

Abstract
The Mediterranean corn borer (Sesamia nonagrioides, Noctuidae, Lepidoptera) is a major pest of maize in Europe and Africa. Here, we report an assembly of the nuclear and mitochondrial genome of a pool of inbred males and females third-instar larvae, based on short- and long-read sequencing. The complete mitochondrial genome is 15,330 bp and contains all expected 13 and 24 protein-coding and RNA genes, respectively. The nuclear assembly is 1021 Mb, composed of 2553 scaffolds and it has an N50 of 1105 kb. It is more than twice larger than that of all Noctuidae species sequenced to date, mainly due to a higher repeat content. A total of 17,230 protein-coding genes were predicted, including 15,776 with InterPro domains. We provide detailed annotation of genes involved in sex determination (doublesex, insulin-like growth factor 2 mRNA-binding protein, and P-element somatic inhibitor) and of alpha-amylase genes possibly involved in interaction with parasitoid wasps. We found no evidence of recent horizontal transfer of bracovirus genes from parasitoid wasps. These genome assemblies provide a solid molecular basis to study insect genome evolution and to further develop biocontrol strategies against S. nonagrioides.
Keywords: genome assembly, Lepidoptera, crop pest, sex determination, alpha-amylase, bracoviruses
Introduction
The Mediterranean corn borer (Sesamia nonagrioides, Noctuidae) is a major pest of maize in Mediterranean regions and in Sub-Saharan Africa (Bosque-Perez and Schulthess 1998; Moyal et al. 2011; Kergoat et al. 2015; Kankonda et al. 2018). The damage it causes to maize is due to the moth’s larval feeding behavior, which involves digging tunnels in the stem of the plants. Strategies to control S. nonagrioides mainly rely on chemical pesticides and transgenic plants such as Bt maize that expresses insecticidal proteins (Farinós et al. 2018). However, as observed in other species, an allele conferring resistance to Bt-toxin has been recently identified in S. nonagrioides (Camargo et al. 2018). Furthermore, most EU countries take positions against genetically modified crops (Farinós et al. 2018). Alternative methods implementing various biological agents such as viruses, pheromones, sterile insects, or RNA interference have been developed to control other pests (Beevor et al. 1990; Moscardi 1999; Cork et al. 2003; Tian et al. 2009; Jin et al. 2013; Alamalakala et al. 2018). In addition, several biological control programs targeting lepidopteran stemborers rely on the use of parasitoid wasps belonging to the genus Cotesia (Kfir et al. 2002; Muirhead et al. 2012; Midingoyi et al. 2016). One species of Cotesia, C. typhae, belonging to the Cotesia flavipes species complex, has recently been described as parasitizing exclusively S. nonagrioides. The potential of C. typhae as a biological control agent against this pest is being currently studied (Kaiser et al. 2017). In this context, and because knowing the genetics and genomics of pest species is essential to develop biocontrol programs (Leung et al. 2020), we assembled the nuclear and mitochondrial genomes of S. nonagrioides using short and long sequencing reads. We provide detailed annotations of genes encoding alpha-amylases, which are likely involved in host recognition, and of genes involved in sex determination, which may be useful in a strategy relying on the release of sterile males. We also report the results of a search for polydnaviral genes that would have been horizontally transferred from Cotesia wasps to S. nonagrioides.
Materials and methods
DNA extraction
We extracted large amounts of high-quality DNA from whole bodies of 10 third-instar larvae of S. nonagrioides, males and females, sampled in our laboratory population. We initiated this population in 2010 with individuals sampled in several localities of the French region Haute Garonne (Longages N43.37; E1.19 and vicinity). Since then, we mixed the population at least every 2 years with individuals collected in several localities and regions of south-west France (Pyrénées Atlantiques, Haute Garonne, Tarn et Garonne, Lot et Garonne, Landes, and Gironde). An analysis of S. nonagrioides population genetics in France revealed weak genetic differentiation over France (Naino-Jika et al. 2020). The laboratory population is reared on a diet adapted from Overholt et al. (1994). Mating and oviposition occur in a cage where we introduce 30 pupae of each sex weekly. The pupae can be sexed by comparing their abdominal characters (Giacometti 1995). The 10 larvae used to extract DNA result from two successive crossings of siblings that we implemented to further reduce heterozygosity. We ground the pool of 10 larvae in liquid nitrogen, amounting to 100 mg of fine dry powder. We then extracted DNA using Nucleobond AXG100 columns and the Buffer Set IV from Macherey Nagel, following the manufacturer’s protocol. We obtained 60 µg of DNA, quantified with QuBit (ThermoFisher Scientific). We checked the integrity of DNA on an agarose gel (Supplementary Figure S1) and we did a spectrophotometer measure (Nanodrop 2000) to check the absence of proteins and other contaminants.
Sequencing and genome assembly
We subcontracted Genotoul (genotoul.fr) to build a paired end library (2 × 150 pb; insert size = 350 bp) for sequencing on an Illumina platform. We performed long-read sequencing using the Oxford Nanopore Technology (ONT) in our lab on six flowcells (R9.4). Sequencing was performed over the course when ONT upgraded ligation kits. Thus, while our three first libraries were prepared with the SQK-LSK108 kit, the three last were prepared with the SQK-LSK109 kit, including one with an additional Bluepippin size selection step (15 kb cutoff). We assembled the genome with the MaSuRCA hybrid assembler v3.3.1 (Zimin et al. 2017). We set all parameters to default, except those related to the location of the data, number of threads (64) and Jellyfish hash size (JF_SIZE = 12,000,000,000). We used all 278,683,802 untrimmed Illumina reads (41.8 Gb) produced by Genotoul, as recommended by Zimin et al. (2017). We filtered Nanopore reads using Nanofilt (De Coster et al. 2018) to only keep reads longer than 7 kb (3,085,942 reads amounting to 45,6 Gb with an N50 of 17 kb). We then purged haplotigs and heterozygous overlaps from the assembly using the purge_dups pipeline described by Guan et al. (2020). We used all the default parameters, except for minimap2, for which we specified that we have ONT reads (xamp-ont), and for get_seqs, where we used the option -e to remove duplications at the ends of the contigs only. We checked for contamination in the assembly using blobtools v1.1 (Laetsch and Blaxter 2017), with default parameters. Blobtools requires three inputs: (1) the assembly, (2) a hit file that we generated using our assembly as a query to perform a blastn search (-task megablast, -max_target_seqs 1, -max_hsps 1, -evalue 1e-25) against the NCBI database NT (downloaded in March 2019), and (3) an indexed BAM file that we generated by mapping the trimmed Illumina reads (Trimmomatic v0.38; Bolger et al. 2014) against the assembly with Bowtie2 v2.3.4.1 (Langmead and Salzberg 2012). We also ran the module “all” of MitoZ v2.3 in order to assemble the mitogenome, annotate it and visualize it (Meng et al. 2019). We used the raw Illumina reads as input as recommended by Meng et al. (2019), we set all parameters to default and we set the genetic code and clade to invertebrate and Arthropoda, respectively. Once assembled, we used the mitogenome as a query to perform a blastn search against the assembly to identify possible nuclear mitochondrial DNA (NUMTs). We validated the largest of these NUMTs by PCR, using primers covering three nuclear-mitochondrial junctions (junction 1F: CAACACCGATGACATATTGGGT; junction 1R: CGCACACATAAACAATAACGCC; junction 2F: TGAGGGAGAAGGTAAGTCGA; junction 2R: TGAGGAGGCGTATTGAGGTT; junction 4F: GCGGCTCCTCCTAGATTAAATC; junction 4R: ACTCTCCACGACCAAACCTC).
