

Abstract
BACKGROUND:
Mitochondrial oxidative stress plays a prominent role in the development of burn-induced cardiac dysfunction. AMP-activated kinase (AMPK), an energy sensor, has a central role in the pathogenesis of heart failure. However, its role in cardiac dysfunction after burn injury is unclear. Our hypothesis is that burn injury acts through the AMPK-SIRT1-PGC1α-NFE2L2-ARE signaling pathway, leading to cardiac mitochondrial impairment, resulting in cardiac dysfunction.
STUDY DESIGN:
Male Sprague Dawley rats underwent sham procedure or 60% total body surface area full-thickness burn. Echocardiograms were performed 24-hour post burn. Heart tissue was harvested at 24 hours post burn for biochemistry/molecular biological analysis. AC16 cardiomyocytes were treated with either sham or burned rat serum (±AMPK inhibitor/AMPK activator/PGC1α activator) for evaluation of cardiomyocyte mitochondrial function by using seahorse in vitro.
RESULTS:
Burn injury induced cardiac dysfunction, measured by echocardiogram. Burn injury suppressed cardiac AMPK, SIRT1 and PGC1 expression, leading to acetylation of cardiomyocyte proteins. In addition, burn injury caused NFE2L2 and NFE2L2 regulated antioxidants (HO-1, NQO1, GCLC, MnSOD and Gpx) to decrease, resulting in cardiac oxidative stress. In vitro, AMPK1 activator and PGC1α agonist treatment improved Ac16 cell mitochondrial dysfunction, while AMPK1 inhibitor treatment worsened Ac16 cellular damage.
CONCLUSIONS:
Burn induced cardiac dysfunction and cardiac mitochondrial damage occur via the AMPK-SIRT1-PGC1α-NFE2L2-ARE signaling pathway. AMPK and PGC1α agonists may be promising therapeutic agents to reverse cardiac dysfunction after burn injury.
Graphical Abstract

Precis
Cardiac dysfunction after severe burn is partially mediated through cardiac mitochondrial damage. The AMPK-SIRT1-PGC1α-NFE2L2-ARE pathway plays a key role in burn-induced cardiac dysfunction. Modulation of this pathway could be useful in the treatment of cardiac dysfunction after burns.
INTRODUCTION
Burns are a significant health problem resulting in over 500,000 people seeking medical treatment, 40,000 hospitalizations, and 4,000 deaths per year in the United States. The annual cost of treating burn patients is estimated to be in excess of $18 billion, including the indirect costs of disability and rehabilitation(1, 2). Major burn injury produces substantial hemodynamic and cardiodynamic derangements which contribute to the development of sepsis, multiple organ failure, and death. Compromised cardiac function results in organ hypoperfusion, impaired peripheral microcirculation, burn zone extension, and increased susceptibility to bacterial infection at the wound site(3). Functionally, myocardial dysfunction following burn injury is characterized by slowed isovolumic relaxation, impaired contractility, and decreased diastolic compliance of the left ventricle (4, 5). Following burn injury, there is a substantial loss in circulating plasma fluid volume due to increased capillary permeability accompanied by decreased cardiac output and compensatory increments in heart rate and peripheral vascular resistance(6). Current investigations indicate that the molecular mechanisms underlying burn-induced cardiac dysfunction are associated with persistent β-Adrenergic receptor stimulation, leading to increased nitric oxide levels(7). However, little is known of the role of the AMPK-SIRT1-PGC1α and NFE2L2-ARE pathways.
Myocardial cells produce energy in mitochondria through oxidative phosphorylation. The homeostasis of mitochondria and energy metabolism are strictly regulated by several key transcriptional factors. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is considered as a main transcription regulator that controls energy metabolism by regulating several processes including mitochondrial biogenesis, respiration and oxidative phosphorylation(8). Recent studies demonstrated that AMP-activated protein kinase (AMPK) and Sirtuin 1 (SIRT1) modulate PGC-1α to regulate energy metabolism. AMPK is one of the major metabolic energy sensors(9). In energy deficient situations, AMPK is activated by enhancement of the cellular AMP/ATP ratio and the expression of genes linked to glucose transport, glycolysis and mitochondrial respiration(10). Additionally, AMPK increases NAD+-dependent deacetylase SIRT1 activity via increasing cellular NAD+ levels(11). Likewise, activation of AMPK results in increased expression of PGC-1α (12). SIRT1 also plays a role by regulating energy metabolism, development and cellular survival. Activation of SIRT1 may increase mitochondrial function, thereby amplifying energy production(13). Moreover, SIRT1 interacts with PGC-1α to increase PGC-1α levels and mitochondrial biogenesis(14).
