric one‐electron transfers while the last peak is a completely irreversible process that happens at the foot of a subsequent oxidation that has not been further investigated herein. The porphyrin 1, at odds with the other two homologue species, shows the first two oxidations as reversible processes whereas the third one is completely irreversible. The E1/2 potentials of the various processes are collected in Table 2.
Figure 2.

Cyclic voltammetric curves of 0.8 mM porphyrin (a) 1, (b) 2 and (c) 3 recorded in TBAH/CH2Cl2 at 298 K; scan rate=1 V s−1; working electrode: platinum disk (diameter 125 μm); reference electrode: SCE.
Table 2.
Half‐wave (E1/2) redox potentials (vs. SCE) of porphyrin compounds recorded in TBAH/CH2Cl2 solution at 25 °C
|
|
E1/2(oxidation) [V] |
|||
|---|---|---|---|---|
|
|
I |
II |
III |
IV |
|
1 |
0.87 |
1.10 |
2.09[a] |
– |
|
2 |
0.88 |
1.11 |
1.57 |
2.23[a,b] |
|
3 |
0.87 |
1.10 |
1.51 |
2.30[a] |
[a] Irreversible process. [b] Peak potential determined by digital simulation of the voltammetric curve.
From an electronic point of view, the three porphyrins can be considered as constituted by two nearly independent moieties: the porphyrinic ring and the polyaromatic fragment, that are mostly arranged in a perpendicular conformation with respect to each other. Therefore, it is possible to localize the electron transfers on the basis of comparing the redox potentials of the various species. Thus, from the values in Table 2, it is evident that the first two oxidations are centered on the porphyrinic moiety for all the compounds whereas the following events relate to the polyaromatic fragment. Moreover, compound 1 shows only one oxidation localized on the naphthalene moiety, at a rather high potential with respect to the corresponding oxidations of the other species bearing anthracene and pyrene, respectively. This is in agreement with the oxidation potentials of the corresponding polyaromatic hydrocarbons. [25] The third oxidation for species 1 and the fourth oxidation for both 3 and 2 are irreversible processes that, upon repeatedly cycling the potential high enough to also include these processes, bring about the deposition onto the electrode surface, forming an observable colored, conducting thin film. The film composition, which is beyond the scope of this work, is currently under investigation.
Among the conditions tested for the thermal oxidation, the time and temperature reported in Table 1 are not optimized to maximize the transformation of the starting materials into fused porphyrin derivatives but to limit the formation of by‐products due to the cleavage or to the addition of phenyl rings on the fused‐porphyrin derivatives. After the pyrolysis reaction, the fused porphyrin derivatives were purified by column chromatography on silica gel to remove the insoluble materials and the unreacted porphyrins, followed by size exclusion chromatography (SEC – BioBeads S−X1 or S−X3) to obtain pure Fused‐1, Fused‐2 and Fused‐3. The unreacted porphyrins represent the majority of the remaining material after the reaction (ca. 50–70 %); they can be recycled and re‐used in new fusion reactions. After the first purification on silica gel, the mass spectra of Fused‐1, Fused‐2 and Fused‐3 revealed the presence of the expected compounds and the presence of peaks at m/z=[M–76] and m/z=[M+76] (Figure S1, Supporting Information). These additional signals are due to the loss or addition of a phenyl ring from or to the porphyrins during the thermal treatment.[ 10 , 27 ] If the transformation of the starting materials was pushed further (either regarding reaction time or temperature), the amount of degraded compounds increased significantly and it became possible to detect compounds at m/z=[M+2*76] in the mass spectra. It is also worth mentioning that the attempts to form π‐extended porphyrins by thermal activation of free‐base porphyrin derivatives 5, 6 and 7 were not successful.
The fused porphyrin derivatives were finally purified by successive SEC using THF as eluent. It was then possible to remove the remaining by‐products (at m/z=+/−76) and obtain pure Fused‐1, Fused‐2 and Fused‐3 porphyrin derivatives (Figure 3). No relevant numbers can be given for the yield of the syntheses of Fused‐1, Fused‐2 and Fused‐3; it is relatively low (5–10 %), small variations can be observed from batch to batch and it is also dependent of the number of SEC steps required to obtain the pure compounds. The HRMS spectra (Figure 3 – right part) show molecular ion peaks at m/z=724.1610 (calcd. 724.1605), 798.1778 (calcd. 798.1762) and 772.1607 (calcd. 772.1605) and the simulated spectra in red for Fused‐1, Fused‐2 and Fused‐3, respectively.
Figure 3.