Genome size estimation
We estimated the genome size of S. nonagrioides using the R packages findGSE and GenomeScope that rely on k-mer frequencies (Vurture et al. 2017; Sun et al. 2018). We counted the number of k-mer on the Illumina reads using Jellyfish, with k equals 17, 21, 25, and 29 (Marçais and Kingsford 2011).
Genome annotation
We annotated genes and repeated elements of S. nonagrioides using Maker v2.31.10 (Holt and Yandell 2011; Campbell et al. 2014). First, we identified repeated elements de novo with RepeatModeler v2.0.1 (https://github.com/Dfam-consortium/RepeatModeler). We then ran a first round of Maker to (1) mask repeated elements and (2) perform a preliminary gene annotation using the transcriptome of S. nonagrioides (Glaser et al. 2015) and the proteomes of three related species: Busseola fusca (Hardwick et al. 2019), Spodoptera litura (Zhu et al. 2018), and Trichoplusia ni (Chen et al. 2019). We merged the outputs of this first round into a GFF3 file, which we used to train SNAP, a gene predictor. We then ran a second round of Maker using this first GFF3 file and SNAP. We then trained Augustus, another gene predictor, with the second GFF3 file, generated by the second round of Maker. Finally, we ran a third and last round of Maker with the second GFF3 file and Augustus. This pipeline led to the final GFF3 file, containing the annotation of S. nonagrioides.
Functional annotation
We identified putative protein functions by blastp search (-evalue 1e-6 -max_hsps 1 -max_target_seqs 1) using the predicted proteins of S. nonagrioides against the nonredundant database UniProtKB/Swiss-Prot that contains unique proteins. In addition, we identified the GO terms and the conserved domains with InterProScan v5.46-81.0. To do this, we ran the 16 analyses proposed by InterProScan, including Pfam.
Comparison with other Noctuidae
We assessed the quality of our S. nonagrioides assembly by comparing its statistics to six other Noctuidae genomes for which all characteristics used in our comparison are available: T. ni (Talsania et al. 2019), S. litura (Cheng et al. 2017), Spodoptera exigua (Zhang et al. 2019), Spodoptera frugiperda (Kakumani et al. 2014), Helicoverpa armigera (Pearce et al. 2017), and Helicoverpa zea (Pearce et al. 2017).
Data availability
The data associated to this article are available on NCBI under the BioProject ID PRJNA680928 and GenBank accession number JADWQK000000000. The BioProject includes the annotated nuclear and mitochondrial assemblies and the raw short and long reads. The data are also available in the DRYAD database at the following address: https://doi.org/10.5061/dryad.dfn2z3515 and on the Bioinformatics Platform for Agrosystem Arthropods (BIPAA) at the following address: https://bipaa.genouest.org/sp/sesamia_nonagrioides/ (last accessed on 05/12/2021). Supplementary material is available at figshare: https://doi.org/10.25387/g3.14185070 (last accessed on 05/12/2021). Supplementary Figure S1 shows the electropherogram and its corresponding gel generated by a fragment analyzer. Supplementary Figure S2 shows the plots generated by GenomeScope, FindGSE, and KAT. Supplementary Figure S3 shows the Blobplot of S. nonagrioides scaffolds. Supplementary Figure S4 is a map of the annotated S. nonagrioides mitogenome generated with mitoZ. Supplementary Figures S5–7 show the structure of the genes involved in sex determination. Supplementary Figure S8 shows the structure of the alpha-amylase gene copies. Supplementary Figure S9 shows the Maximum Likelihood tree of lepidopteran alpha-amylases. Supplementary Table S1 lists size estimates for the S. nonagrioides genome. Supplementary Table S2 lists the name of all 44 scaffolds not assigned to arthropods.
Supplementary material is available at https://doi.org/10.25387/g3.14185070.