The nuclear factor erythroid 2-related factor 2 (NFE2L2)–antioxidant response element (ARE) signaling pathway performs a critical role in maintaining cellular redox balance and metabolism. It can induce an adaptive response against oxidative stress, preventing uncontrolled inflammation. Under higher rates of oxidative metabolism, an increase of intracellular reactive oxygen species can promote the dissociation of NFE2L2-ARE(15) after which NFE2L2 transfers to the nucleus and binds to ARE on target genes, inducing a cascade of events designed to prevent oxidative stress (16, 17). NFE2L2 also regulates multiple antioxidant enzymes (15). While no reports have been made regarding NFE2L2 in myocytes after burn, NFE2L2 deficient transgenic mice were found to be more susceptible to burn-induced intestinal injury(18). Both SIRT1 and PGC1α activate NFE2L2, linking the NFE2L2-ARE and AMPK-SIRT1-PGC1α pathways (19, 20)
Available data indicates that burn-induced myocardial dysfunction may be related to mitochondrial oxidative stress. The AMPK-SIRT1-PGC1α-NFE2L2-ARE pathway provides an important mechanism in the cellular defense against mitochondrial oxidative stress and mitochondrial biogenesis. Therefore, we hypothesized that the AMPK-SIRT1-PGC1α-NFE2L2-ARE pathway may be partially responsible for burn-induced heart dysfunction.
METHODS
Ethics statement
All animal research procedures adhered to the National Institutes of Health guidelines for experimental animal use and were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch (UTMB), Galveston, TX (Protocol number: 1509059).
Rats and cell culture
Male Sprague-Dawley rats (wild-type) were purchased from Harlan Laboratories (Indianapolis, IN). Animals were allowed to acclimate for 1 week before experimentation and received food and water ad libitum throughout the study. Animals were kept on a 12:12 light-dark cycle. Temperature was maintained at ~22°C in all animal housing and procedure rooms. A well-established model for the induction of a 60% total body surface area (TBSA) full-thickness burn was used(21, 22). Briefly, rats (300–350 g) were given analgesia (buprenorphine, 0.05mg/kg, s.c.) and anesthetized with general anesthesia (isoflurane, 3–5%). Rats were placed within a protective mold that exposed ~60% of the TBSA and submerged in 95°C to 100°C water to induce a scald burn. The dorsum was immersed to 10 seconds and the abdomen for 2 seconds, resulting in a 30% TBSA injury on both the dorsum and the abdomen (60% TBSA burn in total). Room temperature Lactated Ringer (LR) solution (40 mL/kg, i.p. ± ZLN005, 15mg/kg body weight) was administered immediately after the burn for resuscitation. Rats received oxygen during the recovery from anesthesia. At pre-determined time points, rats were humanely euthanized, and blood and tissue were collected. Sera/plasma and tissue samples were stored at 4°C and −80°C, respectively. For cell culture, Ac16 cardiomyocytes (Millipore/Sigma, Cat# SCC109, Burlington, MA) were cultured and maintained in Dulbecco’s modified Eagle’s medium/F-12 medium with 12.5% fetal bovine serum according to manufacturer’s instruction. Cardiomyocytes seeded in T25 flask (5 × 105/flask), seahorse 24-well plates (1 ×105/well) were treated with burned rat serum-replaced FBS medium (12.5% serum/plasma) and then incubated for 24 hours.
Echocardiography
Rats were sedated with inhaled anesthesia (1.5% isoflurane/100% O2), placed supine on an electrical heating pad at 37o C and heart rate and respiratory physiology were continuously monitored by electrocardiography. After shaving the chest, warmed ultrasound gel was applied and transthoracic echocardiography was performed using the Vevo® 2100 ultrasound system (VisualSonics, Toronto, Canada) equipped with a high-frequency linear array transducer (MS250 13–24 MHz)(23). All measurements were obtained in triplicate and data was analyzed using the Vevo® 2100 standard measurement package.
Gene Expression Analysis
Analysis of mitochondrial respiration was assessed using the XF24 Extracellular Flux Analyzer (Agilent, Santa Clara, CA). Briefly, human cardiomyocyte (Ac16) cells were seeded on cell culture microplates (105 cells/well) for 24 h prior treatment/analysis. For analysis of mitochondrial respiration, cells were washed twice with low glucose DMEM medium pH 7.4 supplemented with L-Glutamine (2 mM, GIBCO) and sodium pyruvate (0.33 mM, SIGMA). After 1 h incubation at 37 °C in CO2 free incubator, the oxygen consumption rate (OCR) after oligomycin (1.5 µg/mL) was used to assess ATP production rate and the OCR after carbonyl cyanide-4-trifluoromethoxy phenylhydrazone (FCCP, 0.5 µ M) to assess maximal mitocondrial respiratory capacity. Antimycin A (2 µg/mL) and rotenone (2 µ M) were used to inhibit the flux of electrons through complex III and I, to detect residual non-mitochondrial OCR, which is considered to be due to cytosolic oxidase enzymes. To normalize the OCR, cells were resuspended by using 1 x RIPA buffer (Thermo Scientific, Waltham, MA, Cat# 89900) containing protease inhibitor cocktails (Sigma, Cat# S8820), centrifuged at 14,000 x g for 15 min at 4 °C, carefully removed the supernatant and determined the protein concentration with BCA method (Thermo Scientific, Waltham, MA, Cat# BCA1). The unit after normalization for OCR was nmol oxygen/min/mg protein. The oligonucleotides used in this study for qPCR are listed in Table 1.
