

Abstract
Molecular evolution, including nucleotide substitutions, plays an important role in understanding the dynamics and mechanisms of species evolution. Here, we sequenced whole plastid genomes (plastomes) of Quercus fabri, Quercus semecarpifolia, Quercus engleriana, and Quercus phellos and compared them with 14 other Quercus plastomes to explore their evolutionary relationships using 67 shared protein‐coding sequences. While many previously identified evolutionary relationships were found, our findings do not support previous research which retrieve Quercus subg. Cerris sect. Ilex as a monophyletic group, with sect. Ilex found to be polyphyletic and composed of three strongly supported lineages inserted between sections Cerris and Cyclobalanposis. Compared with gymnosperms, Quercus plastomes showed higher evolutionary rates (Dn/Ds = 0.3793). Most protein‐coding genes experienced relaxed purifying selection, and the high Dn value (0.1927) indicated that gene functions adjusted to environmental changes effectively. Our findings suggest that gene interval regions play an important role in Quercus evolution. We detected greater variation in the intergenic regions (trnH‐psbA, trnK_UUU‐rps16, trnfM_CAU‐rps14, trnS_GCU‐trnG_GCC, and atpF‐atpH), intron losses (petB and petD), and pseudogene loss and degradation (ycf15). Additionally, the loss of some genes suggested the existence of gene exchanges between plastid and nuclear genomes, which affects the evolutionary rate of the former. However, the connective mechanism between these two genomes is still unclear.
Keywords: chloroplast genome, evolutionary rate, nonsynonymous substitution, oaks, selective pressure, synonymous substitution
In this paper, our research object is 18 species of Quercus, which is a better forest building tree species. We first sequenced the whole cp genome of Quercus fabri, Q. semecarpifolia, Q. engleriana, and Q. phellosand then compared it with 14 other Quercus cp genomes to explore their evolutionary patterns. We found that most protein‐coding genes have experienced relaxed purifying selection, and discussed the factors related to the evolution rate.
![]()
1. INTRODUCTION
Nucleotide substitution is a driving force of genome evolution, as the production of nonsynonymous substitutions may change protein functions, which may be fatal. Consequently, natural selection tends to delete these harmful mutations, resulting in most species being under negative selective pressure (Susann et al., 2013; Wang et al., 2018). Evolutionary rates of species may be affected by gene or protein function, selective pressure, population size, generation time, DNA‐repair efficiency, species diversity, and individual species size (Bromham et al., 2015; Hao et al., 2018; Wang et al., 2015). Selective pressure on functional genes has been found to be especially significant in determining rates of protein evolution (Minias et al., 2018; Xing & Lee, 2005). For example, selective pressures associated with habitats appear to have caused the rapid evolution of genes involved in cold response in Cardamine (Ometto et al., 2012). Another example are parasitic plant species, which have greater evolutionary rates in their plastid, mitochondrial (mt), and nuclear genomes when compared with other plants as they are deemed to be under greater selective pressure (Bromham et al., 2013). With the rapid development of DNA sequencing technology, it is now possible to study the role of selective pressure in molecular evolution.
Plastids are semiautonomous organelles and include chloroplasts, leucoplasts, chromoplasts, and amyloplasts, which all develop from proplastids of an embryo and have an identical genome. Over the past few decades, plastomes have begun to be used extensively in studying species evolution, migration, identification, and classification (Jose et al., 2015; Moore et al., 2010). Earlier plastome studies mainly focused on comparisons of the absolute rates of evolution of some functional genes, and these results were based on different genes in different species, which may have led to statistical errors (Kenneth et al., 1987). Later, comparisons of evolutionary rates among plastid, mt, and nuclear genomes, as well as between gymnosperms and angiosperms, were conducted (Petersen et al., 2019; Smith et al., 2014). Modern sequencing technology now allows the rapid generation of genomic data, which is essential for a more accurate analysis of factors affecting molecular evolution.
The genus Quercus L. is native to the Northern Hemisphere and contains ~500 species and is widely distributed in Asia, northern Africa, Europe, and America (Aldrich & Cavender‐Bares, 2011). Infrageneric classification of Quercus is controversial mainly because convergent morphological evolution is commonplace within the genus (Denk et al., 2017) and many morphological differences are a result of introgression (Curtu et al., 2007; Moran et al., 2012). Species of Quercus (commonly referred to as oaks) are wind‐pollinated and unable to discriminate among pollen from other species of the same section. Additionally, there is incomplete reproductive isolation among oaks, and as a result, phenotypes of progeny produced by interspecific hybridization are variable and difficult to discern (Williams et al., 2010).
Based on morphological characteristics, Chinese oaks have been classified into two subgenera: Quercus subg. Quercus and Q. subg. Cyclobalanopsis (Oerst.) C.K. Schneid. (Zhou, 1992), or these have been considered as distinct genera in the Flora of China (Huang et al., 1999). More recently, phylogenetic research has been conducted on Quercus and Cyclobalanopsis Oerst. using nuclear DNA (nrDNA) and plastid DNA fragments (Denk et al., 2017) and, most recently, whole plastome, mt, and nuclear genome data (Hipp et al., 2020). In the most recent classification based on the nrDNA and plastid DNA (reviewed in Denk et al., 2017), Quercus is divided into two subgenera, Q. subg. Quercus and Q. subg. Cerris (Oerst.), corresponding to the New‐ and Old‐World oaks, respectively, and with Cyclobalanopsis placed within Q. subg. Cerris Oerst.. This was corroborated by whole plastome, mt, and nuclear genome data (Hipp et al., 2020). However, relationships within the Old‐World oaks of Q. subg. Cerris Oerst. are complex. Phylogenetic relationships established by nrDNA tended to differ from those based on plastid data, especially with regards taxa previously circumscribed in Q. sect. Ilex (reviewed in Denk et al., 2017). Chinese taxonomists continue to accept Cyclobalanopsis as a distinct subgenus or genus (Deng et al., 2014; Pu et al., 2002; Zhou, 1992), and controversy also surrounds whether taxa pertaining to Q. sect. Ilex are a monophyletic lineage (Denk et al., 2017).
In our study, we sequenced the whole plastomes of Quercus fabri. Hence, Quercus semecarpifolia Smith, Quercus engleriana Seem, and Quercus phellos L. Q fabri, of Q. subg. Quercus sect. Quercus, is a deciduous forest species endemic to China. It is widely distributed south of the Yangtze river and grows on hills or mountains between 50 and 1,900 m a.s.l. (Li, Li, & Fang, 2018; Li, Li, Zang, et al., 2018). Q. semecarpifolia and Q. engleriana, of Q. subg. Cerris sect. Ilex, are evergreen forest species endemic to China. Q. semecarpifolia has a narrow distribution, being found only in western China, while Q. engleriana has a comparatively large distribution, with its range covering the Chinese provinces Shanxi, Jiangxi, Fujian, Hunan, Hubei, Guangxi, Sichuan, Guizhou, Yunnan, and Tibet (Tang, 2015). Quercus phellos, of Q. subg. Quercus sect. Lobatae, is a North American deciduous red oak introduced to China and used extensively in landscape gardening (Chen et al., 2013).
All three Chinese endemic oak (Q. fabri, Q. semecarpifolia, and Q. engleriana) species are ecologically and economically important, and their physiology, genetic diversity, breeding, forest management, and food processing are under intense investigation (Wangda & Oshawa, 2006; Wei et al., 2021). Additionally, there are no native species of Q. sect. Lobatae in China, and no plastomes sequenced from this section prior to our study, with Q. phellos critical to our study for including section Lobatae characteristic. The whole plastomes of these four species, coupled with those of 14 other Quercus species retrieved from GenBank, belong to the two subgenera and five of the eight sections currently accepted, that is, Q. subg. Quercus sect. Quercus, sect. Lobatae, and Q. subg. Cerris sect. Cerris, sect. Cyclobalanopsis, sect. Ilex (Denk et al., 2017; Hipp et al., 2020). These species’ distributional ranges and habitats largely differ and thus form ideal material for studying evolution of the Quercus genus.