MALDI‐TOF spectra of Fused‐1 (a), Fused‐2 (b) and Fused‐3 (c); right part: high‐resolution mass spectra and simulation of the HRMS spectra. The simulation was performed using ChemCalc. [28]
The absorption spectra of Fused‐1, Fused‐2 and Fused‐3 are presented in Figure 4; the comparison between the absorption spectra of porphyrins 1–3 and their fused counterparts are shown in Figure S2. The fusion of the PAHs on the porphyrin core results in a bathochromic shift of the absorption bands. The absorption spectra are in agreement with those previously reported for naphthalene‐fused, [5] pyrene‐fused [2] and anthracene‐fused [7] porphyrins. Interestingly, the absorption spectrum of the fused naphthalene derivative (Fused‐1) shows two intense maxima at 412 and 472 nm (Figure 4a). The higher energy band is generally less intense in the compounds reported in the literature;[ 4 , 5 ] however the band with a maximum at 412 nm in the spectrum of Fused‐1 does not correspond to the Soret band of the non‐oxidized porphyrin 1 which appears at 420 nm.
Figure 4.
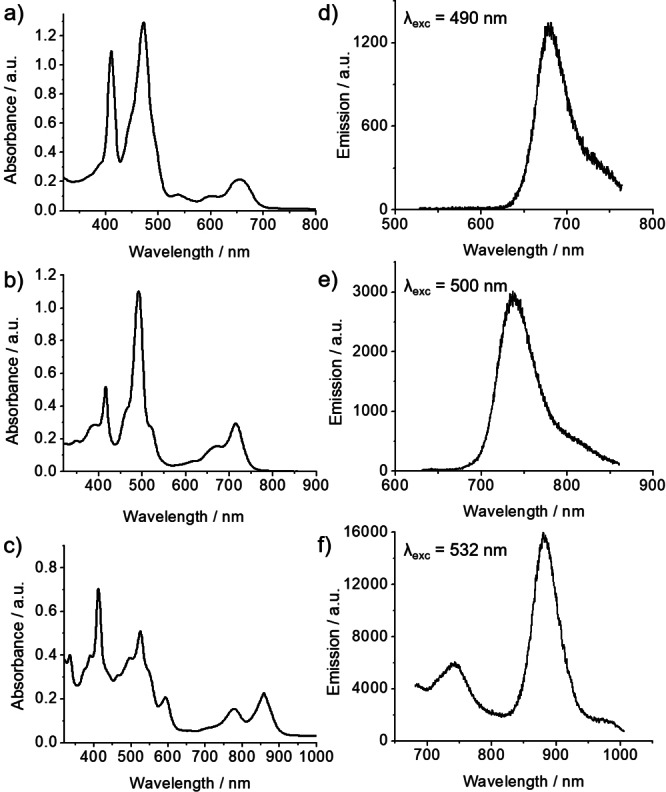
a)‐c) Absorption spectra recorded in CH2Cl2 of Fused‐1, Fused‐2 and Fused‐3, respectively; d)‐f) emission spectra of Fused‐1 (excited at 490 nm), Fused‐2 (excited at 500 nm) and Fused‐3 (excited at 532 nm) recorded in THF.
The photoluminescence spectra of Fused‐1, Fused‐2 and Fused‐3 porphyrins recorded in THF are presented in Figure 4. When excited on the main absorption band at 490 nm, Fused‐1 shows a main emission band at 679 nm. Likewise, Fused‐2 and Fused‐3 compounds, excited at 500 and 532 nm respectively, show main emission bands at 738 nm and 884 nm, respectively. These observations are in line with the bathochromic shift observed for the absorption spectra. Here, one can note that near‐infrared emission is achieved for Fused‐3, which is one of the interests of synthesizing this molecule, since emission in the 800–1000 nm range is not so common for small organic molecules.
Finally, in order to demonstrated the possibility to remove the zinc cation in the porphyrins and thus obtain the free‐base fused porphyrin derivatives, Fused‐1, Fused‐ 2 and Fused‐3 were dispersed in CH2Cl2 and treated with aqueous 4 m HCl solution at room temperature for 1 h. After neutralization and washing, the free‐based porphyrins H2‐Fused‐1, H2‐Fused‐2 and H2‐Fused‐3 were characterized by absorption spectroscopy and mass spectrometry. Unfortunately, the very limited quantity of porphyrin used for the de‐metalation reaction did not permit to characterize the compounds by NMR spectroscopy. The absorption and MALDI‐TOF mass spectra of the free‐base porphyrin derivatives H2‐Fused‐1, H2‐Fused‐2 and H2‐Fused‐3 are given in Figure S3. These compounds are still under investigation and they are presented here as a proof of concept to be fully described later.
