

 This work is licensed under a
This work is licensed under a Abstract
Summary
Familial partial lipodystrophy (FPLD) syndromes are rare heterogeneous disorders especially in women characterized by selective loss of adipose tissue, reduced leptin levels and severe metabolic abnormalities. Here we report a 34-year-old female with a novel heterozygotic c.485 thymine>guanine (T>G) missense variant (p.phenylalanine162cysteine; (Phe162Cys)) in exon 4 of the peroxisome proliferator-activated receptor gamma (PPARG) gene, developing a non-ketotic diabetes and severe hypertriglyceridemia with triglyceride concentrations >50 mmol/L. In this case, a particular interesting feature in comparison to other known PPARG mutations in FPLD is that while glycaemic control could be achieved through standard anti-diabetic medication, hypertriglyceridemia did neither respond to fibrate nor to omega-3-fatty acid therapy. This might suggest a lipid metabolism driven phenotype of the novel PPARG c.485T>G missense variant. Notably, recombinant leptin replacement therapy (metreleptin (Myalepta®)) was initiated showing a rapid and profound effect on triglyceride levels as well as on liver function tests and satiety feeling. Unfortunately, severe allergic skin reactions developed at the side of injection which could be covered by anti-histaminc treatment. We conclude that the heterozygous PPARG c.485T>G variant is a yet undescribed molecular basis underlying FPLD with difficulties predominantly to control hypertriglyceridemia and that recombinant leptin therapy may be effective in affected subjects.
Learning points
Heterozygous c.485T>G variant in PPARG is most likely a cause for FPLD in humans.
This variant results in a special metabolic phenotype with a predominant dysregulation of triglyceride metabolism not responding to standard lipid lowering therapy.
Recombinant leptin therapy is effective in rapidly improving hypertriglyceridemia.
Patient Demographics: Adult, Female, White, Germany
Clinical Overview: Adipose Tissue, Genetics and mutation
Publication Details: Insight into disease pathogenesis or mechanism of therapy, September, 2021
Background
Lipodystrophy (LD) syndromes are rare heterogeneous disorders with diverse pathophysiology characterized by selective deficiency (partial LD) or complete (total LD) loss of adipose tissue with ectopic lipid accumulation, for example, in the liver. Patients are usually suffering from severe metabolic abnormalities, such as non-ketotic diabetes, dyslipidaemia, and metabolic steatohepatitis (MeSH) (1). Familial partial lipodystrophy (FPLD) is one inheritable subtype of these diseases and is caused by mutations in various genes, such as PPARG (NG_011749.1, RefSeqGene) (2), the latter being crucial in the molecular regulation of adipogenesis (1).
Case presentation
A 34-year-old female was presented with manifestation early in life of various severe metabolic abnormalities such as abdominal obesity, diabetes mellitus (initially diagnosed as type 2), and severe hypertriglyceridemia to the outpatient lipid clinic at the University Hospital Schleswig-Holstein (UKSH) on 5 August 2020. The physical examination revealed a height of 184 cm and a weight of 102 kg (BMI 30.1 kg/m2). Besides increased fat mass around her abdomen she had loss of s.c. fat on the extremities. The body composition was assessed using bioelectrical impedance analysis (BIA, seca mBCA 515 device, seca Deutschland, Hamburg, Germany). The patient presented a body fat content of 33.2% with a fat mass index of 8.8 kg/m² (slightly above the normal range of 3.8–8.7 kg/m²). The BIA also showed that the patient presented a total muscle mass content of 28.9 kg at the upper level of the reference range of the device (21.3–30.4 kg). Concisely prominent was her bulging musculature and her veins on the upper and lower extremities (Fig. 1) which was in line with the findings from the BIA. We did not find any signs of acanthosis nigricans. No fat accumulation was found in the face or the neck. The patient showed a blood pressure of 125/80 mmHg without specific medication. Her diabetes at that time was suboptimaly controlled (HbA1c 73 mmol/mol Hb; 8.8% Hb) and the hypertriglyceridemia (first diagnosed in 2018) with blood triglyceride concentrations of 47.9 mmol/L was completely disarranged. Additionally, hepatomegaly and MeSH was documented. The patient also revealed stress eating, sweet eating, and abnormal quantities of eating due to a markedly reduced feeling of satiety. Of importance, allergy against disinfectants had been diagnosed in the past, which might indicate an allergic diathesis. The family history exposed diabetes mellitus and a myocardial infarction of the father. No further information about the family were known by the index patient.