The purposes of this study are to (a) generate a well‐resolved phylogenetic framework based on plastomes; (b) analyze the sequence variation of coding and noncoding plastid gene regions and transfer of plastid genes to understand the diversification patterns of Quercus; and (c) analyze the selective pressure that acts on essential protein‐coding genes to explore the variation and patterns in evolutionary rates of Quercus plastomes.
2. MATERIALS AND METHODS
2.1. Sampling, DNA extraction, Illumina sequencing, and assembly
Fresh leaves were collected from four Quercus species (Q. fabri, Q. semecarpifolia, Q. engleriana, and Q. phellos), kept on ice, and then stored at −80°C until further use. The four oak species were collected from three Chinese provinces (Jiangsu, Yunnan, and Zhejiang; see Table S1 for collection information). Voucher specimens were deposited at the Nanjing Forestry University Herbarium (accession numbers: NJFU‐QF20180501, NJFU‐QS20201001, NJFU‐QE20201002, and NJFU‐QP20201003, respectively).
Genomic DNA was isolated using the modified cetyltrimethylammonium bromide method (Doyle, 1987). A 3 mg of leaf tissue was ground with the addition of 0.35 times the volume of absolute ethanol and inverted several times. Following this, 700 μl of preheated 2% CTAB extract was added to the solution, which was then kept at 65°C in a water bath for 30 min. This solution was then centrifuged at 12,000 r/min for 5 min, after which an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) was added to the supernatant. This was then centrifuged at 12,000 r/min for 10 min, after which an equal volume of chloroform/isoamyl alcohol (24:1) was added to the supernatant. The supernatant was then transferred to an equal volume of prechilled isopropanol and let stand at −20°C for at least 40 min. The precipitate was then collected, 1 mol/L NaCl 400 μl was added for dissolution, then an equal volume of chloroform/isoamyl alcohol was added and the solution was centrifuged at 12,000 r/min for five minutes. Following this, a 1/2 volume of NaCl solution and 2–2.5 times volume of precooled absolute ethanol was added to the supernatant, followed by gentle shaking until white flocculent precipitate appeared. The precipitate was then collected and washed twice with 70% ethanol, before being placed on the clean bench and blow dried. Agarose gel electrophoresis and spectrophotometry (OD‐1000; Shanghai Cytoeasy Biotech Co., Ltd., Shanghai, China) were used to determine DNA integrity and quality.
The DNA was fragmented with a Covaris sonication device and then the fragments were purified, end‐repaired, and A‐tailed. Adapters were lighted through a 3′‐thymine overhang. Next, the fragment size was selected by agarose gel electrophoresis, and the sequencing library was formed by PCR amplification. Finally, the constructed library was inspected. Sequencing was performed on an Illumina HiSeq 2500 platform (Illumina, Nanjing, China), yielding at least 8.18–10 GB of clean reads.
We selected the plastome sequence of Quercus aliena as a reference, and then employed reads to produce two‐way extensions through overlap using NOVOPlasty software (Dierckxsens et al., 2017). When the assembly results were within the expected range, the overlap was greater than 200 bp, and the assembly formed a ring.
2.2. Annotation and analysis of the plastid DNA sequences
Genome annotation was performed using CpGAVAS (Chang et al., 2012), with DOGMA (http://dogma.ccbb.utexas.edu/) and BLAST used to confirm the annotation results. tRNAscanSE was used to identify the tRNAs (Schattner et al., 2005). Circular gene maps of Q. fabri, Q. phellos, Q. engleriana, and Q. semecarpifolia were drawn using the OGDRAWv1.2 program (http://ogdraw.mpimp‐golm.mpg.de/; Lohse et al., 2007). Relative synonymous codon usage was examined using CodonW (Peden, 2000). The annotated plastome sequences of Q. fabri, Q. phellos, Q. engleriana, and Q. semecarpifolia were deposited in the NCBI Sequence Read Archive (Accession numbers MK693136, MZ196210, MZ196209, MZ196211, respectively).
2.3. Genome structure analysis and genome comparisons
The mVISTA program in Shuffle‐LAGAN mode was used to compare the Q. fabri, Q. phellos, Q. engleriana, and Q. semecarpifolia plastomes with the 14 other Quercus plastomes (Q.baronii Skan, Quercus dolicholepis A. Camus, Quercus acutissima Carruth, Quercus variabilis Blume, Quercus tarokoensis Hayata, Quercus edithiae Skan, Quercus glauca Thunb., Quercus sichourensis Hu, Quercus aquifolioides Rehd. and Wils, Quercus spinosa David ex Franchet, Quercus tungmaiensis Y.T. Chang, Q. aliena var. acutiserrata Maximowicz ex Wenzig, Q. aliena Blume, and Quercus rubra L.; Table 1), using the Q. aliena annotation as a reference (Mayor et al., 2000). MAUVE v2.4.0 software was used to compare the genes and sequences of the de novo assembled Q. fabri plastome sequence, with the Q. aliena plastome sequence as the reference (Doose et al., 2017). Geneious Pro v9.1.6 was used to characterize the plastomes of the 18 species and summarize the features into tables (Matthew et al., 2012).
TABLE 1.
Basic features of the Quercus species chloroplast genomes
| Species | Accession No. | Length (bp) | GC content (%) | LSC length (bp) | SSC length (bp) | IR length (bp) | Gene number |
|---|---|---|---|---|---|---|---|
| Quercus acutissima | MH607377 | 161,124 | 36.8 | 90,423 | 19,069 | 25,816 | 135 |
| Quercus aliena | KU240007 | 161,150 | 36.8 | 90,444 | 19,054 | 25,826 | 126 |
| Quercus aliena var. acutiserrata | KU240008 | 161,153 | 36.8 | 90,457 | 19,044 | 25,826 | 126 |
| Quercus aquifolioides | KX911971 | 161,225 | 36.8 | 90,535 | 19,000 | 25,845 | 126 |
| Quercus baronii | KT963087 | 161,072 | 36.8 | 90,341 | 19,045 | 25,843 | 126 |
| Quercus dolicholepis | KU240010 | 161,237 | 36.8 | 90,461 | 19,048 | 25,864 | 126 |
| Quercus edithiae | KU382355 | 160,988 | 36.9 | 90,351 | 18,954 | 25,842 | 128 |
| Quercus fabri | MK693136 | 161,250 | 36.8 | 90,563 | 18,995 | 25,846 | 136 |
| Quercus glauca | NC_036930 | 160,798 | 36.9 | 90,229 | 18,906 | 25,831 | 134 |
| Quercus rubra | JX970937 | 161,304 | 36.8 | 90,542 | 19,025 | 25,869 | 129 |
| Quercus sichourensis | NC_036941 | 160,681 | 36.9 | 90,154 | 18,857 | 25,835 | 134 |
| Quercus spinosa | KX911972 | 161,156 | 36.8 | 90,441 | 18,997 | 25,859 | 126 |
| Quercus tarokoensis | NC_036370 | 161,355 | 36.8 | 90,602 | 19,033 | 25,860 | 134 |
| Quercus tungmaiensis | NC_036936 | 160,702 | 36.9 | 90,113 | 18,939 | 25,825 | 134 |
| Quercus variabilis | KU240009 | 161,077 | 36.8 | 90,387 | 19,056 | 25,817 | 126 |
| Quercus phellos | MZ196210 | 161,331 | 36.8 | 90,550 | 19,061 | 25,860 | 127 |
| Quercus engleriana | MZ196209 | 161,053 | 36.8 | 90,356 | 18,943 | 25,877 | 133 |
| Quercus semecarpifolia | MZ196211 | 161,312 | 36.8 | 90,505 | 19,077 | 25,865 | 133 |
2.4. Phylogenetic analyses
We used 67 shared protein‐coding sequences of the 18 Quercus species and two outgroup species, Malus prunifolia (Willd.) Borkh. (NC_031163) and Ulmus gaussenii W. C. Cheng (NC_037840), to build the phylogenetic trees. Phylogenetic relationships were constructed using the BI analysis in Mrbayes v 3.2.6 (Huelsenbeck & Ronquist, 2001). First, the sequences were aligned in MAFFT (Kazutaka & Standley, 2013). Then, the multiple sequence alignment was visualized and manually adjusted using BioEdit (Hall, 1999). We used JmodelTest2 to evaluate the best‐fitting models of nucleotide substitution (Darriba et al., 2012). GTR +G was selected as the best substitution model for BI analyses.