Table 1:
Oligonucleotides Used in This Study
| Gene | 5’-Forward-3’ | 5’-Reverse-3’ | Amplicon size (bp) | Accession No. |
|---|---|---|---|---|
| AMPK | GCTGACTTCGGACTCTCTAATATG | CATACAGCCTTCCTGAGATGAC | 106 | NM_023991.1 |
| GAPDH | ACTCCCATTCTTCCACCTTTG | CCCTGTTGCTGTAGCCATATT | 105 | NM_017008.4 |
| GCLC | CCTCCTCCTCCAAACTCAGATA | TGGTCAGCAGTACCACAAATAC | 112 | NM_012815.2 |
| HO1 | GATGGCCTCCTTGTACCATATC | AGCTCCTCAGGGAAGTAGAG | 99 | NM_012580.2 |
| MnSOD | AGCGTGACTTTGGGTCTTT | AGCGACCTTGCTCCTTATTG | 111 | NM_017051.2 |
| ND1 | CGCCTGACCAATAGCCATAA | CGACGTTAAAGCCTGAGACTAA | 110 | KF011917.1 |
| NFE2L2 | CACTCTGTGGAGTCTTCCATTT | GAATGTGTTGGCTGTGCTTTAG | 125 | BC061724.1 |
| NQO1 | TGAGAAGAGCCCTGATTGTATTG | CACCTCCCATCCTTTCTTCTTC | 104 | NM_017000.3 |
| PGC1α | GACACGAGGAAAGGAAGACTAAA | GTCTTGGAGCTCCTGTGATATG | 119 | AY237127.1 |
Data analysis
All experiments were conducted with triplicate observations per sample (n =6–8 rats/group) and data were expressed as mean ± standard error mean (SEM). All data were analyzed using GraphPad Prism7 software. Data (linear range or log10 transformed) were analyzed by the Kolmogorov-Smirnov test under Column Statistics to determine if the data are normally distributed. Normally distributed data were analyzed by Student’s t test (comparison of 2 groups) and one-way ANOVA with Tukey’s test (comparison of multiple groups). If data were not normally distributed, then Mann-Whitney (comparison of 2 groups) and Kruskal-Wallis (K-W, comparison of multiple groups) tests were employed. Significance is presented by *burned rats vs. sham rats or &burn/untreated vs. burn/treated) (*,&p<0.05, **,&&p<0.01, ***,&&&p<0.001).
RESULTS
AMPK-SIRT1-PGC1α pathway was potentially involved in pathogenesis of burn-induced cardiac dysfunction
mRNA levels of AMPK, SIRT1 and PGC-1α were tested with real-time qPCR. AMPK mRNA was decreased by 1.45-fold (***p<0.001T-test), SIRT1 mRNA was decreased 1.39-fold (***p<0.001T-test) and PGC-1α was decreased by 3.84-fold (***p<0.001T-test) in burned rats (Fig. 1). SIRT1 Western blot demonstrated a 3.79-fold decrease in myocardium of burned rats (Fig. 2A, ***p<0.001T-test). There was also 46% increase in acetylated protein in myocardium of burned rats (Fig.2B, ***p<0.001T-test). This suggests that the AMPK-SIRT1-PGC1α pathway is potentially involved in burn-induced cardiac dysfunction.
Figure 1.
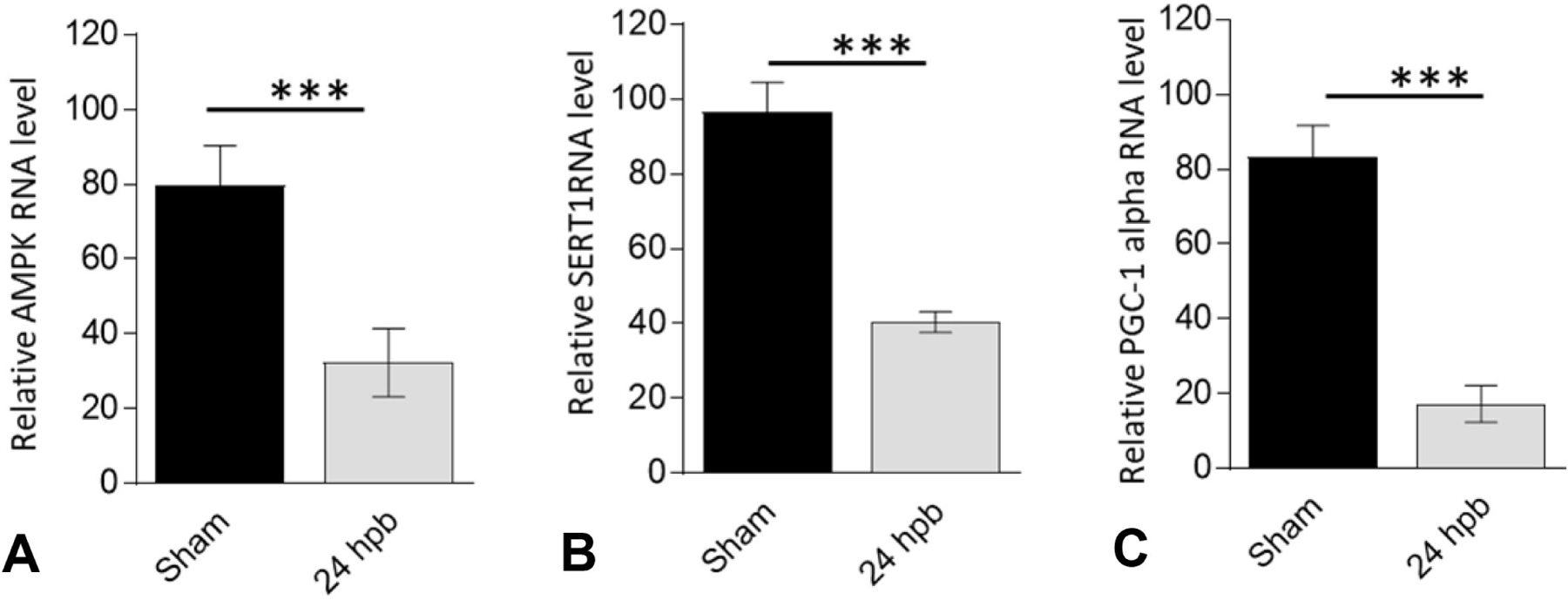
Effect of burn injury on cardiac AMPK-SIRT1-PGC1α pathway in burned rats. Myocardial levels of (A) AMP-activated protein kinase, (B) Sirtuin 1, and (C) Peroxisome proliferator-activated receptor-γ coactivator-1α by quantitative reverse transcription polymerase chain reaction. Data are plotted as mean value ± SEM. Significance is shown as * (burned rats vs matched control) and presented as *p<0.05, **p<0.01, ***p<0.001 (n = ≥6 per group).