3. Conclusion
Here, we described the synthesis and characterization of a series of thermally fused naphthyl‐, pyrenyl‐ and anthracenylporphyrins. The method requires an accurate control of the temperature and of the reaction time and extensive purification but permits the formation of unsubstituted π‐extended porphyrins. We also demonstrated that zinc porphyrin derivatives Fused‐1, Fused‐2 and Fused‐3 can be easily de‐metalated. The anthracenylporphyrin derivatives Fused‐3 and H2‐Fused‐3 are of primary importance and their electronic and optical properties will be further explored. They constitute the first step toward the synthesis of fused tetraanthracenyl derivatives and towards obtaining pure carbon/nitrogen anthracenyl π‐extended porphyrins.
Experimental Section
Techniques. NMR spectra were recorded with a Bruker Avance 400 (400 MHz) instrument with solvent used as internal reference. MS and HRMS MALDI‐TOF spectra were recorded on a Bruker Autoflex maX or on a Bruker UltrafleXtreme. Absorption spectra were recorded in quartz cuvettes on a Perkin Elmer Lambda 900 UV‐Vis‐NIR spectrophotometer. Thin layer chromatography (TLC) was performed on silica gel 60 F254 (Merck) precoated aluminium sheets. Column chromatography was performed on Merck silica gel 60 (0.063–0.200 mm). Size exclusion chromatography was performed on Bio‐Beads S−X1 or Bio‐Beads S−X3 in THF. Thermally induced dehydro‐aromatizations were performed under N2 in a quartz boat in a tubular oven CARBOLITE GERO EHA 12/450B equipped with a 50 mm diameter quartz tube.
Electrochemical Experimental Details. For all the cyclic voltammetric measurements, electrochemical or analytical grade tetrabutylammonium hexafluorophosphate (TBAH) from Sigma‐Aldrich was used as received as a supporting electrolyte. Dichloromethane from Sigma‐Aldrich was purified and dried by refluxing over and successively distilling from B2O3 and activated 4 Å molecular sieves. Afterwards, it was stored in specially designed Schlenk flasks over 3 Å activated molecular sieves, protected from light, and kept under vacuum prior to use, as reported elsewhere. [29] The solvent was distilled in a closed system into a custom designed electrochemical cell containing the supporting electrolyte and the species under examination, immediately before performing the experiment. Electrochemical experiments were carried out in an airtight single‐compartment cell using platinum as working and counter electrodes and a silver spiral as a quasi‐reference electrode. All the E1/2 potentials have been directly obtained from cyclic voltammetric curves as averages of the cathodic and anodic peak potentials and by digital simulation in the case of not Nernstian or overlapping processes. The E1/2 values have been determined by adding ferrocene (FcH) at the end of each experiment, [30] as an internal standard and measuring them with respect to the ferrocinium/ferrocene couple (FcH/[FcH]+) standard potential (+0.42 V vs. SCE [aqueous Saturated Calomel Electrode] at 298 K). [31] Voltammograms were recorded with a custom‐made fast and low‐current potentiostat [32] controlled by an AMEL Mod. 568 programmable function generator. The potentiostat was interfaced to a Nicolet Mod. 3091 digital oscilloscope and the data transferred to a personal computer by the program Antigona. [33]
Photoluminescence Setup. PL spectra are recorded on a home‐made setup. The excitation source is a super continuum source from Fianium company filtered by an AOTF filter leading to a tunable excitation pulsed source. The PL spectra are analyzed in a SP150 spectrometer equipped with a PIXIS 100B (Teledyne) CCD.
Materials. 5‐Bromo‐10,15,20‐triphenylporphyrin (4) was purchased from PorphyChem®. Chemicals were purchased from Aldrich or Fisher Scientific and were used as received. Solvents were purchased from SDS Carlo Erba and were used as received. For synthesis and SEC, CH2Cl2 (CaH2, N2), toluene (K/benzophenone, N2) and THF (K/benzophenone, N2) were distilled before use.