2.5. Gene selective pressure analysis
The 79 shared protein‐coding genes of the 18 Quercus species, which had sequence lengths longer than 300 bp were extracted and aligned separately using Mega 7.0 to analyze synonymous substitution rates (Ds) and nonsynonymous substitution rates (Dn) using the codeml program in PAML package version 4.7.1 under a one‐ratio branch model (Yang & Yang, 2007). We constructed the tree topology using BI as the tree file. After removing outliers, a total of 64 effective genes were included in statistical analyses using SPSS 19.0 software (SPSS, Inc., Chicago, IL, USA). The Ds, Dn, and Dn/Ds ratio of functional gene functional groups were compared by ANOVA. A Mann–Whitney U test of two independent samples was used to determine whether there were significant differences in Ds, Dn, and Dn/Ds among the Quercus species and previously published values of gymnosperms (Wang et al., 2015) based on the calculated P‐values obtained from the double‐tail test (p < .05).
3. RESULTS
3.1. Plastome assembly and characteristics
The Q. phellos complete plastome size was the largest with 161,331 bp (Table 1). The four oak species displayed a typical quadripartite structure, and we chose to describe the Q. fabri plastome, which included a pair of inverted repeats (IRs; 25,846 bp) separated by a single large copy (LSC; 90,563 bp) and a single small copy (SSC; 18,995 bp) region (Figure 1; Table 1). The Q. fabri genome contained the most functional genes (about 136), including 89 protein‐coding genes, 39 tRNA genes, and eight rRNA genes (Tables 1 and S1). Introns occurred in 12 genes, rps16, atpF, rpoC1, ycf3, clpP, petB, petD, rpl16, rpl2, ndhB, ndhA, and rpl2 (Table S2). Of these, the rps12 gene was unique as it contained three exons, one of which occurred in the LSC region, while the other two exons occurred in the IRa and IRb regions. We found 26,756 codons in all the coding sequences (Table S3). Leucine was the most common amino acid, encoded by 10.84% (2,903) of the codons, while cysteine was the least frequent amino acid, encoded by 1.35% (331). The A‐ and U‐ending codons occurred at high frequencies, and all types of preferred synonymous codons (relative synonymous codon usage >1) ended with A or U.
FIGURE 1.
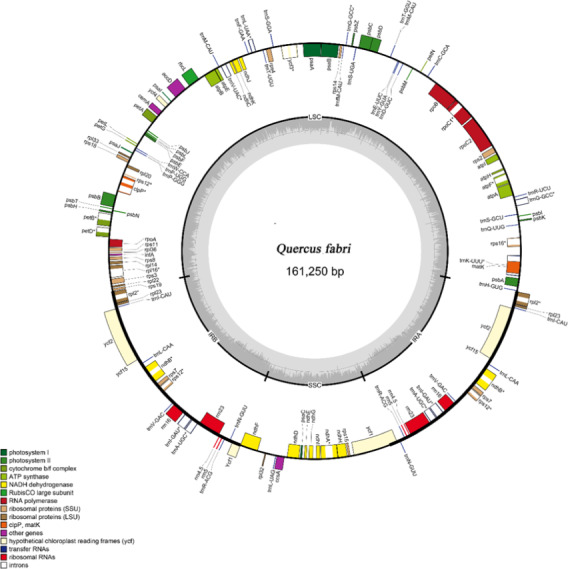
Gene map of the Quercus fabri plastome. Genes displayed outside the circle are transcribed clockwise, while internal transgenes are transcribed counterclockwise. Genes belonging to different functional groups are color‐coded. The dashed line region in the inner circle represents the GC content of the plastome
There was no gene rearrangement among the 18 species during the evolution of Quercus (Figure S1). We compared the details of four junctions (LSC/IRA, LSC/IRB, SSC/IRA, and SSC/IRB) among the plastomes of the 18 species (Figure S2). At the SSC/IRA boundary, the ycf1 gene ranged from 4,600 (Q. rubra) to 4,635 bp (Q. tungmaiensis) in the SSC region. Among the 18 oak species, the length of the ndhF and ycf1 genes of Q. engleriana showed obvious differences from the others. A range of 72–82 simple sequence repeats (SSRs) were detected in the plastomes of the 18 Quercus species (Figure S3).
3.2. Phylogenetic analysis
The phylogenetic relationships of the 18 Quercus species, constructed using 67 shared protein‐coding sequences, were strongly supported and can be seen in Figure 2. Quercus was found to be divided into two major evolutionary clades. The first clade included taxa of Quercus subg. Cerris sections Ilex, Cerris, and Cyclobalanposis, while the second included taxa of Quercus subg. Quercus sections Quercus and Lobatae, as defined by Denk et al., 2017). Quercus subg. Cerris sect. Ilex was not retrieved as a monophyletic group due to the insertion of taxa from sect. Cerris and sect. Cyclobalanposis, which divided sect. Ilex into three strongly supported polyphyletic evolutionary clades. Taxa of Q. subg. Quercus sect. Quercus (Q. aliena, Q. aliena var. acuteserrata, Q. fabri) formed a well‐supported monophyletic clade that was sister to a clade comprising taxa of Q. subg. Quercus sect. Lobatae (Q. rubra and Q. phellos).
FIGURE 2.
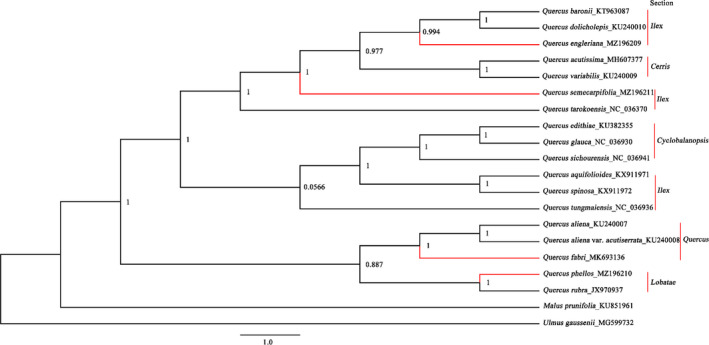
Maximum‐likelihood phylogenetic tree constructed using 67 shared protein‐coding sequences of plastomes of 18 Quercus species. Numbers above the lines indicate the likelihood bootstrap values. Malus prunifolia and Ulmus gaussenii were used as the outgroups
3.3. Deletion and transfer of plastid genes
The variation in plastome length among the 18 oak species studied was small, with plastomes ranging from 160,681bp (Q. sichourensis) to 161,355 bp (Q. tarokoensis; Table 1). Genes were consistently distributed among the different genomic regions across the 18 species. rRNA genes showed the most remarkable conservation, with all species containing eight rRNA genes. The number of protein‐coding genes differed among the Quercus species; however, they shared 79 common genes. In this study, Q. rubra, Q. edithia, and Q. sichourensis lost the psbB, rpl22, and ycf4 genes, respectively. Additionally, only seven species (Q. edithiae, Q. fabri, Q. rubra, and Q. acutissima, Q. phellos, Q. engleriana, and Q. semecarpifolia) contained the ycf15 gene.