Figure 2.

Burn induced cardiac protein acetylation. Shown are (A) Sirtuin 1 protein level by Western Blot, and (B) acetylated proteins by Western Blot. Results were normalized to rat β-actin and represent fold change of density in burned rats as compared to that noted in matched normal controls. Data are plotted as mean value ± SEM. Significance is shown as * (burned vs. matched control) and presented as *p<0.05, **p<0.01, ***p<0.001 (n = ≥6 per group).
Burn interrupted cardiac mitochondrial-biogenesis via the NEF2L2-ARE pathway
ND1, a subunit of NADH dehydrogenase and a critical component of Complex I, demonstrated a 2.23-fold decrease in mRNA levels (***p<0.001T-test) and 7.7-fold decrease in protein levels (***p<0.001T-test) in burned rats (Fig.3Aa and 3Ab). This indicates that burn-induced cardiac oxidative stress is mediated by cardiac mitochondrial damage. We also measured the NFE2L2-ARE pathway-associated gene expressions. A 1.25-fold decline of NFE2L2 mRNA level in burned rats (Fig.3.Ba, vs. sham control) resulted in the decrease of all ARE gene expressions including heme oxygenase 1 (HO-1) (61% decrease, Fig.3Bb, ***p<0.001T-test), NADH quinone oxidoreductase 1 (NQO1) (44% decrease, Fig.3Bc, ***p<0.001T-test), glutamatecysteine ligase catalytic subunit (GCLC) (3.1-fold decrease, Fig.3Bd, ***p<0.001T-test), superoxide dismutase (MnSOD) (4.71-fold decrease, Fig.3Be, ***p<0.001T-test), and glutathione peroxidase (Gpx-1) protein level (4.71-fold decrease, Fig.3C, ***p<0.001T-test) vs. sham control.
Figure 3.

Burn induced the interruption of cardiac mitochondrial biogenesis via NEF2L2-ARE pathway. Shown are the (A) myocardial mRNA level of mtDNA-encoded ND1 gene and ND1 protein level (***p<0.001T-test); (B) myocardial mRNA level of NFE2L2 as well as its down-regulated genes (***p<0.001T-test ); and (C) myocardial levels of Gpx1 protein (***p<0.001T-test). All data are plotted as mean value ± SEM. Significance is shown as * (burned vs matched control) and presented as *<0.05, **p<0.01, ***p<0.001 (n = ≥6 per group).
Mitochondrial biogenesis activator recovered burn-induced heart dysfunction
We used a PGC-1α activator to investigate whether it would restore burn-induced heart dysfunction. ZLN005 is a novel small-molecule PGC-1α transcriptional regulator which has been shown to have beneficial effects on heart dysfunction in other disease processes (24). Transthoracic echocardiography demonstrated left ventricular systolic function was significantly decreased after burn. Ejection fraction (EF) was decreased by 25% (***p<0.001ANOVA, K-W), fractional shortening (FS) was decreased by 26% (***p<0.001ANOVA, K-W), systolic interventricular septum thickness (IVS;s) was decreased by 20% (***p<0.001ANOVA, K-W) and left ventricular systolic posterior wall thickness (LVPW;s) was decreased by 27% (***p<0.001ANOVA, K-W). Left ventricular systolic volume (V;s) was increased by 78% (***p<0.001ANOVA, K-W) and systolic diameter (D;s) was increased by 32% (***p<0.001ANOVA, K-W) in burn/untreated rats (Fig. 4, A-F). ZLN005 treatment during post-burn resuscitation restored left ventricular systolic function (EF, FS vol, s, Dia;s, IVS;s and LVPW;s) when compared to that noted in burn/untreated (Fig. 4, all &&&p<0.001ANOVA).
Figure 4.