Results and discussion
Nuclear genome assembly
The MaSuRCA assembler yielded a preliminary assembly of the S. nonagrioides genome composed of 4300 scaffolds, with a total size of 1162 Mb and an N50 of 955 kb. The completeness of this assembly was good as the BUSCO pipeline (v5.0.0) revealed that it contained 98.7% of the Lepidoptera core genes (n = 5286; Waterhouse et al. 2018). However, given the relatively high amount of duplicated BUSCO genes (7.8%), we deemed that it likely contained haplotigs, heterozygous overlaps and other assembly artifacts. In agreement with this hypothesis, a run of the purge_dup pipeline decreased the amount of duplicated BUSCO genes to 2.7% and removed a large amount of scaffolds (n = 1748) with only minor effects on assembly size and N50. Our purged assembly totals 2552 scaffolds that are 3386–17,305,627-bp long (median length = 66,541 bp). Its N50 is 1105 kb and its size is 1021 Mb, which falls within the range of genome size estimates based on flow cytometry (C-value = 0.97 pg or 951 Mb; Calatayud et al. 2016) and k-mer frequency [971 Mb (FindGSE) to 1406 Mb (GenomeScope); Supplementary Table S1). The average Nanopore and Illumina sequencing depths is 46.3× and 38.9×, respectively, with 95.3% of the Illumina reads mapping to the purged assembly. The level of completeness as assessed by the KAT pipeline was also good as 96.0% of the k-mer identified in the input Illumina reads were included in our purged assembly (Mapleson et al. 2017). The missing 4% k-mer mostly corresponds to usual sequencing errors (Supplementary Figure S2). KAT also estimated a very low level of heterozigosity (0.03%), leading to the absence of a heterozygous peak in the plots of k-mer frequencies (Supplementary Figure S2). It is noteworthy that the genome size inferred by KAT was lower than the ones given by FindGSE and GenomeScope (560–730 vs 960–1600 Mb; Supplementary Table S1), which may be due to the lower ability of KAT to properly estimate the size of genomes containing large amounts of repeated sequences. Related to this, the genome of S. nonagrioides is more than twice bigger than the other Noctuidae genomes sequenced to date (337–438 Mb; Table 1). This difference can be explained by a higher amount of repeated elements (661.6 vs 49.2 to 147.7 Mb), which make up 64.78% of the S. nonagrioides genome, versus only 14% to 33.12% in the other Noctuidae (Figure 1). In fact, as seen in other groups of taxa (Sessegolo et al. 2016; Lower et al. 2017), genome size is correlated to the amount of repeated sequences in Lepidoptera (Talla et al. 2017), a trend that clearly holds among sequenced noctuid genomes included in our comparison (r = 0.98 without S. nonagrioides and 0.99 when it is included). The quality of our S. nonagrioides purged assembly, as measured by its N50 and percent of core Lepidoptera genes, is close to that of the H. armigera genome, the third best assembly of Noctuidae to date (Table 1).
Table 1.
Genome assembly statistics
| Species | Number of fragments | Total size of the assembly (Mb) | N50 (kb) | Ns (%) | Complete BUSCO (duplicated)a |
|---|---|---|---|---|---|
| S. nonagrioides | 2,553 | 1,021 | 1,105 | 0.001 | 98.2% (2.7%) |
| T. ni | 601 | 339 | 894 | 0 | 94.3% (1.5%) |
| S. litura | 2,974 | 438 | 13,592 | 2.488 | 99.1% (0.5%) |
| S.exigua | 301 | 446 | 14,363 | 0.075 | 98.1% (1.2%) |
| S. frugiperda | 37,235 | 358 | 54 | 7.732 | 86.3% (1.2%) |
| H. armigera | 997 | 337 | 1,000 | 11.009 | 98.3% (0.3%) |
| H. zea | 2,975 | 341 | 201 | 10.184 | 96.6% (0.8%) |
Lepidoptera core genes (n = 5,286)
Figure 1.
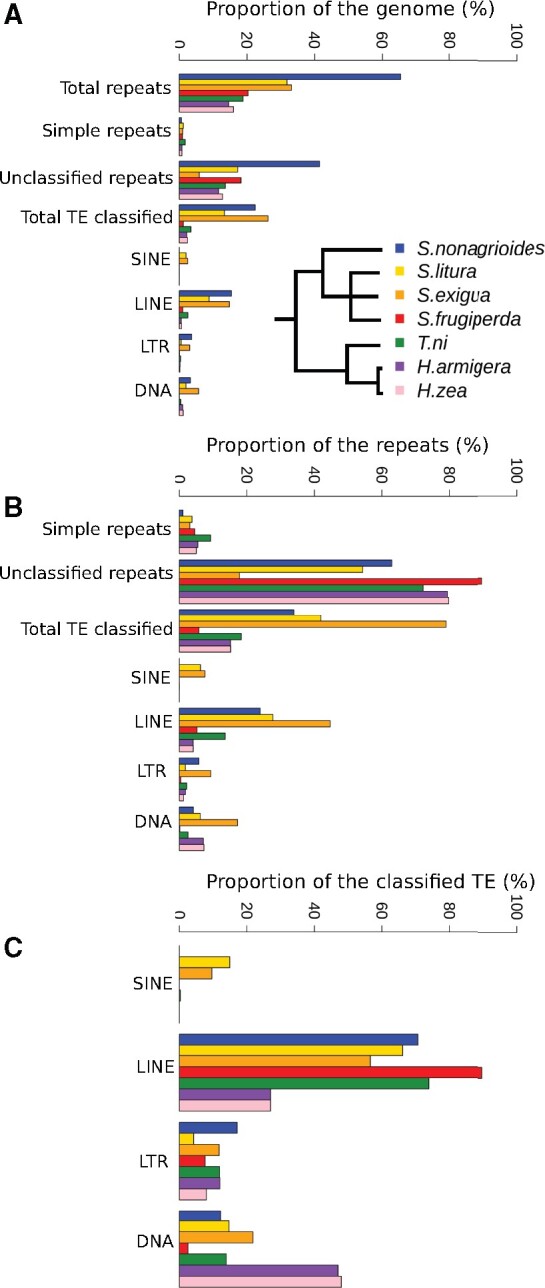
Comparison of repeat contents in sequenced Noctuidae genomes. (A) Genome proportion of each type of repeats as annotated by RepeatModeler v2.0 and RepeatMasker v4.1. (B) Proportion of each type of repeats among all repeated sequences (C). Proportion of each type of TEs among all TEs classified by RepeatModeler V2.0. Phylogenetic relationships among Noctuidae are taken from (Toussaint et al. 2012; Talsania et al. 2019).
Our search for contamination using Blobtools revealed that the amount of contaminating DNA present in our purged assembly is likely low. Among the 2552 scaffolds of our purged assembly, we assigned 2507 scaffolds to arthropods, representing 95.127% of the assembly size. Among the remaining 45 scaffolds, we retrieved no-hit for 25 of them and we assigned the rest to Chordates (2), undefined viruses (15), undefined (2), and Proteobacteria (1). Upon submission of the purged assembly to Genbank, the Proteobacteria scaffold was the only one identified by the NCBI staff as contaminated. It contains an internal 3395-bp fragment showing 95% identity to the genome of Escherichia coli (K-12 strain C3026). This fragment is not covered by any Illumina reads so we removed it from our assembly. We manually placed each of the genome sequences lying upstream and downstream of this contaminant in two new scaffolds, leading to a total of 2553 scaffolds in our final assembly. The sequencing depth and GC content of the remaining 44 scaffolds not assigned to arthropods fall in the range of the arthropod scaffolds, suggesting they may well correspond to S. nonagrioides DNA (Supplementary Figure S3). Thus, we decided not to remove these scaffolds from our final assembly. Instead we listed them in Supplementary Table S2 so that they can be easily retrieved and further studied or removed if needed.