We used the ycf15 gene sequence of Nicotiana tabacum Fischer ex Lehmann. as a reference, as well as those of Olea europaea Linn. and Liriodendron tulipifera Linn., in comparisons with the seven Quercus species (Q. acutissima, Q. fabri, Q. edithiae, Q. rubra, Q. phellos, Q. engleriana, and Q. semecarpifolia) containing ycf15 (Figure 3). In N. tabacum (Z00044) and O. europaea (GU931818), the ycf15 gene is complete, while in L. tulipifera (DQ899947), the ycf15 gene is a pseudogene. Unlike the ycf15 sequence in most species, these seven species employed CTG and TTG start codons instead of ATG and GTG. Compared with the representative species, the ycf15 genes in Q. rubra, Q. phellos, and Q. edithiae were similar to that of L. tulipifera, indicating that they are pseudogenes. The lengths of the ycf15 gene in the remaining four species (Q. fabri, Q. acutissima, Q. engleriana, and Q. semecarpifolia) were only ~50 bp, indicating pseudogene degeneration. These seven species belong to different subgenera and sections, and their ycf15 gene sequences are very different.
FIGURE 3.

Alignment of the ycf15 gene and protein sequences from the 18 Quercus species. a. Alignment of the ycf15 gene sequences; b. Alignment of the ycf15 protein sequences
3.4. Comparative analysis of coding and noncoding regions
Using the mVISTA program (Figure S4), we found that conserved noncoding sequences (containing intron sequences) had greater variation levels than exon sequences. The sequences of noncoding regions which had large variations included trnH_GUG‐psbA, trnK_UUU‐rps16, rps16‐trnQ_UUG, trnS_GCU‐trnG_GCC, atpF‐atpH, atpI‐rps2, psbM‐trnD_GUC, trnM_CAU‐psbD, psbZ‐trnG_UCC, trnfM_CAU‐rps14, trnF_GAA‐ndhJ, ndhC‐trnV_UAC, rbcL‐accD, ycf4‐cemA, ccsA‐ndhD, and rpl32‐trnL_UAG. We analyzed the coding and noncoding regions’ sequences using a sliding window to present the observed variations using digitization. The sliding window analysis showed that protein‐coding genes which had large pi values were: atpF, clpP, infA, ndhA, ndhD, ndhK, ndhH, psbC, rpl20, rpoC2, and rps16 (Figure 4a). Among these genes, those with introns showed the greatest variation. The following genes had sequence variation in their gene interval regions: trnK_UUU‐rps16, petA‐psaJ, rbcL‐accD, trnfM_CAU‐rps14, and trnS_GUC‐trnR_UCU (Figure 4b). The sequence variation in protein‐coding genes and gene interval regions reflected by the sliding window was the same as those determined using the mVISTA software. In general, we confirmed that the variations in noncoding sequences are greater than those of coding sequences through different analyses (Figure 4c).
FIGURE 4.
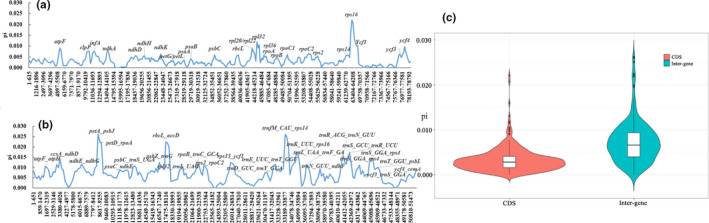
Percentages of variable characters in 18 aligned Quercus plastomes. (a) Coding regions; (b) Noncoding regions; (c) Analysis of the differences between coding and intergenic region. X‐axis: position in a window. Y‐axis: nucleotide diversity of each window
To further explore the sequence variation characteristics of coding and noncoding regions, we counted the shared coding‐protein genes of the 18 Quercus species studied. These species shared 79 protein‐coding genes, 6 of which (ndhB, rpl2, rps12, rps7, ycf2, and rpl23) had duplicate copies. Of the 79 protein‐coding genes, 31 had variable lengths (Figure 5a). We analyzed the lengths of these 31 protein‐coding genes and found 12 had introns, with larger length variations than exons, such as atpF, clpP, ndhA, and rpl2. The intron lengths of the 31 protein‐coding genes also differed. Additionally, Q. edithiae lost intron regions in petB, petD, rps12, rps16, and rpl16, which caused variation in intron lengths (Figure 5b). The sequence lengths of exons of the 31 protein‐coding genes also varied, such as those of rps16, rps12, petD, petB, and rpl16 (Figure 5c). Protein‐coding genes without introns were relatively conserved, with limited variation in gene lengths. Still, there were also several unique genes, such as rpl22, rpoc2, and ycf2, which had lengths that fluctuated greatly among the 18 species.
FIGURE 5.
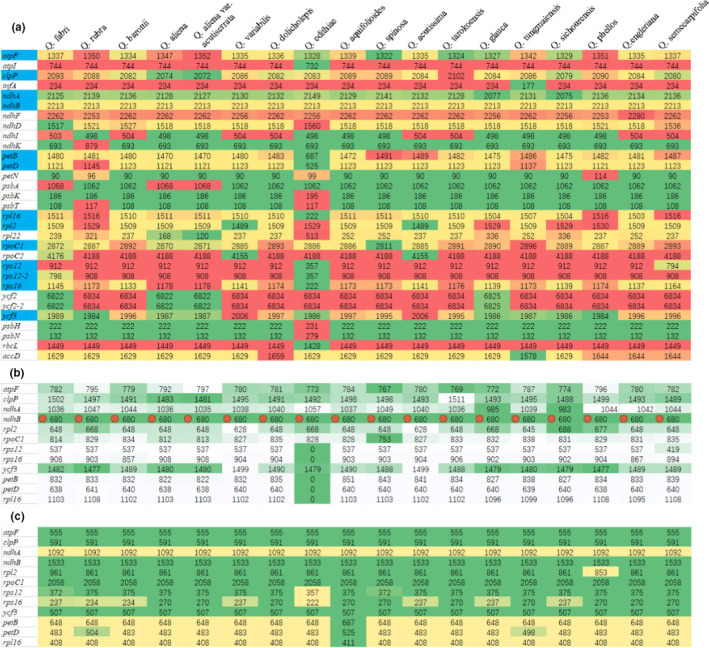
Analysis of variations in protein‐coding gene lengths. (a) A total of 31 protein‐coding genes with variable gene lengths; (b) Length variations of introns (noncoding sequences) in 12 protein‐coding genes; (c) Length variations of exons (coding sequences) in 12 protein‐coding genes. Different colors in the same row represent changes in length
3.5. Gene selective pressure analysis
We divided the 64 protein‐coding genes into four major categories: Photosynthetic metabolism; Photosynthetic apparatus; Gene expression; Other (Table 2). After removing the “other” category, the data presented in Table 2 revealed that “Gene expression” had the highest average synonymous and nonsynonymous substitution rates (Ds = 0.3108 and Dn = 0.2095, respectively), while “Photosynthetic metabolism” had the lowest Ds and Dn values of 0.0412 and 0.0027, respectively. When comparing Ds, Dn, and evolutionary rates (Dn/Ds) of the four gene categories, ANOVA found no significant differences (p > .05, Table 2), which may be due to the small sample size, leading to a lack of statistical validity. To estimate selective pressure, we studied the average evolutionary rates (Dn/Ds) of different genes (Figure 6). The value of Dn or Ds of some genes was equal to 0.0, so Dn/Ds value could not be calculated. The Dn/Ds ratio of the 18 species ranged from 0.0361 to 1.7654, varying by up to a factor of 49 (Figure 6a). With the exception of three genes (ndhJ, psbJ, and rps19), the Dn/Ds ratios of the remaining genes were all less than 1, suggesting that most protein‐coding genes in Quercus are under purifying selection (Figure 6b). Of these, we found the variation of evolutionary rate within gene groups is still considerable, with some genes with unknown functions evolving faster than other genes (Figure 6c). Compared with gymnosperms, Quercus has higher Dn (p < .05) and Dn/Ds (p > .05) ratios (Table S4).
TABLE 2.