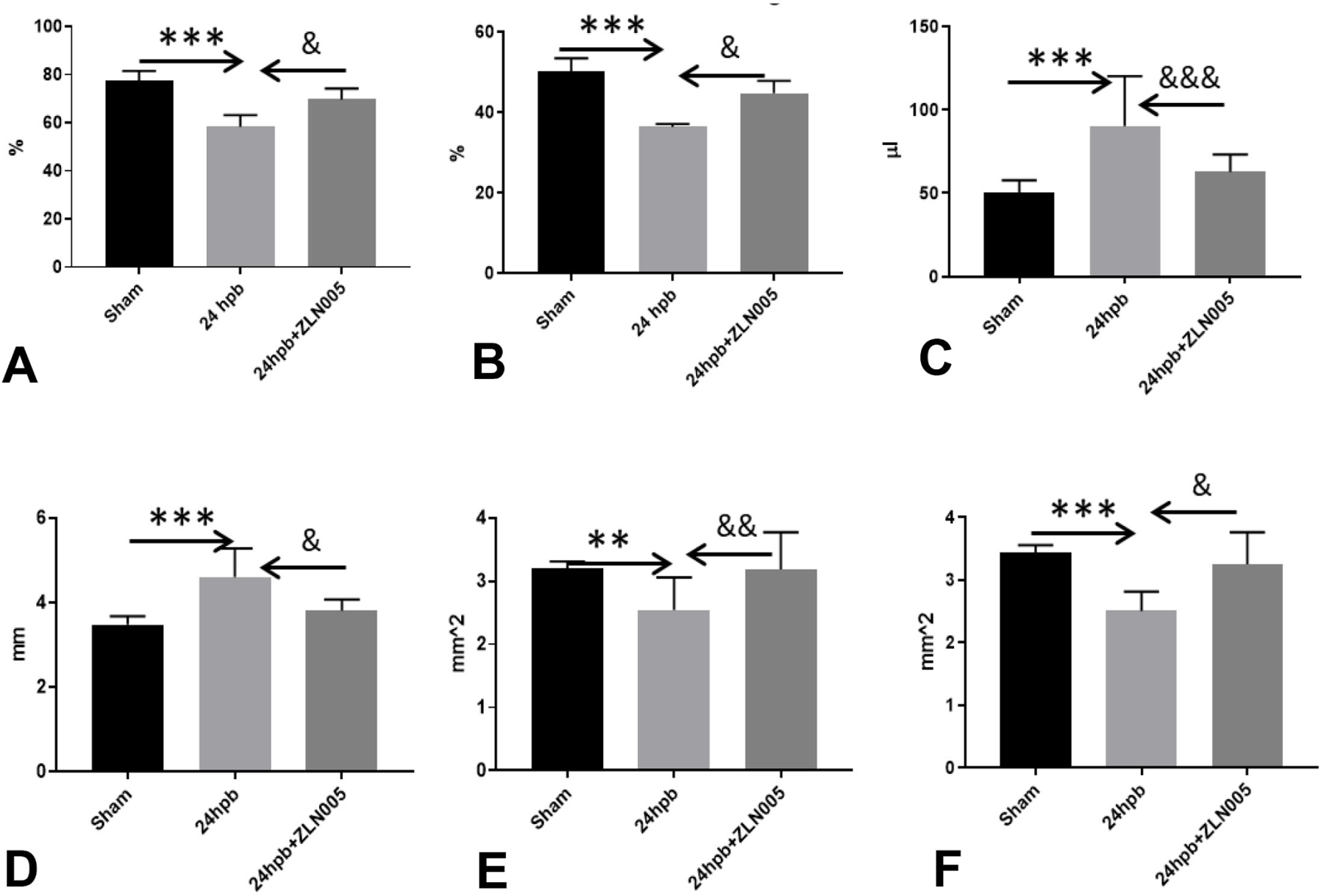
Mitochondrial biogenesis activator recovered burn-induced heart dysfunction. Shown are (A) ejection fraction, (B) fraction shortening, (C) systolic volume, (D) systolic diameter, (E) systolic intraventricular septum width, and (F) systolic left ventricular wall thickness. Data are presented as mean value ± SD shown is * (burned rats vs. matched control) or & (24 burned/untreated vs. 24 burned/treated), and presented as *, &p<0.05, **, &&p<0.01, ***, &&& p<0.001 (n = ≥6 per group).