Figure 1.
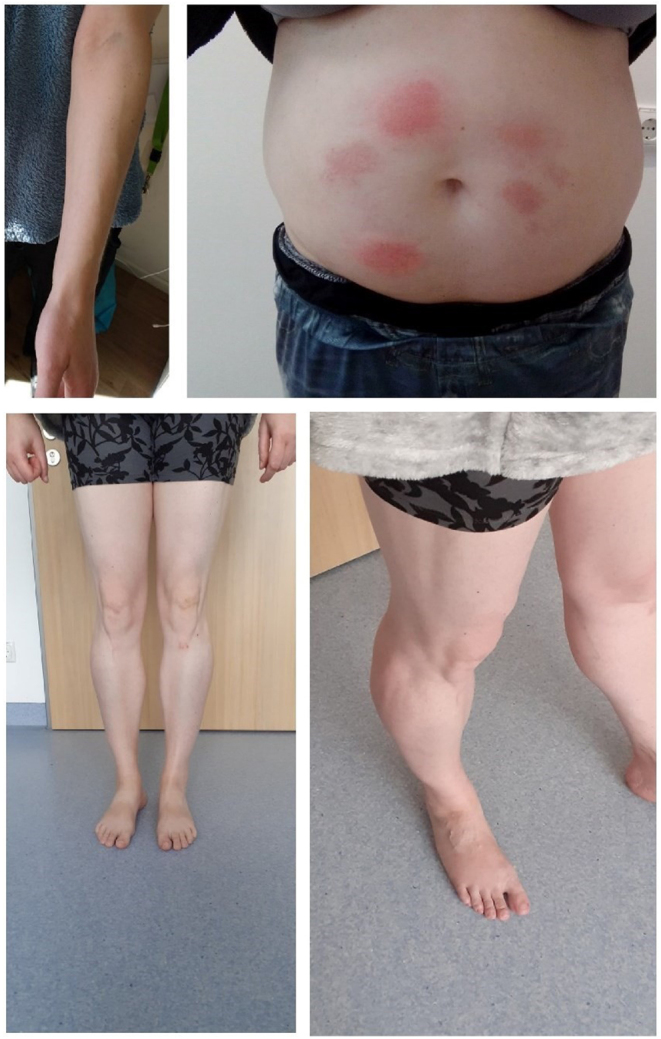
Images showing loss of s.c. adipose tissue at the forearm and legs as well as fat tissue accumulation at the abdomen. In addition, allergic reactions at the injection site after initiation of recombinant leptin therapy can be seen.
For several years, the index patient lives in an assisted living project without any contact to her parents. Thus, it was not possible to contact the parents to obtain further information about a known family history of FPLD. A presented older photography of the mother, however, showed a female with a rather athletic phenotype with similar lower extremities as the index patient. The index patient could not remember the mother having diabetes or lipid abnormalities in earlier times.
The medication of the patient at first visit was as follows: metformin 1000 mg twice daily, insulin glargine once daily 30 IU, and simvastatin 40 mg once daily, as well as an oral contraception. I.v. insulin treatment and lipid-apheresis were not used in this case, since these invasive therapy regimes in Germany are not usually used for outpatient long-term treatment, but only in life-threatening situations of the severe hypertriglyceridemia in hospitals, for example, acute pancreatitis. This severe complication did not occur so far in our patient.
Investigation
Detection of a novel PPARG variant
Next generation sequencing (NGS) revealed the missense variant c.485T>G (p.(Phe162Cys)) in exon 4 of the PPARG gene in our patient, which has not yet been described in the literature. Even in control groups of different ethnicities (genomAD) this variant could not be found. In the context of an implemented in silico analysis, Varsome database was used to classify the variant. All the 12 in Varsome database included prediction programs (BayesDel_addAF, DANN, DEOGEN2, EIGEN, FATHMM-MKL, M-CAP, MVP, MutationAssessor, MutationTaster, PrimateAl, and REVEL und SIFT) classified this variant as pathogenic. Overall, this variety of the variant in the PPARG gene was categorized as a likely pathogenic variant (category 4) according to the American College of Medical Genetics and Genomics (3) and, therefore, is most likely responsible for the observed partial lipodystrophy. Using the latest available mutation databases, no other pathogenic mutations in the other examined genes could be found (1-Acylglycerol-3-Phosphate O-Acyltransferase 2 [AGPAT2], Seipin [BSCL2], Caveolin 1 [CAV1], Caveolae Associated Protein 1 [CAVIN1], Cell Death Inducing DFFA Like Effector C [CIDEC], Lipase E [LIPE], Lamin A/C [LMNA], Phosphoinositide-3-Kinase Regulatory Subunit 1 [PIK3R1], and Perilipin 1 [PLIN1]). The interpretation of the NGS-data permitted a valid copy number variation analysis of the coding section of AGPAT2, BSCL2, CAV1, CAVIN1, CIDEC, LIPE, PIK3R1, PLIN1, and PPARG. The results did not show any deletion or duplication of the examined genes. Also, an additionally implemented gene dosage analysis by use of MLPA exhibited no deletion or duplication in parts of or in the entire LMNA gene.