Substitution and evaluation rates of Quercus plastid genes (standard deviation) using p < .05 as significant criterion
| Categories | Dn | Ds | Dn/Ds |
|---|---|---|---|
| Photosynthetic metabolism | 0.0027 (0.0023) | 0.0412 (0.0281) | 0.0674 (0.0214) |
| Photosynthetic apparatus | 0.1496 (0.5687) | 0.2403 (0.7891) | 0.3302 (0.4131) |
| Gene expression | 0.2095 (0.6143) | 0.3108 (0.9067) | 0.4684 (0.4048) |
| Other | 0.4978 (1.1837) | 0.6953 (1.6252) | 0.5454 (0.2334) |
| Average | 0.1927 (0.6384) | 0.2936 (0.8988) | 0.3793 (0.3747) |
| F | 0.619 | 0.534 | 1.777 |
| p | .606 | .661 | .161 |
FIGURE 6.
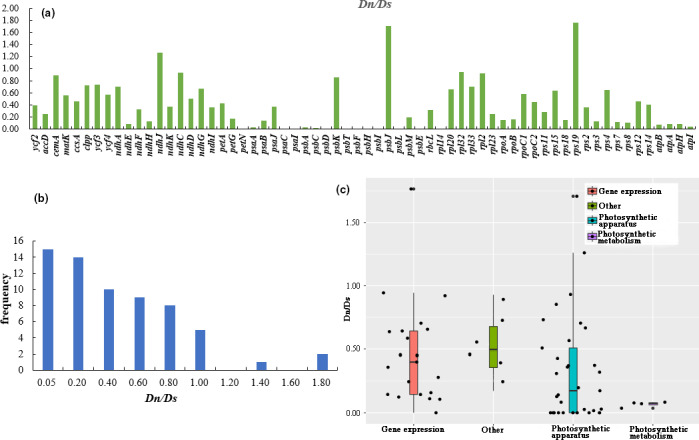
Evaluation of differences in plastid gene substitution rates in Quercus. (a) Dn/Ds distribution map of protein‐coding genes; (b) Dn/Ds frequency distribution of protein‐coding genes; (c) Analysis of Dn/Ds variations in different functional genes
4. DISCUSSION
The genus Quercus has become an established member of the plant communities of the Northern Hemisphere. It is considered to have arisen during the Paleogene (Barrón et al., 2017; 56 Ma according to Hipp et al., 2020) and subsequently underwent deep geographic separation of major clades within the first 15 Myrs after origin of the genus (Hipp et al., 2020). It is then hypothesized to have spread to a diverse range of environments in the later Cenozoic (Barrón et al., 2017), with China now forming the second‐largest diversity center of Quercus (Oldfield & Eastwood, 2007). Their high diversity and remarkable introgression make Quercus a good study‐system for understanding the link between the prevalence of adaptive gene flow and the evolutionary history of forest trees. Over the past five years, much research has focused on reexamining the oak phylogeny and investigating the history of diversification across the genus (Deng et al., 2018; Denk et al., 2017; Hipp et al., 2020; Jiang et al., 2019). The topology of our phylogenomic tree matches previous analyses based on plastid and nrDNA gene fragments and RAD‐seq of whole plastid, mitochondrial, and nuclear genomes (reviewed in Denk et al., 2017; Hipp et al., 2020) for all sections except that of sect. Ilex. In previous analyses (Denk et al., 2017; Hipp et al., 2020), sect. Ilex was retrieved as a monophyletic group, sister to sect. Cerris. However, in our study, sect. Ilex was polyphyletic and divided into three strongly supported evolutionary clades placed in‐between sect. Cyclobalanopsis and sect. Cerris. This may be related to differential rates of introgression, which make it difficult for us to establish completely reliable phylogenomic relationships based on only one genome (plastome) and a relatively small sample size. The focus of future phylogenomic studies of Quercus should be on incorporating a more comprehensive sampling of whole genomes, as well as including other molecular sampling approaches such as nuclear genome skimming, that covers all sections and morphological and biogeographic variability present in the genus. This should include whole plastomes and nrDNA from taxa belonging to Q. subg. Quercus sect. Protobalanus, Ponticae, Virentes, which were lacking in this study.
Compared with gymnosperm genera, Quercus has a much higher species diversity, and thus it is assumed a higher genetic diversity, albeit with extant species having a similar recent evolutionary age (Nagalingum et al., 2011). Thus, the question arises whether differences in the evolutionary patterns of plastomes affect their genetic diversity? Based on our results, we hypothesize that, during its early evolutionary history, Quercus had a considerable Dn value that enabled gene functions to be effectively adjusted to environmental changes occurring over the last c. 56 Ma (Hofmann et al., 2011), with a substantial body of research also supporting this view. For example, there are significant differences in the phenology, flowering biology, and other characteristics of oak species in accordance with their environment. The flowering time of oak species in temperate regions is in the spring, while tropical areas are in the dry season (Ducousso et al., 1993). Northern temperate deciduous forest species also have a higher pollen volume than that of earlier diverging evergreen oak species from warmer and drier climates, with the high pollen volume likely an adaptation evolved to cope with the relatively adverse damper cooler climate which inhibits anemophily (Cao & Zhou, 2002; Nagalingum et al., 2011). Additionally, most protein‐coding genes large Dn/Ds values further indicate that most genes have experienced relaxed purifying selection and that natural selection allowed Dn mutations of these genes. The Dn/Ds values of the three genes, ndhJ, psbJ, and rps19, were greater than 1, indicating positive selection, which is evidence of the proteins’ adaptive evolution. The evolutionary rates of genes which have varied functions are also different. However, the question arises as to why do genes involved in photosynthetic metabolism evolve more slowly than other genes? We speculate that selective constraints may cause differences in gene function and expression. atpA, atpB, and atpE encode the α, β, and ε subunits of plastid ATPase, respectively. ATPase is the critical enzyme in energy metabolism and plays a central role in photosynthesis (Felix et al., 2020). The divergence of Dn is proof that genes go through different degrees of selective constraints, but it is still challenging to discern what degree of selective constraints caused this difference.
Numerous studies of angiosperm plastomes have found that most variation occurred in noncoding sequences, and occasionally in the protein‐coding sequences which evolve more rapidly. Additionally, a large portion of conserved noncoding DNA appears to be under similar selective constraints as protein‐coding DNA but, until now, most studies on molecular evolution have focused on protein‐coding sequences (Chen & Blanchette, 2007; Xie et al., 2018). In this study, the degree of variation in the noncoding sequences (gene spacing regions) was greater than that in the protein‐coding sequences. Insertion and deletion mutations lead to microstructural changes, which can be coded as evolutionary events during phylogenomic analyses. Even in the coding region, introns do not encode proteins, and 20%–68% of introns are species‐specific. Over the past few hundred million years, introns have been gained and lost, although during the evolution of plants, introns have mainly been lost (Roy & Penny, 2007), while in algae abundant introns have been lost and gained recently (Turmel et al., 2017). In our study, we found that Quercus has 12 introns, with only Q. edithiae having lost intron regions in petB, petD, rps12, rps16, and rpl16. This could have occurred as a result from mistakes during gene annotation. However, this scenario could also represent patterns of evolution in Quercus. Differing mutation rate is the main cause of differentiations in evolutionary rate, and most mutation events generally occur in introns, untranslated regions (UTRS), and noncoding regulatory regions. Thus, the number of introns in a genome may reflect the evolutionary rate to a large extent. Species with slower evolutionary rates retain more introns than those with faster evolutionary rates.
