

Abstract
Life history traits underlie the fitness of organisms and are under strong natural selection. A new mutation that positively impacts a life history trait will likely increase in frequency and become fixed in a population (e.g., a selective sweep). The identification of the beneficial alleles that underlie selective sweeps provides insights into the mechanisms that occurred during the evolution of a species. In the global population of Caenorhabditis elegans, we previously identified selective sweeps that have drastically reduced chromosomal-scale genetic diversity in the species. Here, we measured the fecundity of 121 wild C. elegans strains, including many recently isolated divergent strains from the Hawaiian islands and found that strains with larger swept genomic regions have significantly higher fecundity than strains without evidence of the recent selective sweeps. We used genome-wide association (GWA) mapping to identify three quantitative trait loci (QTL) underlying the fecundity variation. In addition, we mapped previous fecundity data from wild C. elegans strains and C. elegans recombinant inbred advanced intercross lines that were grown in various conditions and detected eight QTL using GWA and linkage mappings. These QTL show the genetic complexity of fecundity across this species. Moreover, the haplotype structure in each GWA QTL region revealed correlations with recent selective sweeps in the C. elegans population. North American and European strains had significantly higher fecundity than most strains from Hawaii, a hypothesized origin of the C. elegans species, suggesting that beneficial alleles that caused increased fecundity could underlie the selective sweeps during the worldwide expansion of C. elegans.
Keywords: C. elegans, lifetime fecundity, natural variation, QTL, selective sweeps
Introduction
Life history traits are phenotypic characters that affect the fitness of organisms (Knight and Robertson 1957; Stearns 1976, 1989; Charlesworth et al. 2003; Flatt and Heyland 2011; Flatt 2020). Traits, such as fecundity, size at birth, age at reproductive maturity, and stage- or size-specific rates of survival, interact with each other to affect the fitness of organisms in an ever-changing environment. Genes that affect life history traits should be subject to strong natural selection because they directly affect the fitness of organisms. Adaptive alleles with strong selective advantages in life history-related genes are likely to spread rapidly across a population in a selective sweep (Smith and Haigh 1974; Kaplan et al. 1989; Berry et al. 1991; Stephan 2019). Signatures of selective sweeps include a loss of neutral polymorphism, drastic changes in the site frequency spectrum, and particular patterns of linkage disequilibrium (LD) across the site of selection (Smith and Haigh 1974; Braverman et al. 1995; Fay and Wu 2000; Kim and Nielsen 2004; Stephan et al. 2006; Stephan 2019). Identification of selective sweeps by these signatures provides a key to locate genes under selection and helps to understand the process of adaptation and evolution.
Caenorhabditis elegans is a free-living nematode and a keystone model organism for biological research. The reproductive mode of C. elegans is androdioecy, with predominant self-fertilization of hermaphrodites and rare outcrossing between hermaphrodites and males (Brenner 1974). A single hermaphrodite of the laboratory reference strain N2 lays approximately 300 self-fertilized embryos in standard laboratory conditions (Hodgkin and Doniach 1997; Félix and Braendle 2010). Newly hatched animals develop through four larval stages (L1–L4) into mature reproductive adults after 3 days in favorable conditions at 20° (Frézal and Félix 2015). Under stressful conditions, such as crowding and limited food, C. elegans enters the dauer diapause stage during larval development to enable survival in harsh environments and to facilitate dispersal. C. elegans likely has a boom-and-bust life cycle in the wild because of fluctuating environmental conditions and the spatio-temporal distributed habitats, such as rotting fruits and stems (Félix and Duveau 2012; Frézal and Félix 2015). C. elegans is globally distributed (Kiontke et al. 2011; Andersen et al. 2012; Félix and Duveau 2012; Cook et al. 2017; Crombie et al. 2019; Lee et al. 2021). Although recent studies characterized high genetic diversity of the species in Hawaii and the surrounding Pacific regions (Crombie et al. 2019; Lee et al. 2021), C. elegans exhibits low overall genetic diversity at the global scale (Barrière and Félix 2005; Cutter 2006; Andersen et al. 2012). The metapopulation dynamics, seasonal bottlenecks, predominant selfing, low-outcrossing rate, low-recombination rate, background selection, and recent selective sweeps might all contribute to the low-genetic diversity of the species (Barrière and Félix 2005, 2007; Cutter 2006; Rockman and Kruglyak 2009; Rockman et al. 2010; Andersen et al. 2012). In the genomes of many C. elegans strains sampled in temperate regions, chromosomes I, IV, V, and X exhibit signatures of selective sweeps, such as an excess of rare variants, high LD, and extended haplotype homozygosity over large genomic regions (Andersen et al. 2012). By contrast, the genomes of most Hawaiian C. elegans strains have no such signatures (Andersen et al. 2012; Crombie et al. 2019; Lee et al. 2021). Analyses of C. elegans genetic diversity, population structure, gene flow, and haplotype structure suggest that C. elegans originated from the Pacific region, such as the Hawaii Islands, the western United States, or New Zealand, and expanded worldwide, especially into human-associated habitats (Andersen et al. 2012; Crombie et al. 2019; Lee et al. 2021). The recent positive selective sweeps likely occurred during this expansion, but the beneficial alleles that have driven the sweeps and their fitness advantages are yet unknown.
Here, we measured lifetime fecundity of 121 wild C. elegans strains and compared this trait between swept strains that experienced the recent selective sweeps and divergent strains that avoided these sweeps. We found that swept strains had significantly higher lifetime fecundity than divergent strains, as well as significant geographical differences in lifetime fecundity between strains from the Hawaii Islands and strains from other parts of the world. We then used genome-wide association (GWA) mapping to identify three quantitative trait loci (QTL) on chromosomes I, II, and V that influence the lifetime fecundity of C. elegans. In addition, we identified eight QTL that impact C. elegans fecundity in different laboratory environments using GWA and linkage mappings of previous fecundity data. The 11 QTL reveal the complex genetic architecture of C. elegans fecundity. Furthermore, we discovered that the different alleles at each QTL peak marker and the different haplotypes in each QTL among the 121 strains were strongly correlated with signatures of recent selective sweeps found in each strain. Our results suggest that higher lifetime fecundity could have provided selective advantages for swept strains and the underlying genetic variants might have driven the recent strong sweeps in the C. elegans strains that have colonized the world.