Cardiomyocyte mitochondrial responses to burned rat serum treatments
Human cardiomyocytes were treated by using burned rat serum to test our hypothesis that released factors may be responsible for cardiomyocyte mitochondrial damage. We utilized the Seahorse Analyzer to measure mitochondrial stress after exposure to serum (Fig.5). The oxygen consumption in the burned rat serum group was significantly decreased vs. sham serum group as well as basal respiration (50.4% decline, ***p<0.001T-test), proton leak (58.8% decline, ***p<0.001T-test), ATP-linked respiration (45.9% decline, ***p<0.001T-test) and maximal respiration (58.2% decline, ***p<0.001T-test), non-mitochondrial respiration (60% decline, ***p<0.001T-test) and spare capacity (65.1% decline, ***p<0.001T-test) (Fig.4Aa and Ab, *p<0.001T-test ). Use of an AMPK inhibitor, dorsomorphin, resulted in worse mitochondrial function than the untreated serum (0.23-fold decrease of basal respiration; 0.12-fold decrease of proton leak; 0.29-fold decrease of ATP production; 0.36-fold decrease of maximal respiration; 0.12-fold increase of non-mitochondrial respiration; 0.52-fold decrease of spare capacity) (Fig.4Ba and Ab, &&&p<0.001T-test). Use of an AMPK activator, A768662, completely recovered the cardiac mitochondrial dysfunction to levels seen in the untreated serum group (5.4-fold increase of basal respiration; 6.4-fold increase of proton leak; 4.9-fold increase of ATP production; 5.1-fold increase of maximal respiration; 8.7-fold increase of non-mitochondrial respiration; 4.7-fold increase of spare capacity) (Fig.4Ca and Ab, &&&p<0.001T-test). Use of the PGC1α activator, ZLN005, also resulted in recovery of cardiac mitochondrial function when compared to the untreated group (5.3-fold increase of basal respiration; 7-fold increase of proton leak; 4.4-fold increase of ATP production; 4.87-fold increase of maximal respiration; 5.7-fold increase of non-mitochondrial respiration; 4.4-fold increase of spare capacity) (Fig.4Da and Ab, &&&p<0.001T-test). These results demonstrate that burn-induced circulating factors interfere with cardiomyocyte dysfunction via the AMPK-SIRT1-PGC1α pathway leading to impaired mitochondrial biogenesis.
Figure 5.

Burn-induced cardiac dysfunction via mitochondrial response in vitro. (A) Burn/untreated; (B) burn/APMK inhibitor; (C) burn/APMK activator; (D) burn/PGC1α. Human cardiomyocyte (Ac16 cells) were exposed to control media or burned rat serum (± A769662, ZLN005, Domorsorphin) for 24 hours. Cell oxygen consumptions were measured by seahorse and presented with typical trace images (A1, B1, C1 and, D1) and column graph represents (5A2, 5B2, 5C2, and 5D2). In all figures, data are plotted as mean value ± SEM. Significance is shown as * (burned vs matched control) or $ (burned/untreated vs burned/treated), and presented as *,& p<0.05, **,&& p<0.01, ***,&&& p<0.001 (n = ≥6 per group).
DISCUSSION
In this study, we tested the hypothesis that AMPK-SIRT1-PGC1α-NFE2L2-ARE pathway plays a critical role in burn-induced cardiac dysfunction. Our hypothesis was based on previous work which demonstrated that AMPK and SIRT1 directly affected PGC-1α activity through phosphorylation and deacetylation, SIRT1 and PGC1α activate NFE2L2, and that this pathway might act as an orchestrated network to maintain cardiac homeostasis after burns(25). Our data demonstrated significant protective effects of AMPK and PGC1α activators against burn-induced cardiac dysfunction, especially through restoration of burn-induced cardiac mitochondrial dysfunction. To the best of our knowledge, this is the first study demonstrating that the AMPK-SIRT1-PGC-1α-NFE2L2-ARE pathway plays a very important role in burn-induced heart dysfunction and that both the AMPK activator, A769662, and PGC1α activator, ZLN005, arrest the loss in ATP capacity and mitochondrial function in cardiomyocytes after burns.
The adenosine monophosphate‐activated protein kinase (AMPK) is a αβγ heterotrimeric serine‐threonine kinase. In the myocardium, starvation, ischemia and oxidative stress lead to upregulation of the activity of AMPK, which plays a critical role in alleviating myocardial injury and promoting cardiomyocyte survival (26). The results of our study showed that burns decreased AMPK mRNA level and that administration of an AMPK inhibitor resulted in exaberation of burn-induced cardiac mitochondrial dysfunction. In addition, treatment with an AMPK activator completely recovered burn-induced cardiac mitochondrial dysfunction.
Sirtuins (SIRT), nicotinamide adenine dinucleotide (NAD+)-dependent deacetylating enzymes firstly identified in yeast, belong to the class III histone deacetylases(27). Sirtuins plays a critical role in a variety of cellular processes, including gene silencing, DNA damage repair, and longevity (27). Mammals possess seven sirtuins with different terminal extensions(28). SIRT1 is localized in the nucleus and translocates to the cytosol under specific conditions (29). SIRT1 activity has been shown to be cardioprotective in animal models of heart failure by regulating oxidative stress and antioxidant enzymes through FOXO (Forkhead box O)-dependent mechanisms (30). SIRT1 deficiency increases acetylation of heart proteins and promotes heart dysfunction. In contrast, deacetylating proteins by activating SIRT1 effectively restored heart function. iNOS enhances the burn-induced inflammatory response and apoptotic change in mouse skeletal muscle along with S-nitrosylation of Sirt1, suggesting that Sirt1 S-nitrosylation may play a role in iNOS-mediated enhanced inflammatory response and apoptotic change after burn injury (31). However, there is no information regarding the role of SIRT1 in burn-induced cardiac dysfunction. We demonstrated that burn causes declines of both SIRT1 mRNA and protein levels. Western blot demonstrated a significant increase in acetylated proteins in the myocardium of burned rats, confirming that a decrease in SIRT1 correlates with a decrease in protein deacetylation.