Pseudogenes, that is, sequences lacking coding abilities, may also play essential roles in genome evolution (Wang et al., 2012). Redundant DNA deletion events frequently occur in genomes, significantly reducing the pseudogene contents (Lafontaine & Dujon, 2010). Whether the pseudogene ycf encodes a protein has been controversial (Shi et al., 2013) and only recently was it confirmed that the basal groups of asterids have complete ycf15 gene structures and are assumed to have protein‐coding functions (Curci et al., 2015). However, a small number of eudicots have ycf15 gene structures identified as pseudogenes. These were mainly found in the Ericales, Gentianales, Lamiales, Solanales, and Apiales, which had faster rates of genome evolutionary (Ku et al., 2013). The ycf15 genes of Q. edithiae, Q. phellos, and Q. rubra have structures similar to those of the species mentioned above (L. tulipifera, Gentianopsis paludosa, and Jasminum nudiflorum). We therefore speculate that their pseudogenes are not encoding genes. In addition to the above‐mentioned ycf15 pseudogene exhibiting a complete structure, there are few species, such as those belonging to Poaceae, Ranunculaceae, Caryophyllaceae, Cannabaceae, Moraceae, Asteraceae, Lythraceae, and Melastomataceae, in which the ycf15 gene has undergone genetic degeneration, resulting in its reduction to only c. 50 bp in length (Choi & Park, 2015; Liu et al., 2013; Nie et al., 2012). Pseudogenes are considered to be less influenced by selective pressure during evolution as they can effectively reflect the molecular records of ancestors, which is very helpful in the study of molecular evolution. The question arises as why the fates of the ycf15 genes in the seven species (Q. acutissima, Q. fabri, Q. edithiae, Q. phellos, Q. engleriana, Q. semecarpifolia, and Q. rubra) are so different? This problem may be resolved through the convenience of transcriptome sequencing, which permits mapping the transcriptome reads to the Quercus plastome to identify the pattern of gene expression and especially that of the ycf15 genes. This problem will be gradually solved through whole‐genome sequencing, which will help in understanding the evolution of the Quercus plastome.
Interactions among the three genomes (nuclear, plastid, and mitochondrial) in plant cells may also affect the rate of evolution of Quercus plastomes. Gene transfers among these three genomes may lead to specific correlations between their evolutionary rates (Goremykin et al., 2009; Jansen et al., 2011; Millen et al., 2001). In angiosperms, rpl22, rpl23, rpl32, rpl33, rps16, accD, psaI, ycf4, ycf1, ycf2, and infA genes disappeared in some groups, with the disappearance frequency of infA gene being highest, although the infA gene appeared recently in Quercus (Millen et al., 2001). In this study, we identified some gene losses, such as psbB, rpl22, ycf4, and ycf15. The rpl22 gene occurs in the nuclear genome of Fagaceae plants and appeared approximately 34–37 Ma, which explains the gene exchange between plastid and nuclear genes (Jansen et al., 2011). Additionally, DNA replication and repair mechanisms in mitochondrial and plastid genomes have many similarities (Smith et al., 2014). In conclusion, we believe that these three genomes have related evolutionary rates, but the mechanism behind this is still unclear.
5. CONCLUSIONS
The whole plastome phylogenomic relationships retrieved in our study mostly coincide with previous research (Denk et al., 2017; Hipp et al., 2020) in their support for sect. Quercus and sect. Lobatae of subg. Quercus, and sect. Cerris and sect. Cyclobalanopsis of subg. Cerris. However, our whole plastome topology does not support findings from Denk et al. (2017) and Hipp et al. (2020) that sect. Ilex is a monophyletic group. The polyphyly of sect. Ilex is seen by how sect. Cerris and sect. Cyclobalanposis are inserted between three strongly supported lineages of sect. Ilex taxa. Before taxonomic recircumscription of these lineages can be done, phylogenomic studies incorporating a more comprehensive and varied molecular sampling are needed. This is especially pertinent as different genomes, and regions of the genome, can give largely varied interpretations of the evolutionary history of oaks (Hipp et al., 2020).
Based on the whole plastome sequences of 18 Quercus species, the evolutionary model of nucleotide substitution rates of typical plastomes found that most protein‐coding genes have experienced relaxed purifying selection. Furthermore, the high Dn value indicated that these genes' functions have effectively adjusted to changes in the environment. Noncoding sequences were also found to have more significant variation, including variation in the interpretation of the intergenic region, loss of introns, and loss and degradation of pseudogenes, which are all manifestations of plastid evolution. Additionally, transfers of individual genes between the plastid and nuclear genomes were identified. This allows us to speculate that the evolution of the oak plastome was correlated with that of the other two genomes (nuclear and mitochondrial) to some extent. Continued work focused on a comprehensive molecular sampling of all Quercus species, coupled with integrating new whole‐genome sequencing technologies including those focused on the nuclear genome, will be fundamental in improving our understanding of the underlying mechanisms of evolution of both Quercus and organisms in general.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Xuan Li: Data curation (equal); Formal analysis (equal); Methodology (equal); Resources (equal); Software (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Yongfu Li: Formal analysis (equal); Software (equal). Steven Paul Sylvester: Writing‐review & editing (equal). Mingyue Zang: Formal analysis (equal). Yousry A. El‐Kassaby: Supervision (equal); Writing‐review & editing (equal). Yanming Fang: Funding acquisition (lead); Resources (lead); Supervision (equal).
Supporting information
Appendix S1
ACKNOWLEDGMENTS
This research was supported by the National Natural Science Foundation of China (31770699), the State Scholarship Fund, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Nanjing Forestry University Excellent Doctoral Thesis Fund (2171700124), and the Jiangsu Innovation Engineering Fund (KYCX18_0989). We also thank Xie Lei for his contribution to data processing.
Li, X. , Li, Y. , Sylvester, S. P. , Zang, M. , El‐Kassaby, Y. A. , & Fang, Y. (2021). Evolutionary patterns of nucleotide substitution rates in plastid genomes of Quercus . Ecology and Evolution, 11, 13401–13414. 10.1002/ece3.8063
Xuan Li and Yongfu Li contributed equally.
Contributor Information
Yousry A. El‐Kassaby, Email: y.el-kassaby@ubc.ca.
Yanming Fang, Email: jwu4@njfu.edu.cn.
DATA AVAILABILITY STATEMENT
All the data are available at Genbank. Genbank accessions MK693136, MZ196210, MZ196209, MZ196211.