Treatment
Clinical course before PPARG c.485T>G detection
Initially the patient showed elevated blood glucose levels of 13.5 mmol/L (reference 4.11–5.89 mmol/L) and glycated hemoglobin A1c (HbA1c) levels of 73 mmol/mol Hb (8.8% Hb). Corresponding to the glucose level of 13.5 mmol/L, insulin and C-peptide levels increased to 34.6 mlU/L and 5.86 μg/L, respectively. The levels of thyroid-stimulating hormone, insulin-like growth factor 1, and cortisol were within the reference range. Triglycerides were detected with levels of 47.9 mmol/L as well as blood cholesterol (total) with levels of 12.6 mmol/L. Additionally, the patient showed decreased high-density lipoprotein cholesterol levels of 0.24 mmol/L. Furthermore, elevated aspartate aminotransferase (glutamate-oxaloacetate trasferase (GOT)/ASAT) levels with 102.2 U/L, alanine aminotransferase (glutamate-pyruvate transferase (GPT)/ALAT) levels with 57.2 U/L, and gamma glutamyl transferase (γ-GT) levels with 60 U/L were detected presumably reflecting MeSH. The female patient also showed an increased C-reactive protein levels of 14.4 mg/L in the absence of classical infectious or immunological diseases presumably indicating metabolic inflammation.
Outcome and follow-up
Clinical course after PPARG c.485T>G detection
Since the physical examination showed a classical FPLD phenotype and the molecular diagnostics revealed the novel 485T>G missense variant in exon 4 of the human PPARG gene, the management of the metabolic abnormalities was intensified by the addition of semaglutide (glucagon-like peptide 1; GLP-1 analogue) and dapagliflozin (sodium glucose cotransporter 2 inhibitor; SGLT2-inhibitor) to improve glycemic control as well as switching the statin treatment to the fibrate gemfibrozil in addition to omega-3 fatty acids in order to improve lipid metabolism. Thereby, the HbA1c dropped from 73 mmol/mol Hb (8.8% Hb) to 54 mmol/mol Hb (6.5% Hb) indicating sufficient treatment of non-ketotic diabetes by standard therapy (Table 1). However, despite the initiation of gemfibrozil and omega-3 fatty acids, triglyceride serum concentrations still accounted to >50 mmol/L, reflecting severe disarrangement despite optimal standard pharmacotherapy.
Table 1.
Biochemical analysis before and after standard and recombinant leptin therapy.
| Analyte | Reference | Initial contact | After escalation of standard therapy | After initiation of leptin therapy | Five months after initiation of leptin therapy |
|---|---|---|---|---|---|
| HbA1c | |||||
| IFCC, mmol/mol Hb | 23–43 | 73 | 48 | 55 | 51 |
| NGSP, % Hb | 4.3–6.1 | 8.8 | 6.5 | 7.2 | 6.8 |
| Triglycerides, mmol/L | <2.31 | 47.9 | >50 | 35.1 | 2.3 |
| Total cholesterol, mmol/L | <5.0 | 12.6 | 14.0 | 14.8 | 4.7 |
| HDL cholesterol, mmol/L | >1.29 | 0.24 | 0.28 | 0.47 | 1.09 |
| GPT/ALAT, U/L | <35 | 57.2 | * | 13.2 | 58.3 |
| Leptin, ng/mL | 11–68# | – | 9.3 | 13.0 | 63.0 |
#BMI and sex-specific reference of the laboratory. *Not measurable due to lipidemic serum.
IFCC, International Federation for Clinical Chemistry; NGSP, National Glucose Standardization Program; GPT, glutamate-pyruvate transferase.