Mitochondrial genome assembly
We assembled a complete circular mitogenome of 15,330 bp, which is 79.6% AT rich, and contains all expected 13 coding protein genes, 22 tRNA genes and two rRNA genes (Supplementary Figure S4). We then used this sequence as a query to perform a sequence similarity search against our assembly to identify possible NUMTs (Richly and Leister 2004). This search retrieved five significant alignments scattered on two scaffolds, for a total of 31.10 kb, a quantity falling within the range of what has been previously described in arthropods (Hazkani-Covo et al. 2010). One of the alignments is 735-bp long, it shows 96.19% identity to the mitogenome and it is located on scf7180000016552_1. The four remaining hits are all on the same scaffold (scf7180000018078_1). They are 15,328, 8188, 4637, and 2216-bp long and all show more than 99.8% identity to the mitogenome (Figure 2). The assembly of the cluster, including two mitochondrial breakpoints and four nuclear-mitochondrial junctions, is supported by both Nanopore and Illumina reads (Figure 2). The sequencing depths at the nuclear-mitochondrial junctions (21× to 35× for trimmed Illumina reads and 46× to 55× for Nanopore reads longer than 7 kb) fall in the distribution of sequencing depths for the whole genome (average = 38.9×, SD = 27.3 for trimmed Illumina reads and average = 46.3× for Nanopore reads longer than 7 kb). We also validated the nuclear-mitochondrial junctions by PCR followed by Sanger sequencing (‘see Materials and Methods’). Thus, we conclude that this cluster results from the recent nuclear integration of two copies of the mitochondrial genome, one of which is rearranged in three pieces.
Figure 2.

A large NUMT in the S. nonagrioides genome. (A) Map of the mitogenome assembly. Protein-coding genes are represented by blue rectangles, in which arrows show their directions. The names of the genes are indicated in the blue rectangles or on top of them. The scale at the bottom of the mtDNA applies also for (B). (B). Map of scaffold scf7180000018078_1 showing the coordinates of the four blastn hits obtained using the mitogenome as a query on the Sesamia nonagrioides nuclear assembly. Only the NUMT sequence is drawn to scale, the rest of the scaffold is not, as indicated by broken lines. The scale of the NUMT is the same as in (A). The four hits are located between each coordinate and the orange, green and pink rectangles help to visualize which parts of the mtDNA have been integrated and possibly rearranged in the nuclear DNA. The nuclear-mitochondrial junctions (blue squares) are supported by short reads (depth of the trimmed reads indicated by the top numbers under the junctions). Long reads support both nuclear-mitochondrial junctions (depth of the reads longer than 7 kb indicated by the bottom numbers under the junctions) and mitochondrial breakpoints (red stars). Indeed, we visually checked that some long reads straddle on the mitochondrial breakpoints and the nuclear genome. The coverage at the mitochondrial breakpoints is much higher because these breakpoints are also mapped by all reads originating from the cytoplasmic mitogenomes. The first, second and fourth nuclear–mitochondrial junctions are also supported by PCR.
Genome annotation
Our automatic annotation of the S. nonagrioides genome yielded 17,230 protein-coding genes (average length = 10,570 bp) corresponding to 17.83% of the genome and including 85,919 exons (2.44% of the genome; Table 2). We assigned 33.88% of all repeated sequences to a known superfamily of transposable elements (TEs) and classified another 1.03% of them as simple repeats (Figure 1B). The percentage of unclassified repeats (62.94%) is in the range of the other Noctuidae (17.78–89.79%). Among the classified TEs, S. nonagrioides has mostly LINE elements (70.66%), a similar percentage ofLTR and DNA elements (17.13% and 12.21%, respectively), and no SINE. This landscape, which will have to be refined using manual curation, is very similar to what was found in T. ni (Figure 1C). The two Helicoverpa species display the most different TE landscapes, where almost half of the classified TEs are DNA elements. We assessed the completeness of our annotation based on two metrics, the Annotation Edit Distance (AED) and the percentage of proteins with a Pfam domain, as recommended (Holt and Yandell 2011; Yandell and Ence 2012). The AED varies from 0 to 1, where 0 means a perfect congruence between gene annotation and its supporting evidence (Holt and Yandell 2011; Yandell and Ence 2012). A genome annotation with 90% of its gene models with an AED of 0.5 or better is considered as well annotated (Campbell et al. 2014). Here, we obtained an AED of 0.5 or better for 94.1% of our gene models. Regarding the second metric, it has been shown that the proportion of proteins with a Pfam domain is relatively stable between species, varying between 57% and 75% in eukaryotes (Yandell and Ence 2012). We found that 62.4% of S. nonagrioides proteins have a Pfam domain. Thus, both the AED and Pfam domain metrics indicate a relatively well-supported genome annotation. When compared with the other Noctuidae species, the number of predicted genes in S. nonagrioides is in the range of the other species, although in the upper border (17,230 vs 11,595–17,707; Table 2). We found that 91.56% of these predicted genes have an InterPro domain (71.47–93.2% in other Noctuidae).
Table 2.