REFERENCES
- Aldrich, P. R. , & Cavender‐Bares, J. (2011). Genomics and breeding of oaks and their slightly less‐domesticated wild oak relatives. In Kole C. (Ed.), Wild crop relatives: Genomic and breeding resources (pp. 89–129). Springer. [Google Scholar]
- Barrón, E. , Averyanova, A. , Kvaček, Z. , Momohara, A. , Pigg, K. B. , Popova, S. , Postigo‐Mijarra, J. M. , Tiffney, B. H. , Utescher, T. , & Zhou, Z. K. (2017). The Fossil History of Quercus . In Gil‐Pelegrín E., Peguero‐Pina J., & Sancho‐Knapik D. (Eds.), Tree physiology. Oaks physiological ecology. Exploring the functional diversity of genus Quercus L. (pp. 39–105). Springer. [Google Scholar]
- Bromham, L. , Cowman, P. F. , & Lanfear, R. (2013). Parasitic plants have increased rates of molecular evolution across all three genomes. BMC Evolutionary Biology, 13(1), 126. 10.1186/1471-2148-13-126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromham, L. , Hua, X. , Lanfear, R. , & Cowman, P. F. (2015). Exploring the relationships between mutation rates, life history, genome size, environment, and species richness in flowering plants. The American Naturalist, 185(4), 507–524. 10.1086/680052 [DOI] [PubMed] [Google Scholar]
- Cao, M. , & Zhou, Z. K. (2002). Pollen morphology and systematic significance of Quercus in China. Guangxi Plants, 22(1), 14–18. 10.3969/j.issn.1000-3142.2002.01.005 [DOI] [Google Scholar]
- Chang, L. , Shi, L. , Zhu, Y. , Chen, H. , Zhang, J. , Lin, X. , & Gua, X. J. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics, 13, 715. 10.1186/1471-2164-13-715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , & Blanchette, M. (2007). Detecting non‐coding selective pressure in coding regions. BMC Evolutionary Biology, 7(Suppl 1), 1–13. 10.1186/1471-2148-7-S1-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. T. , Sun, H. J. , Wang, S. F. , & Shi, X. (2013). Growth performances of five North American oak species in Yangzi River Delta of China. Forest Research, 26(3), 344–351. [Google Scholar]
- Choi, K. S. , & Park, S. J. (2015). The complete chloroplast genome sequence of Aster spathulifolius (Asteraceae), genomic features and relationship with Asteraceae. Gene, 572(2), 214–221. 10.1016/j.gene.2015.07.020 [DOI] [PubMed] [Google Scholar]
- Curci, P. , Pasquale, L. , De Paola, D. , Danzi, D. , Vendramin, G. G. , & Sonnante, G. (2015). Complete Chloroplast Genome of the Multifunctional Crop Globe Artichoke and Comparison with Other Asteraceae. PLoS One, 10(3), e0120589. 10.1371/journal.pone.0120589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtu, A. L. , Gailing, O. , & Finkeldey, R. (2007). Evidence for hybridization and introgression within a species‐rich oak (Quercus spp.) community. BMC Evolutionary Biology, 7, 218. 10.1186/1471-2148-7-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G. L. , Doallo, R. , & Posada, D. (2012). JModelTest 2: More models, new heuristics and parallel computing. Nature Methods, 9(8), 772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, M. , Hipp, A. L. , Song, Y. G. , Li, Q. S. , Coombes, A. , & Cotton, A. (2014). Leaf epidermal features of Quercus subgenus Cyclobalanopsis (Fagaceae) and their systematic significance. Botanical Journal of the Linnean Society, 176(2), 224–259. [Google Scholar]
- Deng, M. , Jiang, X. L. , Hipp, A. L. , Manos, P. S. , & Hahn, M. (2018). Phylogeny andbiogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): Insights into the Cenozoic history of evergreen broad‐leaved forestsin subtropical Asia. Molecular Phylogenetics and Evolution, 119, 170–181. [DOI] [PubMed] [Google Scholar]
- Denk, T. , Grimm, G. W. , Manos, P. S. , Deng, M. , & Hipp, A. L. (2017). An updated infrageneric classification of the oaks: Review of previous taxonomic schemes and synthesis of evolutionary patterns. In Gil‐Pelegrin E., Peguero‐Pina J. J., & Sancho‐Knapik D. (Eds.), Tree physiology. Oaks physiological ecology. Exploring the functional diversity of genus Quercus L. (pp. 13–38). Springer. [Google Scholar]
- Dierckxsens, N. , Mardulyn, P. , & Smits, G. (2017). Novoplasty: De novo assembly of organelle genomes from whole genome DNA. Nucleic Acids Research, 45(4), e18. 10.1093/nar/gkw955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doose, D. , Grand, C. , & Lesire, C. (2017). MAUVE runtime: A component‐based middleware to reconfigure software architectures in real‐time. IEEE: Taichung, Taiwan, 1, 208–211. 10.1109/IRC.2017.47 [DOI] [Google Scholar]
- Doyle, J. J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin, 19(1), 11–15. [Google Scholar]
- Ducousso, A. , Michaud, H. , & Lumaret, R. (1993). Reproduction and gene flow in the genus Quercus . Annals Science Forest, 50(Suppl.), 91–106. 10.1051/forest:19930708 [DOI] [Google Scholar]
- Felix, E. B. , Benjamin, B. , & Pierre, J. (2020). ECS‐based investigation of chloroplast ATP synthase regulation (Preprint). 10.1101/2020.04.28.066100 [DOI] [Google Scholar]
- Goremykin, V. V. , Salamini, F. , Velasco, R. , & Viola, R. (2009). Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Genome Biology and Evolution, 26(1), 99–110. 10.1093/molbev/msn226 [DOI] [PubMed] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41(41), 95–98. 10.1021/bk-1999-0734.ch008 [DOI] [Google Scholar]
- Hao, Y. , Washburn, J. D. , Rosenthal, J. , Nielsen, B. , Lyons, E. , Edger, P. P. , Pires, J. C. , & Conant, G. C. (2018). Patterns of population variation in two paleopolyploid eudicot lineages suggest that dosage‐based selection on homeologs is long‐lived. Genome Biology and Evolution, 10(3), 999–1011. 10.1093/gbe/evy061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp, A. L. , Manos, P. S. , Hahn, M. , Avishai, M. , Bodénès, C. , Cavender‐Bares, J. , Crowl, A. A. , Deng, M. , Denk, T. , Fitz‐Gibbon, S. , Gailing, O. , González‐Elizondo, M. S. , González‐Rodríguez, A. , Grimm, G. W. , Jiang, X.‐L. , Kremer, A. , Lesur, I. , McVay, J. D. , Plomion, C. , … Valencia‐Avalos, S. (2020). Genomic landscape of the global oak phylogeny. New Phytologist, 226(4), 1198–1212. 10.1111/nph.16162 [DOI] [PubMed] [Google Scholar]
- Hofmann, C. C. , Mohamed, O. , & Egger, H. (2011). A new terrestrial palynoflora from the Palaeocene/Eocene boundary in the northwestern Tethyan realm (St. Pankraz, Austria). Review of Palaeobotany & Palynology, 166(3–4), 295–310. 10.1016/j.revpalbo.2011.06.003 [DOI] [Google Scholar]
- Huang, C. , Zhang, Y. , & Bartholomew, B. (1999). Fagaceae Dumortier. In Wu Z. Y., & Raven P. H. (Eds.), Flora of China (vol. 4, pp. 314–400). [Google Scholar]
- Huelsenbeck, J. P. , & Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17(8), 754–755. 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Jansen, R. K. , Christopher, S. , Seung‐Bum, L. , Hansen, A. K. , & Henry, D. (2011). Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): Evidence for at least two independent transfers of rpl22 to the nucleus. Genome Biology and Evolution, 28(1), 835–847. 10.1093/molbev/msq261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, X. L. , Hipp, A. L. , Deng, M. , Su, T. , Zho, Z. K. , & Yan, M. X. (2019). East Asianorigins of European holly oaks via the Tibet‐Himalayas. Journal of Biogeography, 46, 2203–2214. [Google Scholar]
- Jose, C. C. , Roberto, A. , Victoria, I. E. , Javier, T. , Manuel, T. , & Joaquin, D. A. (2015). Phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus . Genome Biology and Evolution, 32(8), 2015–2035. 10.1093/molbev/msv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazutaka, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneth, H. W. , Li, W. H. , & Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences of the United States of America, 84(24), 9054–9058. 10.1073/pnas.84.24.9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku, C. , Chung, W. C. , Chen, L. L. , & Kuo, C. H. (2013). The complete plastid genome sequence of Madagascar Periwinkle Catharanthus roseus (L.) G. Don: Plastid genome evolution, molecular marker identification, and phylogenetic implications in Asterids. PLoS One, 8(6), e68518. 10.1371/journal.pone.0068518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine, I. , & Dujon, B. (2010). Origin and fate of pseudogenes in Hemiascomycetes: A comparative analysis. BMC Genomics, 11, 260. 10.1186/1471-2164-11-260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Li, Y. , & Fang, Y. M. (2018). Prediction of potential suitable distribution areas of Quercus fabri in China based on an optimized maxent model. Scientia Silvae Sinicae, 54(8), 153–164. 10.11707/j.1001-7488.20180817 [DOI] [Google Scholar]