Materials and methods
Caenorhabditis elegans strains
All the wild strains were obtained from C. elegans Natural Diversity Resource (CeNDR) (Cook et al. 2017). Animals were cultured at 20° on modified nematode growth medium (NGMA) containing 1% agar and 0.7% agarose to prevent burrowing and fed the Escherichia coli strain OP50.
Swept haplotypes and strains
Haplotype data for 403 C. elegans isotypes, representing 913 wild strains, were acquired from the 20200815 CeNDR release. We compared the total length of each haplotype per chromosome across all isotypes to identify the most common haplotypes on each chromosome. We then searched for the regions of the most common haplotypes in each C. elegans isotype and recorded them if their length was greater than 1 Mb (Crombie et al. 2019; Lee et al. 2021). We classified haplotypes outside of recorded regions as unswept haplotypes. The swept status of some haplotypes was undetermined when no identical-by-descent groups were found, and thus the haplotype information for that region was missing in the CeNDR release.
Signatures of selective sweeps were identified on chromosomes I, IV, V, and X, but not on chromosomes II and III (Andersen et al. 2012). Therefore, we focused on the four chromosomes (I, IV, V, and X) and defined their most common haplotypes as swept haplotypes (Lee et al. 2021). In each C. elegans isotype, chromosomes that contain greater than or equal to 30% of the swept haplotype were classified as swept chromosomes. We classified isotypes with any swept I, IV, V, and X chromosomes as swept isotypes and isotypes without any swept I, IV, V, and X chromosomes as divergent isotypes. Strains that belong to swept isotypes and divergent isotypes were classified as swept strains and divergent strains, respectively (Gilbert et al. 2020).
Genetic relatedness
Genetic variation data for 403 C. elegans isotypes were acquired from the hard-filtered isotype variant call format (VCF) 20200815 CeNDR release. These variants were pruned to the 1,074,596 biallelic single nucleotide variants (SNVs) without missing genotypes. We converted this pruned VCF file to a PHYLIP file using the vcf2phylip.py script (Ortiz 2019). The unrooted neighbor-joining tree was made using the R packages phangorn (v2.5.5) and ggtree (v1.14.6) (Schliep 2011; Yu et al. 2017).
Fecundity measurements
Prior to each assay, strains were grown for three generations without bleaching, entering starvation, or encountering dauer-inducing conditions (Andersen et al. 2014). For each C. elegans strain in the fourth generation, single L4 larval stage hermaphrodites were picked to each of five 3.5 cm NGMA plates with OP50 and were maintained at 20°. For each assay plate, the original hermaphrodite parent was transferred to a fresh plate every 24 hours for 96 hours. A custom-built imaging platform (DMK 23GP031 camera; Imaging Source, Charlotte, NC, USA) was used to collect images for each of the first four assay plates (0, 24, 48, and 72 hour samples) 48 hours after removal of the parent from each plate. Most strains had few offspring after 96 hours. Images of the fifth assay plates were collected 72 hours after the final transfer of the parents. From each image, the total offspring was counted by visual inspection using the Multi-point Tool in ImageJ (v1.8.0_162) (Schneider et al. 2012). The original hermaphrodite parents on the fifth assay plates were excluded from the counts. The number of offspring in each of the first four assay plates corresponds to the daily fecundity. Numbers of offspring on the fifth assay plates contained offspring from 3 days. For each biological replicate of each C. elegans strain, the lifetime fecundity was calculated as the total number of offspring from the five plates. Replicates where the parent died were excluded from the analysis. Only biological replicates with data from all five assay plates were used in the calculations of daily and total fecundity. Daily intrinsic growth rate (r) for each strain was calculated by r = ln(mx)/x, where x is animal age after hatching (2 + day of adulthood) and mx is cumulative fecundity by each age (Vassilieva and Lynch 1999; Anderson et al. 2011).
We collected fecundity data for 557 replicates of 121 C. elegans strains [mean lifetime fecundity (MLF) = 231, standard deviations (SD) = 55]: 84 strains with five replicates (MLF = 232, SD = 55), 28 strains with four replicates (MLF = 229, SD = 52), seven strains with three replicates (MLF = 214, SD = 49), and two strains with two replicates (MLF = 292, SD = 19). These 121 strains were measured in 15 blocks, with 5–10 strains in each block (Supplementary File S3). Six of the 15 blocks (blocks 2, 8, 12, 13, 14, and 15) only contained swept strains. Three of the 15 blocks (blocks 1, 3, and 7) only contained divergent strains. The remaining six blocks (blocks 4, 5, 6, 9, 10, and 11) contained a mix of swept and divergent strains. We performed post hoc analysis to detect potential block effects, using the aov() and the TukeyHSD() functions in the R package stats (v3.5.3) (https://www.R-project.org/). Of the 105 pairwise comparisons of lifetime fecundity among the 15 blocks, only block 13 (with 10 swept strains) showed significantly higher fecundity than four blocks: block 3 (seven divergent strains), block 4 (eight divergent strains and one swept strains), block 6 (eight divergent strains and two swept strains), and block 10 (four divergent strains and six swept strains). Generally, block effects were rare and might be associated with genotypes of strains in blocks. We included all 121 strains of the 15 blocks in the following analysis.
GWA mapping
GWA mapping was performed on the mean fecundity measurements of biological replicates from 121 C. elegans strains, which belong to 121 distinct isotypes. Genotype data for each of the 121 isotypes were acquired from the hard-filtered isotype VCF (20200815 CeNDR release). We performed the mapping using the pipeline cegwas2-nf (https://github.com/AndersenLab/cegwas2-nf) as previously described (Zdraljevic et al. 2019; Na et al. 2020). Briefly, we used BCFtools (Li 2011) to filter variants that had any missing genotype calls and variants that were below the 5% minor allele frequency. We used PLINK v1.9 (Purcell et al. 2007; Chang et al. 2015) to prune the genotypes to 56,878 markers with a LD threshold of r2 < 0.8 and then generated the kinship matrix using the A.mat() function in the R package rrBLUP (v4.6.1) (Endelman 2011). The number of independent tests (Ntest) within the genotype matrix was estimated using the R packages RSpectra (v0.16.0) (https://github.com/yixuan/RSpectra) and correlateR (0.1) (https://github.com/AEBilgrau/correlateR). The eigen-decomposition significance (EIGEN) threshold was calculated as −log10(0.05/Ntest). We used the GWAS() function in the rrBLUP package to perform the genome-wide mapping with the EMMA algorithm (Kang et al. 2008). QTL were defined by at least one marker that was above the Bonferroni-corrected significance (BF) threshold, to locate the best estimate of QTL positions with the highest significance. We used the LD() function from the R package genetics (v1.3.8.1.2) (https://cran.r-project.org/package=genetics) to calculate the LD correlation coefficient r2 among the QTL peak markers associated with C. elegans lifetime fecundity.