Synthesis
5,10,15‐Triphenyl‐20‐(naphthalen‐1‐yl)porphyrin (5). Palladium catalyst Pd2(dba)3 (37 mg, 0.04 mmol, 0.50 eq) with SPhos (66 mg, 0.16 mmol, 1.0 eq), K3PO4 (205 mg, 0.97 mmol, 6.0 eq), 5‐bromo‐10,15,20‐triphenylporphyrin (100 mg, 0.16 mmol, 1.0 eq) and naphthalen‐1‐boronic acid (83.5 mg, 0.48 mmol, 3.0 eq) were dissolved in 80 mL of a degassed solution of toluene and water (1 mL), shielded from ambient light and stirred at 85 °C for 48 h. At the end of the reaction, the mixture was washed with water (3×40 mL), dried over Na2SO4 and the organic solvent was evaporated. The crude mixture was purified by silica column using toluene/heptane (1 : 1, v/v) and the second red fraction was collected. Solvents were removed and final product was recrystallized from CH2Cl2/MeOH to afford 5 (105 mg, 98 %). 1H NMR (400 MHz, (CDCl2)2): δ 8.91 (m, 4H, β‐H pyrroles), 8.80 (d, 2H, J=4.72 Hz, β‐H pyrroles), 8.61 (d, 2H, J=4.72 Hz, β‐H pyrroles), 8.36–8.30 (m, 2H, napthyl), 8.30–8.21 (m, 6H, o‐phenyls), 8.21–8.17 (m, 1H, naphtyl), 7.96–7.88 (m, 1H, napthyl), 7.87–7.72 (m, 9H, m‐phenyls+p‐phenyls), 7.57–7.49 (m, 1H, napthyl), 7.24–7.10 (m, 2H, napthyl), −2.65 (s, 2H, HNH) ppm. 13C{1H} NMR (100 MHz, (CDCl2)2): δ 141.84, 141.67, 139.04, 136.59, 134.48, 132.76, 132.65, 128.60, 128.50, 127.80, 127.66, 126.66, 126.08, 125.55, 124.24, 120.40, 120.06, 117.17 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=419 (5.47), 520 (4.04), 554 (3.58), 594 (3.56), 647 (3.33) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C48H24N4 664.2621, found: 664.2649.
[5,10,15‐Triphenyl‐20‐(naphthalen‐1‐yl)porphyrinato]zinc(II) (1). 5 (90 mg, 0.13 mmol, 1.0 eq) in non‐distilled toluene (150 mL), Zn(acac)2×H2O (106 mg, 0.40 mmol, 3.0 eq), at reflux for 2 h. Recrystallisation from CH2Cl2/MeOH afforded violet‐pink powder as pure target 1 (92 mg, 93 %). 1H NMR (400 MHz, (CDCl2)2): δ 9.01 (m, 4H, β1 pyrroles), 8.89 (d, 2H, J=4.64 Hz, β2 pyrroles), 8.71 (d, 2H, J=4.64 Hz, β3 pyrroles), 8.38–8.22 (m, 8H, 2‐ and 4‐napthyl+o‐phenyls), 8.20–8.15 (m, 1H, 8‐naphtyl), 7.96–7.88 (m, 1H, 3‐napthyl), 7.84–7.73 (m, 9H, m‐phenyls+p‐phenyls), 7.56–7.46 (m, 1H, 7‐napthyl), 7.14–7.06 (m, 2H, 5‐ and 6‐napthyl) ppm. 13C{1H} NMR (100 MHz, (CDCl2)2): δ 150.62, 150.03, 149.93, 149.88, 142.46, 142.35, 139.71, 136.68, 134.43, 134.38, 132.60, 132.56, 132.13, 132.00, 131.90, 131.80, 128.55, 128.36, 127.77, 127.41, 126.50, 125.93, 125.45, 124.19, 121.31, 120.96, 118.11 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=421 (5.54), 552 (4.12) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C48H30N4Zn 726.1756, found: 726.1767.
Fused [5,10,15‐triphenyl‐20‐(naphthalen‐1‐yl)porphyrinato] zinc(II) (Fused‐1). 1 in powder (10 mg) was subjected to pyrolysis at 495 °C for 180 s. The resulting product was dissolved in CH2Cl2 and filtered through a silica pad to remove insoluble products. The crude mixture was purified by silica column eluted with CH2Cl2/cyclohexane (1 : 1, v/v) and two SECs in THF, to afford as pure target Fused‐1. 1H NMR (400 MHz, (CDCl2)2): δ 9.43 (s, 2H, β‐H pyrroles), 9.02 (d, 1H, naphtyl), 8.94–8.87 (m, 5H, 4H β‐H pyrroles and 1H naphtyl), 8.76 (s, 1H, β‐H pyrroles), 8.32–8.25 (m, 6H, 4H naphtyl and 2H o‐phenyls), 8.20–8.15 (m, 3H, p‐phenyls), 7.98–7.81 (m, 10H, o‐phenyls and m‐phenyls) ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=229 (4.49), 304 (4.29), 411 (4.89), 472 (4.97), 656 (4.18) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C48H28N4Zn 724.1605 found: 724.1610.
