

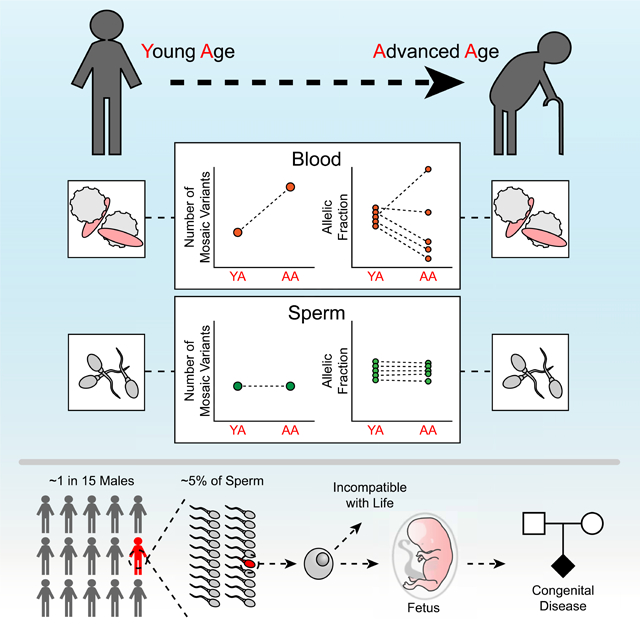
Summary
Throughout development and aging, human cells accumulate mutations, resulting in genomic mosaicism and genetic diversity at the cellular level. Mosaic mutations present in the gonads can affect both the individualand the offspring and subsequent generations. Here we explore patterns and temporal stability of clonal mosaic mutations in male gonads by sequencing ejaculated sperm. Through 300× whole-genome sequencing of blood and sperm from healthy men, we find each ejaculate carries on average 33.3 ± 12.1 (mean ± standard deviation) clonal mosaic variants, nearly all of which are detected in serial sampling, and with the majority absent from sampled somal tissues. Their temporal stability and mutational signature suggest origins during embryonic development from a largely immutable stem cell niche. Clonal mosaicism likely contributes a transmissible, predicted pathogenic exonic variant for 1 in 15 men, representing a life-long threat of transmission for these individuals, and a significant burden on human population health.
Keywords: Clonal mosaicism, Somatic, Sperm, Mutational Signature, Transmission Risk, Embryogenesis
Graphical Abstract
Introduction
New DNA mutations in individual cells can arise during proliferation or metabolism (Alexandrov et al., 2015; Bae et al., 2018). If these mutations occur during embryogenesis they may spread widely across or within tissues at appreciable allelic fractions (AF; i.e. fraction of mutant DNA molecules). Mutations detectable within a tissue or collection of cells are commonly referred to as clonal, whereas those detected only from single cells are considered non-clonal (Machiela and Chanock, 2017), although the distinction can become blurred at the limits of detection. Clonal mosaicism often originates during embryogenesis or follows from later mutations under selection, as observed in clonal hematopoiesis or cancer (Jaiswal and Ebert, 2019). Health consequences of clonal mosaicism have been described in tissues including esophagus, skin, lung, and uterus, with precancerous implications (Martincorena et al., 2018; Martincorena et al., 2015; Moore et al., 2020b; Yoshida et al., 2020).
Mammalian gonads represent a vulnerable tissue, as mutations can predispose both to germ cell tumors, as well as to congenital disease in subsequent generations. While female germ cells do not proliferate beyond embryogenesis, male germ cells are incredibly proliferative, generating around 1,000 new cells per second throughout most of post-adolescent life (Fayomi and Orwig, 2018). Sperm derive from spermatogonial stem cells (SSCs), which have their origin in primordial germ cells (PGCs) (De Felici, 2013). While still a matter of debate, infrequent cell division of Type A SSCs produces Type B SSCs that divide to yield meiotically-active spermatocytes (Fayomi and Orwig, 2018). It has been postulated that the externalization of the testes is an evolutionarily conserved mechanism to restrict mutagenesis (Short, 1997). While most new mutations are neutral or possibly deleterious to sperm, certain mutations seemingly yield selective growth advantage, yet can produce dire consequences in offspring, seen for instance in RAS-pathway mutations leading to achondroplasia (Goriely and Wilkie, 2012). As deleterious mutations are probably present in the testes of all older men, while they contribute to a diminishingly small percent of ejaculated sperm for each individual, they yield significant population-level risk due to their widespread occurrence.
Clonal sperm mosaicism is an important contributor to previously classified “de novo” mutation (DNM) risk in offspring. The number of DNMs in a child doubles for every additional ∼16 years of age of the father at conception, and ∼80% of DNMs in a child phase to the paternal haplotype (Jonsson et al., 2017; Kong et al., 2012; Sasani et al., 2019), suggesting sperm as a major contributor. Sampling blood from parents can identify mosaicism also present in germ cells, and explain 3–8% of DNM monogenic risk (Campbell et al., 2014b; Dou et al., 2017; Krupp et al., 2017; Myers et al., 2018). However, these mutations likely reflect only a small fraction of total clonal sperm mosaicism, and most prior work on sperm mosaicism was limited to studying parental and offspring blood, not sperm directly (Campbell et al., 2014a; Freed et al., 2014; Rahbari et al., 2016). We and others recently showed that clonal sperm mosaicism can be detected for as many as 20% of disease-causing mutations in certain diseases such as autism spectrum disorder (ASD) or early infantile epilepsy due to SCN1A or PCDH19 mutations, (Breuss et al., 2020a; Liu et al., 2018; Yang et al., 2017), but more global clonal sperm mosaicism remains unexplored.
Here, we study the landscape of clonal sperm mosaicism to determine the developmental contribution, as well as time- and age-dependent effects. We recruited cohorts of young- and advanced-age men who underwent single or repeated sperm sampling, coupled with state-of-the-art >300× WGS. We found that clonal sperm mosaicism is remarkably stable over time, both within an individual and between individuals, and impacts disease risk in predictable ways.
Results
Detectable mosaicism is more common within than across tissues
To determine a baseline for clonal mosaicism we recruited 12 males, aged 18–22 years (young age; YA, ID01–12), sampled for blood and sperm (Figure 1A, Figure S1A, Data S1). We further collected multiple (up to 3) sperm samples for 9 subjects at ∼6 months intervals for one year, a relevant timeframe for reproductive decision making. Additionally, for 9 of these subjects, we collected saliva as a third source, since cells derive mostly from buccal epithelia with a small leukocyte contribution (Theda et al., 2018). Finally, we assessed a cohort of 5 older males, aged 48–62 (advanced age; AA, ID13–17), each sampled at a singular timepoint. Together, these approaches revealed the inter-subject, intra-subject, and age-dependent variability of sperm-specific (Sperm), blood-specific (Blood), and tissue-shared (Shared) mosaicism, for both mosaic SNVs (mSNVs) and mosaic INDELs (mINDELs).
Figure 1. Analysis in 12 young aged men uncovers the landscape of sperm clonal mosaicism.

(A) Sampling strategy: 12 healthy males of young age (YA, 18–22 years, blood and up to 3 sperm samples) and 5 healthy males of advanced age (AA, >48 years, blood and 1 sperm sample). Samples subjected to 300× whole-genome sequencing (WGS), then the MSMF computational workflow (see STAR Methods).
(B) Bar charts: number of clonal mosaic variants per individual from each class (sperm-specific: ‘Sperm’, blood-specific: ‘Blood’, tissue-shared: ‘Shared’); Blood typically outnumber Shared or Sperm.
(C) AF distribution (square root-transformed; sqrt-t) of Sperm, Shared, and Blood variants in YA cohort. Shared variants showed higher peak and overall AF compared to Sperm and Blood. sqrt-t: square-root transformed.
(D-E) Rank plot of estimated sperm and blood AFs with 95% exact binomial confidence intervals (CIs) from the YA cohort, grouped by class. Sperm (D) showed steeper initial decay curves, suggesting a relatively lower mutation or higher expansion rate than Shared (E), showing a shallower decay. Norm Sperm AF: sex-chromosome normalized allelic fraction.
(F) Circos histograms for the number of mSNV/INDELs detected from the YA cohort. Variants were evenly distributed across the genome. Colors distinguish classes of variants.
(G) Mosaic SNV/INDELs and the corresponding allelic fractions (AFs) detected from the YA cohort, colors are the same as B. Inner circle: AFs in the blood; outer circle: AFs in the sperm. Colors distinguish classes of variants. Note that Shared variants in brown will be represented in both circles as they are—by definition—detected within both tissues.
We employed a combination of state-of-the-art mosaic variant callers termed MSMF (derived from variant calls from Mutect2, Strelka2, and MosaicForecast, Figure S1, STAR Methods) (Breuss et al., 2020a; Dou et al., 2020; Wang et al., 2021). This approach demonstrated sensitivity to ∼1% AF and a validation rate of 97.6% on benchmarked data (Figure S1C-D).
We found that each YA male harbored between 9–38 Sperm (mean ± SD: 23.1 ± 9.0; total: 277), 1–16 Shared (10.3 ± 5.5; total: 123), and 23–54 Blood (39.4 ± 9.1; total: 473) variants (Figure 1B, Figure S2A, Data S1, STAR Methods), for a total of 873 across all 12 YA males. Together, 10–50 (33.3 ± 12.1; total: 400) variants with an average AF of 4.8% were detected per sperm sample. Two-thirds of sperm-detected (i.e. Sperm and Shared) variants were not found in WGS from blood, and 80% of variants detected from blood were not identified in sperm, justifying separate samplings. The AFs of Shared were, in general, higher than Sperm or Blood variants (Figure 1C-E, Figure S2B-D) and correlated tightly in separate sperm and blood samples (Figure S2D), suggesting an origin prior to gonadal specification. Clonal mosaic variants were distributed evenly across the chromosomes and did not evidence mutational hotspots (Figure 1F-G). These results suggest an early developmental origin of Shared variants and a separate origin of sperm- and blood-specific variants after lineage separation, with higher AFs of Shared likely reflecting an earlier origin.
Sperm mosaicism remains stable across repeated sampling within an individual
For 9 YA individuals, we obtained repeated sperm samples (up to 3 over the course of 1 year) to measure clonal sperm mosaicism stability (Figure 2A). First, to assess whether new mosaic variants appeared over time, we performed 300× WGS and MSMF on two additional sperm samples ∼6 months apart from each of ID04 and ID12 (Figure 2B). The AFs of variants that were mosaic in sperm at time point 1 correlated tightly across subsequent time points (Figure S2E). For a number of variants that were close to the detection limit of WGS, we observed some that were absent in one or two of the datasets, likely reflecting a limitation of binomial sampling, but in general, new somatic variants did not appear or dropout, suggesting relative stability.
Figure 2. Sperm clonal mosaicism shows temporal stability within an individual.

(A) Available blood, sperm, and saliva samples for ID01–12 and their WGS status. ID04 and ID12 (underlined) had three samples subjected to WGS to assess whether new mutations appear over time.
(B) Analysis strategy for ID04 and ID12. 300× WGS was used on 3 independent sperm sample time points (t1, 2, 3).
(C) WGS-discovery of sperm mosaicism variants in each male at one time point, followed by >5000× read depth targeted amplicon sequencing (TAS) in all available samples for all individuals, allowing for accurate assessment of AFs at each time point.
(D) Scatter plot showing pair-wise AF comparison across the YA cohort by TAS. All validated variants were detected in all available sperm samples (i.e. new variants did not appear, nor did existing variants disappear). Number of variants per plot: upper left: 84, upper right and lower left: 71, lower right: 103, with Spearman’s ρ and P-values.