We also performed GWA mapping using fecundity data in DMSO control conditions from a previous study (Hahnel et al. 2018), where 236 C. elegans wild strains were cultured and phenotyped using the high-throughput fitness assays (HTA) as previously described. Briefly, L4 larval stage hermaphrodites were cultured to gravid adult stage on plates and were bleached to obtain synchronized offspring. The embryos were grown to L4 larval stage in liquid (K medium) (Boyd et al. 2012) and fed an E. coli HB101 lysate (García-González et al. 2017) in 96-well plates. A large-particle flow cytometer (COPAS BIOSORT; Union Biometrica, Holliston, MA, USA) was used to sort three L4 larvae into each well of new 96-well plates containing K medium, E. coli HB101 lysate, and 1% DMSO. Animals in the 96-well plates were incubated at 20° for 96 hours to allow animals to grow and produce offspring, followed by measurements of various fitness parameters, including fecundity. Raw fecundity data were pruned, normalized, and regressed using the R package easysorter (v1.0) (Shimko and Andersen 2014; Hahnel et al. 2018). The processed fecundity, norm.n, of each strain was used here for GWA mapping.
Statistical analysis
Statistical significance of fecundity and intrinsic growth rate differences between swept strains (groups) and divergent strains (groups), and fecundity differences among different sampling locations, were tested using the Wilcoxon test and P-values were adjusted for multiple comparisons (Holm method) using the compare_means() function in the R package ggpubr (v0.2.4) (https://github.com/kassambara/ggpubr/). Broad-sense heritability of C. elegans lifetime fecundity was calculated using the lmer() function in the R package lme4 (v1.1.21) with the model phenotype ∼ 1+ (1|strain) (Bates et al. 2015).
Linkage mapping
We performed linkage mapping using fecundity data from a large panel of recombinant inbred advanced intercross lines (RIAILs) derived from QX1430 and CB4856 (Andersen et al. 2015). The fecundity (norm.n) of the RIAILs and the parents were measured using the HTA as described above, under three conditions: 1% H2O (402 RIAILs), 1% DMSO (417 RIAILs), and 0.5% DMSO (432 RIAILs). Linkage mapping was performed on each trait using the R package linkagemapping (v1.3) (https://github.com/AndersenLab/linkagemapping) and the single-nucleotide variation data of the RIAILs in the package as described previously (Evans and Andersen 2020). Briefly, logarithm of the odds (LOD) scores for each genetic marker and each trait were calculated using the function fsearch(). The QTL threshold for significant LOD scores in each mapping was defined by permuting trait values 1000 times, mapping the permuted trait data, and taking the 95th quantile LOD score as the 5% genome-wide error rate. 95% confidence intervals of each QTL were determined using the function annotate_lods.
Data availability
The datasets, code for generating all figures, and Supplementary figures can be found at https://github.com/AndersenLab/swept_broods. Supplementary File S1 contains the haplotype data of 403 C. elegans isotypes from CeNDR release 20200815. Supplementary File S2 contains genetic relatedness of 403 C. elegans isotypes. Supplementary File S3 contains lifetime fecundity of 121 C. elegans strains, their classification of swept strains and divergent strains, and the assay blocks of these strains. Supplementary File S4 contains daily fecundity and daily intrinsic growth rate of 121 C. elegans strains. Supplementary File S5 contains GWA results on lifetime fecundity of 121 C. elegans strains. Supplementary File S6 contains genotype and phenotype data of 121 C. elegans strains at the peak markers of GWA mapping. Supplementary File S7 contains the sampling locations of 121 C. elegans strains. Supplementary File S8 contains the GPS coordinates of sampling locations of 121 C. elegans strains. Supplementary File S9 contains lifetime fecundity and swept and divergent classifications of each of the four swept chromosomes for each of the 121 C. elegans strains. Supplementary File S10 contains LD results among the three QTL of GWA using 121 C. elegans strains. Supplementary File S11 contains the shared haplotypes of the 121 strains within the QTL of GWA mapping. Supplementary File S12 contains GWA results on fecundity data of 236 strains from a previous study (Hahnel et al. 2018). Supplementary File S13 contains genotype and phenotype data of 236 strains at the peak marker of GWA mapping. Supplementary File S14 contains the shared haplotypes of the 236 strains within the QTL of GWA mapping. Supplementary File S15 contains the linkage mapping results for the 402 RIAILs in 1% water condition. Supplementary File S16 contains genotype and phenotype data of the 402 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results. Supplementary File S17 contains the linkage mapping results for the 417 RIAILs in 1% DMSO condition. Supplementary File S18 contains genotype and phenotype data of the 417 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results. Supplementary File S19 contains the linkage mapping results for the 432 RIAILs in 0.5% DMSO condition. Supplementary File S20 contains genotype and phenotype data of the 432 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results.
Results
Chromosome-scale sweeps shape C. elegans strain relationships
Genomic information of 913 wild C. elegans strains, grouped into 403 genetically distinct isotypes, are currently available in CeNDR (Cook et al. 2017). The latest CeNDR haplotype data, inferred from identical-by-descent groups among the 403 isotypes, include 22,859 distinct haplotypes across the genome. The number of haplotypes on each chromosome ranged from 2567 to 5199. We identified 11 most common haplotypes found in the majority of wild strains. Of the 403 C. elegans isotypes, 331 share more than 1 Mb of regions with at least one of the 11 most common haplotypes, particularly on chromosomes I, IV, V, and X (Figure 1A, Supplementary File S1). The haplotype structure of shared haplotypes over large regions across 403 isotypes further supported the selective sweeps identified previously (Andersen et al. 2012).
Figure 1.