5,10,15‐triphenyl‐20‐(pyren‐1‐yl)porphyrin (6). Palladium catalyst Pd2dba3 (37 mg, 0.04 mmol, 0.50 eq), SPhos (66 mg, 0.16 mmol, 1.0 eq), K3PO4 (205 mg, 0.97 mmol, 6.0 eq), 5‐bromo‐10,15,20‐triphenylporphyrin (100 mg, 0.16 mmol, 1.0 eq) and pyrene‐1‐boronic acid (119.6 mg, 0.48 mmol, 3.0 eq) were dissolved in a mixture of toluene (80 mL) and water (1 mL), shielded from ambient light and stirred at 85 °C for 48 h. At the end of the reaction, the mixture was washed with water (3×40 mL), dried over Na2SO4 and the organic solvent was evaporated. The crude mixture was purified by silica column using toluene/heptane (1 : 1, v/v) to collect the second red fraction. Solvents were removed and final product was recrystallized from CH2Cl2/MeOH to afford 6 (116 mg, 96 %). 1H NMR (400 MHz, (CDCl2)2): δ 8.94 (m, 4H, β‐H pyrroles), 8.86 (d, 1H, J=7.72 Hz, pyrenyl), 8.81 (d, 2H, J=4.76 Hz, β‐H pyrroles), 8.57 (d, 1H, J=7.84 Hz, pyrenyl), 8.54 (d, 2H, J=4.80 Hz, β‐H pyrroles), 8.45 (d, 1H, J=9.08 Hz, pyrenyl), 8.40–8.34 (m, 2H, pyrenyl), 8.33–8.24 (m, 6H, o‐phenyls), 8.18–8.08 (m, 2H, pyrenyl), 7.87–7.81 (m, 3H, p‐phenyls), 7.81–7.74 (m, 6H, m‐phenyls),7.51 (d, 1H, J=9.28 Hz, pyrenyl), −2.58 (s, 2H, HNH) ppm. 13C{1H} NMR (100 MHz, (CDCl2)2): δ 141.84, 141.65, 136.78, 134.48, 133.25, 132.70, 131.41, 131.18, 130.53, 128.00, 127.72, 127.67, 127.48, 127.12, 126.67, 126.37, 125.63, 125.27, 124.31, 123.90, 122.84, 120.44, 120.20, 117.54 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=244 (4.65), 268 (4.36), 276 (4.47), 327 (4.29), 340 (4.36), 421 (5.43), 519 (4.14), 555 (3.74), 595 (3.60), 648(3.44) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C54H34N4 738.2778, found: 738.2787.
[5,10,15‐Triphenyl‐20‐(pyren‐1‐yl)porphyrinato]zinc(II) (2). 6 (85 mg, 0.11 mmol, 1.0 eq) in non‐distilled toluene (150 mL), Zn(acac)2×H2O (91 mg, 0.34 mmol, 3.0 eq), at reflux for 2 h. Recrystallization from CH2Cl2/MeOH afforded violet‐pink powder as pure target 2 (80 mg, 87 %). 1H NMR (400 MHz, (CDCl2)2): δ 9.01 (m, 4H, β1 pyrroles), 8.92–8.83 (m, 3H, 1H pyrenyl+2H β2 pyrroles), 8.61 (d, 2H, J=4.68 Hz, β3 pyrroles), 8.57 (d, 1H, J=7.80 Hz, pyrenyl), 8.45 (d, 1H, J=9.08 Hz, pyrenyl), 8.38–8.23 (m, 8H, 2H pyrenyl+o‐phenyls), 8.14–8.05 (m, 2H, pyrenyl), 7.86–7.79 (m, 3H, p‐phenyls), 7.79–7.73 (m, 6H, m‐phenyls), 7.71 (d, 1H, J=9.40 Hz, pyrenyl), 7.51 (d, 1H, J=9.32 Hz, pyrenyl) ppm. 13C{1H} NMR (100 MHz, (CDCl2)2): δ 150.83, 150.11, 149.95, 149.91, 142.51, 142.39, 137.61, 134.47, 134.45, 134.39, 133.23, 132.66, 132.22, 132.02, 131.94, 131.44, 131.01, 130.54, 127.78, 127.75, 127.44, 127.40, 127.29, 127.25, 126.49, 126.28, 125.52, 125.17, 124.35, 123.87, 122.71, 121.33, 121.06, 118.42 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=244 (4.71), 268 (4.37), 276 (4.49), 327 (4.33), 341 (4.34), 422 (5.56), 552 (4.27) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C54H32N4Zn 800.1913, found: 800.1921.
Fused [5,10,15‐triphenyl‐20‐(pyren‐1‐yl)porphyrinato] zinc(II) (Fused‐2). 2 in powder (10 mg) was subjected to pyrolysis at 495 °C for 120 s. The resulting product was dissolved in CH2Cl2 and filtered through a silica pad to remove insoluble products. The crude mixture was purified by silica column eluted with CH2Cl2/cyclohexane (1 : 1, v/v) and two SECs in THF, to afford as pure target Fused‐2. 1H NMR (400 MHz, (CDCl2)2): δ 9.58 (s, 1H, β1 pyrrole or 9‐pyrenyl), 9.54 (d, 1H, J=4.8 Hz, β2 pyrrole), 9.47 (s, 1H, β1 pyrrole or 9‐pyrenyl), 9.38 (d, 1H, J=8.0 Hz, 2‐pyrenyl), 8.97 (d, 1H, J=4.8 Hz, β3 pyrrole), 8.86–8.77 (m, 3H, β4, β5, β6 pyrrole), 8.71 (d, 1H, J=8.4 Hz, 3‐pyrenyl), 8.46 (d, 1H, J=7.2 Hz, pyrenyl), 8.35–8.27 (m, 6H, o‐phenyls), 8.25–8.19 (m, 3H, pyrenyl), 8.11 (t, 1H, J=7.6 Hz, pyrenyl), 7.93–7.87 (m, 3H, p‐phenyls), 7.86–7.72 (m, 8H, 6H m‐phenyls+2H pyrenyl) ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=389 (3.50), 413 (3.57), 492 (4.14), 665 (3.29), 703 (3.48) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C54H30N4Zn 798.1762 found: 798.1778.