(E) Modified scatter plot showing absolute sperm AF changes for each variant tested across the three time points was typically below 0.02 (i.e. 2%) AF. Violin plot: maximal, absolute change for each variant.
(F) Heatmap of AF variation relative to t1 for variants with three available samples did not show a clear linear increase or decrease.
Second, we performed targeted amplicon sequencing (TAS), with an average read-depth of >5000× (see STAR Methods), to validate variants and more accurately assess AFs (Figure 2C, Data S2). We examined blood, sperm, and saliva on all YA males at all timepoints, for ∼15% of all variants, focusing on representative variants detected in one tissue and at lower AFs. Mosaic variants and AFs in blood and saliva were tightly correlated, with a Spearman’s ρ=0.904 (P<2.20e-16; Figure S2F). All mutations observed in sperm through TAS were detectable and correlated tightly across all sperm samples within a subject (Figure 2D). Absolute AF changes were typically under 2%, tending to fluctuate around a mean rather than drifting in a particular direction (Figure 2E). The variation across time points was imperfectly correlated with the initially observed AF, and fold-changes were higher for variants with AFs below 5% (Figure S2G-I). We found no evidence of significant positive or negative selection (Figure 2F), suggesting that progenitors contribute roughly equally to ejaculates over at least this time course.
Age-dependent changes in blood-specific but not sperm-specific variants
We next applied the same computational pipeline to the 5 AA males. Each AA male harbored between 15–34 Sperm (mean ± SD: 26.0 ± 6.9; total: 130), 8–15 Shared (11.0 ± 2.6; total: 55), and 63–454 Blood (217.4 ± 186.7; total: 1087) variants (Figure 3A-B, Figure S3A-G, Data S1). Notably, AA individuals did not harbor a greater burden of Sperm or Shared variants or show changes in AFs (Mann-Whitney U-test P=0.4866 for Sperm, and P=0.9764 for Shared, Figure 3C, Figure S3H-I). Instead, AA individuals harbored a greater burden of Blood variants compared to YA individuals (P=0.0003) as reported (Zink et al., 2017). In particular, ID14 and ID17 had a further 5-fold increase in Blood variants, consistent with age-dependent clonal hematopoiesis (CH, Figure 3B) (Catlin et al., 2011; Jaiswal et al., 2014). None of these variants overlapped with known leukemia or CH drivers (Bick et al., 2020), thus the observed clonality likely represents driver-independent CH (Zink et al., 2017) (Data S3, see STAR Methods). The sensitivity of our methods to detect CH suggests loss of clonal diversity in blood but not sperm progenitors with age.
Figure 3. Blood but not Sperm clonal mosaic mutations increase with advanced age (AA).

(A) Both blood and sperm in 5 AA men were subjected to 300× WGS.
(B) Number of clonal mosaic variants detected in the 5 AA men, with Sperm and Shared clonal variants comparable to the YA cohort, whereas Blood variants showed dramatic accumulation especially in ID14 and ID17.
(C) Scatter plot, regression lines, and 95% prediction intervals showing the number of mosaic variants from YA (n=12) and AA (n=5) cohort. Left: stability of the number of Sperm and Shared variants, but a dramatic age-dependent accumulation of Blood variants (orange). Right: combined boxplot of all data points (black: median, box: quartiles, whiskers: total data extent). Mann-Whitney U: Sperm 23, Shared 29.5, Blood 0; Two-tailed P-value: Sperm 0.4866, Shared 0.9764, Blood 0.0003).
(D-E) Histogram of the AF distribution of individuals without (D; ID13, ID15, and ID16) or with (E; ID14 and ID17) clonal hematopoiesis compared to YA (ID01–12) individuals. Both subgroups of the AA cohort exhibited similar differences compared to the YA cohort despite their difference in Blood variant numbers.
Consistent with a positive selection for new somatic clones and variants in hematopoietic lineages, we found a shift of Blood AFs towards lower abundance with age, suggesting newly emerging clones during CH can dilute AFs of existing mosaicism in the blood (Figure 3D-E). Unexpectedly, this was independent of whether the number of Blood variants was slightly (ID13, ID15, ID16) or greatly (ID14, ID17) increased, suggesting that changes in blood mosaicism diversity can precede CH, identified here with MSMF as early as the 5th or 6th decade.
Mutational features of clonal mosaicism in sperm and blood
To increase the number of mosaic mutations available for aggregated analysis, given that deep (>200×) WGS datasets of sperm are rare, we added data from our previous sperm sequencing study of 8 men (REACH, F01-F08) (Brandler et al., 2018; Brandler et al., 2016; Breuss et al., 2020a) that were processed identically. We found similar numbers of mosaic variants and AF distributions in each cohort, with some differences in sensitivity as expected from a lower read depth (Figure S4); we thus combined the two cohorts, yielding 522 Sperm and 251 Shared variants from 25 individuals. As CH-derived mosaic variants may differ in origins, we divided Blood mosaicism into 473 ‘Blood-Y’ (YA) and 1673 ‘Blood-A’ (AA, REACH) variants (Figure 4A). Of note, the latter class was heavily biased towards the three AA individuals displaying dramatic CH (i.e. ID14, ID17, and F02, Data S1). These four aggregated classes were then used for a combined analysis of mutational features.
Figure 4. Distinct early developmental signatures distinguish Shared and tissue-specific clonal mosaicism.

(A) Combination of the YA (n=12), AA (n=5), and REACH (n=8) cohorts. Blood-Y from YA, Blood-A from AA and REACH (n=13) were analyzed separately for SNVs and INDELs. Sperm and Shared variants were combined across all cohorts (n=25).
(B) Bar charts show the base substitution profiles of variant classes from panel A. All mosaic classes showed depletion of the aging T>C substitution supporting their origin during embryogenesis. Grey: 95% CI from 10,000 permutations of Simons Simplex Cohort Control de novo mutations (Simons DNMs). Asterisks: data points outside of the 95% permutation CI.
(C-E) Relative contribution of 6-category variant base substitution profiles. (C) C>T predominance and an additional T>G enrichment only in sperm samples with AF < 5%. (D) After distinguishing the cohorts into different sequencing groups, the higher read depths used in ID01–17 (i.e. 300×) likely accounted for the greater sensitivity to detect this T>G signature. (YA: ID01–12, AA: ID13–17, REACH: F01–08). (E) After distinguishing cohorts into those with and without evidence of clonal hematopoiesis, C>T relative contribution correlated with stronger clonal collapse in blood. nCH, non-clonal hematopoiesis (ID13, ID15, and ID16), CH, clonal hematopoiesis (ID14 and ID17).
(F) Scatter plot showing the fraction of variants located across genomic regions for the six categories based on tissue distribution. H3k27ac/H3k27me3/H3K4me1 (H1/Mrg): H3k27ac/H3k27me3/H3K4me1 acetylation peak regions measured in human H1esc or merged from 9 different cell lines; Top2a/b: topoisomerase binding regions; Early and Late replication: measured DNA replication timing; Nucleosome (high/low): nucleosome occupancy tendency; Enhancers: annotated enhancer regions; DNase I: DNase I hypersensitive regions; TF Binding: Transcription factor binding sites. 95% permutation CIs were calculated from 10,000 random permutations of the same number of variants of Simons Simplex Consortium de novo mutations (if a data point is outside of the permutation interval it is colored red). Blood-A showed the most deviations from expectations.
(G) Rank plot of estimated sperm and blood AF with 95% confidence intervals for all 773 gonadal mosaic variants detected as mosaic in sperm (Sperm and Shared). Lower plot shows the log10 transformed ratio of sperm and blood AFs (0 replaced by 1e-8) and the rolling average of over 20 data points to display the local trend. Sperm variants reached maximal AF of 15% and showed a relatively lower average AF.
First, we contrasted base substitution patterns of these four classes with matched permutations of variants from de novo mutations from WGS of the Simons Simplex Collection (Turner et al., 2017) and variants from gnomAD (Karczewski et al., 2020). We found that gonadal mosaic variants showed significantly distinct mutational patterns (Figure 4B, Figure S5A-B, STAR Methods). For instance, C>G and T>C were depleted in all mosaic classes, whereas Shared variants additionally had higher levels of T>A and lower levels of C>T, thought to result from cytosine deamination (Tubbs and Nussenzweig, 2017). We observed a relative increase of T>G in Sperm, particularly at lower AFs (Figure 4C-D). Advanced age was associated with increased relative contribution of C>G and T>C in the blood, where T>G was depleted, an effect amplified by CH (Figure 4E). Collectively, these data suggest that T>C mutations are depleted during the early stages of male embryonic development, an effect supported by prior mutational profiling in trios (Jonsson et al., 2018). However, T>G mutations appear to be enriched during germ cell-specific development, a potentially novel signature for this process.
Next, we assessed mutation enrichment within genomic features (Figure 4F; Data S3; STAR Methods). Shared variants did not show a significant difference compared with permutations, other than an increase in late replication timing. Blood-A showed the most significant deviation across all genomic features, specifically depletions in age-related epigenetic marks, in early replication timing, and in gene-body regions (both intronic and exonic), as well as enrichment in high nucleosome occupancy. These Blood-A-specific genomic features, supported by previous literature in CH and leukemia (Adelman et al., 2019; Bochkis et al., 2014; Du et al., 2019; Rivera-Mulia et al., 2019), demonstrated our ability to detect evidence of selection during CH and aging. Sperm variants, however, showed no evidence of selective pressure. They instead showed significant enrichment in transcription factor binding sites and depletion in areas bound by topoisomerases. Both Sperm and Blood-Y variants were increased in DNase I hypersensitive sites. These findings suggest a correlation in Sperm and Blood-Y between open chromatin and mutagenic stress as previously described (Makova and Hardison, 2015).
Rank plots of the AFs of more than 700 Sperm and Shared mosaic mSNV/mINDELs across the 25 individuals showed a long tail of low AF mutations that were predominantly sperm-specific (Figure 4G, Figure S5C). While mutations mainly accumulate as a function of the cell cycle, models have suggested that this process is accelerated in early post-zygotic phases (Huang et al., 2018; Ye et al., 2018), which would correlate with higher AFs. To assess this, we developed a quantitative metric termed ‘Mutation Factor’ (MF), defined by the rate of mutation accumulation during the exponential expansion of progenitor cells (i.e. per cell cycle). We determined this metric by fitting a step-wise exponential regression with minimal loss to rank plots of mosaic variants (Figure S6A-F, STAR Methods).
We found almost identical MFs for Shared variants in blood and sperm, suggesting transmission to both tissues at stable fractions. Sperm and Blood-Y variants also had comparable MFs, supporting a similar accumulation of mutations that was more dependent on the developmental time rather than the fate of the progenitors. Shared variants have a higher MF than the MFs measured in Sperm or Blood-Y, suggesting a faster accumulation of mutations, as postulated to result from DNA damage repair differences in early development (Gao et al., 2019; Huang et al., 2018; Ye et al., 2018). Blood-A, however, had an even higher MF than the other classes, likely reflecting the dynamic changes in clonal proportions with aging. These observations, together with quantile analysis of AF distributions (Figure S6G), support the expected increase in mutational burden in early development and revealed similar mutational patterns for sperm and somatic progenitors.