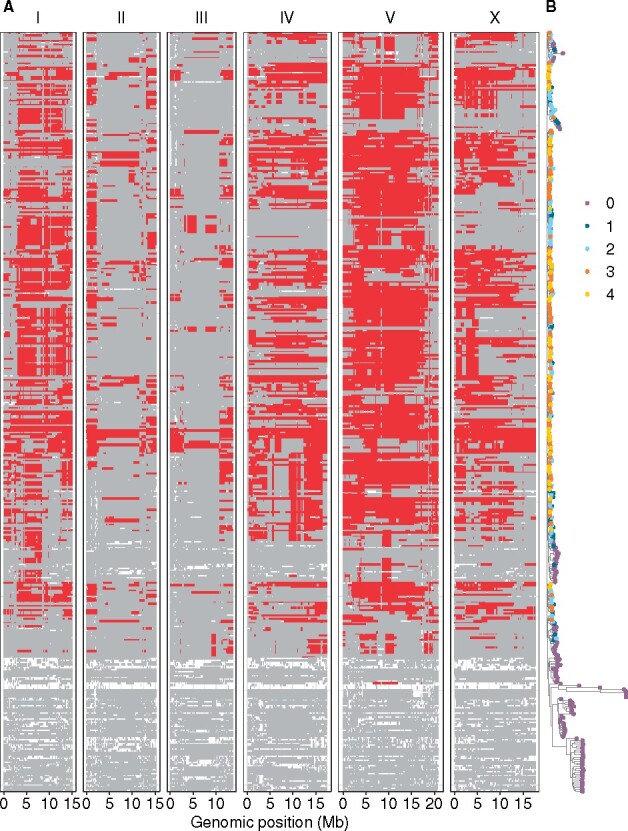
Swept chromosomes and genetic relatedness of wild C. elegans isotypes. (A) Sharing of the most common haplotypes (red) across the genome of C. elegans for 403 isotypes is shown. Genomic regions of unswept haplotypes (haplotypes other than the most common haplotypes) are colored gray. White segments are undetermined haplotypes in regions where no identical-by-descent groups were found (Crombie et al. 2019). The genomic position is plotted on the x-axis. Each row on the y-axis represents one of the 403 isotypes, ordered as their positions in (B). (B) A tree showing genetic relatedness of the 403 C. elegans isotypes, using 1,074,596 biallelic segregating sites, is shown. The tips of the tree are colored by the number of swept chromosomes (purple for zero, deep blue for one, light blue for two, orange for three, and gold for four) in each C. elegans isotype.
The shared fraction of the most common haplotypes per chromosome varies in each C. elegans isotype. Among chromosomes with shared regions in the 331 isotypes, chromosomes I, II, III, IV, V, and X have mean shared fractions and SD of 0.45 ± 0.25, 0.21 ± 0.19, 0.22 ± 0.17, 0.52 ± 0.28, 0.60 ± 0.27, and 0.43 ± 0.28, respectively. We focused on swept haplotypes, the most common haplotypes on chromosomes I, IV, V, and X, where evidence of selective sweeps were identified (Andersen et al. 2012). The chromosomal sharing of swept haplotypes contributes substantially to the genetic relatedness of C. elegans isotypes (Figure 1B, Supplementary File S2). Isotypes with swept chromosomes, which contain greater than or equal to 30% of swept haplotypes, clustered together. Of the 331 isotypes noted above, 281 have at least one swept chromosome (Figure 1B). We classified these 281 C. elegans isotypes as swept isotypes. We found that 244 swept isotypes have at least two swept chromosomes. By contrast, most of the 122 divergent isotypes with no swept chromosomes clustered together (Figure 1B). Previous analyses on genome-wide average nucleotide diversity (π), Tajima’s D, and genome-wide Hudson’s FST between 43 Hawaiian isotypes (most are divergent isotypes) and 233 non-Hawaiian isotypes (most are swept isotypes) also revealed a high degree of divergence, the highest of which were found in genomic regions impacted by the selective sweeps (Crombie et al. 2019). The high degree of genetic relatedness across the species is driven by the selective sweeps, but the fitness advantage causing the strong selective sweeps is yet unknown.
Natural variation in fecundity among swept and divergent strains
To compare the fitness between swept and divergent isotypes, we measured lifetime fecundity of 121 wild C. elegans strains sampled across the globe (Supplementary Figure S1 and File S8). Single fourth larval stage hermaphrodites were transferred daily for 5 days and maintained under normal laboratory conditions. We manually counted the viable offspring from images of assay plates. The results showed large variation in lifetime fecundity among wild C. elegans strains (Figure 2A, Supplementary File S3). The MLF ranged from 106 to 335 offspring among the 121 strains. We observed the species reproductive peak in the second day of the assay, with a median peak number of 109 offspring (Figure 2B, Supplementary File S4).
Figure 2.

Natural variation in C. elegans fecundity. (A) A bar plot for lifetime fecundity (y-axis) of 121 wild C. elegans strains is shown. Strains on the x-axis are sorted by their MLF of two to five biological replicates. Error bars show standard errors of lifetime fecundity among replicates. The lab reference strain N2 and the Hawaii strain CB4856 are colored orange and blue, respectively; other strains are colored gold for swept strains and purple for divergent strains. (B) Comparisons of lifetime and daily fecundity between 68 swept strains (gold) and 53 divergent strains (purple) are shown as Tukey box plots. Statistical significance was calculated using the Wilcoxon test and was corrected for multiple comparisons (Holm method). Significance of each comparison is shown above each comparison pair (ns: adjusted P-value > 0.05; *: adjusted P-value ≤ 0.05; **: adjusted P-value ≤ 0.01; ***: adjusted P-value ≤ 0.001; ****: adjusted P-value ≤ 0.0001).