Genome annotation statistics
| Species | Predicted genes | InterPro domains (% of predicted genes) | GO terms (% of predicted genes) | Pfam domain (% of predicted genes) | Number of exons in predicted genes/count per predicted gene |
|---|---|---|---|---|---|
| S. nonagrioides | 17,230 | 15,776 (91.56) | 8,472 (49.17) | 10,751 (62.40) | 85,919/4.99 |
| T. ni | 14,101 | 13,143 (93.2) | 8,680 (61.56) | 10,846 (76.91) | 105,550/7.48 |
| S. litura | 15,317 | 13,637 (89.03) | 11,440 (74.69) | NA | NA/6.64 |
| S. exigua | 17,707 | 13,234 (74.74) | 8,814 (49.78) | NA | NA/5.88 |
| S. frugiperda | 11,595 | NA | 7,743 (66.79) | NA | 64,725/5.58 |
| H. armigera | 17,086 | 12,212 (71.47) | 11,324 (66.28) | 10,700 (62.62) | NA |
| H. zea | 15,200 | 11,061 (72.77) | 10,221 (67.24) | 9,795 (64.44) | NA |
Sex-determination genes
A good knowledge of sex determination in a pest species could be useful in the context of the sterile insect technique. It could help developing genetic sexing strains, in turn facilitating the mass production and release of sterile males (Marec and Vreysen 2019). We set out to provide a detailed annotation of genes likely involved in sex determination in S. nonagrioides. Sex is chromosomally determined in lepidopterans, all species studied so far displaying a form of female-heterogamety (i.e., Z0/ZZ or a ZW/ZZ; Traut et al. 2007). At the gene level, sex determination is best understood in Bombyx mori, which females carry a W dominant gene called Feminizer (Fem). Fem is the precursor of a piwi-interacting RNA (piRNA) that downregulates the expression of a Z-linked gene: Masculinizer (Masc; Kiuchi et al. 2014; Katsuma et al. 2014). In males, Masc splices doublesex (dsx) into its male isoform (dsxM). In females, fem piRNA inhibits Masc, leaving dsx in its default form, the female isoform (dsxF; Nagaraju et al. 2014; Xu et al. 2017; Wang et al. 2019). In addition, the product of IMP (Insulin-like growth factor 2 mRNA-binding protein), a gene located on the Z chromosome, binds to PSI (P-element somatic inhibitor) in males. This interaction increases the binding activity of PSI to dsx, allowing PSI to participate with Masc in dsx mRNA splicing to its male isoform (Suzuki et al. 2010; Xu et al. 2017). Our automatic annotation coupled to alignments using B. mori genes as queries retrieved bona fide orthologs of dsx, IMP and PSI in our assembly of S. nonagrioides, the structure and genomic coordinates of which are given in Supplementary Figures S5–S7. The exons of S. nonagrioides dsx (Sndsx) align over the entire length of the female and male isoforms of Bmdsx (NP_001036871.1 and NP_001104815). The automatic annotation of Sndsx is incomplete as both the 5′ and 3′ UTRs of the gene are missing. Our similarity search for SnPSI retrieved all 14 coding exons of BmPSI. Its automatic annotation also includes predicted 5′ and 3′ UTRs. For IMP, we also found a complete ortholog gene, with a predicted 3′ UTR. Finally, our annotation of the S. nonagrioides ortholog of Masc is less complete, in agreement with the fact that this gene is less conserved among lepidopterans (Harvey-Samuel et al. 2020). The BmMasc gene encodes a 588 amino acid (aa) protein (NP_001296506). Using this protein as a query to perform a similarity search against the Plutella xylostella genome, Harvey-Samuel et al. (2020) identified two sequences encompassing a 7-aa long highly conserved motif of Masc which includes a cysteine domain necessary for promoting male-specific splicing of dsx. One sequence was annotated as a zing finger CCCH domain-containing protein 10-like and the other as a cytokinesis protein SepA-like. An RNAi experiment allowed them to identify the second one as PxyMasc. Here, our similarity search returned 11 hits between 60 and 143 aa long, all on different scaffolds. Only one hit (positions 210,793 to 211,113 of scaffold scf7180000016834_1) overlaps with the highly conserved cysteine-cysteine domain of Masc. This hit is 113 aa long and has 31.86% identity with the BmMasc protein.
Amylases
Obonyo et al. (2010) found that soluble materials deposited on the host caterpillar cuticle were important chemical cues for the proper recognition of the host by the female wasp in the host-parasitoid system Chilo partellus (Lepidoptera: Crambidae)/C. flavipes (Hymenoptera: Braconidae). Bichang’a et al. (2018) identified that the protein alpha-amylase from the oral secretions of the host caterpillar played an important role in antennation and oviposition behaviors prior to egg-laying. Therefore, we investigated alpha-amylase genes in more details in the S. nonagrioides genome. Our similarity search using the H. armigera amylase protein sequence XP_021188243 as a query returned three different gene copies, hereafter named SnAmy1 to SnAmy3, located on two scaffolds: scf7180000017447_1 (SnAmy1 and SnAmy2) and scf7180000016148_1 (SnAmy3; Supplementary Figure S8). SnAmy1 and SnAmy2 are tandemly arranged in inverted orientation, 55 kb apart. SnAmy1 is 5882-bp long; SnAmy2 is 8753-bp long. Both encode exactly 500-aa long proteins. They share 97.6% nucleotide identity. SnAmy3 is 7198-bp long and diverges by 25% from the two other copies. The three genes have seven introns each. We found a subterminal intron located before the last three codons, as noticed in other Lepidopteran amylase genes and in some Hymenopteran amylase genes (Da Lage et al. 2011). For example, in SnAmy2 we found the last three codons downstream of ca. 4 kb of intronic sequence. In SnAmy3, we showed by RT-PCR that two isoforms are transcribed through alternative splicing, with one isoform leading to the presence of a 42-aa long C-terminal tail to the protein through reading in-frame codons in the last intron up to the first stop found. Indeed, two isoforms are also found in the orthologous gene in T. ni. To date, it is not known whether the longer isoform is translated. We also found SnAmy1 and SnAmy3 transcripts in salivary glands and in the midgut (not shown). Amylase genes often form multigene families in insects, with varying levels of divergence among copies (Da Lage 2018). We identified three amylase types in Lepidoptera, named types A, B, and C. Upon inspection of the phylogenetic tree (Supplementary Figure S9), SnAmy1 and SnAmy2 belong to type A and may result from a recent duplication since there is only one copy in H. armigera, whereas SnAmy3 belongs to type B. The type C copy, which is ancestral to butterflies and moths, was lost in S. nonagrioides. Synteny comparison with H. armigera indicates that this type C copy was neighbor to the type A copies (not shown).