One in 15 men harbors a predicted pathogenic transmissible mutation in sperm
We next assessed the likelihood that sperm mosaic variants could contribute towards disease in offspring, most relevant for genes in which mutation of one copy is not compatible with healthy outcomes (i.e. haploinsufficient, HI; defined by pLI>0.9) (Samocha et al., 2014) (see STAR Methods). Across all 25 individuals, we found that men harbored an average total of 30.9 sperm mosaic variants (Sperm: 20.9, Shared: 10.0) (Figure 5A). Of these, 1.6 (Sperm: 1.1, Shared: 0.5) were exonic (Figure 5B), and 0.3 (Sperm: 0.2, Shared: 0.1) were ‘high-impact, i.e. with a CADD score, a clinically relevant metric that summarizes deleteriousness of a mutation (Kircher et al., 2014), above 25 or predicted loss-of-function (C-LoF; Figure 5C, Data S3). Comparisons with alternative prediction tools yielded similar results (Data S3). As a consequence of harboring these high-impact mutations, across 100 men, 28 (i.e. ∼1/3) are predicted to harbor a C-LoF variant in sperm at measurable AFs. Of these, 7.2 (i.e. ∼1/15) occurred in an HI gene (Figure 5D). Comparisons of HI genes with ‘disease gene lists’ can thus be used to determine the ‘transmissible burden’ for any given disorder. For instance, we found that 1.3 in 100 men are estimated to demonstrate a mutation that increases risk for monogenetic autism spectrum disorder (ASD, Figure 5D). Because most of these ASD-associated genes show high penetrance (Iossifov et al., 2012; Sanders et al., 2015), AFs likely would correlate closely with risk of disease. For genes and alleles with a lower penetrance, the odds ratio of the disease (i.e. risk of disease when the gene is mutated) needs to be also specified (Deciphering Developmental Disorders, 2017). Similarly, we estimate that 0.35 in 100 men carry risk mutations for congenital heart disease (STAR Methods) that can be detected as clonal sperm mosaicism.
Figure 5. Clonal sperm mosaicism represents a life-long transmission risk with 1 in 15 males carrying a predicted high-impact pathogenic mutation.

(A) Number of detectable mosaic variants in each category from 2909 total variants; shown are numbers of variants from each individual and the population mean with the 95% CI.
(B) Number of detectable mosaic variants in each category for exonic variants. Shown are individual data points and mean with a 95% confidence interval.
(C) Number of Sperm and Shared variants with a CADD score >25 or a loss-of-function prediction (C-LoF); shown are numbers of variants from each individual and the population mean with the 95% CI.
(D) Estimated number of males per 100 (with 95% CI) with a detectable C-LoF variant in any gene (All), a haploinsufficient (HI) gene, or in a HI gene in the SFARI gene list (SFARI/HI).
(E) Kernel density estimation of the AF distribution of all sperm mosaic variants. The 95% prediction interval for AF is 1–26%.
(F) Stacked bar charts show the relative frequency of AF categories, binned at 5% increments or above 25% for Sperm and Shared variants. The majority of mutations were <5% AF, and most of these were not shared with blood.
(G) Scatter plot and regression lines show the inaccuracy of transmissible mosaicism detection from blood increases with age (YA and AA cohort). Based on the number of blood detectable mosaic variants and their presence in sperm, blood-only detection produces a high false-positive rate that further increases with age due to CH (blue). Blood-only detection produces a consistent 66% false-negative rate (red) for the prediction of transmission across different age groups.
See also Data S3.
Most variants in the combined cohort were at AFs between 1–26% (Figure 5E), with the majority of sperm AFs—and thus likely transmission risk—below 5% (Figure 5F). However, ∼1 in 5 variants had higher AFs (i.e. AF > 5%), with the majority detectable in both blood and sperm. Adjusted for relative frequency and AF, Sperm and Shared variants represented a similar total transmissible burden (Figure 5C and F), the latter fewer in number but higher in AF. Assessment of sperm mosaicism directly when considering risk to offspring could be critical, because using blood as a surrogate can produce false-negatives due to sperm-specific variants. This could also produce false-positives due to blood-specific variants, increasing substantially as a function of age due to CH (Figure 5G).
Discussion
Here we provide an overview of the landscape of clonal sperm mosaicism through assessment of sperm and blood using deep WGS across multiple men, multiple sample types, multiple time points, and multiple ages. We conclude that every man’s semen harbors clonal mosaic variants that likely originate in embryonic development. The mutations we identified were temporally stable across serial samples and age groups, supporting a distribution of early developmental clones across the gonads and relative temporal stability of the stem cell niche during aging. As a consequence, the subset of mutations that are predicted to impact a conceptus’s health represents a life-long threat of transmission.
Most de novo mutations in offspring are thought to have their origin in parental germ cells or the fertilized zygote (Jonsson et al., 2017; Kong et al., 2012). Our data suggest that some of these instead originate when the father was an embryo. Such mutations may occur prior or after to primordial germ cell specification, are clonal, and are stable throughout life. Consistent with this idea, we observed a depletion of T>C mutations, reported to correlate with gonadal aging (Jonsson et al., 2018; Jonsson et al., 2017). Thus, clonal mosaicism appears to differ from non-clonal mosaicism, the latter accumulating with age and having a low likelihood of recurring in two or more offspring. Distinguishing between clonal and non-clonal mosaicism at a finer scale will likely require single-cell sperm sequencing. This method currently suffers from poor detection accuracy for single nucleotide variants, but recent advances suggest non-clonal detectable karyotype defects in up to 3% of sperm (Bell et al., 2020).
When do clonal mutations arise during embryonic development? Mutations that were shared in both blood and sperm showed higher AFs than those only detected in a single tissue, likely representing earlier embryonic origins. Primordial germ cells separate from somatic progenitors before the third post-conception week in humans (De Felici, 2013) (Figure 6A), whereas hematopoietic progenitors arise later from mesoderm (Dzierzak and Speck, 2008). Due to this early separation, clonal mutations detected in sperm are less likely to be shared with other tissues, whereas those detected in blood were often also detected in saliva. In aged, however, clonal hematopoiesis may amplify blood-specific clonal mutations.
Figure 6. Developmental origin and transmission of clonal and non-clonal mosaicism in sperm.

(A) Mosaic variants occur throughout development and are typically Shared if they occur prior to germ cell specification. For instance mutation a (resulting in genotype A/a) occurs during the 4 cell stage, is present in ∼25% of cells (i.e. ∼12.5% AF), and is shared across blood and sperm. B/b, which occurs later, is also shared in sperm and blood and are clonal. C/c and D/d occur in specific tissues and are present as clonal mosaicism, whereas E/e, and F/f occur later and are non-clonal at young age (i.e. not detectable from bulk sequencing). This schematic shows male development; however, due to the similarity of early germ cell development between sexes, female mosaicism likely exhibits similar patterns.
(B) Relative contributions of variants to cellular diversity detected in blood and sperm, and changes with age. Variants occurring during early embryogeneis (a and b) are shared in young and aged in both sperm and blood. A group of sperm specific (c) or blood specific (d) variants arise during embryogenesis and are stable during aging. A group of blood specific variants (f) arise to the level of clonal during aging. Gray: unmarked clones.
(C) Sperm mosaicism subtypes. Clonal mosaicism is present in primordial germ cells (green bolt); non-clonal mutations arise in spermatogonial stem cells (gray bolt) and sperm (white bolt). Note that the mutations accumulate within sperm, and ultimately the fetus which harbors all as de novo mutations.
(D) Absolute contribution of clonal sperm mosaicism (green) is stable as men age whereas non-clonal sperm mosaicism increases with age (gray). As a result, the relative contribution of clonal mosaic SNVs or INDELs to the number of de novo mutations in an offspring decreases with age (red line).
Spermatogonial stem cells, despite proliferating throughout reproductive life, unlike blood, do not appear to exhibit detectable clonal collapse or expansion, likely a reflection of the anatomical constraints of the testicular stem cell niche. Certain mutations appear to provide a proliferative growth advantage (known as ‘selfish sperm’ or ‘paternal age effect’), particularly those impacting RAS signaling (Goriely and Wilkie, 2012), but show no evidence of negatively impacting clonal diversity like in clonal hematopoiesis (Arends et al., 2018). We also found some individuals of advanced age harbor an order of magnitude more blood-specific mutations, without similar findings of clonal collapse in sperm (Genovese et al., 2014). Together this supports that clonal hematopoiesis may represent a spectrum of clonality of the blood rather than a collection of discrete clonal events (Zink et al., 2017).
While clonal mosaicism makes a substantial contribution to the total pool of sperm mutations potentially impacting the health of offspring, our data suggest that as men age, their relative contribution to disease risk actually declines. This may be attributable to the age-dependent accumulation of non-clonal mosaicism that accompanies the massive proliferation during spermatogenesis. Yet, clonal mosaicism contributes an absolute disease threat to offspring that remains stable throughout a man’s life (Figure 6B-D). These ideas are consistent with recurrence risk estimates from population analysis that factor in age (Campbell et al., 2014b; Jonsson et al., 2018). As early germ cell development is thought to be similar if not identical between men and women, female clonal mosaicism—unlike non-clonal mosaicism—will likely mirror that found in men (De Felici, 2013). This is supported by population and family studies on DNMs, which demonstrate that recurrent (i.e. sibling shared) mutations are as likely to be located on the maternal as the paternal haplotype (Jonsson et al., 2018; Rahbari et al., 2016). This is in contrast to the typically observed male dominance that considers the sum of all DNMs (Kong et al., 2012).
De novo mutations represent a major contributor to congenital human disease (Acuna-Hidalgo et al., 2016; Deciphering Developmental Disorders, 2017; Veltman and Brunner, 2012). We provide the first estimate of the burden of clonal sperm mosaicism in healthy men contributing to this risk. Assuming that transmission of variants follows observed abundance in sperm, we predict that approximately 1 in 300 concepti (see STAR Methods) harbors one or more such variants that is predicted pathogenic, likely contributing to miscarriage or congenital disease. This is a direct result from our observation that 1 in 15 males carries such a mutation, and the observed average AF across all clonal sperm variants. Consequently, for the monogenetic component of a well-studied disorder like autism (∼4 in 1000 children), we estimate that ∼15% could be attributed to clonal mosaicism; this would apply similarly to congenital heart disease or other severe de novo mutation-related disease (see STAR Methods). This relative estimated contribution varies with parental age, and it is higher in younger fathers and—to some degree—mothers. Future studies assessing the presence of DNMs in sperm could better clarify risks for particular diseases.
Limitations of the study
There are several limitations of our study. We restricted our study to clonal sperm mosaicism, and thus we did not attempt to identify non-clonal sperm-specific mosaicism. Such mutations may account for the paternal age-dependent increase in de novo mutations in offspring, but because they likely occur in individual SSCs or sperm cells, our bulk sperm sequencing approach was not designed for their identification. A different sampling method (laser capture of testicular stem cell niche) demonstrated an age-dependent mutation accumulation of mutations in SSCs (Moore et al., 2020a), but whether these appear in sperm is still an open question. Another limitation resulted from our use of WGS rather than targeted sequencing, which increased the number of detectable somatic variants but resulted in a trade-off due to the limited number of samples and the limited read depth we could achieve. Although 250–300× WGS is emerging as a standard for clonal mosaicism analysis from bulk (Bizzotto et al., 2021; Breuss et al., 2020b; Rodin et al., 2021; Wang et al., 2021), this nevertheless limits sensitivity to around 1% AF, as these methods typically require multiple supporting reads. Moreover, while we were able to determine the stability of mosaicism found in sperm, the variability of genome-wide blood mosaicism rendered it challenging to accurately describe its changes with age. Future studies may incorporate larger cohort sizes or resampling timepoints, to yield a clearer definition of the phenomenon of CH on a genome-wide level. Finally, while our results allow speculation on the biological basis of the observed difference between sperm and blood progenitors during aging, our approach is unable to provide direct mechanistic insights into this problem. More specialized models or targeted post-mortem sampling will be necessary to elucidate this problem in more detail.