5‐(Anthracen‐9‐yl)‐10,15,20‐triphenylporphyrin (7). Palladium catalyst Pd2(dba)3 (9.1 mg, 0.01 mmol, 0.50 eq) with SPhos (16.4 mg, 0.04 mmol, 1.0 eq), K3PO4 (51.5 mg, 0.54 mmol, 6.0 eq), 5‐bromo‐10,15,20‐triphenylporphyrin (25 mg, 0.04 mmol, 1.0 eq) and anthracen‐9‐boronic acid (27 mg, 0.12 mmol, 3.0 eq) were dissolved in 30 mL of preliminary degassed solution of toluene and water (1 mL), shielded from ambient light and stirred at 85 °C for 48 h. At the end of the reaction, the mixture was washed with water (3×40 mL), dried over Na2SO4 and the organic solvent was evaporated. The crude mixture was purified by silica column using toluene/heptane (1 : 1, v/v) to collect the second red fraction. Solvents were removed and final product was recrystallized from CH2Cl2/MeOH to afford 7 (15 mg, 51 %). 1H NMR (400 MHz, (CDCl2)2): δ 8.96 (s, 1H, anthracenyl), 8.93–8.88 (m, 4H, β pyrroles), 8.69 (d, 2H, J =4.4 Hz, β pyrroles), 8.32 (d, 2H, J =4.8 Hz, β pyrroles), 8.29–8.20 (m, 8H, 2H anthracenyl+6H o‐phenyls), 7.85–7.79 (m, 3H, p‐phenyls), 7.78–7.71 (m, 6H, m‐phenyls), 7.52–7.46 (m, 2H, anthracenyl), 7.13–7.07 (m, 2H, anthracenyl), 7.07–7.01 (m, 2H, anthracenyl), −2.55 (s, 2H, HNH) ppm. 13C{1H} NMR (125 MHz, (CDCl2)2, 333 K): δ 141.85, 141.55, 135.60, 135.02, 134.48, 134.25, 130.64, 128.46, 128.21, 128.02, 127.76, 127.68, 126.68, 126.68, 125.70, 124.95, 120.63, 120.07, 114.98 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=231 (4.19), 257 (4.95), 352 (4.11), 373 (4.20), 419 (5.34), 515 (4.06), 550 (3.61), 589 (3.53), 647 (3.24) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C52H34N4 714.2778, found: 714.2775.
[5‐(Anthracen‐9‐yl)‐10,15,20‐triphenylporphyrinato]zinc(II) (3). 7 (10 mg, 0.01 mmol, 1.0 eq) in non‐distilled toluene (100 mL), Zn(acac)2×H2O (8 mg, 0.03 mmol, 3.0 eq), at reflux overnight. Recrystallization from CH2Cl2/MeOH afforded violet‐pink powder as pure target 3 (7 mg, 87 %). 1H NMR (400 MHz, (CDCl2)2): δ 9.06–8.97 (m, 4H, β1 pyrroles), 8.94 (s, 1H, 10‐anthracenyl), 8.80 (d, 2H, J =4.4 Hz, β2 pyrroles), 8.36 (d, 2H, J =4.8 Hz, β3 pyrroles), 8.34–8.20 (m, 8H, 4,5‐anthracenyl+o‐phenyls), 7.87–7.79 (m, 3H, p‐phenyls), 7.78–7.69 (m, 6H, m‐phenyls), 7.52–7.43 (m, 2H, 3,6‐anthracenyl), 7.11–6.97 (m, 4H, 2,7‐ and 1,8‐anthracenyl) ppm. 13C{1H} NMR (125 MHz, (CDCl2)2, 333 K): δ 151.12, 150.09, 149.90, 149.81, 142.41, 142.19, 136.37, 134.98, 134.46, 132.57, 132.16, 132.01, 131.71, 130.58, 128.56, 128.20, 127.78, 127.52, 127.45, 126.59, 126.56, 125.59, 124.91, 121.58, 120.95, 115.93 ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=257 (4.75), 352 (3.71), 421 (5.20), 548 (3.90), 586 (3.00) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C52H34N4Zn 776.1913, found: 776.1926.