Investigation of horizontal transfer of bracoviruses
In its native range in Eastern Africa, S. nonagrioides is naturally parasitized by the braconid wasp C. typhae which is sister to C. sesamiae within the C. flavipes species complex (Kaiser et al. 2017). During oviposition, braconid wasps inject their eggs in host caterpillars together with bracoviruses. These bracoviruses contain circular DNA molecules (DNA circles) many of which typically become integrated into somatic host genomes. Integration of DNA circles will ensure proper persistence and expression of wasp genes during the development of wasp embryos (Beck et al. 2011; Chevignon et al. 2018). In addition, ancient events of horizontal transfer of bracoviral genes from wasps to various lepidopteran species have been reported, suggesting that integration of these genes has also occurred in the germline of lepidopterans (Gasmi et al. 2015; Di Lelio et al. 2019). Here, we investigated whether the S. nonagrioides genome contains traces of wasp DNA circles resulting from recent events of HT from wasp to moth. Given that the circles of C. typhae have not been sequenced, we used the 26 DNA circles of the sister species C. sesamiae (Jancek et al. 2013; NCBI BioProject PRJEB1050) as queries to perform similarity searches on our assembly. Our results revealed no evidence for recent events of HT of DNA circles from Cotesia wasps to S. nonagrioides. Specifically, we retrieved significant alignments only for three circles (2, 28, and 32,) and they all covered <2% of the circle length. Interestingly however, a region of circle 32 (HF562927.1, positions 18,762–19,959) yielded 46 hits longer than 500 bp (up to 678 bp) showing 95.4–99.4% nucleotide identity. We used this 1197-bp sequence as a query to perform a similarity search against GenBank non-redundant proteins and against a custom TE protein database, which yielded no significant alignment. However, this region yielded a 209-bp significant alignment showing 88.7% identity to a B. mori helitron (Helitron-N1_BM, 266-bp long). Given the high nucleotide identity between the wasp and moth sequences (95.4–99.4%) and the deep divergence time between hymenopterans and lepidopterans (>300 million years; Misof et al. 2014), we infer that this helitron-like sequence has been recently transferred between S. nonagrioides and C. sesamiae. This event adds up to the list of helitrons reported to have undergone HT between parasitoid wasps and lepidopterans (Thomas et al. 2010; Guo et al. 2014; Coates 2015; Heringer et al. 2017; Han et al. 2019). Whether these transfers were facilitated by the integration of wasp DNA circles in germline genomes of lepidopterans larvae during parasitism is an interesting possibility that deserves further investigation.
Conclusions and perspectives
We have assembled the complete mitochondrial genome and a draft nuclear genome of S. nonagrioides. The nuclear genome is remarkable in that it is the largest noctuid genome sequenced by far, being two to three times larger than the 10 other noctuid genomes available in GenBank as of January 2021. This difference merely stems from a higher repeat content in S. nonagrioides, in line with the known correlation between genome size and the amount of repeated sequences. It will be interesting to decipher the causes of this higher repeat content, by comparing population sizes, mutation rates and the dynamics of TE activity between the various noctuid species. We found no sign of recent HT from the bracovirus circles of C. sesamiae, which is sister to C. typhae, to S. nonagrioides. However, it will be necessary to repeat this analysis using the bracovirus circles from C. typhae, the very species that parasitizes S. nonagrioides. Finally, given the N50 of the nuclear genome assembly and the high percent of core Lepidoptera genes it contains, we predicted that the vast majority of S. nonagrioides genes are present in one scaffold and can be easily retrieved. This genome thus provides a solid tool to further study the evolutionary history of Noctuidae and it represents an interesting new asset to develop biocontrol strategies against S. nonagrioides.
Acknowledgments
We are grateful to Frabrice Legeai and Anthony Bretaudeau for making the Sesamia nonagrioides nuclear genome available on the BioInformatics Platform for Agroecosystem Arthropods (BIPAA) platform.
Funding
This study was funded by Agence Nationale de la Recherche (ANR CoteBio ANR-17-CE32-0015).
Conflicts of interest
None declared.
Literature Cited
- Alamalakala L, Parimi S, Patel N, Char B.. 2018. Insect RNAi: integrating a New Tool in the Crop Protection Toolkit. In: Kumar D, Gong C, editors. Trends in Insect Molecular Biology and Biotechnology. Cham: Springer International Publishing. p. 193–232. [Google Scholar]
- Beck MH, Zhang S, Bitra K, Burke GR, Strand MR.. 2011. The encapsidated genome of microplitis demolitor bracovirus integrates into the host pseudoplusia includens. J Virol. 85:11685–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beevor PS, David H, Jones OT.. 1990. Female sex pheromones of Chilo spp. (Lepidoptera: Pyralidae) and their development in pest control applications. Int J Trop Insect Sci. 11:785–794. [Google Scholar]
- Bichang’a G, Da Lage J-L, Capdevielle-Dulac C, Zivy M, Balliau T, et al. 2018. α-Amylase mediates host acceptance in the braconid parasitoid Cotesia flavipes. J Chem Ecol. 44:1030–1039. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque-Perez AM, Schulthess F.. 1998. Maize: west and central Africa. In: Polaszek A, editor. African Cereal Stem Borers: Economic Importance, Taxonomy, Natural Enemies and Control. England: CAB International. https://www.cabi.org/cpc/abstract/19981108334. [Google Scholar]
- Calatayud P-A, Petit C, Burlet N, Dupas S, Glaser N, et al. 2016. Is genome size of Lepidoptera linked to host plant range? Entomol Exp Appl. 159:354–361. [Google Scholar]
- Camargo AM, Andow DA, Castañera P, Farinós GP.. 2018. First detection of a Sesamia nonagrioides resistance allele to Bt maize in. Sci Rep. 8:3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Holt C, Moore B, Yandell M.. 2014. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics. 48:4.11.1–4.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Yang X, Tetreau G, Song X, Coutu C, et al. 2019. A high-quality chromosome-level genome assembly of a generalist herbivore, Trichoplusia ni. Mol Ecol Resourc. 19:485–496. [DOI] [PubMed] [Google Scholar]
- Cheng T, Wu J, Wu Y, Chilukuri RV, Huang L, et al. 2017. Genomic adaptation to polyphagy and insecticides in a major East Asian noctuid pest. Nat Ecol Evol. 1:1747–1756. [DOI] [PubMed] [Google Scholar]