STAR+METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for samples and data should be directed to and will be fulfilled by the lead author, Joseph G. Gleeson (jogleeson@health.ucsd.edu).
Materials Availability
This study did not generate new unique materials. All reagents and kits used in this study are described in STAR Methods.
Data and Code Availability
Raw whole genome sequencing and targeted amplicon sequencing BAM files used in this study are available on SRA (accession number: PRJNA660493 and PRJNA588332). Summary tables of the data are included as supplementary tables. Codes for data analysis pipelines as well as codes to generate the figures are freely available on GitHub at https://github.com/shishenyxx/Sperm_control_cohort_mosaicism. Other materials or software are detailed in STAR Methods.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Subject recruitment
17 healthy ethnically diverse males (Figure S1A) were enrolled according to approved human subjects protocols from the Institutional Review Board (IRB) of the University of California for blood, saliva, and semen sampling (140028, 161115). All participants signed informed consents according to the IRB requirement, and the study was performed in accordance with Health Insurance Portability and Accountability Act (HIPAA) Privacy Rules. None of the participants reported severe psychological conditions or showed significant signs of neurological disorders, infectious diseases, or cancer. Semen and blood samples were collected for all subjects (ID01–17). ID01–08 and ID11 further provided saliva samples. ID05 further provided a second semen sample approximately half a year after the first collection; ID02–04, ID06-S08, ID11, and ID12 provided a total of 3 samples within ∼12 months. Ages for all individuals are included in Data S1.
METHOD DETAILS
DNA extraction for blood and saliva
Genomic DNA was extracted from peripheral blood and saliva samples containing buccal cells using the Puregene kit (Qiagen, 158389) following the manufacturers’ recommendations.
Sperm extraction
Extraction of sperm cell DNA from fresh ejaculates was performed as described (Breuss et al., 2020a; Wu et al., 2015). In short, sperm cells were isolated by centrifugation of the fresh (up to 2 days) ejaculate over an isotonic solution (90%) (Sage/Origio, ART-2100; Sage/Origio, ART-1006) using up to 2 mL of the sample. Following a washing step, quantity and quality were assessed using a cell counting chamber (Sigma-Aldrich, BR717805–1EA). Cells were pelleted and lysis was performed by addition of RLT lysis buffer (Qiagen, 79216), Bond-Breaker TCEP solution (Thermo Scientific, 77720), and 0.2 mm stainless steel beads (Next Advance, SSB02) on a Disruptor Genie (Scientific Industries, SI-238I). The lysate was processed using reagents and columns from an AllPrep DNA/RNA Mini Kit (Qiagen, 80204). Concentration of the final eluate was assessed employing standard methods. Concentrations ranged from ∼0.5–300 ng/μl.
WGS of sperm and blood samples
WGS sequencing was performed as described (Breuss et al., 2020a). A total of 1.0 μg of extracted DNA was used as the starting material for PCR-free library construction (KAPA HyperPrep PCR-Free Library Prep kit; Roche, KK8505); libraries were then mechanically sheared (Covaris microtube system; Covaris, SKU 520053) to obtain ∼400 base pairs (bp) fragments. Then Illumina dual index adapters were ligated to these DNA fragments. Following beads-based double size selection (300–600 bp), the concentration of ligated fragments in each library was quantified (KAPA Library Quantification Kits for Illumina platforms; Roche/KAPA Biosystems, KK4824). Libraries with concentrations of more than 3 nM and fragments with peak size 400 bp were sequenced on an Illumina NovaSeq 6000 S4 and/or S2 Flow Cell (FC), in 6–8 independent pools. The target for WGS with high quality sequencing raw data was 120 GB or greater with a Q30 >90% per library per sequencing run. In case the first sequencing runs generated insufficient reads, additional sequencing was performed by sequencing the same library. Raw data was processed through an Illumina FPGA-based platform to generate BAM files.
WGS data processing and germline variant calling
Raw data were aligned to the GRCh37d5 reference genome, sorted, and PCR duplicates were removed by an Illumina FPGA-based platform. Reads aligned to the INDEL regions were realigned with GATK’s (v3.8.1) RealignerTargetCreator and IndelRealigner following the GATK best practice. Base quality scores were recalibrated using GATK’s (v3.8.1) BaseRecalibrator and PrintReads. Read groups were renamed by Picard’s (v2.20.7) AddOrReplaceReadGroups command. Germline SNVs and INDELs were detected by GATK’s (v3.8.1) HaplotypeCaller. The distribution of library DNA insertions was assessed by Picards’ (v2.20.7) CollectInsertSizeMetrics. The depth of coverage was analyzed by GATK’s (v3.8.1) DepthOfCoverage command.
Principal component analysis (PCA) of genetic origins of the assessed individuals
In order to determine the origins of the included individuals, heterozygous variants generated by GATK’s (v3.8.1) HaplotypeCaller, genomic VCF format were used as output and genotyped across all samples by using the GATK’s (v3.8.1)’s GenotypeGVCFs and CombineGVCFs; in addition, all variants from dbSNP (v137) were added. The VCF file was reformatted by BCFtools (v1.10.32) and converted to bfiles by PLINK (v1.90b6.16). Single nucleotide polymorphisms (SNPs) were extracted from both the samples in this study and samples from the 1000 Genomes phase 3 (Genomes Project et al., 2015) and merged. SNPs overlapping with the repeat mask region were removed. PCA was carried out by PLINK (v1.90b6.16) and the results were plotted in R (v3.5.1).
Mosaic SNV/INDEL detection pipeline in WGS data (MSMF)
Mosaic single nucleotide variants/mosaic small (typically below 20 bp) INDELs were called by using a combination of four different computational methods based on previous published and adapted pipelines (Breuss et al., 2020a; Breuss et al., 2020b): the intersection of variants from the paired-mode of GATK’s (v4.0.4) Mutect2 (Cibulskis et al., 2013) (paired mode) and Strelka2 (Kim et al., 2018) (v 2.9.2) (set on ‘pass’ for all variant filter criteria) for sample-specific variants; or single-mode of Mutect2 (with an in-house panel of normal) followed by MosaicForecast (v 8–13-2019) for sample-specific or tissue-shared variants. For the YA cohort, the panel of normal is generated using a “leave one out” strategy, by excluding samples from each individual; for the AA and REACH cohort, all samples from the YA were used to generate the panel of normal. Variants were excluded if they 1] resided in segmental duplication regions as annotated in the UCSC genome browser (UCSC SegDup) or RepeatMasker regions, 2] resided within a homopolymer or dinucleotide repeat with more than 3 units, 3] overlapped with annotated germline INDELs, 4] did not show a minimum of 3 alternative reads, or 5] were detected more than once across multiple individuals. We further removed variants with an overall population allele frequency >0.001 in gnomAD (Karczewski et al., 2020) (v 2.1.1) or >0 for variants only detected by MosaicForecast (Dou et al., 2020) to exclude false positive calls from population-level polymorphisms. To avoid binomial sampling bias and false positive signal from copy number/structural variations or non-annotated repetitive regions, we randomly chose 1600 single nucleotide polymorphism from dbSNP (v137), estimated the 95% confidence interval of all those variants in each sample respectively, and excluded variants whose coverage is not within this CI. Finally, variants with an AF>0.35 in both sperm and blood (or >0.7 for sex chromosomes) were considered likely germline variants and removed. Variants with a lower CI of AF<0.001 were also removed. Fractions of mutant alleles for variants called in one sample were calculated in the other sample with the exact binomial confidence intervals using scripts described below. If a variant was only detected in one tissue, mosaicism in the second tissue was confirmed if a minimum of 3 alternative reads were present. Scripts for variant filtering and annotations are provided on GitHub (https://github.com/shishenyxx/Sperm_control_cohort_mosaicism).
Simulation analysis to determine the sensitivity of MSMF
To determine the sensitivity for detecting mosaic variants, we created simulated datasets that contained known mosaic variants at low frequencies. We first randomly generated 10,000 variants from chromosome 22 based on GRCh37d5 as our set of mosaic variants. We then used Pysim(Xia et al., 2017) to simulate Illumina paired-end sequencing reads with a NovaSeq 6000 error model from the GRCh37d5 reference chromosome 22 and a version of chromosome 22 that contained the alternate alleles from our 10,000 mosaic variants. These two sets of reads were then combined to create a series of datasets with mosaic variants at 1, 2, 3, 4, 5, 10, 15, 20, 25, and 50% AF, at coverages at 50×, 100×, 200×, 300×, 400×, and 500× depth. Reads were mapped to GRCh37d5 using BWA (v0.7.8) mem, processed with Picard’s (v2.20.7) MarkDuplicates, and INDELs were realigned and base quality scores recalibrated as described above. We applied our somatic variant calling pipelines containing GATK’s (v4.0.4) Mutect2 (single mode and paired mode), Strelka2 (v 2.9.2), and MosaicForecast (v 8–13-2019) to detect mosaic variants at each AF and each depth. We further applied the same filters we used for the genomic regions; as we excluded the repetitive and segmental duplication regions, only ∼75% of the genomic region remained valid. The sensitivity and recovery rate of the pipeline was then determined through these data.
Visualization of genomic distribution of mosaic variants
The genomic distribution pattern of mosaic variants and the allelic fractions of different variants across the genome was presented using Circos (Krzywinski et al., 2009) (v0.69–6).
Targeted amplicon sequencing (TAS) and experimental benchmark of the SNV/INDEL calling pipeline
TAS analysis was first applied to 82 variants from the previously published 200× WGS sequencing results (Breuss et al., 2020a), to experimentally confirm the validation rate of the new pipeline. PCR products for sequencing were designed with a target length of 160–190 bp with primers being at least 60 bp away from the base of interest. Primers were designed using the command-line tool of Primer3 (Untergasser et al., 2012; Untergasser et al., 2007) with a Python (v3.7.3) wrapper (Breuss et al., 2020a). PCR was performed according to standard procedures using GoTaq Colorless Master Mix (Promega, M7832) on sperm, blood, and an unrelated control (>20 ng input per reaction; >6000 sperm genome equivalents). Amplicons were enzymatically cleaned with ExoI (NEB, M0293S) and SAP (NEB, M0371S) treatment. Following normalization with the Qubit HS Kit (ThermFisher Scientific, Q33231), amplification products were processed according to the manufacturer’s protocol with AMPure XP beads (Beckman Coulter, A63882) at a ratio of 1.2x. Library preparation was performed according to the manufacturer’s protocol using a Kapa Hyper Prep Kit (Kapa Biosystems, KK8501) and barcoded independently with unique dual indexes (IDT for Illumina, 20022370). The libraries were sequenced on an Illumina HiSeq 4000 platform with 100 bp paired-end reads. After determining the validation rate of the new pipeline, TAS was further performed for a subset of called variants on the different sperm time points, blood, saliva, and unrelated control sample to quantify the AFs and to extend analysis to tissues that were not subjected to WGS.