Of the 121 C. elegans strains, 68 strains were classified as “swept” strains and 53 strains were classified as “divergent” strains (see Materials and Methods, Figure 2B, Supplementary Figure S1 and File S3). MLF of swept strains was significantly higher than divergent strains (Wilcoxon test, adjusted P = 9.1E-6) (Figure 2B). Because different strains could have different swept chromosomes, we extended the comparisons to chromosome levels (Supplementary Figure S2 and File S9). We assigned strains into swept groups or divergent groups in each swept chromosome, depending on whether isotypes had a specific swept chromosome. Although the numbers of strains in the two groups were different across swept chromosomes, swept groups always showed significantly higher lifetime fecundity than divergent groups (Wilcoxon test, adjusted P < 0.0001) (Supplementary Figure S2). The striking differences in lifetime fecundity suggested that swept strains have higher fitness than divergent strains under normal laboratory conditions. A later switch from spermatogenesis to oogenesis during the development of C. elegans could lead to the generation of more sperm and thus higher lifetime fecundity. This later switch would likely be associated with a trade-off of lower fecundity in early reproduction days. Surprisingly, we found that swept strains showed significantly higher daily fecundity than divergent strains in the first 3 days of the assays (Wilcoxon test, adjusted P = 0.0016, adjusted P = 1.7E-6, and adjusted P = 0.014, respectively) (Figure 2B). Swept strains also showed significantly higher intrinsic growth rate (r, maximum r was found at day two of adulthood for most strains) than divergent strains (Wilcoxon test, adjusted P = 9.9E-6) (Supplementary Figure S3 and File S4). This significant difference of fecundity between swept and nonswept groups provided an opportunity to dissect the genetic basis of the natural variation in lifetime fecundity. We calculated the broad-sense heritability and found a substantial heritable genetic component (H2 = 0.63) of the phenotypic variance across these strains.
Three QTL are associated with natural variation in C. elegans lifetime fecundity
To identify genomic loci that underlie fecundity variation, we performed a marker-based GWA mapping using MLF data from 121 C. elegans strains and the whole-genome variant data from CeNDR. We identified three distinct QTL (Figure 3A, Supplementary File S5). The first QTL, located on the right arm of chromosome I, has a peak-marker at position 13,917,228 and explains 21% of the phenotypic variation among the 121 strains. The second QTL located on the left arm of chromosome II has a peak-marker position at 543,326 and explains 22% of the phenotypic variation. The third QTL spans the center of chromosome V with the peak marker located at 14,534,671 and explains 30% of the phenotypic variation. Because of the strong LD within and between chromosomes in C. elegans (Andersen et al. 2012), linked regions might be falsely discovered as QTL even though they have no variants that underlie the phenotypic variation. To test the independence of the three QTL, we calculated the pairwise LD among their peak markers (Supplementary Figure S4 and File S10). The results showed moderate levels of LD (ranged from 0.387 to 0.512) for all three pairs, suggesting that they might not be independent. Notably, at all QTL peak markers, most swept strains have the reference alleles and most divergent strains have the alternative alleles (Figure 3B, Supplementary File S6). We further compared the sharing of haplotypes among the 121 strains within each QTL region (Supplementary Figure S5 and File S11). The majority of the strains with the reference alleles at the peak markers have the most common haplotypes in the QTL regions. By contrast, few strains with alternative alleles have the most common haplotypes in the QTL regions. Taken together, these results suggest that the genetic variants and different haplotypes underlying lifetime fecundity variation might be linked to the selective sweeps in the global population of C. elegans.
Figure 3.

Three QTL were identified in GWA mapping of lifetime fecundity variation in 121 C. elegans wild strains. (A) Manhattan plot indicating GWA mapping results. Each point represents an SNV that is plotted with its genomic position (x-axis) against its −log10(p) value (y-axis) in mapping. SNVs that pass the genome-wide EIGEN threshold (the dotted gray horizontal line) and the genome-wide BF threshold (the solid gray horizontal line) are colored pink and red, respectively. (B) Tukey box plots showing lifetime fecundity between strains with different genotypes at the peak marker position in each QTL. Each point corresponds to a C. elegans strain and is colored gold for swept strains and purple for divergent strains. On the x-axis, REF represents strains with the N2 reference allele and ALT represents strains with the alternative allele.
Hawaiian C. elegans exhibit lower lifetime fecundity than strains sampled across the globe
Most of the 121 C. elegans strains were originally sampled from three geographically isolated locations: 50 from the Hawaiian Islands, 22 from North America, and 41 from Europe (Supplementary Figure S1). Of the 50 Hawaiian C. elegans strains, 46 were classified as divergent, and the other four strains have no more than two swept chromosomes (Figure 4, Supplementary Figure S1 and File S7). Most C. elegans strains from North America and Europe were classified as swept strains (Figure 4, Supplementary Figure S1 and File S7). We compared lifetime fecundity of strains isolated from these three locations (Figure 4). Compared to strains from North America and Europe, Hawaiian strains had significantly lower lifetime fecundity (Wilcoxon test, adjusted P = 0.0013 and adjusted P = 2.2E-5, respectively). The difference in lifetime fecundity between strains from North America and strains from Europe was not significant. These data suggested that the selective sweeps that occurred outside Hawaii contribute substantially to the geographical lifetime fecundity difference.
Figure 4.
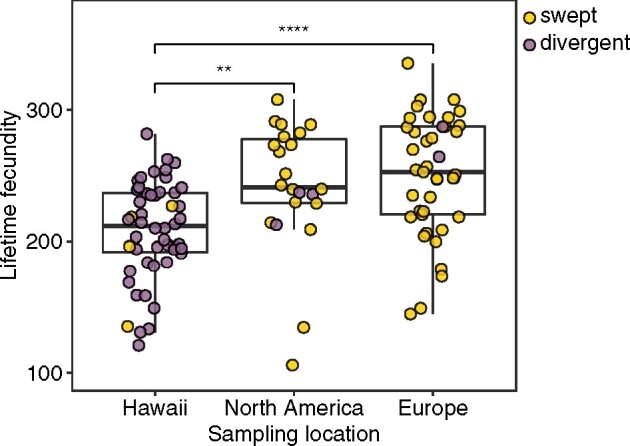
Lifetime fecundity comparisons in wild C. elegans strains among different sampling locations. Comparisons of lifetime fecundity among strains collected from Hawaii (50 strains), North America (22 strains), and Europe (41 strains). Each point corresponds to a strain and is colored gold for swept strains and purple for divergent strains. Statistical significance was calculated using the Wilcoxon test. Significance of each comparison is shown above each comparison pair (**: adjusted P-value ≤ 0.01; ****: adjusted P-value ≤ 0.0001). The difference of lifetime fecundity between North American and European strains is not significant.