In the search for additional therapy options, serum leptin concentrations were analyzed since this adipocyte derived hormone is often reduced in FPLD and recombinant leptin therapy is approved for this orphan disease. Indeed, the analysis exhibited reduced leptin levels of 9.3 ng/mL (physiological levels for women at that age with a BMI of 30 are 10.6–58.3 ng/mL). Thus, recombinant leptin replacement therapy with metreleptin (Myalepta®) in the dose of 5 mg once daily via s.c. injection was initiated. Two weeks after the leptin therapy was started, biochemical analysis revealed leptin levels in the normal range (13.0 ng/mL) as well as markedly reduced triglyceride levels of 35.1 mmol/L (coming from >50 mmol/L) (Table 1). In addition, the patient reported a dramatic effect on her appetite regulation with a marked improvement of the feeling of satiety and the liver function test (GPT/ALAT) reduced dramatically (Table 1). Indeed, we found a slight increase in the HbA1c levels, which is not explained by adjustment/reduction of the insulin doses, since these were similar in the 2 weeks interval between the 2 measurements. We would not suggest the HbA1c increase as a side effect of the initiation of the leptin treatment but would expect the HbA1c to drop in a metabolic steady state situation in the future. Unfortunately, the patient developed severe allergic reactions at the injection sites. In that respect it should be mentioned that the patient reported to have allergic reactions to disinfectant agents priorly. Currently, the leptin therapy is reduced to 5 mg every other day with additionally anti-histaminic local skin therapy after injection.
Five months after initiation of the leptin replacement therapy the patient reported an improved satiety feeling accompanied by a weight loss of 7 kg (actual BMI 26.6 kg/m²). The triglyceride levels dropped to the normal range, the insulin glargine could be stopped without significant change in HbA1c levels and the leptin concentration increased into the age- and sex-adjusted normal range (Table 1). However, the GPT levels increased toward the slightly elevated levels before initiation of the leptin replacement therapy, which might suggest chronic damage of the liver due to severe metabolic arrangement in the past.
Discussion
Our results indicate that PPARG heterozygous c.485T>G (p.(Phe162Cys)) missense variant is most likely a yet undescribed molecular basis for a FPLD in humans. Our patient had loss of s.c. fat from the extremities and accumulation of truncal adipose tissue. A patient described by Agarwal and Garg (2) with heterozygous PPARG R425C mutation had a similar phenotype with additional loss of s.c. fat from the face and neck. She was a 64-year-old non-Hispanic White woman with diabetes and hypertriglyceridemia since the age of 32, accompanied by hirsutism. The onset of lipodystrophy occurred when she was 50 years old. Thus, our patient may have the same subtype of FPLD but with slightly different anthropometric features due to another mutation from the amino acid phenylalanine to cysteine of the PPARG protein.
In our subject, diabetes but not hypertriglyceridemia responded to standard pharmacotherapy which might indicate a specificity of the negative effects of c.485T>G on lipid vs glucose metabolism. Due to the markedly elevated triglycerides, our patient was first advised to take gemfibrozil, which importantly belongs to the class of fibrates (see Gevilon® (pfizer.de), cited 18 March 2021). These activate PPARA, which in addition to PPARG is also involved in metabolism of carbohydrates and lipids as well as adipose tissue differentiation, resulting in an increase of the activity of extrahepatic lipoprotein lipase (4). Interestingly, the triglyceride levels of our patient were still higher than 50 mmol/L despite gemfibrozil treatment indicating lipid-pronounced metabolic consequences of to the novel heterozygous c.485T>G variant with no response of the hypertriglyceridemia to standard PPARA-based pharmacotherapy.