Data analysis for TAS
Reads from TAS were mapped to the GRCH37d5 reference genome by BWA mem and processed according to GATK (v3.8.2) best practices without removing PCR duplicates. Putative mosaic sites were retrieved using SAMtools (v1.9) mpileup and pileup filtering scripts described in previous TAS pipelines (Breuss et al., 2020a). Variants were considered mosaic if 1] their lower 95% exact binomial CI boundary was above the upper 95% CI boundary of the control; 2] their AF was >0.5%. For the validation of the mosaic variant calling pipeline, 82 variants from the benchmark data (REACH) detected by the MSMF pipeline were subjected to TAS. Candidates were randomly selected from all detected variants, and 80 (97.6%) of them were considered mosaic based on the above criteria. Sperm samples from the YA cohort were labeled as time point 1 (t1), t2, and t3, based on the data of sample collection. t1 was used as an anchor to determine absolute and relative (i.e. fold change) AF differences of the same variant measured across samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mutational signature analysis
Mutational signatures were determined for each variant by retrieving the tri-nucleotide sequence context using Python (v3.5.4) with pysam (v 0.11.2.2) and plotting the transversion or transition based on the pyrimidine base of the original pair similar to previous studies (Alexandrov et al., 2013). Mutational signatures from de novo mutations in the Simons Simplex Consortium cohort (from healthy siblings) and mutations from gnomAD were obtained by retrieving SNVs present in their respective, publicly available VCFs. In order to obtain a 95% band of expectation, an equivalent number of variants was randomly chosen from the Simons Simplex Consortium or gnomAD VCF. This process was performed for a total of 10,000 times to obtain a distribution and the 2.5th and 97.5th percentile of the simulated mutational signatures. Significance was reported if a mutational signature was outside the permuted 95% bands.
Step-wise exponential regression model for the burden of variants
In order to model the exponential decay of the variants, a step-wise exponential regression model was made based on the following assumptions: 1] variants happening at roughly the same cell division during early embryonic development have similar allelic fractions in different individuals; 2] during early embryonic development the number of cells are growing exponentially but at different rates across tissues due to varying growth rates and cell death; 3] the spontaneous mutation rate is stable within each category; 4] the number of mosaic variants occurring in each cell generation is in proportion with the number of cells in that generation. For each group of ranked variants from an already developed tissue (sperm or blood), during the tth cell division, we assume that all variants came from a starting population of variants, and AF0 is estimated from the exact binomial CI of the highest AFs found in each group. Based on assumption 2 the mutation is accumulated at a speed of θ (θ ≥ 1 and θ ≤ 2). For the tth cell division, the average , and the number of expected variants with this , we rank the AFt to get an estimated rank vector
to get the best estimation of ET towards the observed ranked AF vector 0T, we defined the loss .
By minimizing L, we obtained the best estimation of the ranked AF curve. We finally defined a Mutation Factor which ranges from 1 to +∞ and is a measure of mutational accumulation speed relative to cellular proliferation. Thus, lower MFs are found if the rank plot is more concave, i.e. the mutation rate is relatively lower (e.g. Sperm); and higher MFs are found if the rank plot is less concave or shallow, i.e. the mutation rate is relatively higher (e.g. Shared). Note that MF includes both mutations derived from the proliferation itself and those acquired during homeostasis, which potentially uncouples mutational accumulation from proliferation.
Assessment of mosaic variants overlap with genomic features
In order to assess the distribution of mosaic variants and their overlap with genomic features, an equal number of variants (mSNV/INDELs that were Sperm, Shared, Blood-Y, and Blood-A) was randomly generated with the BEDTools (v2.27.1) shuffle command within the region from Strelka 2 without the subtracted regions (e.g. repeat regions). This process was repeated 10,000 times to generate a distribution and their 95% CI. Observed and randomly subsampled variants were annotated with whole-genome histone modifications data for H3k27ac, H3k27me3, H3k4me1, and H3k4me3 from ENCODE v3 downloaded from the UCSC genome browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/)—specifically for the overlap with peaks called from the H1 human embryonic cell line (H1), as well as peaks merged from 9 different cell lines (Mrg; Gm12878, H1, Hmec, Hsmm, Huvec, K562, Nha, Nhek, and Nhlf). Gene region, intronic, and exonic regions from NCBI RefSeqGene (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/refGene.txt.gz); 10 Topoisomerase 2A/2B (Top2a/b) sensitive regions from ChIP-seq data (Canela et al., 2017) (Samples: GSM2635602, GSM2635603, GSM2635606, and GSM2635607); CpG islands: data from the UCSC genome browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/); genomic regions with annotated early and late replication timing (Hansen et al., 2010); nucleosome occupancy tendency (high/>0.7 or low/0.0–0.7 as defined in the source) from GM12878, for which all non-zero values were extracted and merged (Valouev et al., 2011); enhancer genomic regions from the VISTA Enhancer Browser (https://enhancer.lbl.gov/); and DNase I hypersensitive regions and transcription factor binding sites from Encode v3 tracks from the UCSC genome browser (wgEncodeRegDnaseClusteredV3 and wgEncodeRegTfbsClusteredV3, respectively). 74 Leukemogenic driver mutations, 4938 clonal hematopoiesis of indeterminate potential (CHIP) mutations identified in TOPMed participants, 28,594 SNV/INDELs associated with CHIP, 619 rare loss of function regions associating with CHIP, 27 rare enhancer regions associating with CHIP, as well as 476 common variants associating with CHIP identified from 97,691 individuals (Bick et al., 2020) were extracted and annotated with bedtools annotate to find the co-occurance of CH identified in this study and CH identified from large-scale population study.
Annotation of variant function
A variant was annotated as loss-of-function if it was annotated as frameshift, nonsense, canonical splice site, or start loss with a gnomAD allele frequency <0.0001. For genomic features, dbSNP annotations were carried out with version 150 (Sherry et al., 1999). All variants were further annotated with a CADD score, and values >25 were considered likely pathogenic for our classification (Kircher et al., 2014). The following genome-wide metrics were used to determine deleteriousness for additional tools to compare to our CADD annotation: Eigen score >1.7 (Ionita-Laza et al., 2016); FATHMM score >0.98 (Shihab et al., 2013); DeFine predicted pathogenic probability >0.95 (Wang et al., 2018); raw phastCons 100 way score for vertebrates >0.7 (Siepel et al., 2005); or raw phyloP 100 way score for all organisms >1.9 (Pollard et al., 2010). For coding mutations only we used the following: SIFT predicted ‘D’, LRT score predicted ‘D’ or ‘U’ (Liu et al., 2016); MutationTaster predicted ‘H’ or ‘M’ (Reva et al., 2007); PROVEAN predicted ‘D’ (Choi and Chan, 2015); iFish predicted ‘deleterious’ (Wang and Wei, 2016); or GERP_RS score >2 (Davydov et al., 2010).
Burden estimation
Using the observed fraction of variants that are classified as C-LoF, we calculated a 95% estimation interval of the true fraction using SciPy (v1.3.1) stats’s t-interval and multiplied by the chosen number of men (n=100). This fraction was further modified by taking into account the subset of genic regions that are annotated to belong to a haploinsufficient gene (HI) with pLI higher than 0.9, or that belong to an HI gene which is annotated as a likely autism spectrum disorder gene by SFARI (Level 1, 2, 3, and S, with pLI higher than 0.9). A gene list of likely disease-causing candidates for congenital heart disease was obtained by merging the candidate gene list from four recent large scale studies (Alankarage et al., 2019; Ellesoe et al., 2018; Jin et al., 2017; Li et al., 2017); genes reported by at least two studies were further filtered by their pLI (>0.9). Genomic regions of those genes were extracted from http://www.openbioinformatics.org/annovar/download/hg19_refGene.txt.gz.
Estimation of disease impact conveyed by clonal mosaicism
For transmission risk we assume that 1] expression of the disrupted gene does not impact a sperm cell’s fertility; 2] AFs estimated in purified sperm directly reflect the percentage of sperm cells carrying the mutation and determine the average transmission risk θ. For any disease with incident rate I and a fraction P, which are caused by de novo HI-C-LoF SNV/INDELs within a set of genes HI – C – LoF ∩ Disease gene set (monogenetic, autosomal dominant contribution), we can calculate the percentage of the relevant genome by comparing . Taking μ into account, which is the fraction of men predicted to carry a C-LoF mutation, we can estimate the explained risk for a specific disease/phenotype with
Taking ASD as an example, exonic de novo C-LoF SNV/INDELs contribute to P = 21% of ASD diagnoses (Iossifov et al., 2014). According to the CDC, in 2020, approximately I = 1/54 children in the US is diagnosed with ASD (https://www.cdc.gov/ncbddd/autism/data.html). Roughly I ∙ P = 3.89/1000 children are born with ASD caused by de novo C-LoF SNV/INDELs. Our data determines an average θ = 0.047 and a μ = 0.27, and thus , assuming that ASD HI-C-LoF mutations do not increase miscarriage rates. Therefore, clonal mosaicism described in this manuscript contributes an estimated E ≈ 1/6 of de novo SNV/INDELs underlying ASD diagnosis. As those mutations are of early embryonic origin, prior to sex divergence, this contribution should be similar in both parents (Dou et al., 2017), suggesting that overall, parental gonadal mosaicism contributes 1/3 of de novo ASD SNV/INDELs. This approach can be extended to other diseases or phenotypes with known monogenetic architecture, such as epilepsy, intellectual disabilities, or congenital heart disease (Homsy et al., 2015; Yang et al., 2017). Similarly, for congenital heart disease, P = 8% were caused by de novo deleterious mutations (Alankarage et al., 2019; Jin et al., 2017). According to the CDC and previously studies, approximately I ≈ 1/100 (https://www.cdc.gov/ncbddd/heartdefects/data.html), roughly I ∙ P ≈ 0.8/1000. According to the candidate gene list we determined, we calculated E ≈ 0.12/1000 for de novo SNV/INDELs underlying congenital heart disorder, suggesting that overall, paternal clonal mosaicism could explain 15% of congenital heart disease diagnoses. Note that the HI-C-LoF themselves, based on the data and considerations outlined above, will be transmitted to ∼1 in 300 concepti, likely leading to a miscarriage or congenital disease.
Data processing
Data analysis and plotting were performed using R (v 3.5.1) with ggplot2 (v 3.3.1) and Rcpp (v 1.0.3) packages; or with Python (v3.6.8) with pandas (v 0.24.2), matplotlib (v 3.1.1), NumPy (v1.16.2) SciPy (v 1.3.1) and seaborn (v 0.9.0) packages.
Statistical analyses
Statistical analyses were performed with R (Spearman, exact binomial confidence intervals, quantile analysis, and Kolmogorov-Smirnov test), GraphPad Prism (Mann-Whitney Test), and Python with pandas (95% confidence interval determination). The distribution of number of variants in Sperm, Shared and Blood did not differ significantly from a normal distribution using the Kolmogorov-Smirnov tests of normality.
ADDITIONAL RESOURCES
There are not additional resources used for this manuscript.
Supplementary Material
The sequencing summary information contains peak insert size values and depth of coverage measurement where 85% of genomic positions are sequenced above this depth. Raw mosaic SNV/INDEL calls from the 300× WGS. Information includes the genomic position, reference and alternative alleles, subject group information, genomic annotations, as well as estimated AFs of each variants in each sample, related to Figure 1 and 3.
TAS quantification in blood, saliva, and sperm samples collected at different timepoints, related to Figure 2.
Genomic annotations and predicted impact for each variant, see STAR Methods, related to Figure 4 and 5
(A) Principal component analysis revealing ancestry of study donors (YA and AA cohort), compared with the data from the 1000 Genomes project phase3 release. Shown are PC2 and PC3, as they provided best separation of the diverse populations. AFR: African; AMR: Admixed American; EAS: East Asian; EUR: European; SAS: South Asian.