More QTL underlying lifetime fecundity of C. elegans
We also mapped the fecundity data in the 1% DMSO control condition from one of our published studies that used the high-throughput fitness assays (HTA) (see Materials and Methods) to measure various fitness parameters of 236 strains (209 swept strains and 27 divergent strains) (Hahnel et al. 2018). Here, we performed GWA mapping using the fecundity measurements (norm.n) and identified a QTL on chromosome X (from 3.9 to 5.4 Mb, with the peak marker at 4,831,537) (Figure 5, Supplementary Figure S6A and File S12). Divergent strains showed no enrichment with either genotype at the peak marker (Supplementary Figure S6B and File S13). However, most strains with the reference allele have the most common haplotypes and most strains with the alternative allele have unswept haplotypes (Supplementary Figure S6C and File S14). These results suggest that the genetic variants in this region might also be linked to the recent selective sweeps in wild C. elegans populations.
Figure 5.
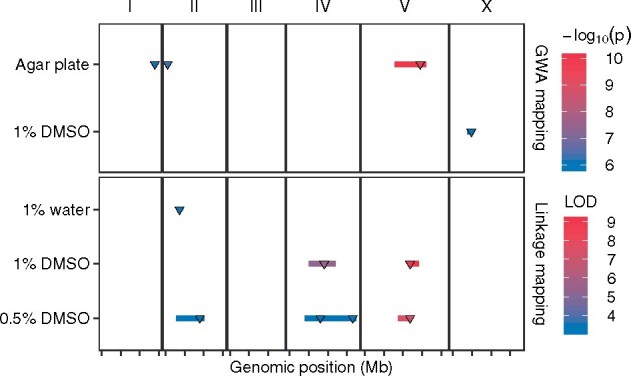
Multiple QTL impacting C. elegans lifetime fecundity in different conditions. Four GWA mapping QTL of two conditions (121 strains cultured in agar plate and 236 strains cultured in liquid with 1% DMSO) and seven linkage mapping QTL of three conditions (C. elegans RIAILs cultured in liquid with 1% water, 1% DMSO, and 0.5% DMSO, respectively) are plotted. Each condition is plotted on the y-axis against the genomic position of its QTL on the x-axis separated by chromosomes with tick marks denoting every 5 Mb. Each QTL is plotted as a line with a triangle indicating the peak marker and colored by the −log10(p) value (GWA QTL) or the logarithm of the odds (LOD) score (for linkage mapping QTL), increasing in significance from blue to red.
Also using HTA as above, we measured fecundity in liquid culture using the C. elegans RIAILs derived from QX1430 (a derivative strain of N2 with replacement of the N2 npr-1 allele with the counterpart version from the CB4856 strain and a transposon insertion into the peel-1 gene) and CB4856 (Andersen et al. 2015) under three conditions: 1% water, 1% DMSO, and 0.5% DMSO (see Materials and Methods). By contrast to the fecundity variation of C. elegans strains cultured in agar plates, the QX1430 strain showed lower fecundity than the CB4856 strain using HTA (Supplementary Figures S7B, S8B, and S9B and Files S16, S18, and S20), indicating that the gene npr-1 or environmental factors can have drastic effects on C. elegans fecundity (Andersen et al. 2014). We found seven QTL for fecundity on chromosomes II, IV, and V under the three conditions (Figure 5, Supplementary Figures S7A, S8A, and S9A). In 1% water, linkage mapping identified a single QTL confidence interval (II: 3.4–4 Mb) on the left arm of chromosome II (Figure 5, Supplementary Figure S7A and File S15). In 1% DMSO, linkage mapping identified two QTL located on chromosomes IV (5–11.9 Mb) and V (11.8–14.2 Mb), respectively (Figure 5, Supplementary Figure S8A and File S17). In 0.5% DMSO, the four QTL on chromosomes II (2.9–10.2 Mb), IV (two loci, 3.9–17.5 Mb), and V (8.7–12.3 Mb) recapitulated the three QTL detected in 1% water and 1% DMSO, respectively (Figure 5, Supplementary Figure S9A and File S19). Furthermore, the QTL on chromosome V in both DMSO conditions overlapped with the GWA QTL on chromosome V using the 121 wild strains in agar plates (Figure 5). Because linkage mapping using this set of C. elegans RIAILs can only find QTL in the CB4856 strain, overlapping of QTL between linkage mapping and GWA mapping suggests that the CB4856 strain carries the common alternative alleles among wild C. elegans strains in the shared regions. Altogether, these results suggest that C. elegans might have shared and separated loci controlling fecundity in agar cultures and in liquid cultures with slightly different concentrations of DMSO.
Discussion
In this study, we report natural variation of lifetime fecundity for 121 wild C. elegans strains and found that the previously reported chromosome-scale selective sweeps play a key role in the different fecundity among strains. We defined swept haplotypes, swept isotypes, and swept strains, using the latest C. elegans haplotype data from CeNDR. Swept strains that have at least one chromosome with equal or greater than 30% of swept haplotypes showed significantly higher lifetime fecundity than divergent strains that have avoided the sweeps. We identified three QTL that underlie differences in lifetime fecundity among the 121 C. elegans strains using single-marker based GWA mappings. Remarkably, across all three QTL, swept strains tend to have shared haplotypes and the reference alleles at peak markers. By contrast, divergent strains tend to have unswept haplotypes and the alternative alleles at peak markers. We also observed significant geographical differences in lifetime fecundity between Hawaiian strains and strains from other parts of the world, likely because of the selective sweeps. We further mapped previous data using GWA mapping and linkage mapping and identified eight QTL underlying C. elegans fecundity in different environments. Taken together, our results showed the diverse genetic basis of C. elegans fecundity and suggest that higher fecundity in most C. elegans strains could be caused by alleles that have recently swept throughout the world population.