The transcriptional activator PGC-1α is master regulator of oxidative phosphorylation and fatty acid oxidation gene expression. PGC-1α is a central regulator of energy metabolism and mitochondrial function in heart as well as a therapeutic target for heart failure(32). Heart failure is associated with repressed PGC-1α gene expression. Maintaining expression of PGC-1α preserves contractile function in response to a pathological increase in workload(33). PGC-1α regulates the expression of genes via coactivation of the transcription factors NFE2L2, TFAm (mitochondrial transcription factor A), and ERR (estrogen-related receptor)-α (34). While there are no other studies examining the role of PGC-1α in burn-induced cardiac dysfunction, other studies have reported inhibition of PGC-1α human skeletal muscle(35) and liver(22) after burn. In this study, we found that burns decreased PGC1α mRNA levels in the heart and that administration of a PGC1α activator restored burn-induced heart dysfunction and cardiac mitochondrial dysfunction.
Nuclear factor (erythroid-derived 2)–like 2 (NFE2L2) is a basic leucine zipper transcription factor that principally defends against oxidative stress and also plays a unique role in severe sepsis. The transcription factor NFE2L2 belongs to the cap ‘n’ collar family that contains a conserved basic leucine zipper structure. NFE2L2 can protect cells against environmental and oxidative stress(36) and maintains the cellular redox balance by regulating endogenous antioxidants, phase II detoxification enzymes, and other defensive proteins via antioxidant response elements in the promoters of its target gene(37). Well-characterized NFE2L2-dependent genes include heme oxygenase-1 (HO-1), NAD(P)H dehydrogenasequinine-1 (NQO1), glutamate-cysteine ligase (GCL), and glutathione S-transferase A1(37). In this role, NFE2L2 activity has been shown to be an important disease modifier in many oxidative/inflammatory diseases, such as asthma, sepsis, and pulmonary fibrosis, in which decreased NFE2L2 activity exacerbates disease progression(38). It was demonstrated that mice lacking NFE2L2 were more susceptible to burn-induced intestinal injury, had more systemic inflammation, and a lower survival rate(18). Our study demonstrated that NFE2L2 mRNA level as well as NFE2L2-regulated genes including HO1, NQO1, GCLC, MnSOD, and GpX1 were significantly decreased after burn.
Recent studies showed that AMPK and SIRT1 play major roles through PGC-1α to transcriptionally regulate energy metabolism. AMPK is one of the major metabolic energy sensors(9). AMPK could enhance NAD+-dependent deacetylase SIRT1 activity by increasing cellular NAD+ levels(11). Moreover, activation of AMPK leads to increased expression of PGC-1α(12). Furthermore, SIRT1 has been shown to interact with PGC-1α to increase the expression level of PGC-1α and mitochondrial biogenesis(14, 39). This crucial energy regulation pathway is perturbed under ischemic conditions and may serve as pharmacological targets for the treatment of cardiovascular disease(40). Our study confirmed that the AMPK-SIRT1-PGC1α pathway also has a key critical role in burn-induced heart dysfunction. We are also the first to demonstrate that the NFE2L2-ARE pathway was linked to the AMPK-SIRT1-PGC1α pathway in burn-induced heart dysfunction resulting in burn-induced cardiac mitochondrial dysfunction.
CONCLUSIONS
Our findings provide the first evidence that burn-induced heart dysfunction occurs via the AMPK-SIRT1-PGC1α-NFE2L2-ARE pathway. Use of an AMPK inhibitor resulted in worsening of cardiac mitochondrial function after burn. In contrast, use of an AMPK activator or a PGC1α activator has a powerful cardio-protective effect against cardiac mitochondrial dysfunction and left ventricular dysfunction after burn. We surmise that further development of currently available AMPK-SIRT1-PGC1α targeting drugs could potentially be useful in the treatment of patients with cardiac dysfunction following severe burns.
Abbreviations and Acronyms:
- AMPK
AMP-activated protein kinase
- ARE
antioxidant response element
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazone
- EF
ejection fraction
- GCLC
glutamatecysteine ligase catalytic subunit
- GCLM
glutamyl cystine ligase modulatory subunit
- Gpx
glutathione peroxidase
- NFE2L2
nuclear factor erythroid 2-related factor 2
- NQO1
NADH quinone oxidoreductase 1
- PGC-1α
Peroxisome proliferator-activated receptor-γ coactivator-1α
- SIRT1
Sirtuin 1
- SOD
superoxide dismutase
- TBSA
total body surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Information: Nothing to disclose.
Presented at the Southern Surgical Association 131st Annual Meeting, Hot Springs, VA, December 2019.