Due to the increased triglycerides despite standard pharmacotherapy, from our point of view in the year 2020 leptin replacement therapy was the only option despite lipid-apheresis, the latter being invasive, time consuming and exhibiting marked side effects. Efficacy and safety of recombinant leptin in individuals with PPARG pathogenic variants have been demonstrated (5, 6, 7). However, it has to be mentioned that these trials included only ten patients and showed contradictory results regarding the effects on hypertriglyceridemia. While improvements in glycaemic parameters could be observed in all individuals, significant triglyceride-lowering effects by recombinant leptin occurred only to a few of them with one patient even having a 30% increase in serum triglycerides after 1 year of therapy (7). From a mechanistic point of view, recombinant leptin in patients with lipodystrophy has the potential to reduce de novo lipogenesis and enhance hepatic insulin sensitivity resulting in improvements of hepatic steatosis and dyslipidemia. Of importance, former studies demonstrated that within 6 months of metreleptin (Food and Drug Administration and European Medicines Agency approved recombinant leptin) treatment the availability of glycerol and free fatty acids to the liver decreases, so that there are less substrates for triglyceride synthesis which might indicate indirect effects on hepatic lipogenesis (8). In contrast, in our patient carrying the novel heterozygous c.485T>G variant, a dramatic reduction of triglyceride levels was observed not after month of treatment but as early as within 2 weeks. Given the mixed findings from former studies that recombinant leptin therapy does not lower triglycerides regularly in all FPLD patients carrying other PPARG pathogenic variants and that it takes often months of treatment to observe marked lipid lowering effects, it can be assumed that the novel variant described here could be a plausible predictor of a rather unique lipid phenotype.
The intake of recombinant human leptin is known to also have favorable effects on appetite and satiety regulation and thereby on the homoeostatic control of eating behavior (9). The female patient described here reported a marked effect on her satiety feeling after a few days of treatment, which has been reported before.
Furthermore, it has been shown that metreleptin treatment also has a positive impact on hepatic steatosis and MeSH due to increased fatty acid oxidation and reduced intrahepatocellular lipid accumulation (10). Although we do not have ultrasound or even histological data available, it has to be mentioned that former elevated GPT/ALAT decreased profoundly after initiation of metreleptin therapy (Table 1), which might suggest positive effects on MeSH also in our patient.
The authors would like to point out that the clinical case description and the corresponding clinical data presented in this study are not a proof of causality of the novel mutation inducing FPLD.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient.
Author contribution statement
Fiona Melzer and Corinna Geisler analyzed the data and wrote the manuscript. Dominik M Schulte and Matthias Laudes took care of the patient, guided the diagnostics and the therapy and participated in writing the manuscript.
Acknowledgements
The authors thank our patient for her collaboration and for letting us publish her case in scientific journals.
References
- 1.Garg A.Acquired and inherited lipodystrophies. New England Journal of Medicine 2004. 350 1220–1234. ( 10.1056/NEJMra025261) [DOI] [PubMed] [Google Scholar]
- 2.Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gene in a patient with familial partial lipodystrophy. Journal of Clinical Endocrinology and Metabolism 2002. 87 408–411. ( 10.1210/jcem.87.1.8290) [DOI] [PubMed] [Google Scholar]
- 3.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector Eet al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015. 17 405–424. ( 10.1038/gim.2015.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snow EK.AHFS Drug Information. Bethesda, MD: American Society of Health-System Pharmacists, 2009. [Google Scholar]
- 5.Sekizkardes H, Cochran E, Malandrino N, Garg A, Brown RJ. Efficacy of metreleptin treatment in familial partial lipodystrophy due to PPARG vs LMNA pathogenic variants. Journal of Clinical Endocrinology and Metabolism 2019. 104 3068–3076. ( 10.1210/jc.2018-02787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guettier JM, Park JY, Cochran EK, Poitou C, Basdevant A, Meier M, Clément K, Magré J, Gorden P. Leptin therapy for partial lipodystrophy linked to a PPAR-gamma mutation. Clinical Endocrinology 2008. 68 547–554. ( 10.1111/j.1365-2265.2007.03095.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia 2010. 53 27–35. ( 10.1007/s00125-009-1502-9) [DOI] [PubMed] [Google Scholar]
- 8.Baykal AP, Parks EJ, Shamburek R, Syed-Abdul MM, Chacko S, Cochran E, Startzell M, Gharib AM, Ouwerkerk R, Abd-Elmoniem KZet al. Leptin decreases de novo lipogenesis in patients with lipodystrophy. JCI Insight 2020. 5 e137180. ( 10.1172/jci.insight.137180) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schlögl H, Müller K, Horstmann A, Miehle K, Püschel J, Villringer A, Pleger B, Stumvoll M, Fasshauer M. Leptin substitution in patients with lipodystrophy: neural correlates for long-term success in the normalization of eating behavior. Diabetes 2016. 65 2179–2186. ( 10.2337/db15-1550) [DOI] [PubMed] [Google Scholar]
- 10.Simha V, Szczepaniak LS, Wagner AJ, DePaoli AM, Garg A. Effect of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes Care 2003. 26 30–35. ( 10.2337/diacare.26.1.30) [DOI] [PubMed] [Google Scholar]