Genetically divergent strains have substantially lower fecundity than swept strains
We measured lifetime fecundity in 121 genetically distinct C. elegans strains. In our measurements (Figure 2A), the laboratory reference strain N2 (known as the Bristol strain) and a frequently used wild strain CB4856 (known as the Hawaii strain) had lifetime fecundity of 308 and 237, respectively, with similar fecundity values as reported previously (Hodgkin and Doniach 1997; Wegewitz et al. 2008; Andersen et al. 2014; Poullet et al. 2015). The CB4856 strain had been considered the most genetically distant strain from the N2 strain for decades. In the last 5 years, researchers have collected and identified many genetically divergent C. elegans strains, some of which are more divergent from the N2 strain than the CB4856 strain is (Cook et al. 2017; Crombie et al. 2019; Lee et al. 2021). Most of these divergent strains were from Hawaii and showed none or rare evidence of the globally distributed swept haplotypes (Figure 1, Supplementary Figure S1) (Crombie et al. 2019; Lee et al. 2021). In our fecundity assays, we included many of these divergent strains. Divergent strains showed significantly lower fecundity than swept strains that have large blocks of swept haplotypes, suggesting that divergent strains have lower fitness than swept strains under normal laboratory conditions. The disadvantage in fecundity of divergent strains was present from the beginning of the reproductive period throughout the peak. This lower fitness of divergent strains could have at least two possible explanations. First, laboratory conditions might favor swept strains over divergent strains. Standard laboratory conditions to culture C. elegans have been designed, modified, and improved based on the growth of the N2 strain (Brenner 1974), which is a swept strain. Most swept strains were from temperate zones (Andersen et al. 2012; Félix and Duveau 2012; Petersen et al. 2014; Richaud et al. 2018), such as Western Europe, whereas most divergent strains were isolated in the high elevation and cool temperature niches in the Hawaiian Islands (Crombie et al. 2019). The conditions of the natural habitats and the microenvironments in the niches of swept strains could be drastically different from niches of divergent strains. The closer the natural niche condition is to the laboratory condition, the higher fitness a strain might have (Volkers et al. 2013). For example, compared to N2, the strain CB4856 showed a clear thermal preference of approximately 17°, which is lower than the canonical and the most typical C. elegans culture temperature of 20° in the laboratory (Brenner 1974; Stiernagle 2006; Anderson et al. 2007). In a competition assay between two swept strains that were isolated from locations with distinct climates, CX11314 (isolated at 20.9°) showed higher fitness than JU847 (isolated at 11.3°) at both 15° and 25°, but JU847 grew better at 15° than at 25° (Evans et al. 2017). Divergent strains that were isolated from cool regions might exhibit higher fitness at temperatures lower than 20°.
The second explanation is that genetic variants at unknown loci directly caused differences in lifetime fecundity between swept strains and divergent strains. The environmental factors in our assays might have similar or minor influences on the fecundity for both swept strains and divergent strains. The major differences in fecundity between swept strains and divergent strains could be attributed to their genetic differences. For instance, because a C. elegans hermaphrodite produces 200–300 sperm in the late L4 stage before irreversibly switching to oogenesis to produce up to 1000 oocytes, the number of sperm limits fecundity of self-fertilized hermaphrodites (Ward and Carrel 1979; Cutter 2004; Félix and Braendle 2010). Alleles at unknown loci in swept strains might lead to an increased number of sperm and thus a higher fecundity than divergent strains. It is also possible that swept strains and divergent strains produce similar numbers of sperm, but divergent strains have reduced sperm fertility, defects in oogenesis, or higher embryonic lethality than swept strains (Poullet et al. 2015). Because we quantified the viable offspring from each of the C. elegans strains as their fecundity (see Materials and Methods), defects related to fertilization or higher embryonic lethality could have caused the lower daily fecundity in the first 3 days of the reproductive period and the lower lifetime fecundity observed in divergent strains. The higher fecundity of swept strains in the first 3 days might also be caused by a shorter duration of L4 larval stage and/or an earlier or more efficient germline development (e.g., earlier onset or faster development of spermatogenesis and/or oogenesis) than divergent strains. We picked L4 stage animals to start each assay, so those L4 larvae of swept strains might be more mature than divergent strains. Swept strains might start laying embryos and enter into reproductive peak faster than divergent strains, demonstrating the earlier advantages. Although our GWA results might have mapped genomic regions underlying spermatogenesis, oogenesis, fertilization success, or embryonic lethality, future efforts to quantify developmental timing, the numbers of sperm, and fertilized embryos among wild C. elegans strains will help to further elucidate the differences in fecundity among strains. Moreover, some divergent strains continued to produce many offspring in the last few assay days, at a time when most swept strains gradually reduce offspring production. It is possible that swept strains have a shortened but accelerated reproductive period. By contrast, divergent strains could have a prolonged but slow reproductive period. To investigate the variation of reproductive schedules and underlying genetic basis, further work to quantify fecundity should proceed until the full depletion of self-sperm.
Diverse QTL for lifetime fecundity in different environments
We performed GWA mapping and identified three QTL on chromosomes I, II, and V for lifetime fecundity of C. elegans, which were grown on agar plates and fed E. coli OP50. The split of strains by genotypes at peak markers and the haplotypes of each strain in each QTL strongly suggest that the three QTL could be the genetic basis of different lifetime fecundity between swept strains and divergent strains. The reference alleles and the most common haplotypes in each QTL, which provided the selective advantage of higher fecundity, could have swept through the C. elegans population as these strains spread throughout the world. Under similar conditions, a previous study using linkage mapping and a large panel of RIAILs derived from the N2 and CB4856 strains have mapped fecundity to QTL on chromosomes II (2.6–3.6 Mb) and X (4.6–7.7 Mb) (Andersen et al. 2014). A laboratory-derived mutation in the gene npr-1 from N2 was identified to have driven the QTL on chromosome X (McGrath et al. 2009; Andersen et al. 2014).
In liquid culture and fed the E. coli strain HB101, a new panel of C. elegans RIAILs with QX1430 and CB4856 was used to map fecundity to a QTL on chromosome IV (10.7–12.8 Mb) using linkage mapping (Andersen et al. 2015). Using the same RIAIL panel but under three different liquid conditions (1% H2O, 1% DMSO, and 0.5% DMSO), we mapped fecundity to seven QTL on chromosomes II, IV, and V. In both DMSO conditions, the three QTL on chromosome IV recapitulated the QTL in the above study (Andersen et al. 2015); the two overlapping QTL on chromosome V overlapped with the QTL using our 121 wild strains grown in agar plates. We further used GWA to map previously published wild strain fecundity data from liquid culture and 1% DMSO. A QTL linked to the selective sweeps located on the left arm of chromosome X was identified. Although npr-1 is in the region of this QTL, the laboratory-derived N2 npr-1 allele that is only found in the N2 strain could not underlie this QTL because it is not found in wild strains. Distinct QTL were detected in the two GWA mappings. The bleaching method to synchronize animals, liquid cultures, and a different bacterial diet (E. coli HB101) might have affected fecundity and the mapping results.