- Chevignon G, Periquet G, Gyapay G, Vega-Czarny N, Musset K, et al. 2018. Cotesia congregata bracovirus circles encoding PTP and ankyrin genes integrate into the DNA of parasitized manduca sexta hemocytes. J Virol. 92:e00438-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates BS. 2015. Horizontal transfer of a non-autonomous Helitron among insect and viral genomes. BMC Genomics. 16:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork A, Kamal NQ, Alam SN, Choudhury JCS, Talekar NS.. 2003. Pheromones and their applications to insect pest control. Bangladesh J Entomol. 13:1–13. [Google Scholar]
- Da Lage J-L. 2018. The amylases of insects. Int J Insect Sci. 10:1179543318804783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Lage J-L, Maczkowiak F, Cariou M-L.. 2011. Phylogenetic distribution of intron positions in alpha-amylase genes of bilateria suggests numerous gains and losses. PLoS One. 6:e19673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C.. 2018. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34:2666–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lelio I, Illiano A, Astarita F, Gianfranceschi L, Horner D, et al. 2019. Evolution of an insect immune barrier through horizontal gene transfer mediated by a parasitic wasp. PLoS Genet. 15:e1007998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinós GP, Hernández-Crespo P, Ortego F, Castañera P.. 2018. Monitoring of Sesamia nonagrioides resistance to MON 810 maize in the European Union: lessons from a long-term harmonized plan. Pest Manag Sci. 74:557–568. [DOI] [PubMed] [Google Scholar]
- Gasmi L, Boulain H, Gauthier J, Hua-Van A, Musset K, et al. 2015. Recurrent domestication by Lepidoptera of genes from their parasites mediated by bracoviruses. PLoS Genetics. 11:e1005470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacometti R. 1995. Rearing of Sesamia nonagrioides Lefevre on a meridic diet (Lepidoptera, Noctuidae). Redia. 78:19–27. [Google Scholar]
- Glaser N, Gallot A, Legeai F, Harry M, Kaiser L, et al. 2015. Differential expression of the chemosensory transcriptome in two populations of the stemborer Sesamia nonagrioides. Insect Biochem Mol Biol. 65:28–34. [DOI] [PubMed] [Google Scholar]
- Guan D, McCarthy SA, Wood J, Howe K, Wang Y, et al. 2020. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36:2896–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Gao J, Li F, Wang J.. 2014. Evidence of horizontal transfer of non-autonomous Lep 1 Helitrons facilitated by host-parasite interactions. Sci Rep. 4:5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G, Zhang N, Xu J, Jiang H, Ji C, et al. 2019. Characterization of a novel Helitron family in insect genomes: Insights into classification, evolution and horizontal transfer. Mobile DNA. 10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KM, Ojwang’ AME, Stomeo F, Maina S, Bichang’a G, et al. 2019. Draft genome of Busseola fusca, the maize stalk borer, a major crop pest in Sub-Saharan Africa. Genome Biol Evol. 11:2203–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey-Samuel T, Norman VC, Carter R, Lovett E, Alphey L.. 2020. Identification and characterization of a Masculinizer homologue in the diamondback moth, Plutella xylostella. Insect Mol Biol. 29:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W.. 2010. Molecular Poltergeists: mitochondrial DNA copies (NUMTs) in sequenced nuclear genomes. PLOS Genet. 6:e1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heringer P, Dias GB, Kuhn GCS.. 2017. A horizontally transferred autonomous helitron became a full polydnavirus segment in Cotesia vestalis. G3 (Bethesda). 7:3925–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancek S, Bézier A, Gayral P, Paillusson C, Kaiser L, et al. 2013. Adaptive selection on bracovirus genomes drives the specialization of cotesia parasitoid wasps. PLoS One. 8:e64432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Walker AS, Fu G, Harvey-Samuel T, Dafa’alla T, et al. 2013. Engineered female-specific lethality for control of pest lepidoptera. ACS Synth. Biol. 2:160–166. [DOI] [PubMed] [Google Scholar]
- Kaiser L, Fernandez-Triana J, Capdevielle-Dulac C, Chantre C, Bodet M, et al. 2017. Systematics and biology of Cotesia typhae sp. n. (Hymenoptera, Braconidae, Microgastrinae), a potential biological control agent against the noctuid Mediterranean corn borer. Zookeys. 682:105–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakumani PK, Malhotra P, Mukherjee SK, Bhatnagar RK.. 2014. A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics. 104:134–143. [DOI] [PubMed] [Google Scholar]
- Kankonda OM, Akaibe BD, Sylvain NM, Ru B-PL.. 2018. Response of maize stemborers and associated parasitoids to the spread of grasses in the rainforest zone of Kisangani, DR Congo. Agr Forest Entomol. 20:150–161. [Google Scholar]
- Katsuma S, Kawamoto M, Kiuchi T.. 2014. Guardian small RNAs and sex determination. RNA Biol. 11:1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kergoat GJ, Toussaint EFA, Capdevielle-Dulac C, Clamens A-L, Ong’amo G, et al. 2015. Integrative taxonomy reveals six new species related to the Mediterranean corn stalk borer Sesamia nonagrioides (Lefèbvre) (Lepidoptera, Noctuidae, Sesamiina). Zool J Linn Soc. 175:244–270. [Google Scholar]
- Kfir R, Overholt WA, Khan ZR, Polaszek A.. 2002. Biology and management of economically important Lepidopteran cereal stem borers in Africa. Annu Rev Entomol. 47:701–731. [DOI] [PubMed] [Google Scholar]
- Kiuchi T, Koga H, Kawamoto M, Shoji K, Sakai H, et al. 2014. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature. 509:633–636. [DOI] [PubMed] [Google Scholar]
- Laetsch DR, Blaxter ML.. 2017. BlobTools: interrogation of genome assemblies. F1000Res. 6:1287. [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K, Ras E, Ferguson KB, Ariëns S, Babendreier D, et al. 2020. Next-generation biological control: the need for integrating genetics and genomics. Biol Rev Camb Philos Soc. 95:1838.–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower SS, Johnston JS, Stanger-Hall KF, Hjelmen CE, Hanrahan SJ, et al. 2017. Genome size in North American fireflies: substantial variation likely driven by neutral processes. Genome Biol Evol. 9:1499–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J, Clavijo BJ.. 2017. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics. 33:574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marec F, Vreysen MJB.. 2019. Advances and challenges of using the sterile insect technique for the management of pest Lepidoptera. Insects. 10:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G, Li Y, Yang C, Liu S.. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midingoyi S-K. G, Affognon HD, Macharia I, Ong’amo G, Abonyo E, et al. 2016. Assessing the long-term welfare effects of the biological control of cereal stemborer pests in East and Southern Africa: evidence from Kenya, Mozambique and Zambia. Agric Ecosyst Environ. 230:10–23. [Google Scholar]
- Misof B, Liu S, Meusemann K, Peters RS, Donath A, et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science. 346:763–767. [DOI] [PubMed] [Google Scholar]