REFERENCES
- 1.Colohan SM. Predicting prognosis in thermal burns with associated inhalational injury: a systematic review of prognostic factors in adult burn victims. J Burn Care Res 2010. Jul-Aug;31(4):529–39. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence BA, Zaloshnja E, Miller TR, Jones PR. Estimates of the Incidence and Costs Of Fire-Related Injuries Calveston, MD: the U.S. Consumer Product Safety Commission; 2009. [Google Scholar]
- 3.Hoesel LM, Niederbichler AD, Schaefer J, et al. C5a-blockade improves burn-induced cardiac dysfunction. J Immunol 2007. June 15;178(12):7902–10. [DOI] [PubMed] [Google Scholar]
- 4.Adams HR, Baxter CR, Izenberg SD. Decreased contractility and compliance of the left ventricle as complications of thermal trauma. American heart journal 1984. December;108(6):1477–87. [DOI] [PubMed] [Google Scholar]
- 5.Adams HR, Baxter CR, Parker JL. Contractile function of heart muscle from burned guinea pigs. Circulatory shock 1982;9(1):63–73. [PubMed] [Google Scholar]
- 6.Wolfe RR, Miller HI. Cardiovascular and metabolic responses during burn shock in the guinea pig. The American journal of physiology 1976. September;231(3):892–7. [DOI] [PubMed] [Google Scholar]
- 7.Guillory AN, Clayton RP, Herndon DN, Finnerty CC. Cardiovascular Dysfunction Following Burn Injury: What We Have Learned from Rat and Mouse Models. Int J Mol Sci 2016. January 2;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Austin S, St-Pierre J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 2012. November 1;125(Pt 21):4963–71. [DOI] [PubMed] [Google Scholar]
- 9.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007. October;8(10):774–85. [DOI] [PubMed] [Google Scholar]
- 10.Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol (1985) 2000. June;88(6):2219–26. [DOI] [PubMed] [Google Scholar]
- 11.Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009. April 23;458(7241):1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun 2002. August 16;296(2):350–4. [DOI] [PubMed] [Google Scholar]
- 13.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience 2009. March 31;159(3):993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aquilano K, Vigilanza P, Baldelli S, Pagliei B, Rotilio G, Ciriolo MR. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: possible direct function in mitochondrial biogenesis. J Biol Chem 2010. July 9;285(28):21590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med 2009. November 1;47(9):1304–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee OH, Jain AK, Papusha V, Jaiswal AK. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J Biol Chem 2007. December 14;282(50):36412–20. [DOI] [PubMed] [Google Scholar]
- 17.Yates MS, Kensler TW. Keap1 eye on the target: chemoprevention of liver cancer. Acta Pharmacol Sin 2007. September;28(9):1331–42. [DOI] [PubMed] [Google Scholar]
- 18.Chen Z, Zhang Y, Ma L, Ni Y, Zhao H. Nrf2 plays a pivotal role in protection against burn trauma-induced intestinal injury and death. Oncotarget 2016. April 12;7(15):19272–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baldelli S, Aquilano K, Ciriolo MR. Punctum on two different transcription factors regulated by PGC-1alpha: nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim Biophys Acta 2013. August;1830(8):4137–46. [DOI] [PubMed] [Google Scholar]
- 20.Ren Z, He H, Zuo Z, Xu Z, Wei Z, Deng J. The role of different SIRT1-mediated signaling pathways in toxic injury. Cell Mol Biol Lett 2019;24:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mascarenhas DD, Elayadi A, Singh BK, et al. Nephrilin peptide modulates a neuroimmune stress response in rodent models of burn trauma and sepsis. Int J Burns Trauma 2013;3(4):190–200. [PMC free article] [PubMed] [Google Scholar]
- 22.Bohanon FJ, Nunez Lopez O, Herndon DN, et al. Burn Trauma Acutely Increases the Respiratory Capacity and Function of Liver Mitochondria. Shock 2018. April;49(4):466–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhan A, Sirker A, Zhang J, et al. High-frequency speckle tracking echocardiography in the assessment of left ventricular function and remodeling after murine myocardial infarction. Am J Physiol Heart Circ Physiol 2014. May;306(9):H1371–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang LN, Zhou HY, Fu YY, et al. Novel small-molecule PGC-1alpha transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 2013. April;62(4):1297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 2009. April;20(2):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi D, Young LH. AMPK: energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol Metab 2015. August;26(8):422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 2010;5:253–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell 2009. April;8(2):100–12. [DOI] [PubMed] [Google Scholar]
- 29.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem 2007. March 2;282(9):6823–32. [DOI] [PubMed] [Google Scholar]
- 30.Hsu CP, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010. November 23;122(21):2170–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakazawa H, Chang K, Shinozaki S, et al. iNOS as a Driver of Inflammation and Apoptosis in Mouse Skeletal Muscle after Burn Injury: Possible Involvement of Sirt1 S-Nitrosylation-Mediated Acetylation of p65 NF-kappaB and p53. PLoS One 2017;12(1):e0170391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schilling J, Kelly DP. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol 2011. October;51(4):578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riehle C, Abel ED. PGC-1 proteins and heart failure. Trends Cardiovasc Med 2012. May;22(4):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 2004. February 15;18(4):357–68. [DOI] [PubMed] [Google Scholar]
- 35.Tzika AA, Mintzopoulos D, Mindrinos M, Zhang J, Rahme LG, Tompkins RG. Microarray analysis suggests that burn injury results in mitochondrial dysfunction in human skeletal muscle. Int J Mol Med 2009. September;24(3):387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 2013;53:401–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 2004. May 15;36(10):1199–207. [DOI] [PubMed] [Google Scholar]
- 38.Kikuchi N, Ishii Y, Morishima Y, et al. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res 2010. March 18;11:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 2005. April 22;280(16):16456–60. [DOI] [PubMed] [Google Scholar]
- 40.Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res 2010. October 1;107(7):825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]