(B) Computational benchmark for the detection sensitivity of the MSMF workflow. x-axis: expected AFs; y-axis: sensitivity of detection; colors: simulated read depths. Among the 10,000 simulated variants, 75% fall in non-repetitive regions. The 25% repetitive regions were excluded (see STAR Methods). For low-AF variants (i.e. 1–2%), 300–500× sequencing show similar sensitivity, outcompeting the sensitivity of lower read depth of 200×, 100×, and 50×. For high-AF variants (i.e. >20%), all depths show similar sensitivity.
(C) UpSet plot show reanalysis of data from the REACH cohort (Breuss et al., 2020a). Yield and TAS validation rate for each individual variant caller from MSMF, amounting to an overall 97.6% (80/82) validation rate.
(D) UpSet plot for the YA and AA cohort show similar relative mosaic variant detection by each method, although MosaicForecast had a higher relative yield.
(A) Bar charts show the number of mosaic SNVs and INDELs detected from the YA cohort. 266 of 376 (∼71%) of the clonal SNVs in sperm were only detectable in sperm samples.
(B) Violin plots show the AF distribution (square root-transformed; sqrt-t; for orientation 0.2 AF is highlighted with a dotted line) of Sperm, Shared, and Blood variants within each individual. Shared variants show higher peak and overall AF compared to Sperm and Blood.
(C) Rank plot of estimated blood AFs with 95% confidence intervals of Blood variants.
(D) Scatter plot show correlation between the square root-transformed (sqrt-t) AFs from WGS in blood and sperm of the YA cohort for all detected variants.
(E) Scatter plots show pair-wise AF comparisons of sperm mosaic variants from ID04 and ID12 analyzed by WGS. Sperm samples show high correlation, but some variants were only detectable in one sperm sample. n=125 variants.
(F) Scatter plot shows correlation between sqrt-t AFs from TAS in blood and saliva in YA. n=36 variants.
(G) Scatter plot shows correlation of maximum AF from TAS in any of the 3 sperm samples and the maximum differences between the 3 sperm AFs. The positive correlation suggests increased variation of absolute differences with increased AF. n=84 variants. Max Sperm AF: maximum AF of a one variant measured from all collected sperm samples of same individual.
(H) Scatter plot shows correlation of maximum AF in any of the 3 sperm samples and the maximal fold change between the 3 sperm AFs from TAS. No significant correlation was observed, suggesting that smaller AF variants may experience outsized, relative variability. n=84 variants.
(I) Modified scatter and violin plots show fold change for each tested variant. Variation reached up to 1.5–2-fold. n=84 variants.
Panels (D-H) show Spearman’s ρ and P-value across all variants.
(A) Bar charts show the number of mosaic SNVs and INDELs detected from the AA cohort. Compared with the YA cohort the number of Blood mSNV/INDELs was elevated, in line with CH due to aging.
(B-D) Rank plot of the estimated sperm and blood AF with 95% confidence intervals (exact binomial CIs) from AA. Other than the number of Blood variants, results replicate observations in the YA cohort.
(E-F) Violin plots of the AF distribution of Sperm, Shared, and Blood variants within each individual (E) and the entire AA cohort (F). Consistent with the YA cohort, Shared variants show higher peak and overall AF compared to both Sperm and Blood.
(G) Scatter plot showing the significant correlation between sqrt-t AFs from WGS in blood and sperm of AA, similar to Figure S2D. Also shown are Spearman’s ρ and P-value across all variants.
(H) Scatter plot and regression lines show AF in AA and YA cohort relative to age. Each data point represents one variant. y-axis: sqrt-t AFs; x-axis: age at (initial) sample collection. Variants grouped based on the tissues in which they were detected from 300× WGS: AFs of Sperm variants detected in sperm samples, AFs of Shared variants detected in sperm samples, AFs of Shared variants detected in blood samples, and AFs of Blood variants detected in blood samples. Regression lines with 95% prediction intervals are shown for each panel. Results show that the AFs of mosaic variants detected in this study were stable within and between cohorts across a time span of several decades.
(I) Scatter plot and regression lines show combined analysis of the average AF of AA and YA mosaic variants relative to the age of individuals.
(A) Bar plot showing combined number of mosaic variants detected in the 8 individuals from the REACH cohort.
(B) Bar plot showing per-individual number of mosaic variants detected in the 8 individuals from the REACH cohort. Compared with the YA and AA cohort the number of mSNV/INDELs detected from the REACH cohort was lower likely due to the lower sequencing depth. Clear evidence for CH was observed in one individual with advanced age (F02; 70 years old).
(C-E) Rank plot of the estimated sperm and blood AF with 95% confidence intervals (exact binomial CIs) from the AA cohort. Data reveal similar patterns as for the YA and AA cohorts.
(F-G) Violin plots of the AF distribution of Sperm, Shared, and Blood variants within each individual (F) and the entire REACH cohort (G). Consistent with the YA and AA cohorts, Shared variants showed higher peak and overall AF compared to both Sperm and Blood.
(H-H) Comparison of the number of mosaic variants and their AF distribution between the three different cohorts. While numbers are similar overall across the three cohorts, due to the lower read depth in the REACH cohort, total number of variants are slightly reduced in each category; likewise, the lowest detectable AFs are higher in this cohort.
(A) Base substitution profiles of the 4 different variant groups (tri-nucleotide level).
(B) Bar plots show the signature of the six possible base substitutions of the 4 different variant groups (single nucleotide level). Grey bands: 95% permutation intervals calculated from 10,000 random permutations of gnomAD (v2.0.1) variants using the same number of variants as found from each variant group. Mosaic variants differ from permutations and each other in several categories. Color scheme same as Figure 4A.
(C) Scatter plot showing the correlation between sqrt-t AFs from WGS in blood and sperm in each of the four aggregated groups. Panel shows Spearman’s ρ and P-value across all variants.
(A) Mutation Factor (MF) represents the mutational rate relative to cellular proliferation. Shown are two rank plots that result from mutational accumulation (green bolts) within a proliferating population of cells. Lower rates of accumulation of mutations relative to cell proliferation result in a lower MF (light blue). Higher rates of accumulation of mutations result in a higher MF (dark blue). Note that MF represents an aggregated measure of mutation rate relative to proliferation speed.
(B-F) Rank plot of the sex-chromosome normalized AFs measured for variants from each aggregated class; observed values use a color scheme as in Figure 4A. A step-wise exponential regression described in (A) is used to model the distribution of the rank plot with minimum L1-norm loss (see STAR Methods); the estimated values are shown in blue. AF0, MF, Spearman’s correlation coefficients (ρ) and P-values are shown for each variant group. All estimations with Spearman’s ρ≥0.995. A rank plot of variants in Sperm (B) was estimated with an MF=6.54, very similar to MF=6.33 from variants measured in Blood-Y (E), indicating similar mutation burden accumulation during tissue development. Rank plot of variants in Shared measured in sperm (C) were estimated with an MF=9.09, similar to the same variants measured in blood (D, MF=10.00), indicating a similar but relatively increased (compared to Sperm and Blood-Y) mutation accumulation speed during early embryonic development. A rank plot of variants in Blood-A (F) was estimated with MF=18.87, different from all other observed variant groups. Note that B-F show different maximal values on the x-axis. df: degrees of freedom.
(G) Quantile-quantile plots (qq-plots) show the comparison of AF distributions between variant groups (x and y title; indicates whether AFs from sperm or blood are used for Shared category). D statistic and P-value from a Kolmogorov–Smirnov test for each comparison pair.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Human sperm, blood, and saliva samples | See Experimental Model and Subject Details | ID01-ID17 |
| Chemicals, peptides, and recombinant proteins | ||
| AMPure XP Beads | Beckman Coulter | A63882 |
| PureCeption™ 100% Isotonic Solution | Sage/Origio | ART-2100 |
| QUINN’S™ Sperm Washing Medium | Sage/Origio | ART-1006 |
| RLT lysis buffer | Qiagen | 40724 |
| Bond-Breaker TCEP solution | Thermo Scientific | 77720 |
| stainless steel beads with 0.2 mm diameter | Next Advance | SSB02 |
| GoTaq Colorless Master Mix | Promega | M7832 |
| Exonuclease I | New England Biolabs | M0293S |
| Shrimp Alkaline Phosphatase | New England Biolabs | M0371S |
| Critical Commercial Assays | ||
| AllPrep DNA/RNA Mini Kit | Qiagen | 80204 |
| KAPA HyperPrep PCR-Free Library Prep kit | Roche | KK8505 |
| KAPA Library Quantification Kits for Illumina platforms | Roche/KAPA Biosystems | KK4824 |
| Illumina SBS kits | Illumin | 20012866 |
| Qubit dsDNA High Sensitivity kit | Thermo Fisher Scientific | Q33231 |
| Deposited Data | ||
| 200× WGS and TAS data | Sequence Read Archive (SRA) | PRJNA588332 |
| 300× WGS and TAS data | Sequence Read Archive (SRA) | PRJNA660493 |
| Software and Algorithms | ||
| Picard v 2.20.7 | “Picard Toolkit.” 2019. Broad Institute, GitHub Repository. | https://broadinstitute.github.io/picard/ |
| BWA v 0.7.8 | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| Circos v0.69–6 | (Krzywinski et al., 2009) | http://circos.ca/ |
| SAMtools v 1.7 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| GATK v 3.8–1, v4.0.4 | (McKenna et al., 2010) | https://gatk.broadinstitute.org/hc/en-us |
| BCFtools v 1.10.32 | (Li et al., 2009) | http://samtools.github.io/bcftools/ |
| BEDTools v 2.27.1 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| MosaicForecast v 8–13-2019 | (Dou et al., 2020) | https://github.com/parklab/MosaicForecast |
| Mutect2 v4.0.2 | (Benjamin et al., 2019) | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 |
| Strelka2 v 2.9.2 | (Kim et al., 2018) | https://github.com/Illumina/strelka |
| Pysam v 0.11.2.2 | (Li et al., 2009) | https://github.com/pysam-developers/pysam |
| Pysim | (Xia et al., 2017) | https://github.com/aldebjer/pysim |
| PLINK v 1.90b6.16 | (Purcell et al., 2007) | http://zzz.bwh.harvard.edu/plink/ |
| gnomAD v 2.1.1 | (Karczewski et al., 2020) | https://gnomad.broadinstitute.org/ |
| Python v 3.6.8 and v 3.7.3 | Python Software Foundation | https://www.python.org/downloads/ |
| SciPy v 1.3.1 | Community library project | https://www.scipy.org/ |
| sklearn v 0.20.1 | scikit-learn project | https://scikit-learn.org/stable/ |
| R v 3.5.1 | R Core Team | https://www.r-project.org/ |
| FSA v 0.8.30 | http://derekogle.com/FSA/authors.html | https://cran.r-project.org/web/packages/FSA/index.html |
| pingouin v 0.3.5 | https://pingouin-stats.org/ | |
| pandas v 0.24.2 | the pandas development team | https://pandas.pydata.org/ |
| seaborn v 0.9.0 | Michael Waskom | https://seaborn.pydata.org/ |
| NumPy v 1.16.2 | the NumPy project | http://numpy.org/ |
| matplotlib v 3.1.1 | https://ieeexplore.ieee.org/document/4160265 | https://maplotlib.org/ |
| Primer 3 | (Untergasser et al., 2012; Untergasser et al., 2007) | http://primer3.org/manual.html |
| Other | ||
| Cell counting chamber | Sigma-Aldrich | BR717805-1EA |
| Disruptor Genie | Scientific Industries | SI-238I |
| 1.5 ml microcentrifuge tube | USA Scientific | 1615-5500 |
| Covaris microtube system | Covaris | SKU 520053 |
| Bio-IT platform | Illumina | DRAGEN |
| Next generation sequencer | Illumina | NovaSeq 6000 |
| Next generation sequencer | Illumina | HiSeq 4000 |
| focused-ultrasonicator | Covaris | E220 |
| Plate reader | Eppendorf | PlateReader AF2200 |
| Qubit 3 Fluorometers | Thermo Fisher | Q33216 |
| Deposited Data | ||
| Raw and analyzed data | This paper | SRA: PRJNA660493 |
| Raw and analyzed data | (Breuss et al., 2020a) | SRA: PRJNA588332 |
Highlights.
Mosaic variants are stably present across serial ejaculates.
In sperm, unlike in blood, clonal mosaicism does not change with age.
Mutational origins and temporal stability suggest an embryonic origin.
Clonal sperm mosaicism is predicted to cause adverse outcomes in 1:300 concepti
Sequencing of sperm from healthy men identifies clonal mosaic mutations that are likely embryonic in origin and unlike blood, stable over age. Further, clonal mosaic mutations likely contribute to transmissible pathogenic mutations in 1 of 15 men.
Acknowledgments
We thank the participants in this study for their contribution. We also thank Drs. Yong E. Zhang, Johannes C. M. Schlachetzki, Bruce Hamilton, Hannah Carter, Amit R. Majithia, and Alan Packer for feedback and suggestions. M.W.B. was supported by an EMBO Long-Term Fellowship (ALTF 174-2015), which is co-funded by the Marie Curie Actions of the European Commission (LTFCOFUND2013, GA-2013-609409), and an Erwin Schrödinger Fellowship by the Austrian Science Fund (FWF): J 4197-B30. This study was supported by grants to J.G.G. from the NIH (U01MH108898, R01NS083823), Howard Hughes Medical Institute, and the Simons Foundation Autism Research Initiative (SFARI, 571583). Sequencing support was provided by the Rady Children’s Institute for Genomic Medicine. We thank R. Sinkovits, A. Majumdar, S. Strande, and the San Diego Supercomputer Center for hosting computing infrastructure. Data was generated using NIH SIG grant (#S10 OD026929) for an Illumina sequencer, and supported by NIH CTSA UL1TR001442.
Footnotes
Declaration of Interests
M.W.B., D.A., K.N.J., J.S., and J.G.G. are inventors on a patent (PCT/US2018/024878, WO2018183525A1) filed by UC, San Diego that is titled “Methods for assessing risk of or diagnosing genetic defects by identifying de novo mutations or somatic mosaic variants in sperm or somatic tissues”.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acuna-Hidalgo R, Veltman JA, and Hoischen A. (2016). New insights into the generation and role of de novo mutations in health and disease. Genome biology 17, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman ER, Huang HT, Roisman A, Olsson A, Colaprico A, Qin T, Lindsley RC, Bejar R, Salomonis N, Grimes HL, et al. (2019). Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov 9, 1080–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alankarage D, Ip E, Szot JO, Munro J, Blue GM, Harrison K, Cuny H, Enriquez A, Troup M, Humphreys DT, et al. (2019). Identification of clinically actionable variants from genome sequencing of families with congenital heart disease. Genet Med 21, 1111–1120. [DOI] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, and Stratton MR (2015). Clock-like mutational processes in human somatic cells. Nat Genet 47, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jager M, Yoshida K, Seemann R, Noerenberg D, Waldhueter N, Fleischer-Notter H, et al. (2018). Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 32, 1908–1919. [DOI] [PubMed] [Google Scholar]
- Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, Franjic D, Pletikos M, Pattni R, Chen BJ, Venturini E, et al. (2018). Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 359, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AD, Mello CJ, Nemesh J, Brumbaugh SA, Wysoker A, and McCarroll SA (2020). Insights into variation in meiosis from 31,228 human sperm genomes. Nature 583, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, and Lichtenstein L. (2019). Calling Somatic SNVs and Indels with Mutect2. bioRxiv 10.1101/861054 [DOI]
- Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X, Leventhal MJ, Nasser J, et al. (2020). Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzotto S, Dou Y, Ganz J, Doan RN, Kwon M, Bohrson CL, Kim SN, Bae T, Abyzov A, Network NBSM, et al. (2021). Landmarks of human embryonic development inscribed in somatic mutations. Science 371, 1249–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkis IM, Przybylski D, Chen J, and Regev A. (2014). Changes in nucleosome occupancy associated with metabolic alterations in aged mammalian liver. Cell reports 9, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, et al. (2018). Paternally inherited cis-regulatory structural variants are associated with autism. Science 360, 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandler WM, Antaki D, Gujral M, Noor A, Rosanio G, Chapman TR, Barrera DJ, Lin GN, Malhotra D, Watts AC, et al. (2016). Frequency and Complexity of De Novo Structural Mutation in Autism. Am J Hum Genet 98, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss MW, Antaki D, George RD, Kleiber M, James KN, Ball LL, Hong O, Mitra I, Yang X, Wirth SA, et al. (2020a). Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat Med 26, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuss MW, Yang X, Antaki D, Schlachetzki JCM, Lana AJ, Xu X, Chai G, Stanley V, Song Q, Newmeyer TF, et al. (2020b). Somatic mosaicism in the mature brain reveals clonal cellular distributions during cortical development. bioRxiv 10.1101/2020.08.10.244814. [DOI]
- Campbell IM, Stewart JR, James RA, Lupski JR, Stankiewicz P, Olofsson P, and Shaw CA (2014a). Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet 95, 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wisniowiecka-Kowalnik B, et al. (2014b). Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet 95, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Maman Y, Jung S, Wong N, Callen E, Day A, Kieffer-Kwon KR, Pekowska A, Zhang H, Rao SSP, et al. (2017). Genome Organization Drives Chromosome Fragility. Cell 170, 507–521 e518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlin SN, Busque L, Gale RE, Guttorp P, and Abkowitz JL (2011). The replication rate of human hematopoietic stem cells in vivo. Blood 117, 4460–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, and Chan AP (2015). PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, and Batzoglou S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS computational biology 6, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felici M. (2013). Origin, migration, and proliferation of human primordial germ cells. In Oogenesis G. Coticchio Albertini, David F, De Santis Lucia ed. (Springer; ), pp. 19–37. [Google Scholar]
- Deciphering Developmental Disorders S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Kwon M, Rodin RE, Cortes-Ciriano I, Doan R, Luquette LJ, Galor A, Bohrson C, Walsh CA, and Park PJ (2020). Accurate detection of mosaic variants in sequencing data without matched controls. Nature biotechnology 10.1038/s41587-019-0368-8. [DOI] [PMC free article] [PubMed]
- Dou Y, Yang X, Li Z, Wang S, Zhang Z, Ye AY, Yan L, Yang C, Wu Q, Li J, et al. (2017). Postzygotic single-nucleotide mosaicisms contribute to the etiology of autism spectrum disorder and autistic traits and the origin of mutations. Hum Mutat 38, 1002–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Q, Bert SA, Armstrong NJ, Caldon CE, Song JZ, Nair SS, Gould CM, Luu PL, Peters T, Khoury A, et al. (2019). Replication timing and epigenome remodelling are associated with the nature of chromosomal rearrangements in cancer. Nature communications 10, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierzak E, and Speck NA (2008). Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol 9, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellesoe SG, Workman CT, Bouvagnet P, Loffredo CA, McBride KL, Hinton RB, van Engelen K, Gertsen EC, Mulder BJM, Postma AV, et al. (2018). Familial co-occurrence of congenital heart defects follows distinct patterns. Eur Heart J 39, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayomi AP, and Orwig KE (2018). Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res 29, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed D, Stevens EL, and Pevsner J. (2014). Somatic mosaicism in the human genome. Genes 5, 1064–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Moorjani P, Sasani TA, Pedersen BS, Quinlan AR, Jorde LB, Amster G, and Przeworski M. (2019). Overlooked roles of DNA damage and maternal age in generating human germline mutations. Proc Natl Acad Sci U S A 116, 9491–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely A, and Wilkie AO (2012). Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 90, 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, and Stamatoyannopoulos JA (2010). Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 107, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AY, Yang X, Wang S, Zheng X, Wu Q, Ye AY, and Wei L. (2018). Distinctive types of postzygotic single-nucleotide mosaicisms in healthy individuals revealed by genome-wide profiling of multiple organs. PLoS Genet 14, e1007395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, McCallum K, Xu B, and Buxbaum JD (2016). A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet 48, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, and Ebert BL (2019). Clonal hematopoiesis in human aging and disease. Science 366, eaan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson H, Sulem P, Arnadottir GA, Palsson G, Eggertsson HP, Kristmundsdottir S, Zink F, Kehr B, Hjorleifsson KE, Jensson BO, et al. (2018). Multiple transmissions of de novo mutations in families. Nat Genet 10.1038/s41588-018-0259-9. [DOI] [PubMed]
- Jonsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, et al. (2017). Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, Riviere JB, Fombonne E, and O’Roak BJ (2017). Exonic Mosaic Mutations Contribute Risk for Autism Spectrum Disorder. Am J Hum Genet 101, 369–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AH, Hanchard NA, Furthner D, Fernbach S, Azamian M, Nicosia A, Rosenfeld J, Muzny D, D’Alessandro LCA, Morris S, et al. (2017). Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Yang X, Yang X, Wu Q, Zhang J, Sun D, Yang Z, Jiang Y, Wu X, Wei L, et al. (2018). Mosaicism and incomplete penetrance of PCDH19 mutations. J Med Genet 10.1136/jmedgenet-2017-105235. [DOI] [PMC free article] [PubMed]
- Liu X, Wu C, Li C, and Boerwinkle E. (2016). dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum Mutat 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, and Chanock SJ (2017). The ageing genome, clonal mosaicism and chronic disease. Current opinion in genetics & development 42, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, and Hardison RC (2015). The effects of chromatin organization on variation in mutation rates in the genome. Nature reviews Genetics 16, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, et al. (2018). Somatic mutant clones colonize the human esophagus with age. Science 10.1126/science.aau3879. [DOI] [PMC free article] [PubMed]
- Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, et al. (2015). Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore L, Cagan A, Coorens T, Neville MD, Sanghvi R, Sanders MA, Oliver TR, Leongamornlert D, Ellis P, and Noorani A. (2020a). The mutational landscape of human somatic and germline cells. bioRxiv 10.1101/2020.11.25.398172. [DOI] [PubMed]
- Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, Dawson KJ, Butler T, Rahbari R, Mitchell TJ, et al. (2020b). The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646. [DOI] [PubMed] [Google Scholar]
- Myers CT, Hollingsworth G, Muir AM, Schneider AL, Thuesmunn Z, Knupp A, King C, Lacroix A, Mehaffey MG, Berkovic SF, et al. (2018). Parental mosaicism in “de novo” epileptic encephalopathies. The New England journal of medicine 378, 1646–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, et al. (2016). Timing, rates and spectra of human germline mutation. Nat Genet 48, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, and Sander C. (2007). Determinants of protein function revealed by combinatorial entropy optimization. Genome biology 8, R232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Mulia JC, Sasaki T, Trevilla-Garcia C, Nakamichi N, Knapp D, Hammond CA, Chang BH, Tyner JW, Devidas M, Zimmerman J, et al. (2019). Replication timing alterations in leukemia affect clinically relevant chromosome domains. Blood Adv 3, 3201–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodin RE, Dou Y, Kwon M, Sherman MA, D’Gama AM, Doan RN, Rento LM, Girskis KM, Bohrson CL, Kim SN, et al. (2021). The landscape of somatic mutation in cerebral cortex of autistic and neurotypical individuals revealed by ultra-deep whole-genome sequencing. Nat Neurosci 24, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasani TA, Pedersen BS, Gao Z, Baird L, Przeworski M, Jorde LB, and Quinlan AR (2019). Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. Elife 8, e46922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward M, and Sirotkin K. (1999). dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 9, 677–679. [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, and Gaunt TR (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short RV (1997). The testis: the witness of the mating system, the site of mutation and the engine of desire. Acta Paediatr Suppl 422, 3–7. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theda C, Hwang SH, Czajko A, Loke YJ, Leong P, and Craig JM (2018). Quantitation of the cellular content of saliva and buccal swab samples. Scientific reports 8, 6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, and Nussenzweig A. (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, Kronenberg ZN, Hormozdiari F, Raja A, Pennacchio LA, et al. (2017). Genomic Patterns of De Novo Mutation in Simplex Autism. Cell 171, 710–722 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, and Rozen SG (2012). Primer3--new capabilities and interfaces. Nucleic acids research 40, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, and Leunissen JA (2007). Primer3Plus, an enhanced web interface to Primer3. Nucleic acids research 35, W71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson SM, Boyd SD, Smith CL, Fire AZ, and Sidow A. (2011). Determinants of nucleosome organization in primary human cells. Nature 474, 516–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, and Brunner HG (2012). De novo mutations in human genetic disease. Nature reviews Genetics 13, 565–575. [DOI] [PubMed] [Google Scholar]
- Wang M, Tai C, E W, and Wei L. (2018). DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic acids research 46, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, and Wei L. (2016). iFish: predicting the pathogenicity of human nonsynonymous variants using gene-specific/family-specific attributes and classifiers. Scientific reports 6, 31321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bae T, Thorpe J, Sherman MA, Jones AG, Cho S, Daily K, Dou Y, Ganz J, Galor A, et al. (2021). Comprehensive identification of somatic nucleotide variants in human brain tissue Genome biology 22, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, de Gannes MK, Luchetti G, and Pilsner JR (2015). Rapid method for the isolation of mammalian sperm DNA. Biotechniques 58, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Liu Y, Deng M, and Xi R. (2017). Pysim-sv: a package for simulating structural variation data with GC-biases. BMC bioinformatics 18, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Liu A, Xu X, Yang X, Zeng Q, Ye AY, Yu Z, Wang S, Huang AY, Wu X, et al. (2017). Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Scientific reports 7, 15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye AY, Dou Y, Yang X, Wang S, Huang AY, and Wei L. (2018). A model for postzygotic mosaicisms quantifies the allele fraction drift, mutation rate, and contribution to de novo mutations. Genome Res 28, 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Gowers KHC, Lee-Six H, Chandrasekharan DP, Coorens T, Maughan EF, Beal K, Menzies A, Millar FR, Anderson E, et al. (2020). Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The sequencing summary information contains peak insert size values and depth of coverage measurement where 85% of genomic positions are sequenced above this depth. Raw mosaic SNV/INDEL calls from the 300× WGS. Information includes the genomic position, reference and alternative alleles, subject group information, genomic annotations, as well as estimated AFs of each variants in each sample, related to Figure 1 and 3.