- Moscardi F. 1999. Assessment of the application of baculoviruses for control of Lepidoptera. Annu Rev Entomol. 44:257–289. [DOI] [PubMed] [Google Scholar]
- Moyal P, Tokro P, Bayram A, Savopoulou-Soultani M, Conti E, et al. 2011. Origin and taxonomic status of the Palearctic population of the stem borer Sesamia nonagrioides (Lefèbvre) (Lepidoptera: Noctuidae). Biol J Linn Soc. 103:904–922. [Google Scholar]
- Muirhead KA, Murphy NP, Sallam N, Donnellan SC, Austin AD.. 2012. Phylogenetics and genetic diversity of the Cotesia flavipes complex of parasitoid wasps (Hymenoptera: Braconidae), biological control agents of lepidopteran stemborers. Mol Phylogenet Evol. 63:904–914. [DOI] [PubMed] [Google Scholar]
- Nagaraju J, Gopinath G, Sharma V, Shukla JN.. 2014. Lepidopteran sex determination: a cascade of surprises. Sex Dev. 8:104–112. [DOI] [PubMed] [Google Scholar]
- Naino-Jika AK, Ru BL, Capdevielle-Dulac C, Chardonnet F, Silvain JF, et al. 2020. Population genetics of the Mediterranean corn borer (Sesamia nonagrioides) differs between wild and cultivated plants. PLoS One. 15:e0230434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obonyo M, Schulthess F, Le Ru B, van den Berg J, Silvain J-F, et al. 2010. Importance of contact chemical cues in host recognition and acceptance by the braconid larval endoparasitoids Cotesia sesamiae and Cotesia flavipes. Biol Control. 54:270–275. [Google Scholar]
- Overholt WA, Ochieng JO, Lammers P, Ogedah K.. 1994. Rearing and field release methods for Cotesia flavipes cameron (Hymenoptera: Braconidae), a parasitoid of tropical gramineous stem borers. Int J Trop Insect Sci. 15:253–259. [Google Scholar]
- Pearce SL, Clarke DF, East PD, Elfekih S, Gordon KHJ, et al. 2017. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biol. 15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richly E, Leister D.. 2004. NUMTs in sequenced eukaryotic genomes. Mol Biol Evol. 21:1081–1084. [DOI] [PubMed] [Google Scholar]
- Sessegolo C, Burlet N, Haudry A.. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol Lett. 12:20160407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Ding J, Piednoël M, Schneeberger K.. 2018. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics. 34:550–557. [DOI] [PubMed] [Google Scholar]
- Suzuki MG, Imanishi S, Dohmae N, Asanuma M, Matsumoto S.. 2010. Identification of a male-specific RNA binding protein that regulates sex-specific splicing of bmdsx by increasing RNA binding activity of BmPSI. Mol Cell Biol. 30:5776–5786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talla V, Suh A, Kalsoom F, Dincă V, Vila R, et al. 2017. Rapid increase in genome size as a consequence of transposable element hyperactivity in wood-white (Leptidea) Butterflies. Genome Biol Evol. 9:2491–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talsania K, Mehta M, Raley C, Kriga Y, Gowda S, et al. 2019. Genome assembly and annotation of the Trichoplusia ni Tni-FNL insect cell line enabled by long-read technologies. Genes. 79.10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J, Schaack S, Pritham EJ.. 2010. Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol. Evol. 2:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Peng H, Yao Q, Chen H, Xie Q, et al. 2009. Developmental control of a Lepidopteran pest Spodoptera exigua by ingestion of bacteria expressing dsRNA of a non-midgut gene. PLoS One. 4:e6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint EFA, Condamine FL, Kergoat GJ, Capdevielle-Dulac C, Barbut J, et al. 2012. Palaeoenvironmental shifts drove the adaptive radiation of a noctuid stemborer tribe (Lepidoptera, Noctuidae, Apameini) in the miocene. PLoS One. 7:e41377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut W, Sahara K, Marec F.. 2007. Sex chromosomes and sex determination in Lepidoptera. Sex Dev. 1:332–346. [DOI] [PubMed] [Google Scholar]
- Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, et al. 2017. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33:2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-H, Chen X-E, Yang Y, Xu J, Fang G-Q, et al. 2019. The Masc gene product controls masculinization in the black cutworm, Agrotis ipsilon. Insect Sci. 26:1037–1044. [DOI] [PubMed] [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, et al. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 35:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chen S, Zeng B, James AA, Tan A, et al. 2017. Bombyx mori P-element Somatic Inhibitor (BmPSI) is a key auxiliary factor for silkworm male sex determination. PLoS Genet. 13:e1006576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M, Ence D.. 2012. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet. 13:329–342. [DOI] [PubMed] [Google Scholar]
- Zhang F, Zhang J, Yang Y, Wu Y.. 2019. A chromosome-level genome assembly for the beet armyworm (Spodoptera exigua) using PacBio and Hi-C sequencing. bioRxiv 2019.12.26.889121. [Google Scholar]
- Zhu J-Y, Xu Z-W, Zhang X-M, Liu N-Y.. 2018. Genome-based identification and analysis of ionotropic receptors in Spodoptera litura. Sci Nat. 105:38. [DOI] [PubMed] [Google Scholar]
- Zimin AV, Puiu D, Luo M-C, Zhu T, Koren S, et al. 2017. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27:787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data associated to this article are available on NCBI under the BioProject ID PRJNA680928 and GenBank accession number JADWQK000000000. The BioProject includes the annotated nuclear and mitochondrial assemblies and the raw short and long reads. The data are also available in the DRYAD database at the following address: https://doi.org/10.5061/dryad.dfn2z3515 and on the Bioinformatics Platform for Agrosystem Arthropods (BIPAA) at the following address: https://bipaa.genouest.org/sp/sesamia_nonagrioides/ (last accessed on 05/12/2021). Supplementary material is available at figshare: https://doi.org/10.25387/g3.14185070 (last accessed on 05/12/2021). Supplementary Figure S1 shows the electropherogram and its corresponding gel generated by a fragment analyzer. Supplementary Figure S2 shows the plots generated by GenomeScope, FindGSE, and KAT. Supplementary Figure S3 shows the Blobplot of S. nonagrioides scaffolds. Supplementary Figure S4 is a map of the annotated S. nonagrioides mitogenome generated with mitoZ. Supplementary Figures S5–7 show the structure of the genes involved in sex determination. Supplementary Figure S8 shows the structure of the alpha-amylase gene copies. Supplementary Figure S9 shows the Maximum Likelihood tree of lepidopteran alpha-amylases. Supplementary Table S1 lists size estimates for the S. nonagrioides genome. Supplementary Table S2 lists the name of all 44 scaffolds not assigned to arthropods.
Supplementary material is available at https://doi.org/10.25387/g3.14185070.