Fused [5‐(anthracen‐9‐yl)‐10,15,20‐triphenylporphyrinato] zinc(II) (Fused‐3). 3 in powder (10 mg) was subjected to pyrolysis at 495 °C for 165 s. The resulting product was dissolved in CH2Cl2 and passed through a silica pad to remove insoluble products. The crude mixture was purified by silica column eluted with CH2Cl2/cyclohexane (1 : 1, v/v) and two successive SECs, in which the red fraction was collected to afford as pure target Fused‐3. 1H NMR (400 MHz, (CDCl2)2): δ 8.72 (s, 4H, β1 pyrroles), 8.31 (s, 2H, β2 pyrroles), 8.23 (d, 2H, 1,7‐anthracenyl), 8.08 (s, 1H, 4‐anthracenyl), 7.88 (m, 6H, phenyls), 7.67 (t, 1H, 2‐anthracenyl or 6‐anthracenyl), 7.56 (t, 1H, 2‐anthracenyl or 6‐anthracenyl), 7.44–7.30 (m, 3H, phenyls), 7.16 (s, 1H, 5‐anthracenyl or 3‐anthracenyl), 6.99 (s, 1H, 5‐anthracenyl or 3‐anthracenyl) ppm. UV‐Vis‐NIR (CH2Cl2): λmax (log ϵmax)=408 (4.45), 526 (4.32), 593 (3.93), 769 (3.80), 851 (3.97) nm. HRMS (MALDI‐TOF) m/z [M]+ calcd for C52H28N4Zn 772.1605 found: 772.1607.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was partly funded by the ANR project MAGMA (ANR‐16‐CE29‐0027‐01), the ANR‐DFG project GRANAO (ANR‐19‐CE09‐0031‐02), the FLAG‐ERA Joint Transnational Call OPERA (FLAG‐ERA JTC 2019) and by a public grant overseen by the French National Research Agency (ANR) as part of the “Investissements d'Avenir” program (Labex NanoSaclay, reference: ANR‐10‐LABX‐0035). University of Bologna is also gratefully acknowledged.
J. Pijeat, L. Chaussy, R. Simoës, J. Isopi, J.-S. Lauret, F. Paolucci, M. Marcaccio, S. Campidelli, ChemistryOpen 2021, 10, 997.
References
- 1. Mori H., Tanaka T., Osuka A., J. Mater. Chem. C 2013, 1, 2500–2519. [Google Scholar]
- 2. Yamane O., Sugiura K.-I., Miyasaka H., Nakamura K., Fujimoto T., Nakamura K., Kaneda T., Sakata Y., Yamashita M., Chem. Lett. 2004, 33, 40–41. [Google Scholar]
- 3. Kurotobi K., Kim K. S., Noh S. B., Kim D., Osuka A., Angew. Chem. Int. Ed. 2006, 45, 3944–3947; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4048–4051. [Google Scholar]
- 4. Tanaka M., Hayashi S., Eu S., Umeyama T., Matano Y., Imahori H., Chem. Commun. 2007, 2071. [DOI] [PubMed] [Google Scholar]
- 5. Hayashi S., Tanaka M., Hayashi H., Eu S., Umeyama T., Matano Y., Araki Y., Imahori H., J. Phys. Chem. C 2008, 112, 15576–15585. [Google Scholar]
- 6. Lewtak J. P., Gryko D., Bao D., Sabai E., Vakuliuk O., Scigaj M., Gryko D. T., Org. Biomol. Chem. 2011, 9, 8178–8181. [DOI] [PubMed] [Google Scholar]
- 7. Davis N. K. S., Pawlicki M., Anderson H. L., Org. Lett. 2008, 10, 3945–3947. [DOI] [PubMed] [Google Scholar]
- 8. Davis N. K. S., Thompson A. L., Anderson H. L., Org. Lett. 2010, 12, 2124–2127. [DOI] [PubMed] [Google Scholar]
- 9. Davis N. K. S., Thompson A. L., Anderson H. L., J. Am. Chem. Soc. 2011, 133, 30–31. [DOI] [PubMed] [Google Scholar]
- 10. Diev V. V., Schlenker C. W., Hanson K., Zhong Q., Zimmerman J. D., Forrest S. R., Thompson M. E., J. Org. Chem. 2012, 77, 143–159. [DOI] [PubMed] [Google Scholar]
- 11. Diev V. V., Hanson K., Zimmerman J. D., Forrest S. R., Thompson M. E., Angew. Chem. Int. Ed. 2010, 49, 5523–5526; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5655–5658. [Google Scholar]
- 12. Chen Q., Brambilla L., Daukiya L., Mali K. S., De Feyter S., Tommasini M., Müllen K., Narita A., Angew. Chem. Int. Ed. 2018, 57, 11233–11237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11403–11407. [Google Scholar]