- Li, X. , Li, Y. F. , Zang, M. Y. , Li, M. Z. , & Fang, Y. M. (2018). Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima . International Journal of Molecular Sciences, 19(8), 2443. 10.3390/ijms19082443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Huo, N. X. , Dong, L. L. , Wang, Y. , Zhang, S. X. , Young, H. A. , Feng, X. X. , & Gu, Y. Q. (2013). Complete chloroplast genome sequences of Mongolia Medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS One, 8(2), e57533. 10.1371/journal.pone.0057533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse, M. , Drechsel, O. , & Bock, R. (2007). Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high‐quality custom graphical maps of plastid and mitochondrial genomes. Current Genetics, 52(5–6), 267–274. 10.1007/s00294-007-0161-y [DOI] [PubMed] [Google Scholar]
- Matthew, K. , Richard, M. , Amy, W. , Steven, S. H. , Matthew, C. , Shane, S. , Simon, B. , Alex, C. , Sidney, M. , Chris, D. , Tobias, T. , Bruce, A. , Peter, M. , & Alexei, D. (2012). Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics, 28(12), 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor, C. , Brudno, M. , Schwartz, J. R. , Poliakov, A. , Rubin, E. M. , Frazer, K. A. , Pachter, L. S. , & Dubchak, I. (2000). VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics, 16(11), 1046–1047. 10.1093/bioinformatics/16.11.1046 [DOI] [PubMed] [Google Scholar]
- Millen, R. S. , Olmstead, R. G. , Adams, K. L. , Palmer, J. D. , Lao, N. T. , Heggie, L. , Kavanagh, T. A. , Hibberd, J. M. , Gray, J. C. , Morden, C. W. , Calie, P. J. , Jermiin, L. S. , & Wolfe, K. H. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. The Plant Cell, 13(3), 645–658. 10.2307/3871412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minias, A. , Minias, P. , Czubat, B. , & Dziadek, J. (2018). Purifying selective pressure suggests the functionality of a vitamin B12 biosynthesis pathway in a global population of Mycobacterium tuberculosis . Genome Biology and Evolution, 10(9), 2336–2337. 10.1093/gbe/evy153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, M. J. , Soltis, P. S. , Bell, C. D. , Gordon, J. B. , & Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of Eudicots. Proceedings of the National Academy of Sciences of the United States of America, 107(10), 4623–4628. 10.1073/pnas.0907801107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, E. V. , John, W. , & Clark, J. S. (2012). Genetic evidence for hybridization in red oaks (Quercus Sect. Lobatae, Fagaceae). American Journal of Botany, 99(1), 92–100. [DOI] [PubMed] [Google Scholar]
- Nagalingum, N. S. , Marshall, C. R. , Quental, T. B. , Rai, H. S. , Little, D. P. , & Mathews, S. (2011). Recent synchronous radiation of a living fossil. Science, 334(6057), 796–799. 10.1126/science.1209926 [DOI] [PubMed] [Google Scholar]
- Nie, X. J. , Lv, S. Z. , Zhang, Y. X. , Du, X. H. , Wang, L. , Biradar, S. S. , Tan, X. F. , Wan, F. H. , & Song, W. N. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS One, 7(5), e36869. 10.1371/journal.pone.0036869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield, S. , & Eastwood, A. (2007). The red list of oaks. Fauna and Flora International. [Google Scholar]
- Ometto, L. , Li, M. G. , Bresadola, L. , & Varotto, C. (2012). Rates of evolution in stress‐related genes are associated with habitat preference in two Cardamine lineages. BMC Evolutionary Biology, 12, 7. 10.1186/1471-2148-12-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden, J. F. (2000). Analysis of codon usage. University of Nottingham, 90(1), 73–74. 10.1006/expr.1997.4185 [DOI] [Google Scholar]
- Petersen, G. , Darby, H. , Lam, V. K. Y. , Pedersen, H. Æ. , Merckx, V. S. F. T. , Zervas, A. , Seberg, O. , & Graham, S. W. (2019). Mycoheterotrophic Epirixanthes (Polygalaceae) has a typical angiosperm mitogenome but unorthodox plastid genomes. Annals of Botany, 124(5), 791–807. 10.1093/aob/mcz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu, C. X. , Zhou, Z. K. , & Luo, Y. (2002). A cladistic analysis of Quercus (Fagaceae) in China based on leaf epidermis and architecture. Acta Botanica Yunnanica, 24(6), 689–698. [Google Scholar]
- Roy, S. W. , & Penny, D. (2007). Patterns of intron loss and gain in plants: Intron loss‐dominated evolution and genome‐wide comparison of O‐sativa and A‐thaliana . Molecular Biology and Evolution, 24(1), 171–181. 10.1093/molbev/msl159 [DOI] [PubMed] [Google Scholar]
- Schattner, P. , Brooks, A. N. , & Lowe, T. M. (2005). The tRNAscan‐SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research, 33(2), W686. 10.1093/nar/gki366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, C. , Liu, Y. , Huang, H. , Xia, E. H. , Zhang, H. B. , & Gao, L. Z. (2013). Contradiction between plastid gene transcription and function due to complex posttranscriptional splicing: An exemplary study of ycf15 function and evolution in angiosperms. PLoS One, 8(3), e59620. 10.1371/journal.pone.0059620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D. R. , Jackson, C. J. , & Prieto, R. A. (2014). Nucleotide substitution analyses of the glaucophyte Cyanophora suggest an ancestrally lower mutation rate in plastid vs mitochondrial DNA for the Archaeplastida. Molecular Phylogenetics & Evolution, 79, 380–384. 10.1016/j.ympev.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Susann, W. , Schäferhoff, B. , Depamphilis, C. W. , & Muller, K. F. (2013). Disproportional plastome‐wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Molecular Biology and Evolution, 31(3), 529–545. 10.1093/molbev/mst261 [DOI] [PubMed] [Google Scholar]
- Tang, C. Q. (2015). Plant distribution, diversity and ecology. In The subtropical vegetation of southwestern China. Plant and vegetation (vol. 11, pp. 113–125). [Google Scholar]
- Turmel, M. , Otis, C. , & Lemieux, C. (2017). Divergent copies of the large inverted repeat in the chloroplast genomes of ulvophycean green algae. Scientific Reports, 7, 994. 10.1038/s41598-017-01144-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Jiang, B. , Zhou, Y. , Su, Y. J. , & Wang, T. (2015). Higher substitution rates and lower dN/dS for the plastid genes in Gnetales than other gymnosperms. Biochemical Systematics and Ecology, 59, 278–287. 10.1016/j.bse.2015.02.009 [DOI] [Google Scholar]
- Wang, L. , Si, W. N. , Yao, Y. F. , Tian, D. C. , Araki, H. , & Yang, S. H. (2012). Genome‐Wide survey of pseudogenes in 80 Fully re‐sequenced arabidopsis thaliana Accessions. PLoS One, 7(12), e51769. 10.1371/journal.pone.0051769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. M. , Shi, X. , Chen, S. , Ma, C. , & Xu, S. B. (2018). Evolutionary origin, gradual accumulation and functional divergence of heat shock factor gene family with plant evolution. Frontiers in Plant Science, 9, 71. 10.3389/fpls.2018.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangda, P. , & Ohsawa, M. (2006). Structure and regeneration dynamics of dominant tree species along altitudinal gradient in a dry valley slopes of the Bhutan Himalaya. Forest Ecology & Management, 230, 136–150. 10.1016/j.foreco.2006.04.027 [DOI] [Google Scholar]
- Wei, G. M. , Li, X. , & Fang, Y. M. (2021). Sympatric genome size variation and hybridization of four oak species as determined by flow cytometry genome size variation and hybridization. Ecology and Evolution, 00, 1–12. 10.1002/ece3.7163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. H. , Boecklen, W. J. , & Howard, D. J. (2010). Reproductive processes in two oak (Quercus) contact zones with different levels of hybridization. Heredity, 87, 680–690. [DOI] [PubMed] [Google Scholar]
- Xie, J. B. , Qian, K. C. , Si, J. N. , Xiao, L. , Ci, D. , & Zhang, D. Q. (2018). Conserved noncoding sequences conserve biological networks and influence genome evolution. Heredity, 120(5), 437–451. 10.1038/s41437-018-0055-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, Y. , & Lee, C. (2005). Evidence of functional selection pressure for alternative splicing events that accelerate evolution of protein subsequences. Proceedings of the National Academy of Sciences of the United States of America, 102(38), 13526–13531. 10.1073/pnas.0501213102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , & Yang, Z. H. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution, 24(8), 1586–1591. 10.1093/molbev/msm088 [DOI] [PubMed] [Google Scholar]
- Zhou, Z. K. (1992). Origin, phylogeny and dispersal of Quercus for China. Plant Diversity, 14(3), 227–236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
All the data are available at Genbank. Genbank accessions MK693136, MZ196210, MZ196209, MZ196211.