As a complex life history trait, lifetime fecundity could be influenced by many loci (Houle 1992). Under different conditions, GWA mappings identified QTL on chromosomes I, II, V, and X; linkage mappings identified QTL on chromosomes II, IV, V, and X. Because swept haplotypes shared among C. elegans strains might have driven all the QTL in GWA mappings, genetic variants in these swept haplotypes might be the beneficial alleles that swept through the C. elegans population. Natural habitats of C. elegans are likely quite different from both the laboratory standard conditions and liquid cultures with DMSO. Our results of GWA and linkage mappings suggest that shared and separate loci in the C. elegans genome control fecundity in different environmental conditions in the laboratory. We do not know how those environments relate to the wild, but it is possible that similar conditions could occur (e.g., swimming or crawling in environments with ample bacteria). Our results also suggest that fecundity of C. elegans is sensitive to environmental changes in cultures (agar plates vs liquid cultures; with or without DMSO) or diet (E. coli OP50 and HB101). Larval and germline development of C. elegans were previously found to be sensitive to food availability, diet, and temperature (Poullet et al. 2015; Filina et al. 2020). Lifetime fecundity of C. elegans might also be sensitive to changes in these environmental factors. To deepen our understanding of the influence of genetic factors, environmental factors, and gene-environment interactions on fecundity, future efforts should include more strains and compare their fecundity in diverse environments.
Potential adaptive alleles for C. elegans in temperate zones
The QTL for lifetime fecundity using the 121 C. elegans strains also shared genomic regions with QTL on weather and climate variables related to natural habitats of 149 wild C. elegans strains (Evans et al. 2017). Two of the GWA mapping QTL for relative humidity were on chromosomes II and V, which overlapped with our QTL on chromosomes II and V, respectively. GWA mappings for 3-year average temperature also located the same QTL just right of the center of chromosome V. We showed that C. elegans strains sampled from Europe and North America had similar lifetime fecundity, which was significantly larger than fecundity of Hawaiian C. elegans strains. Because Hawaii is in the tropical zone, C. elegans isolated from high elevation areas in Hawaii could have experienced high humidity and low temperatures in a much more stable climate in the long term than C. elegans in temperate zones. Alleles of swept strains in the shared QTL underlying lifetime fecundity and climate variables could have enhanced the adaptability of C. elegans to variable humidity and temperatures in temperate zones along the C. elegans expansion out of the Pacific region (Andersen et al. 2012; Crombie et al. 2019; Lee et al. 2021). It is possible that, because of these adaptive alleles, the N2 strain showed no preference at these temperatures (Anderson et al. 2007).
Some Hawaiian strains, exclusively isolated at lower elevations closer to the coasts, exhibited admixture with non-Hawaiian populations, which might come from gene flow from outcrossing with immigrating swept strains from outside to Hawaii (Crombie et al. 2019). But compared to most non-Hawaiian strains, Hawaiian strains only contain, if any, small fractions of swept haplotypes. Of the 50 Hawaiian C. elegans strains used in this study, four strains are classified as swept strains, who have no more than two swept chromosomes (Supplementary Figure S1). The alleles that increase lifetime fecundity in swept strains might not contribute to higher fitness for C. elegans strains in Hawaii. In fluctuating environments in temperate zones, the randomly distributed and limited habitats might select for C. elegans that have higher fecundity, although the high density of animals also facilitates dauer formation, which could limit population growth but underlie future survival success. Moreover, C. elegans populations in temperate zones also undergo bottlenecks in winter, from which dauer larvae are more likely to survive. By contrast, Hawaiian C. elegans might not need to enter and stay in the dauer stage as often and long as non-Hawaiian C. elegans in temperate zones. Habitats hypothesized to have more ample bacterial food (e.g., rotting fruits) and a stable environment in Hawaii could lead to a higher survival rate and lower fecundity as a trade-off (Stearns 1989; Marshall and Sinclair 2010). Two genotypes of the gene srg-37 were found to coexist in the wild population and associate with different niches (Lee et al. 2019). The deletion in srg-37, which likely originated outside of Hawaii, reduces dauer formation and promotes reproduction in niches hypothesized to promote rapid growth. C. elegans strains without deletion of srg-37 could have higher fitness during the dispersal phase in nutrient-poor environments (Lee et al. 2019). Among the 121 strains we studied, none of the divergent strains have the srg-37 deletion. However, other QTL might exist among divergent strains to reduce dauer formation (Green et al. 2013, 2014). Future dauer formation assays, such as responses to ascaroside pheromones, among divergent strains could help dissect the interactions of traits that affect fitness of C. elegans in the wild.
However, the QTL we found that underlie higher fecundity in swept strains might not directly underlie the selective advantages during the expansion of the species. Loci in C. elegans that affect other fitness traits (e.g., dauer formation, response to natural food source of different bacteria, or resistance to natural pathogens) might be under direct selective pressures in the wild. Alleles that provided higher fitness in these traits might underlie the selective sweeps in C. elegans population. The QTL for fecundity variation in our results might be in LD with genomic regions that affect these other fitness traits mentioned above and maintained by linked selection. The swept strains are widely distributed in different environments around the world, so the effects of the interaction between genotype and environment could have also influenced this expansion. To find the direct targets of selection and the principal drivers of selective sweeps, multiple abiotic and biotic factors in natural habitats of C. elegans should be measured. Then, several fitness traits under different conditions could be measured in the laboratory. For instance, fecundity and viral load could be measured at different temperatures at the same time (Félix et al. 2011; Samuel et al. 2016).
Acknowledgments
The authors would like to thank members of the Andersen Lab for helpful comments on the manuscript.
Funding
G.Z. and E.C.A. received support from the NSF-Simons Center for Quantitative Biology at Northwestern University (awards Simons Foundation/SFARI 597491-RWC and the National Science Foundation 1764421). J.D.M received support from a Northwestern Undergraduate Research Grant.
Conflicts of interest
The authors declare no conflicts of interest.
Literature cited
- Andersen EC, