

Abstract
Botrytis cinerea is a fungal pathogen that causes necrotic disease on more than a thousand known hosts widely spread across the plant kingdom. How B. cinerea interacts with such extensive host diversity remains largely unknown. To address this question, we generated an infectivity matrix of 98 strains of B. cinerea on 90 genotypes representing eight host plants. This experimental infectivity matrix revealed that the disease outcome is largely explained by variations in either the host resistance or pathogen virulence. However, the specific interactions between host and pathogen account for 16% of the disease outcome. Furthermore, the disease outcomes cluster among genotypes of a species but are independent of the relatedness between hosts. When analyzing the host specificity and virulence of B. cinerea, generalist strains are predominant. In this fungal necrotroph, specialization may happen by a loss in virulence on most hosts rather than an increase of virulence on a specific host. To uncover the genetic architecture of Botrytis host specificity and virulence, a genome-wide association study (GWAS) was performed and revealed up to 1492 genes of interest. The genetic architecture of these traits is widespread across the B. cinerea genome. The complexity of the disease outcome might be explained by hundreds of functionally diverse genes putatively involved in adjusting the infection to diverse hosts.
Keywords: plant–pathogen interactions, Botrytis cinerea, fungus, host specificity, generalist pathogen, polygenic virulence, plant domestication
Introduction
Plants and their pathogens exist within ecosystems that create diverse combinatorial abiotic and biotic pressures (Glazebrook and Roby 2018). The additivity of these various pressures opens the question: how does it translate into the genome of species and impact specific interactions to ecosystems? Answering this question remains a challenge, especially given the diversity of interactions, ranging from within (e.g., plant–plant) to across kingdom (e.g., plant–pathogen) interactions and from mutualism to predation. When focusing on plant–pathogen interactions, various levels of complexity are found in nature. Historically, the predominant focus of research has been on pairwise interactions (e.g., one plant, one pathogen). However, more complex plant–pathogen networks wherein a plant or pathogen interacts with a broader range of organisms are now trending (Zhang et al. 2019; Delplace et al. 2020).
The range of plant–pathogen interactions (Möller and Stukenbrock 2017) has created a myriad of pathogen virulence mechanisms (mechanisms with detrimental effects to a host) to attack and/or interfere with the plant resistance (the host's capacity to fight back against the pathogen; Lannou 2012). Virulence mechanisms vary across the continuum of pathogen lifestyles and host specificity (Barrett et al. 2009; Cowger and Brown 2019; Frantzeskakis et al. 2020). At one end of the continuum, specialist biotrophs develop intricate relationships, living and feeding within the host tissues. At the other end, generalist necrotrophs attack, kill and feed on degraded tissues. The pressure on pathogens to specialize also varies across the continuum of lifestyles. Some pathogens develop specific and intricate responses to their host (e.g., biotrophs), while other pathogen interactions are broad ranged (e.g., saprophytes) (Leggett et al. 2013; Krah et al. 2018). Pathogens also cover a large range from specialists attacking one or few hosts to generalists that can have multiple hosts, taxonomically related or not. Both lifestyle and host-specificity can influence the genetic architecture of the plant–pathogen interaction (Barrett et al. 2009; Morris and Moury 2019). This leads to diverse genetic architectures that can explain the specificity and virulence of pathogens (Möller and Stukenbrock 2017; Morris and Moury 2019). In pathosystems following the co-evolution model such as Magnaporthe oryzae and Phytophtora infestans, few avirulence factors in the pathogen and resistance proteins in the host dictate the interactions (Brown and Tellier 2011; Białas et al. 2018; Upson et al. 2018). Pathogens such as Fusarium oxysporum and Alternaria alternata comprise individual strains that display host specialization linked to accessory chromosomes (van der Does and Rep 2007; Plissonneau et al. 2017; Bertazzoni et al. 2018). Other fungal plant pathogens such as Ustilago maydis have compartmentalized genomic regions with rapidly evolving gene clusters (Möller and Stukenbrock 2017; Plissonneau et al. 2017). Large-effect genes dominate such qualitative genetic architecture with two-speed genome architectures being frequent (Dong et al. 2015; Plissonneau et al. 2017). Finally, pathogens such as Sclerotina sclerotiorum and Microbotryum lychnidis‐dioicae rely on quantitative genetic architectures based on hundreds of small effect loci (Poland et al. 2009; Lannou 2012; Corwin and Kliebenstein 2017; Cowger and Brown 2019). As advanced models of quantitative architectures, recent transcriptomic studies show that necrotrophs have intricate interactions with hosts by altering gene networks involved in signaling, regulation of cellular processes, transport, metabolism, and proteolysis (Zhang et al. 2019; Delplace et al. 2020).
As a host, a plant can be exposed to multiple pathogens with various attack strategies. As counter-measures to the diversity of pathogens, plants have developed multi-layered resistance mechanisms ranging from constitutive physical defenses to inducible responses coordinated by the plant immune system upon the recognition of danger (Glazebrook 2005; Herman and Williams 2012; Wilkinson et al. 2019). The individual components in these multi-layered defense systems have complex and diverse evolutionary histories. Defense components such as resistance genes (Jacob et al. 2013) or specialized defense compounds (Chae et al. 2014) are frequently specific to limited lineages or even individual plant species. In contrast, other components such as the cell wall (Sørensen et al. 2010), defense hormone signaling (Berens et al. 2017), or reactive oxygen species (Inupakutika et al. 2016) are widely shared across plant lineages. Crop domestication further complicates the pattern of plant resistance through artificial selection. Crop domestication can create a loss of genetic diversity and canonical reductions in plant defense compounds (Karasov et al. 2014; Chen et al. 2015; De Gracia et al. 2015; Whitehead et al. 2017). It remains to clarify how these various evolutionary histories of plant resistance contribute to plant–pathogen interactions.
In this study, we use the necrotrophic fungus, Botrytis cinerea (gray mold) to estimate the relative contribution of variation across multiple hosts in shaping the host-strain interactions. B. cinerea causes billions/annum of crop damage both pre- and post-harvest to various crops from ornamentals to vegetables and fruits (Fillinger and Elad 2015; Veloso and van Kan 2018). It has a broad host range from mosses to gymnosperms to eudicots, and is classically considered as a generalist fungal pathogen. An analysis of known (Elad et al. 2016) hosts showed that the eudicots contain 71% of the plant species with documented Botrytis disease symptoms, currently 996 species are widely spread across orders (Figure 1). B. cinerea has extensive standing genetic variation in both local and global populations (Ma and Michailides 2005; Calpas et al. 2006; Atwell et al. 2015, 2018; Walker et al. 2015; Bardin 2018). In addition, B. cinerea spores are airborne and widespread within the environment ranging from presence on plants to presence on nonplant substrates including within rain, indicating an ability to spread rapidly and widely (Leyronas et al. 2015a; Bardin 2018). In the Botrytis-Arabidopsis pathosystem, genome-wide association studies (GWAS) detected complex genetic architectures in both Arabidopsis thaliana and Botrytis cinerea. In Arabidopsis, resistance genes accounted only a small proportion of candidate genes that were connected to disease resistance, signaling, reactive oxygen species, and other developmental processes such as light signal transduction (Corwin et al. 2016; Fordyce et al. 2018). In B.cinerea, genes part of networks associated with vesicular transport, degradation enzymes, metabolism, and cellular processes are frequently identified (Soltis et al. 2019; Zhang et al. 2019; Soltis et al. 2020).
Figure 1.

Disease symptoms caused by B. cinerea observed on plants from the core Eudicots. The list of plant species with disease symptoms was compiled from Elad et al. (2016). The tree represents major orders from the basal Eudicots (in green), Asterids (in orange), and Rosids (in pink). The size of the circle at the end of the branches is proportional to the species diversity of the order. The species diversity was extracted from the Missouri Botanical garden angiosperm phylogeny (http://www.mobot.org/MOBOT/Research/APweb/ [last accessed June 2021]). For each order, the number of species with known disease symptom is indicated.
Because B. cinerea strains infect a large array of species (Figure 1), it is possible to empirically assess how host resistance and strain virulence contribute to the disease outcome. We generated infectivity matrices for 90 plant genotypes from eight eudicot species against 98 diverse B. cinerea strains under experimental conditions in a standardized environment to ask a set of questions about this generalist pathogen. First, how do genetic diversity among and within eight hosts and among Botrytis’ strains contribute to the disease outcome? Second, do crop domestication and phylogenetic distances consistently influence the interactions with this generalist pathogen? And finally, how do the genomic architecture of virulence and host specificity relate to this generalist pathogen?
Materials and methods
Plant material and growth condition
The experimental design of this study (Figure 2) was chosen to assess the effect of genetic diversity in and between crops in interaction with B. cinerea. It considers four Asterales, one Solanales, one Fabales, and two Brassicales, covering various phylogenetic distances, centered on the Asterales, a family with large species diversity and frequently hosting B. cinerea (Figure 1). While the chosen species partially represent the eudicot phylogeny, they were also chosen to represent the diversity of plant domestication syndromes and geographical origins (Meyer et al. 2012) (Supplementary Figure S1). Helianthus annuus and Glycine max were domesticated for seeds, Solanum for fruit (Lin et al. 2014) while Lactuca, Cichorium intybus, and Cichorium endivia were domesticated for leaf and root (Dempewolf et al. 2008). All of these species underwent a single domestication event while Brassica rapa domestication is more complex, being domesticated multiple times for multiple traits including seed, leaf, and root characteristics (Bird et al. 2017).
Figure 2.
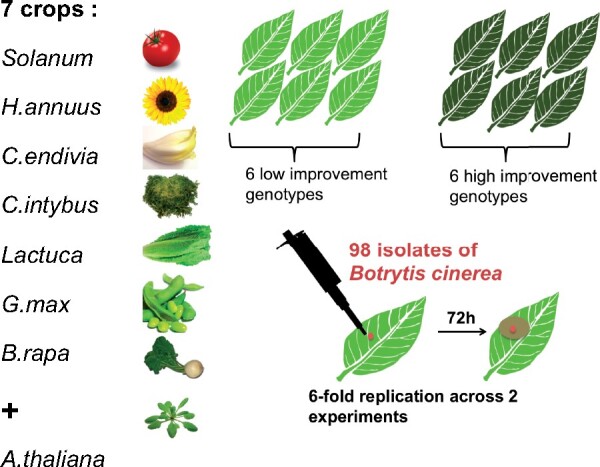
Experimental design. We tested the virulence of 98 B. cinerea strains on seven eudicot crop species and the plant model A. thaliana using randomized complete block design detached leaf assays with sixfold replication.
Within each plant species, six genotypes were chosen to represent the genetic diversity in the high improvement germplasm (cultivar and inbred lines) while six genotypes represented the low improvement germplasm (wild, landraces) (Figure 2). Twelve genotypes per host were chosen as a compromise between the ability to conduct the experiment while testing the Botrytis strain diversity and allowing independent biological replication. The genotypes were chosen within the seed collections of the US department of agriculture (Soybean NAM, Sunflower, and brassica mapping populations), the UC Davis Tomato Genetics Resource Center and the Center for Genetic Resources Netherland (CGN), with the help of experts on these collections to maximize genetic diversity, as known in 2014 at the beginning of this project. For simplicity in the language, we refer to the different crops as “species” although referring to taxa would be more appropriate for tomato and lettuce for which we selected sister species for wild and domesticated genotypes. When referring to Lactuca (lettuce), Lactuca sativa was sampled for domesticated varieties and Lactuca serriola for wild accessions (Walley et al. 2017). When referring to Solanum (tomato), Solanum lycopersicum was sampled for domesticated varieties and Solanum pimpenellifolium for wild accessions native from Ecuador and Peru (Lin et al. 2014). Landrace genotypes were selected for C. endivia for which no wild relative is known and two genotypes of B. rapa (Figure 2, Supplementary Figure S1). For soybean, the comparison was within G. max as the growth behavior, vining, and growth conditions, tropical, for wild soybean, G. soja, was sufficiently different as to unnecessarily confound the comparison. While none of the crop species have been explicitly bred for resistance to B. cinerea, domestication of lettuce and chicory did shift their growing conditions toward leaves densely compacted and adaptation to colder and wetter environments, two traits associated with increased B. cinerea prevalence (De Vries 1997; Dempewolf et al. 2008; Mou 2011). Domestication in all of these species has also been associated with significant changes in their interactions with insects and specialist pathogens (Karasov et al. 2014; Chen et al. 2015; De Gracia et al. 2015; Whitehead et al. 2017).
A. thaliana was used as a reference for genotypes with single gene alterations in major plant defense. This reference dataset is composed of Col-0 and five knockout mutants altering plant immunity (Zhang et al. 2017; Atwell et al. 2018), through the jasmonic pathway (anac055 and coi1), salicylic pathway (npr1 and tga3) and camalexin pathway (pad3), an anti-fungal defense compound in Arabidopsis.
C. endivia, C. intybus, B. rapa, G. max, and A. thaliana seeds were directly sowed in standard potting soil. Solanum and Lactuca seeds were bleach-sterilized and germinated on wet paper in the growth chamber using flats covered with humidity domes. After 7 days, the seedlings were transferred to soil. Seed surface sterilization and scarification was used for H. annuus to increase seed germination. Seeds were surface sterilized in 30% bleach for 12 minutes, followed by rinsing with sterilized distilled water, and then soaked in sterilized water for 3 hours. One-fourth of the seeds were cut off from the cotyledon end, then placed in 100 mg/L gibberellic acid for 1 hour, followed by rinsing several times with sterilized distilled water. Treated seeds were then put in covered sterilized Petri dishes with wet sterilized germination disks at 4°C for 2 weeks and sowed.
All plants were grown in growth chambers in pots containing Sunshine Mix #1 (Sun Gro Horticulture, Agawam, MA, USA) horticulture soil at 20°C with 16 hours photoperiod at 100–120 mE light intensity. All plants were watered every 2 days with deionized water for the first 2 weeks and then with nutrient solution (0.5% N-P-K fertilizer in a 2-1-2 ratio; Grow More 4-18-38). All infection experiments were conducted on mature (nonjuvenile) fully developed leaves collected on adult plants that grew in these conditions for 4–8 weeks (Supplementary Table S2) to account for the different developmental rates. As it is challenging to fully compare developmental stages across species (soybean stages are defined by nodes, sunflower by leaf size and so on), all leaves for the assays were collected on plants in the vegetative phase at least several weeks before bolting initiation to minimize ontogenetic effects.
Botrytis collection
This study is based on a collection of 98 strains of B. cinerea that samples 14 plant hosts and to a smaller degree geographical origins. Ninety percent of the strains were isolated in California, largely in vineyards on the UC Davis campus, while the remaining 10% of the collection are worldwide strains (Supplementary Table S3). The spore collection is maintained for long-term preservation as conidial suspension in 60% glycerol at −80°C. The strains were grown for each experiment from spores on peach at 21°C for 2 weeks.
All strains were previously whole-genome sequenced (WGS) to an average 164-fold coverage (Atwell et al. 2015, 2018). All reads were aligned to B05.10 genome assembly ASM83294v1 (Van Kan et al. 2017) and subsequent analyses were done with 271,749 SNPs at MAF 0.20 and less than 20% of missing calls (Soltis et al. 2019) as determined by using GATK’s UnifiedGenotyper. These thresholds were chosen due to the high genetic diversity detected, on average a genetic variant every 27 bp (Atwell et al. 2018), and the potential role of structural variations in generating missing data in this haploid fungi.
In California, no evidence of host-specialization or local population structure has previously been found (Ma and Michailides 2005; Saito and Xiao 2018; Cosseboom et al. 2019; Soltis et al. 2019). To further confirm absence of population structure in the collection that also includes the worldwide samples, we estimated the strain relatedness and calculated the genetic differentiation (Fst) between Californian and worldwide strains. The SNP table was converted into phylip format using PGD-spider (Lischer and Excoffier 2012) and plotted in Splitstree (Huson and Bryant 2006). For Fst estimation, the vcf file was imported with the R package vcfR and converted to the genind format. The Fst values were calculated using all SNPs with the hierfstat package (Goudet 2005) following Weir and Cockerham (1984).
Detached leaf assay
To maximize the comparability across the diverse collection of wild relatives and crop species domesticated for different traits, leaves were chosen as a common plant organ. Detached leaf assays were conducted on adult leaves (Denby et al. 2004; Corwin et al. 2016). In brief, leaves were cut and added to trays filled with 1 cm of 1% phyto-agar. The phyto-agar provided water and nutrients to the leaf that maintained physiological functions during the course of the experiment. Botrytis spores were extracted in sterile water, counted with a hemacytometer and sequentially diluted with 50% grape juice to 10 spores/ul. Drops of 4ul (40 spores of Botrytis) were used to inoculate the leaves. From spore collection to inoculation, Botrytis spores were maintained on ice to stop spore germination. Spores were maintained under agitation while inoculating leaves to keep the spore density homogeneous and decrease technical error. The inoculated leaves were maintained under humidity domes under constant light. For each host tested, the experiment included three replicates for each strain × plant genotype in a randomized complete block design and were repeated over two experiments for a total of six replicates per strain × plant genotype.
The lesion area is a quantitative measurement of the interaction between the fungus and the host plant. To render a project of this size possible, a single time point was chosen to measure the plant-Botrytis interaction. At 72 hours post infection (hpi), the lesions are well defined on all targeted species but have not reached the edges of the leaves and thus are not tissue limited. Spore germination assays in 50% grape juice showed that strains germinate within the first 12 hours. On all species, the same pattern of growth was observed: after 48 hours, most strains are visible within the leaf with the beginning of lesion formation. From 36 hours onward, Botrytis growth is largely linear and grows until the entire leaf is consumed (Rowe et al. 2010).
Image analysis
Each experimental tray, a unit of the randomized complete block design, was photographed in full at a fixed distanced using a camera stand and an 18 Mp T3i Canon camera outfitted with an EF-S 10–22 mm f/3.5–4.5 USM ultra-wide angle lens. Two lateral lamps and a white background reflecting light were used to minimize shading. The images were saved in jpeg format and analyzed with an R pipeline (Fordyce et al. 2018). In short, the images were transformed into hue/saturation/value (hsv) color space and threshold accounting for leaves color and intensities were defined for each species. Masks marking the leaves and lesions were created by the script and further confirmed manually. The lesions were measured by counting the number of pixels of the original pictures within the area covered by the lesion mask. The numbers of pixels were converted into centimeters using a reference scale within each image.
Data quality control
A dataset of 51,920 lesions was generated in this project, but not all leaves inoculated with Botrytis developed a visible lesion at 72 hpi. These “failed lesions” can be explained either by technical or biological failures. Technical failures, such as failed spore germination, stochastic noise, or other nonvirulence-related issues can bias the true estimates of the mean. To partition biological from technical failures, the lesion area distribution was analyzed for each species, and thresholds were fixed at the top of the first bell curve (Supplementary Table S4). These small areas are either small lesions or the spread of the inoculation drop without lesions, considered as a technical error. Therefore, a lesion below that size threshold was considered a technical error only if the median of lesion area for a plant genotype—strain pair was larger than the threshold. The rationale is the following: when most lesions within a host-Botrytis comparison are of small size, the likelihood of biological reasons for such small lesion areas is high. In contrast, when the majority of lesion areas in a host-Botrytis comparison are large, it is likely that any outlier small lesions are due to technical errors like pipetting issues is high. A total of 6395 lesions (13% of all lesions) were considered as technical failures and removed from the dataset. All statistical analyses and modeling were run on both original and filtered datasets. The removal of technical failures does not impact the effect size of the estimates but simply allowed for more variance to be attributed to biological terms in the model and less in the random error term. This is as expected when partitioning out predominantly technical failures.
Statistical analysis
All data handling and statistical analyses were conducted in R. Lesion area was modeled independently for each species using linear mixed models with the lme4 package. Variance estimates were converted into a percentage of total variance to ease the comparison of the different models.
To test the respective contribution of host resistance and pathogen virulence into the disease outcome for each host species individually, we modeled the lesion area according to the host (PlantGeno) and pathogen (Strain) genotypic interactions while correcting for experimental error. The plant genotypes were nested within their improvement (Improv) status (high or low) for each species.
Linear mixed model: Lesion.Area ∼ Strain + Improv + Improv/PlantGeno + Strain*Improv + Strain*Improv/PlantGeno + (1|Exp.Replicate) + (1|Exp.Tray) + (1|Indiv.Plants).
Experimental replicate (experiment 1 or 2) and trays (micro-environment with a subset of infected leaves within the randomized complete block design) as well as the individual plants from which the leaves were collected for the detached leaf assay are considered as random factors. Plant genotypes and Botrytis strains were coded as fixed factors, although they represent random sampling of the plant and fungal species. This simplification of the model was done because previous research (Corwin et al. 2016; Fordyce et al. 2018) showed that this does not influence the effect size or significance of the estimates, and dramatically decreases the computational requirements to run the model.
To check the potential effect of leaf area on the development of the lesion area, linear models including the strains, the host genotypes, experimental factors, and leaf area were run. The percentages of variance of lesion area explained by the leaf size were not significant and linked to a low fraction of variance (range: 0.09–0.44% of variance). Therefore, that leaf area was not included in the final model.
To incorporate the data across the host species and test the respective contributions of the host diversity, crop domestication, and pathogen virulence into the disease outcome, a meta-analysis linear model was run. The improvement status or plant genotypes were nested within the plant species to account for their common evolutionary history and possibly shared resistance traits of genotypes associated with the domestication bottleneck.
Linear model: Mean.Lesion.Area ∼ Strain + Species + Species/Improv + Species/PlantGeno + Strain*Species+ Strain*Species/Improv.
This modeling was performed on the mean lesion area of replicates rather than individual lesions. Given that the experimental design allows controlling for three random factors, model corrected least-square mean (LS-mean) of each genotype infected with each strain were calculated with the emmeans function using the Satterwaite approximation (Lenth et al. 2018).
To visualize the disease outcome resulting from the individual interactions of the 91 Botrytis strains and with the 90 plant genotypes (including Arabidopsis), a clustered heatmap was constructed on standardized LS-means with iheatmapr (Schep and Kummerfeld 2017). Seven isolates did not grow or sporulate in time for some of the detached leaf assays. It resulted in missing data for some of the eight species tested. These isolates were dropped for the following analyses, following concerns about the sensitivity of hierarchical clustering to missing data. The LS-means were standardized (z-score) over each plant genotype by centering the mean to zero and fixing the standard deviation to one to overcome the large variation on lesion area across species and large variation in variance linked to the lesion area. Species with low lesion area had also small variance while species with large lesion area presented large variance. The unsupervised hierarchical clustering was run with the “complete” agglomerative algorithm on Euclidean distances. The significance of the dendrogram was estimated with pvclust (Suzuki and Shimodaira 2006) over 20,000 bootstraps. The significance of branches was fixed at α = 0.95. For the plant genotypes dendrogram, branches were consistently assigned across hierarchical clustering methods (both “complete” and “average” algorithms were ran) and bootstrapping while in the Botrytis strains dendrogram, none of the branches showed consistency.
Using our large infectivity matrix, we estimated the host specificity and overall virulence of the B. cinerea strains in our collection. The global virulence of each strain was calculated as the mean lesion area across the eight eudicot species. Host specificity is usually calculated qualitatively based on the broadness of presence/absence of symptoms on hosts. As B. cinerea is a necrotroph with quantitative disease symptoms, we estimated host specificity as the variation in disease symptom across hosts. With such estimates, generalist strains will tend to have equal virulence on various host, while more specialized strains will have increased virulence on a host relative to the other hosts. To account for variations in resistance among hosts, we used the coefficient of variation (standard deviation corrected by the mean σ/μ) of least-square mean of lesion area across the eight eudicot species (Poisot et al. 2012). Low host specificity indicates that strains grew consistently across the eight species, while high host specificity indicates large variation in lesion area across species, therefore higher virulence on some species.
Genome-wide association study
To estimate the respective genetic architectures of the virulence and host specificity of the strains in our collection, we performed a GWAS using two different statistical models. The first model was a univariate linear model modeling either virulence or host specificity to the SNPs matrix as implemented in GEMMA (Zhou et al. 2013), which implement both effect size and P-values (Wald test). To account for multiple testing, a Bonferroni approach was used to establish the significance threshold (P < 1.29 × 10−5). Instead of correcting on the number of SNPs in the dataset, we used the genetic type I error calculator to estimate the effective number of SNPs that account for linkage disequilibrium (Li et al. 2012). We also tested the false discovery rate associated with this 1.29 × 10−5 significance threshold by running 10 permutation linear models on both traits. By permuting the phenotypes, it allows testing how often a P-value smaller than the threshold happens in random associations. The second model was a Bayesian sparse linear mixed model (BSLMM) using Markov chain Monte Carlo algorithm implemented in GEMMA (Zhou et al. 2013). The BSLMM model conducts the marker association tests while accounting for population structure and implementing a polygenic approach. The output of the BSLMM includes estimates of the effect size and the significance, calculated as the posterior inclusion probability (PIP). The standardized relatedness matrix was calculated in GEMMA based on SNP data in the binary ped format. For each trait, 20 separate runs with 500000 burn-in and 5000000 iterations with recording every 10 steps were performed. We restricted the priors used to estimate the PVE to 0.1 < h < 0.9 to increase convergence. For each SNP, the median of the PIP distribution of the 20 runs was used for subsequent analyses. To estimate the null distribution of the PIP, we performed 10 random permutations of both host specificity and virulence. PIP larger than 8.2 x10-5 (equivalent to 1% chance of false-positive above this threshold) was considered significant based on the null distribution from the 20 randomizations.
For the identification of candidate genes, the significant SNPs were filtered based on their potential effect. SNPs were annotated based on their location in the B05.10 genome ASM83294v1 assembly (Van Kan et al. 2017) with SnpEff (Cingolani et al. 2012). SNPs annotated as missense variants (modification of the amino acid in the coding region), 5’ UTR premature start codon gain, missense in the splicing area, stop gained or start lost by SnpEff and having higher probability of functional effect than intergenic gene variants are discussed in priority. The complete list of genes identified by LM and/or BSLMM models for virulence and/or host specificity was analyzed for estimation of the individual gene effects. The mean of the P-value and effect sizes of all SNPs within the genes or with 500 bp downstream or upstream were calculated. The gene functional annotations were extracted from the fungal genomic resources portal (fungidb.org). Gene ontology and metabolic enrichment were also implemented on the online portal, however, none of the enrichment analyses were significant after correction for multiple testing.
Data availability
Correspondence and requests for materials should be addressed to Kliebenstein@ucdavis.edu. R codes and datasets are available on Dryad https://doi.org/10.25338/B8Z91N. Supplementary material is available at figshare: https://doi.org/10.25387/g3.14591145.
Results
Population structure in the strain collection
Although genetic differentiation follows trends of isolation by distance for most species, we observed that our strain collection contains no significant genetic stratification by either geographic or host species origin based on genome-wide SNPs (Figure 3). If genetic differentiation were structuring our strain collection, the worldwide strains would be on different branches than the B. cinerea strains collected in California’s vineyard. Similarly, if strains had adapted specifically to growth on grapes, there would be stratification between the grape and nongrape isolates. This is confirmed by low Fst values between strains isolated in Europe and California with Fst = 0.03 and between grape and other hosts averaging at Fst= 0.02 (Fst range: 0–0.13). Because the isolates behave genomically as a single large population, we combined all the isolates to maximize statistical power in the ensuing analysis.
Figure 3.

Population structure and genetic distances in the Botrytis strain collection. Color represents the host on which the strains were collected. Dashed lines represent strains collected outside of California.
Parsing the effects of host resistance and B. cinerea virulence
All 90 plant genotypes were infected with 98 B. cinerea strains (Figure 2) resulting in an infectivity matrix containing 51,920 independent lesion measurements. Mean lesion areas ranged from no visible damage to over 4 cm2 of necrotic leaf tissue after 72 hours of infection (Figure 4). A. thaliana has the lowest susceptibility while H. annuus is the most susceptible species (Figure 4). To answer how much of the disease outcome is explained by genetic diversity in the host and the pathogen, we modeled the lesion area both within individual species and at large scale, as a meta-analysis across species. This approach revealed that within hosts, B. cinerea genetic diversity matters more than the variation in host resistance. Indeed, the effects of the genetic diversity among B. cinerea strains explain 40–71% of the variance in the lesion area while the pathogen interaction with the host genotypes explains 15–35% of the variance (Figure 5B, Supplementary Figure S2B and Table S5). In contrast, the genetic variation within the plant genotypes explains between 2.8 and 8% of the variance in the lesion area (Figure 5B). To test the impact of the experimental design on these percentages of variance, we conducted a power test by random selection of isolates or plant genotypes from the dataset. We randomly selected 3–8 genotypes of each plant species with a 10-fold replication and ran all the statistical modeling on independent bootstraps. Modeling on the number of Botrytis strains was done by random sampling of 5–50 different strains with a 10-fold replication. Both of those bootstraps revealed that the percentage of variance explained by the species, the isolates, the plant genotype or the interaction mean variance is stable when changing the sampling design. As expected, the variance around the mean is affected. It should be noted that the variations based on bootstraps of the number of isolates are larger than variations based on plant genotypes, confirming the design of this experiment favors a large number of Botrytis strains.
Figure 4.

Lesion areas at 72 hours post inoculation on seven crop species shows small and inconsistent effect of domestication on the B. cinerea interaction. Half-violins and boxplots (median and interquartile range) represent the mean lesion area distribution of high (black, n = 588) and low (gray, n = 588) improvement genotypes for all 98 Botrytis strains. Lactuca refers to L. sativa for high and L. serriola for low improvement genotypes. Solanum refers to S. lycopersicum for high and S. pimpenellifolium for low improvement genotypes. As reference, virulence on A. thaliana wild-type (Col-0, n = 98) and jasmonic acid signaling mutant (coi1, n = 98) are presented. The nonscaled tree represents the phylogenetic relationship between eudicot species. A lesion example on an A. thaliana leaf is provided.
Figure 5.

Host variation predominates the outcomes of plant-Botrytis interactions across species while Botrytis variation predominates within plant species. (A) Multi-host linear model estimating the contribution of plant species, plant genotypes, improvement status, Botrytis strains and their interaction on the percentage of variance in lesion area. (B) Species-specific linear mixed models that estimate the percentage of variance in lesion area. In gray are the experimental parameters classed as random factors. Two-tailed t-test: *P < 0.05, **P < 0.01, ***P < 0.005.
When looking at the disease outcome across the phylogeny, the resistance of the various hosts matters the most. The multi-host analysis showed that differences in host resistance between plant species account for 52% of the total variance in lesion area (P < 2.2 × 10−16; Figure 5A, Supplementary Table S5). Furthermore, the interaction between the host species and B. cinerea strains (16% of total variance; P < 2.2 × 10−16) matters more than the B. cinerea strains alone (12% of total variance; P < 2.2 × 10−16). Finally, the host of origin for each strain (1.7% of total variance; P < 2.2 × 10−16) and the geographical origin (0.2% of total variance; P = 0.0015) have small effects on the lesion area, confirming the minimal influence of population structure observed at the genetic level (Figure 3).
Botrytis cinerea and crop domestication
When infected with B. cinerea, tomato and lettuce wild accessions were more resistant than the domesticated ones (Figure 4). The trend was reversed in the other five species (sunflower, endive, chicory, Brassica, and soybean), with wild or landrace (low improvement) genotypes being less resistant than cultivars or inbred lines (high improvement, Figure 4). While the effect of crop improvement on lesion area is statistically significant (P < 2.2 × 10−16), this effect is exceedingly small, 0.6% of the total variance across all plant species and 2–4% of the variance within specific species models (Figure 5, Supplementary Table S5). The use of 12 genotypes per species is a limitation to the ability to identify rare outlier genotypes associated with domestication and local adaptation. However, given that domestication is associated with only a small component of the host-pathogen interaction across seven crop species, it is valid to consider the domesticated/wild genotypes as statistically equivalent leading to 12 genotypes per species complex. The bi-directional small effect of domestication on this generalist pathogen is potentially an indirect effect of other selective pressures in domestication and illustrates the challenge of predicting resistance to a generalist pathogen.
Botrytis cinerea and the eudicots
Unsupervised clustering showed that variation in host-Botrytis interactions identifies genotype groupings for all species, i.e., the virulence of the strains across the host genotypes is most similar within a host species. However, beyond the individual species level, the evolutionary relatedness between host species did not correlate with the similarity between the measured host-pathogen interactions (Figure 6). For example, phylogenetically distant crops such as B. rapa and Lactuca have a similar susceptibility pattern to the 98 B. cinerea strains. In contrast, the two sister species, C. endivia and C. intybus, have highly divergent susceptibility patterns.
Figure 6.

Variation of plant susceptibility in the Botrytis pathosystem is species specific and does not track plant evolution. Heatmap of standardized (z-scored) least-squares means of lesion area (n = 6) for Botrytis strains (x-axis) interacting with 90 plant genotypes (y-axis). The strains were isolated largely in California (light blue in the origin bar) and on grape (light purple in the host bar). For A. thaliana, five single gene knockout mutants and the corresponding wild-type Col-0 are presented. For the seven crop species, six genotypes with low (gray) and high (black) improvement are presented. The seven crop species were chosen to represent a wide spectrum of phylogenetic distances across Rosids (Brassicales and Fabales) and Asterids (Asterales and Solanales). Branches in the dendrogram that are supported with 95% certainty after bootstrapping are indicated in blue. No branches in the Botrytis strain dendrogram were significant.
To provide a mechanistic benchmark for how the host-pathogen relationships within crops compare to single gene alterations in plant resistance, we included data for A. thaliana Col-0 and five single gene knockout mutants (coi1, anac055, npr1, tga3, and pad3). These single gene mutants have a larger effect on plant susceptibility (39% of the variance in lesion area; Supplementary Figure S2 and Table S5) in comparison to variation between crop genotypes (4–8.8% of the variance within a species; Figure 5B). While these mutants abolish major sectors of plant resistance, the Botrytis-host interactions still cluster these mutants to wild-type Arabidopsis (Figure 6) with branch lengths comparable to variation between genotypes within crop species. Altogether, this infectivity matrix shows that the interaction of B. cinerea with host plants is predominantly defined by variation at the species level with little comparability between species of a particular family or genus.
Host specificity vs virulence
The B. cinerea strains cover a range of host specificity and virulence, from moderate host specificity with high virulence to high host specificity strains with low virulence (Figure 7). In B. cinerea, virulence and host specificity are not independent and are best explained by a quadratic relation. The six strains that are outliers for high host specificity are also outliers for low virulence (Figure 7A). This is illustrated by Davis Navel, a strain collected on orange that has low virulence on six hosts, moderate virulence on Solanum sp. (tomato), and high virulence on C.endivia (Figure 6B). B05.10, the reference strain for the B. cinerea genome and the strain used in >90% of all papers to assess host susceptibility, is the strain with the lowest host-specificity/highest generalist behavior (Figure 7). The B05.10 strain infects all hosts with constant and average virulence. Finally, the strains with the highest virulence have average host specificity (Figure 7A). Katie Tomato, a strain collected on tomato that develops large lesions on all hosts is an example of high virulence strain (Figure 7B). To check whether these trends were an artifact due to a potential relation between standard deviation and mean of lesion area on which the calculation of virulence and host specificity are based, we investigated host specificity as the lesion area of each strain's optimal host versus the strain's average lesion area on the other 7 hosts (Supplementary Figure S3). Each strain's deviation from theoretical generalism (equal virulence on all hosts, Supplementary Figure S3B) revealed a quadratic relation matching the relation between coefficient of variation based on host specificity and virulence (Figure 7). Both metrics revealed that strains with increased host specificity were rare in the strain collection. In combination, this suggests that B. cinerea is under pressure to maintain broad host ranges and moderate virulence within individual strains.
Figure 7.

Strains with low virulence have high host specificity. (A) Estimates of virulence (average lesion area) and host specificity (coefficient of variation) across eight eudicots for the Botrytis strains are shown. The strains are colored according to the plant host from which they were collected. A quadratic relationship was the optimal description of the relationships between specificity and virulence and is shown with a gray confidence interval (R2=0.44, P = 6.36 × 10−13). (B) Mean lesion area and standard error (n = 12, except A. thaliana n = 6) across the eight eudicot species is provided for two strains at the extremes of the host specificity/virulence distribution.
The genetic architecture of virulence and host specificity
Using the above mean virulence across all hosts and the virulence across these hosts (host specificity) as two separate traits with GWAS via two statistical methods (linear model and Bayesian sparse linear mixed models) revealed that both virulence across eight eudicots and host specificity are quantitative traits linked to a large number of candidate loci. To verify the risk of false discovery associated to the significance threshold with the linear model approach, we performed 10 independent permutations. These randomizations resulted in the calculation of an empirical threshold with a false discovery rate of 0.001% (Supplementary Figure S5). Significantly associated SNPs are spread across 16 of the 18 chromosomes (Figure 8, Supplementary Figure S4 and Table S6). None of the SNPs on chromosomes 17 and 18, hypothesized to be potential accessory chromosomes were associated with virulence or host specificity in this dataset (Figure 8, Supplementary Table S6). The spread of significant SNPs across 16 chromosomes and regions (Supplementary Table S6) observed with both GWAS methods suggests that the genetic architecture of virulence and host specificity is not based on specific genomic areas.
Figure 8.

Botrytis virulence and host specificity genetic architecture. GWAS significance of 271,749 SNPs estimated as posterior inclusion probability (PIP) of Bayesian sparse linear mixed model for host specificity (in red) and virulence (in green). The dashed lines represent a probability of false-positive P < 0.01 based on the tail of the PIP distribution of 20 random permutation tests.
Filtering for SNPs-based annotation as potential functional variants revealed 128 genes associated with virulence and 119 genes associated with host specificity (Supplementary Table S7). The candidate genes putatively associated with virulence and host specificity are different as only nine genes are significantly associated with both traits (Figure 8, Supplementary Table S7). Furthermore, the effect size estimates of genes associated with these two traits are negatively correlated (Supplementary Figure S6). Among the genes of interest, Bcin11g05830 is a ferroportin with iron transmembrane transporter activity. Bcin07g05630 is a transcription factor with Zn2/Cys6 DNA-binding domain and Bcin11g05860 is a cytochrome P450. Four of these candidate genes (Bcin03g01060, Bcin06g04770, Bcin11g05830, and Bcin12g00340) have computed gene ontology tagging them as integral components of the membrane. Candidate genes for virulence include 24 genes that are integral components of the membrane. Genes of interest also include a polygalacturonase known to modulate virulence (BcPG1 and Bcin14g00850), two ABC transporters (Bcin02g06110 and Bcin13g04870), an MSF transporter (Bcin10g01450), an ATPase involved in microtubule interaction and transport (Bcin02g08530). A thioesterase (Bcpks20 and Bcin04g00640) active in the secondary metabolism and five genes involved in fumarate reductase complex (Bcin02g03080, Bcin03g02070, Bcin06g06520, Bcin09g04570, and Bcin13g00840) were also identified. Candidate genes for host specificity include a cellulase catalyzing the decomposition of cellulose (Bcin02g04440) and 22 genes that are integral component of the membrane. Among the candidate genes are two major facilitator superfamily (MFS) transporters (Bcin01g01640 and Bcin03g03460) and two cytochrome P450s (Bcin12g00010 and Bcin15g02240). The peak at the beginning of chromosome 12 (Figure 7) is one of those cytochrome P450s (Bcin12g00010). Among the genes with potential functional variants identified for host specificity and virulence, 95 genes have unknown functions (Supplementary Table S7). Filtering on variants with potential functional effect reveals only a partial view of the genetic architecture of the studied traits. A list of 1492 genes including information on the effect sizes and significance in the linear and BSLMM models is provided in Supplementary Table S8. No large effect genes were found but rather a continuous distribution of small to moderate effect size genes (Supplementary Figure S6) detected by both GWAS models.
Discussion
Generating a matrix of B. cinerea disease symptoms across multiple genotypes from eight plant species across four plant orders (Figure 2) illustrates the complexity of quantitative host-pathogen interactions (Figure 6). The disease outcome is very different between hosts and cannot be predicted based on phylogenetic distances. Indeed, this study presents B. cinerea as a pathogen harboring large phenotypic diversity sensitive to the resistance mechanisms of its various hosts and with a possible advantage to remain a generalist (Figure 7).
The genetic architecture of Botrytis
Many Botrytis virulence factors are known such as enzymes contributing to the breakdown of host tissues, transporters detoxifying the host defenses and toxicity factors killing the host cells (Nakajima and Akutsu 2014). More than 100 genes have been successfully validated for their effect on the virulence in different hosts species as documented in the Phi-base database (Urban et al. 2017). One of the first functionally validated genes for B. cinerea virulence (ten Have et al. 1998), the polygalacturonase 1 (Bcin14g00850) active in the breakdown of pectin was identified by GWAS for virulence (Supplementary Table S7) in agreement with its previously seen selective signatures (Rowe and Kliebenstein 2007). Multiple candidate genes for host specificity and virulence reveal the potential role of the membrane-associated proteins and also have the exact or similar functions as functionally validated genes. Similar to previous identification of virulence genes influencing redox processes was the identification of candidate SNPs linked to enzymes of the fumarate reductase respiratory complex and a globin-like ferredoxin as potential candidates active in the redox battle with the host (Viefhues et al. 2014). Similarly, numerous candidate genes were associated with ion and small metabolite transport as previously found with copper, MSF transporters and ABC transporters having been previously shown as virulence factors in Botrytis (Schoonbeek et al. 2001; Saitoh et al. 2010; Vela-Corcía et al. 2019) (Supplementary Tables S7 and S8). The complex architecture of host specificity and virulence (Figure 8, Supplementary Figure S4 and Tables S7 and S8) can be interpreted as a response to the diverse multi-layer components of plant resistance at large phylogenetic scale. The similarity in the known virulence factors and the candidate genes identified suggests that this generalist pathogen might be best equipped to overcome diverse host resistance mechanisms using a large and diverse set of virulence genes that through their net effects enable virulence. It remains to be tested if this is truly a polygenic structure, massive number of genes of small effect, or a more oligogenic structure, moderate number of genes but with larger effect size.
Botrytis, a jack-of-all-trades pathogen
The B. cinerea strains in our collection are generalists (Figure 7, Supplementary Figure S3) that infected all hosts tested in our experimental approach. Only a few strains showing a propensity to prefer an individual host. This pattern does not fit well with the patterns of plant–pathogen interactions generally described by co-evolutionary arms-race scenarios (Figure 7 and Supplementary Tables S7 and S8). In the arms-race model, defense innovations in the host select counter mechanisms in the pathogen driving by evolution of large effect genes linked to specific interactions. As such, the arms-race model shows strong directional pressure in pathogens to become specialists (Barrett et al. 2009; Leggett et al. 2013) while host resistance is expected to increase with the evolutionary distances (Gilbert and Webb 2007; Schulze-Lefert and Panstruga 2011; Gilbert and Parker 2016). Other qualitative models such as the inversed gene-for-gene, matching alleles or inversed matching alleles (Morris and Moury 2019) do not fit B. cinerea well either. This may indicate that B. cinerea may track with a model wherein frequent opportunistic host switches lead to a more oligogenic additive quantitative model. There is some evidence for linking Botrytis genetic population structures to quantitative host preference within France and Tunisia (Karchani-Balma et al. 2008; Walker et al. 2015; Mercier et al. 2019). Furthermore, Botrytis strains obtained from closed environments like intensive greenhouse systems were also shown to display a reduced genetic diversity associated with an increase in clonal distribution (Walker et al. 2015; Diao et al. 2019; Mercier et al. 2019). As such, it is possible that the specific model shaping B. cinerea host interactions could vary depending on environment and geography (Calpas et al. 2006; Decognet et al. 2009; Leyronas et al. 2015). More work is needed to assess what environmental factors may shape specialization versus generalization in Botrytis.
One factor influencing this choice suggested by our data is that generalist strains may have an advantage as they maintain moderate virulence across diverse hosts. Specialization within our collection was better explained as a loss in virulence on most hosts rather than an increase of virulence on a specific host (Figures 6 and 7). This contrasts to the classic model of specialization where specialization occurs by a gain of virulence on a specific host (Woolhouse et al. 2001; Leggett et al. 2013). If Botrytis virulence is largely a net effect summed across a large number of virulence loci of unequal effects on different hosts, this could explain how specialization occurs by loss of virulence mechanisms. If an environmental or geographic factor limits Botrytis from switching hosts and constrains it to a single host, this may lead to the loss of virulence factors that have little to no benefit on the predominant host while maintaining the factors that help with infection on that specific host. Testing what shapes host generalization versus specialization and the corresponding genetic causality within broad-spectrum pathogens like B. cinerea will help to better understand how host-pathogen interactions evolve.
Acknowlegments
Seeds used in this study were provided by Laura Marek and Kathleen Reitsma at the US department of agriculture, Chris Pires, the UC Davis Tomato Genetics Resource Center, the Center for Genetic Resources Netherland (CGN) through Guy Barker and Graham Teakle. The experiments were performed with the help of Dihan Gao, Aysha Shafi, David Kelly, Matisse Madrone, Melissa Wang, Josue Vega, Aleshia Hopper, and Ayesha Siddiqui.
Conceptualization and supervision: D.J.K.; Data analysis: C.C.; Funding acquisition: D.J.K., Investigation: G.S., C.C., N.S., R.G., and J.C.; Resources: S.A.; Writing: C.C. and D.J.K.
Contributor Information
Celine Caseys, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA.
Gongjun Shi, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA; Department of Plant Pathology, North Dakota State University, Fargo, ND 58102, USA.
Nicole Soltis, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA; Plant Biology Graduate Group, University of California, Davis, Davis, CA 95616 USA.
Raoni Gwinner, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA; Embrapa Amazonia Ocidental, Manaus 69010-970, Brazil.
Jason Corwin, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA; Department of Ecology and Evolution Biology, University of Colorado, Boulder, CO 80309-0334, USA.
Susanna Atwell, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA.
Daniel J Kliebenstein, Department of Plant Sciences, University of California, Davis, Davis, CA 95616, USA; DynaMo Center of Excellence, University of Copenhagen, Frederiksberg C DK-1871, Denmark.
Funding
National science foundation (USA), division of integrative organismal system, grants IOS1339125 and IOS1021861 to Daniel J. Kliebenstein.
Conflicts of interest
None declared.
Literature cited
- Atwell S, Corwin JA, Soltis N, Zhang W, Copeland D, et al. 2018. Resequencing and association mapping of the generalist pathogen Botrytis cinerea. bioRxiv. 489799. [Google Scholar]
- Atwell S, Corwin JA, Soltis NE, Subedy A, Denby KJ, et al. 2015. Whole genome resequencing of Botrytis cinerea isolates identifies high levels of standing diversity. Front Microbiol. 6:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin M. 2018. Striking similarities between Botrytis cinerea from non-agricultural and from agricultural habitats. Front Plant Sci. 9:1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LG, Kniskern JM, Bodenhausen N, Zhang W, Bergelson J. 2009. Continua of specificity and virulence in plant host-pathogen interactions: causes and consequences. New Phytol. 183:513–529. [DOI] [PubMed] [Google Scholar]
- Berens ML, Berry HM, Mine A, Argueso CT, Tsuda K. 2017. Evolution of hormone signaling networks in plant defense. Annu Rev Phytopathol. 55:401–425. [DOI] [PubMed] [Google Scholar]
- Bertazzoni S, Williams AH, Jones DA, Syme RA, Tan K-C, et al. 2018. Accessories make the outfit: accessory chromosomes and other dispensable DNA regions in plant-pathogenic fungi. Mol Plant Microbe Interact. 31:779–788. [DOI] [PubMed] [Google Scholar]
- Białas A, Zess EK, De la Concepcion JC, Franceschetti M, Pennington HG, et al. 2018. Lessons in effector and NLR biology of plant-microbe systems. Mol Plant Microbe Interact. 31:34–45. [DOI] [PubMed] [Google Scholar]
- Bird KA, An H, Gazave E, Gore MA, Pires JC, et al. 2017. Population structure and phylogenetic relationships in a diverse panel of Brassica rapa L. Front Plant Sci. 8:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JKM, Tellier A. 2011. Plant-parasite coevolution: bridging the gap between genetics and ecology. Annu Rev Phytopathol. 49:345–367. [DOI] [PubMed] [Google Scholar]
- Calpas JT, Konschuh MN, Toews CC, Tewari JP. 2006. Relationships among isolates of Botrytis cinerea collected from greenhouses and field locations in Alberta, based on RAPD analysis. Can J Plant Pathol. 28:109–124. [Google Scholar]
- Chae L, Kim T, Nilo-Poyanco R, Rhee SY. 2014. Genomic signatures of specialized metabolism in plants. Science. 344:510–513. [DOI] [PubMed] [Google Scholar]
- Chen YH, Gols R, Benrey B. 2015. Crop domestication and its impact on naturally selected trophic interactions. Annu Rev Entomol. 60:35–58. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corwin JA, Copeland D, Feusier J, Subedy A, Eshbaugh R, et al. 2016. The quantitative basis of the Arabidopsis innate immune system to endemic pathogens depends on pathogen genetics. PLoS Genet. 12:e1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corwin JA, Kliebenstein DJ. 2017. Quantitative resistance: more than just perception of a pathogen. Plant Cell. 29:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosseboom SD, Ivors KL, Schnabel G, Bryson PK, Holmes GJ. 2019. Within-season shift in fungicide resistance profiles of Botrytis cinerea in California strawberry fields. Plant Dis. 103:59–64. [DOI] [PubMed] [Google Scholar]
- Cowger C, Brown JKM. 2019. Durability of quantitative resistance in crops: greater than we know? Annu Rev Phytopathol. 57:253–277. [DOI] [PubMed] [Google Scholar]
- Decognet V, Bardin M, Trottin-Caudal Y, Nicot P. 2009. Rapid change in the genetic diversity of Botrytis cinerea populations after the introduction of strains in a tomato glasshouse. Phytopathology. 99:185–193. [DOI] [PubMed] [Google Scholar]
- De Gracia M, Cascales M, Expert P, Bellanger M-N, Le Cam B, et al. 2015. How did host domestication modify life history traits of its pathogens? PLoS One. 10:e0122909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delplace F, Huard-Chauveau C, Dubiella U, Khafif M, Alvarez E, et al. 2020. Robustness of plant quantitative disease resistance is provided by a decentralized immune network. Proc Natl Acad Sci USA. 117:18099–18109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempewolf H, Rieseberg LH, Cronk QC. 2008. Crop domestication in the Compositae: a family-wide trait assessment. Genet Resour Crop Evol. 55:1141–1157. [Google Scholar]
- Denby KJ, Kumar P, Kliebenstein DJ. 2004. Identification of Botrytis cinerea susceptibility loci in Arabidopsis thaliana. Plant J. 38:473–486. [DOI] [PubMed] [Google Scholar]
- ΘDe Vries I. 1997. Origin and domestication of Lactuca sativa L. Genetic Res Crop Evol. 44:165–174. [Google Scholar]
- Diao Y Larsen, M, Kamvar, Z. N, Zhang, C, Li, S, Wang, W, Lin, D, Peng, Q, Knaus, B. J, Foster, Z. S. L, Grunwald, N. J. and Liu, X. L. 2019. Genetic differentiation and clonal expansion of Chinese Botrytis cinerea populations from tomato and other crops in China. Phytopathology. [DOI] [PubMed] [Google Scholar]
- Dong S, Raffaele S, Kamoun S. 2015. The two-speed genomes of filamentous pathogens: waltz with plants Curr Opin Genet Dev. 35:57–65. [DOI] [PubMed] [Google Scholar]
- Elad Y, Pertot I, Cotes PA, Stewart A. 2016. Plant Hosts of Botrytis spp. In: Fillinger S, Elad Y, editors. Botrytis – the Fungus, the Pathogen and Its Management in Agricultural Systems. Cham: Springer International Publishing. p. 413–486. [Google Scholar]
- Fillinger S, Elad Y. 2015. Botrytis – the Fungus, the Pathogen and Its Management in Agricultural Systems. In: Fillinger S, Elad Y, editors. Switzerland: Springer International Publishing. pp. 189–216. [Google Scholar]
- Fordyce RF, Soltis NE, Caseys C, Gwinner R, Corwin JA, et al. 2018. Digital imaging combined with genome-wide association mapping links loci to plant-pathogen interaction traits. Plant Physiol. 178:1406–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantzeskakis L, Di Pietro A, Rep M, Schirawski J, Wu C-H, et al. 2020. Rapid evolution in plant–microbe interactions – a molecular genomics perspective. New Phytol. 225:1134–1142. [DOI] [PubMed] [Google Scholar]
- Gilbert GS, Webb CO. 2007. Phylogenetic signal in plant pathogen-host range. Proc Natl Acad Sci U S A. 104:4979–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert GS, Parker IM. 2016. The Evolutionary Ecology of Plant Disease: A Phylogenetic Perspective. Annu Rev Phytopathol. 54:549–578. [DOI] [PubMed] [Google Scholar]
- Glazebrook J. 2005. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu Rev Phytopathol. 43:205–227. [DOI] [PubMed] [Google Scholar]
- Glazebrook J, Roby D. 2018. Plant biotic interactions: from conflict to collaboration. Plant J. 93:589–591. [DOI] [PubMed] [Google Scholar]
- Goudet J. 2005. Hierfstat, a package for R to compute and test hierarchical F‐statistics. Mol Ecol Notes. 5:184–186. [Google Scholar]
- Herman M, Williams M. 2012. Fighting for their lives: plants and pathogens. Plant Cell. 24:tpc.112.tt0612. [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23:254–267. [DOI] [PubMed] [Google Scholar]
- Inupakutika MA, Sengupta S, Devireddy AR, Azad RK, Mittler R. 2016. The evolution of reactive oxygen species metabolism. J Exp Bot. 67:5933–5943. [DOI] [PubMed] [Google Scholar]
- Jacob F, Vernaldi S, Maekawa T. 2013. Evolution and conservation of plant NLR functions. Front Immunol. 4:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasov TL, Horton MW, Bergelson J. 2014. Genomic variability as a driver of plant–pathogen coevolution? Curr Opin Plant Biol. 18:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karchani-Balma S, Gautier A, Raies A, Fournier E. 2008. Geography, plants, and growing systems shape the genetic structure of Tunisian Botrytis cinerea populations. Phytopathology. 98:1271–1279. [DOI] [PubMed] [Google Scholar]
- Krah F-S, Bässler C, Heibl C, Soghigian J, Schaefer H, et al. 2018. Evolutionary dynamics of host specialization in wood-decay fungi. BMC Evol Biol. 18:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannou C. 2012. Variation and selection of quantitative traits in plant pathogens. Annu Rev Phytopathol. 50:319–338. [DOI] [PubMed] [Google Scholar]
- Leggett HC, Buckling A, Long GH, Boots M. 2013. Generalism and the evolution of parasite virulence. Trends Ecol Evol. 28:592–596. [DOI] [PubMed] [Google Scholar]
- Lenth R, Singmann H, Love J, Buerkner P, Herve M. 2018. Emmeans: Estimated Marginal Means, Aka Least-Squares Means. R package version 1, 3.
- Leyronas C, BryoneF, DuffaudM, TrouletC, Nicot PC. 2015. Assessing host specialization of Botrytis cinerea on lettuce and tomato by genotypic and phenotypic characterization. Plant Pathol. 64:119–127. [Google Scholar]
- Leyronas C, Bardin M, Duffaud M, Nicot PC. 2015a. Compared dynamics of grey mould incidence and genetic characteristics of Botrytis cinerea in neighbouring vegetable greenhouses. J Plant Pathol. 97:439–447. [Google Scholar]
- Leyronas C, Bryone F, Duffaud M, Troulet C, Nicot PC. 2015b. Assessing host specialization of Botrytis cinereaon lettuce and tomato by genotypic and phenotypic characterization. Plant Pathol. 64:119–127. [Google Scholar]
- Li M-X, Yeung JMY, Cherny SS, Sham PC. 2012. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 131:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T, Zhu G, Zhang J, Xu X, Yu Q, et al. 2014. Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 46:1220–1226. [DOI] [PubMed] [Google Scholar]
- Lischer HEL, Excoffier L. 2012. PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics. 28:298–299. [DOI] [PubMed] [Google Scholar]
- Ma Z, Michailides TJ. 2005. Genetic structure of Botrytis cinerea populations from different host plants in California. Plant Dis. 89:1083–1089. [DOI] [PubMed] [Google Scholar]
- Mercier A, Carpentier F, Duplaix C, Auger A, Pradier J-M, et al. 2019. The Polyphagous plant pathogenic fungus Botrytis cinerea encompasses host-specialized and generalist populations. Environ Microbiol. 21:4808–4821. [DOI] [PubMed] [Google Scholar]
- Meyer RS, DuVal AE, Jensen HR. 2012. Patterns and processes in crop domestication: an historical review and quantitative analysis of 203 global food crops. New Phytologist. 196:29–48. [DOI] [PubMed] [Google Scholar]
- Möller M, Stukenbrock EH. 2017. Evolution and Genome Architecture in Fungal Plant Pathogens. Nature Reviews Microbiology. 15:756. [DOI] [PubMed] [Google Scholar]
- Morris CE, Moury B. 2019. Revisiting the concept of host range of plant pathogens. Annu Rev Phytopathol. 57:63–90. [DOI] [PubMed] [Google Scholar]
- Mou B. 2011. Mutations in lettuce improvement. Int J Plant Genomics. 2011:723518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Akutsu K. 2014. Virulence factors of Botrytis cinerea. J Gen Plant Pathol. 80:15–23. [Google Scholar]
- Plissonneau C, Benevenuto J, Mohd-Assaad N, Fouché S, Hartmann FE, et al. 2017. Using population and comparative genomics to understand the genetic basis of effector-driven fungal pathogen evolution. Front Plant Sci. 8:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisot T, Canard E, Mouquet N, Hochberg ME. 2012. A comparative study of ecological specialization estimators. Methods Ecol Evol. 3:537–544. [Google Scholar]
- Poland JA, Balint-Kurti PJ, Wisser RJ, Pratt RC, Nelson RJ. 2009. Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 14:21–29. [DOI] [PubMed] [Google Scholar]
- Rowe HC, Kliebenstein DJ. 2007. Elevated genetic variation within virulence-associated Botrytis cinerea polygalacturonase loci. Mol Plant Microbe Interact. 20:1126–1137. [DOI] [PubMed] [Google Scholar]
- Rowe HC, Walley JW, Corwin J, Chan EK-F, Dehesh K, et al. 2010. Deficiencies in jasmonate-mediated plant defense reveal quantitative variation in Botrytis cinerea pathogenesis. PLoS Pathog. 6:e1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Xiao CL. 2018. Fungicide resistance in Botrytis cinerea populations in California and its influence on control of gray mold on stored Mandarin fruit. Plant Dis. 102:2545–2549. [DOI] [PubMed] [Google Scholar]
- Saitoh Y, Izumitsu K, Morita A, Tanaka C. 2010. A copper-transporting ATPase BcCCC2 is necessary for pathogenicity of Botrytis cinerea. Mol Genet Genomics. 284:33–43. [DOI] [PubMed] [Google Scholar]
- Schep AN, Kummerfeld SK. 2017. iheatmapr: interactive complex heatmaps in R. J Open Source Soft. 2:359. [Google Scholar]
- Schoonbeek H Del Sorbo G De Waard MA. 2001. The ABC transporter BcatrB affects the sensitivity of Botrytis cinerea to the phytoalexin resveratrol and the fungicide fenpiclonil. Mol Plant Microbe Interact. 14:562–571. 10.1094/MPMI.2001.14.4.562 11310744 [DOI] [PubMed] [Google Scholar]
- Schulze-Lefert P, Panstruga R. 2011. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 16:117–125. [DOI] [PubMed] [Google Scholar]
- Soltis NE, Atwell S, Shi G, Fordyce R, Gwinner R, et al. 2019. Crop domestication and pathogen virulence: interactions of tomato and Botrytis genetic diversity. Plant Cell. 31:502–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis NE, Caseys C, Zhang W, Corwin JA, Atwell S, et al. 2020. Pathogen genetic control of transcriptome variation in the Arabidopsis thaliana–Botrytis cinerea pathosystem. Genetics. 215:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen I, Domozych D, Willats WG. 2010. How have plant cell walls evolved? Plant Physiol. 153:366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Shimodaira H. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 22:1540–1542. [DOI] [PubMed] [Google Scholar]
- ten Have A, Mulder W, Visser J, van Kan JAL. 1998. The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol Plant Microbe Interact. 11:1009–1016. [DOI] [PubMed] [Google Scholar]
- Upson JL, Zess EK, Białas A, Wu C-H, Kamoun S. 2018. The coming of age of EvoMPMI: evolutionary molecular plant–microbe interactions across multiple timescales. Curr Opin Plant Biol. 44:108–116. [DOI] [PubMed] [Google Scholar]
- Urban M, Cuzick A, Rutherford K, Irvine A, Pedro H, et al. 2017. PHI-base: a new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 45:D604–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Does HC, Rep M. 2007. Virulence genes and the evolution of host specificity in plant-pathogenic fungi. Mol Plant Microbe Interact. 20:1175–1182. [DOI] [PubMed] [Google Scholar]
- Van Kan JAL, Stassen JHM, Mosbach A, Van Der Lee TAJ, Faino L, et al. 2017. A gapless genome sequence of the fungus Botrytis cinerea. Mol Plant Pathol. 18:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela-Corcía D, Aditya Srivastava D, Dafa-Berger A, Rotem N, Barda O, et al. 2019. MFS transporter from Botrytis cinerea provides tolerance to glucosinolate-breakdown products and is required for pathogenicity. Nat Commun. 10:2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloso J, van Kan JAL. 2018. Many shades of grey in Botrytis–Host plant interactions. Trends Plant Sci. 23:613–622. [DOI] [PubMed] [Google Scholar]
- Viefhues A, Heller J, Temme N, Tudzynski P. 2014. Redox systems in Botrytis cinerea: impact on development and virulence. Mol Plant Microbe Interact. 27:858–874. [DOI] [PubMed] [Google Scholar]
- Walker A-S, Gladieux P, Decognet V, Fermaud M, Confais J, et al. 2015. Population structure and temporal maintenance of the multihost fungal pathogen Botrytis cinerea: causes and implications for disease management. Environ Microbiol. 17:1261–1274. [DOI] [PubMed] [Google Scholar]
- Walley PG, Hough G, Moore JD, Carder J, Elliott M, et al. 2017. Towards new sources of resistance to the currant-lettuce aphid (Nasonovia ribisnigri). Mol Breed. 37:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution. 38:1358–1370. [DOI] [PubMed] [Google Scholar]
- Whitehead SR, Turcotte MM, Poveda K. 2017. Domestication impacts on plant–herbivore interactions: a meta-analysis. Philos Trans R Soc Lond B Biol Sci. 372:20160034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson SW, Magerøy MH, López Sánchez A, Smith LM, Furci L, et al. 2019. Surviving in a hostile world: plant strategies to resist pests and diseases. Annu Rev Phytopathol. 57:505–529. [DOI] [PubMed] [Google Scholar]
- Woolhouse ME, Taylor LH, Haydon DT. 2001. Population biology of multihost pathogens. Science. 292:1109–1112. [DOI] [PubMed] [Google Scholar]
- Zhang W, Corwin JA, Copeland D, Feusier J, Eshbaugh R, et al. 2017. Plastic transcriptomes stabilize immunity to pathogen diversity: the jasmonic acid and salicylic acid networks within the Arabidopsis/Botrytis Pathosystem. Plant Cell. 29:2727–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Corwin JA, Copeland DH, et al. 2019. Plant-necrotroph co-transcriptome networks illuminate a metabolic battlefield. eLife. 8:e44279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Carbonetto P, Stephens M. 2013. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 9:e1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Correspondence and requests for materials should be addressed to Kliebenstein@ucdavis.edu. R codes and datasets are available on Dryad https://doi.org/10.25338/B8Z91N. Supplementary material is available at figshare: https://doi.org/10.25387/g3.14591145.