- 13. Jiao C., Huang K.-W., Chi C., Wu J., J. Org. Chem. 2011, 76, 661–664. [DOI] [PubMed] [Google Scholar]
- 14. Diev V. V., Femia D., Zhong Q., Djurovich P. I., Haiges R., Thompson M. E., Chem. Commun. 2016, 52, 1949–1952. [DOI] [PubMed] [Google Scholar]
- 15. Fox S., Boyle R. W., Chem. Commun. 2004, 1322–1323. [DOI] [PubMed] [Google Scholar]
- 16. Röckert M., Franke M., Tariq Q., Ditze S., Stark M., Uffinger P., Wechsler D., Singh U., Xiao J., Marbach H., Steinrück P., Lytken O., Chem. Eur. J. 2014, 20, 8948–8953. [DOI] [PubMed] [Google Scholar]
- 17. Weingarten A., Lloyd J. A., Seufert K., Reichert J., Auwärter W., Han R., Ducan D. A., Allegretti F., Fischer S., Oh S. C., Saglam Ö., Jiang L., Vijayaraghavan S., Ecija D., Papageorgiou A. C., Barth J. V., Chem. Eur. J. 2015, 21, 12285–12290. [DOI] [PubMed] [Google Scholar]
- 18. Ruggieri C., Rangan S., Bartynski R. A., Galoppini E., J. Phys. Chem. C 2016, 120, 7575–7585. [Google Scholar]
- 19. Li J., Merino-Díez N., Carbonell-Sanromà E., Vilas-Varela M., de Oteyza D. G., Peña D., Corso M., Pascual J. I., Sci. Adv. 2018, 4, eaaq0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun Q., Mateo L. M., Robles R., Ruffieux P., Lorente N., Bottari G., Torres T., Fasel R., J. Am. Chem. Soc. 2020, 142, 18109–18117. [DOI] [PubMed] [Google Scholar]
- 21. Zhao Y., Jiang K., Li C., Liu Y., Xu C., Zheng W., Guan D., Li Y., Zheng H., Liu C., Luo W., Jia J., Zhuang X., Wang S., J. Am. Chem. Soc. 2020, 142, 18532–18540. [DOI] [PubMed] [Google Scholar]
- 22. Mateo L. M., Sun Q., Liu S.-X., Bergkamp J. J., Eimre K., Pignedoli C. A., Ruffieux P., Decurtins S., Bottari G., Fasel R., Torres T., Angew. Chem. Int. Ed. 2020, 59, 1334–1339; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1350–1355. [Google Scholar]
- 23. Mateo L. M., Sun Q., Eimre K., Pignedoli C. A., Torres T., Fasel R., Bottari G., Chem. Sci. 2021, 12, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yen T. F., The role of trace metals in petroleum, Ann Arbor Science Publishers, 1975. [Google Scholar]
- 25. Davis A. P., Fry A. J., J. Phys. Chem. A 2010, 114, 12299–12304. [DOI] [PubMed] [Google Scholar]
- 26.J. Pijeat, PhD thesis, Université Paris-Saclay (France), 2019.
- 27. Shukla B., Koshi M., Phys. Chem. Chem. Phys. 2010, 12, 2427–2437. [DOI] [PubMed] [Google Scholar]
- 28. Pantigny L., Borel A., J. Chem. Inf. Model. 2013, 53, 1223–1228. [DOI] [PubMed] [Google Scholar]
- 29. Bruno C., Marcaccio M., Paolucci D., Castellarin-Cudia C., Goldoni A., Streletskii A. V., Drewello T., Barison S., Venturini A., Zerbetto F., Paolucci F., J. Am. Chem. Soc. 2008, 130, 3788–3796. [DOI] [PubMed] [Google Scholar]
- 30. Burley G. A., Avent A. G., Boltalina O. V., Gol′dt I. V., Guldi D. M., Marcaccio M., Paolucci F., Paolucci D., Taylor R., Chem. Commun. 2003, 148–149. [DOI] [PubMed] [Google Scholar]
- 31. Bruno C., Benassi R., Passalacqua A., Paolucci F., Fontanesi C., Marcaccio M., Jackson E. A., Scott L. T., J. Phys. Chem. B 2009, 113, 1954–1962. [DOI] [PubMed] [Google Scholar]
- 32. Amatore C., Lefrou C., J. Electroanal. Chem. 1992, 324, 33–58. [Google Scholar]
- 33.Mottier, L. Antigona. Simulation and analysis program (University of Bologna, Bologna, Italy, 1999).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information