TAS quantification in blood, saliva, and sperm samples collected at different timepoints, related to Figure 2.
Genomic annotations and predicted impact for each variant, see STAR Methods, related to Figure 4 and 5
(A) Principal component analysis revealing ancestry of study donors (YA and AA cohort), compared with the data from the 1000 Genomes project phase3 release. Shown are PC2 and PC3, as they provided best separation of the diverse populations. AFR: African; AMR: Admixed American; EAS: East Asian; EUR: European; SAS: South Asian.
(B) Computational benchmark for the detection sensitivity of the MSMF workflow. x-axis: expected AFs; y-axis: sensitivity of detection; colors: simulated read depths. Among the 10,000 simulated variants, 75% fall in non-repetitive regions. The 25% repetitive regions were excluded (see STAR Methods). For low-AF variants (i.e. 1–2%), 300–500× sequencing show similar sensitivity, outcompeting the sensitivity of lower read depth of 200×, 100×, and 50×. For high-AF variants (i.e. >20%), all depths show similar sensitivity.
(C) UpSet plot show reanalysis of data from the REACH cohort (Breuss et al., 2020a). Yield and TAS validation rate for each individual variant caller from MSMF, amounting to an overall 97.6% (80/82) validation rate.
(D) UpSet plot for the YA and AA cohort show similar relative mosaic variant detection by each method, although MosaicForecast had a higher relative yield.
(A) Bar charts show the number of mosaic SNVs and INDELs detected from the YA cohort. 266 of 376 (∼71%) of the clonal SNVs in sperm were only detectable in sperm samples.
(B) Violin plots show the AF distribution (square root-transformed; sqrt-t; for orientation 0.2 AF is highlighted with a dotted line) of Sperm, Shared, and Blood variants within each individual. Shared variants show higher peak and overall AF compared to Sperm and Blood.
(C) Rank plot of estimated blood AFs with 95% confidence intervals of Blood variants.
(D) Scatter plot show correlation between the square root-transformed (sqrt-t) AFs from WGS in blood and sperm of the YA cohort for all detected variants.
(E) Scatter plots show pair-wise AF comparisons of sperm mosaic variants from ID04 and ID12 analyzed by WGS. Sperm samples show high correlation, but some variants were only detectable in one sperm sample. n=125 variants.
(F) Scatter plot shows correlation between sqrt-t AFs from TAS in blood and saliva in YA. n=36 variants.
(G) Scatter plot shows correlation of maximum AF from TAS in any of the 3 sperm samples and the maximum differences between the 3 sperm AFs. The positive correlation suggests increased variation of absolute differences with increased AF. n=84 variants. Max Sperm AF: maximum AF of a one variant measured from all collected sperm samples of same individual.
(H) Scatter plot shows correlation of maximum AF in any of the 3 sperm samples and the maximal fold change between the 3 sperm AFs from TAS. No significant correlation was observed, suggesting that smaller AF variants may experience outsized, relative variability. n=84 variants.
(I) Modified scatter and violin plots show fold change for each tested variant. Variation reached up to 1.5–2-fold. n=84 variants.
Panels (D-H) show Spearman’s ρ and P-value across all variants.
(A) Bar charts show the number of mosaic SNVs and INDELs detected from the AA cohort. Compared with the YA cohort the number of Blood mSNV/INDELs was elevated, in line with CH due to aging.
(B-D) Rank plot of the estimated sperm and blood AF with 95% confidence intervals (exact binomial CIs) from AA. Other than the number of Blood variants, results replicate observations in the YA cohort.
(E-F) Violin plots of the AF distribution of Sperm, Shared, and Blood variants within each individual (E) and the entire AA cohort (F). Consistent with the YA cohort, Shared variants show higher peak and overall AF compared to both Sperm and Blood.
(G) Scatter plot showing the significant correlation between sqrt-t AFs from WGS in blood and sperm of AA, similar to Figure S2D. Also shown are Spearman’s ρ and P-value across all variants.
(H) Scatter plot and regression lines show AF in AA and YA cohort relative to age. Each data point represents one variant. y-axis: sqrt-t AFs; x-axis: age at (initial) sample collection. Variants grouped based on the tissues in which they were detected from 300× WGS: AFs of Sperm variants detected in sperm samples, AFs of Shared variants detected in sperm samples, AFs of Shared variants detected in blood samples, and AFs of Blood variants detected in blood samples. Regression lines with 95% prediction intervals are shown for each panel. Results show that the AFs of mosaic variants detected in this study were stable within and between cohorts across a time span of several decades.
(I) Scatter plot and regression lines show combined analysis of the average AF of AA and YA mosaic variants relative to the age of individuals.
(A) Bar plot showing combined number of mosaic variants detected in the 8 individuals from the REACH cohort.
(B) Bar plot showing per-individual number of mosaic variants detected in the 8 individuals from the REACH cohort. Compared with the YA and AA cohort the number of mSNV/INDELs detected from the REACH cohort was lower likely due to the lower sequencing depth. Clear evidence for CH was observed in one individual with advanced age (F02; 70 years old).
(C-E) Rank plot of the estimated sperm and blood AF with 95% confidence intervals (exact binomial CIs) from the AA cohort. Data reveal similar patterns as for the YA and AA cohorts.
(F-G) Violin plots of the AF distribution of Sperm, Shared, and Blood variants within each individual (F) and the entire REACH cohort (G). Consistent with the YA and AA cohorts, Shared variants showed higher peak and overall AF compared to both Sperm and Blood.
(H-H) Comparison of the number of mosaic variants and their AF distribution between the three different cohorts. While numbers are similar overall across the three cohorts, due to the lower read depth in the REACH cohort, total number of variants are slightly reduced in each category; likewise, the lowest detectable AFs are higher in this cohort.
(A) Base substitution profiles of the 4 different variant groups (tri-nucleotide level).
(B) Bar plots show the signature of the six possible base substitutions of the 4 different variant groups (single nucleotide level). Grey bands: 95% permutation intervals calculated from 10,000 random permutations of gnomAD (v2.0.1) variants using the same number of variants as found from each variant group. Mosaic variants differ from permutations and each other in several categories. Color scheme same as Figure 4A.
(C) Scatter plot showing the correlation between sqrt-t AFs from WGS in blood and sperm in each of the four aggregated groups. Panel shows Spearman’s ρ and P-value across all variants.
(A) Mutation Factor (MF) represents the mutational rate relative to cellular proliferation. Shown are two rank plots that result from mutational accumulation (green bolts) within a proliferating population of cells. Lower rates of accumulation of mutations relative to cell proliferation result in a lower MF (light blue). Higher rates of accumulation of mutations result in a higher MF (dark blue). Note that MF represents an aggregated measure of mutation rate relative to proliferation speed.
(B-F) Rank plot of the sex-chromosome normalized AFs measured for variants from each aggregated class; observed values use a color scheme as in Figure 4A. A step-wise exponential regression described in (A) is used to model the distribution of the rank plot with minimum L1-norm loss (see STAR Methods); the estimated values are shown in blue. AF0, MF, Spearman’s correlation coefficients (ρ) and P-values are shown for each variant group. All estimations with Spearman’s ρ≥0.995. A rank plot of variants in Sperm (B) was estimated with an MF=6.54, very similar to MF=6.33 from variants measured in Blood-Y (E), indicating similar mutation burden accumulation during tissue development. Rank plot of variants in Shared measured in sperm (C) were estimated with an MF=9.09, similar to the same variants measured in blood (D, MF=10.00), indicating a similar but relatively increased (compared to Sperm and Blood-Y) mutation accumulation speed during early embryonic development. A rank plot of variants in Blood-A (F) was estimated with MF=18.87, different from all other observed variant groups. Note that B-F show different maximal values on the x-axis. df: degrees of freedom.
(G) Quantile-quantile plots (qq-plots) show the comparison of AF distributions between variant groups (x and y title; indicates whether AFs from sperm or blood are used for Shared category). D statistic and P-value from a Kolmogorov–Smirnov test for each comparison pair.
Data Availability Statement
Raw whole genome sequencing and targeted amplicon sequencing BAM files used in this study are available on SRA (accession number: PRJNA660493 and PRJNA588332). Summary tables of the data are included as supplementary tables. Codes for data analysis pipelines as well as codes to generate the figures are freely available on GitHub at https://github.com/shishenyxx/Sperm_control_cohort_mosaicism. Other materials or software are detailed in STAR Methods.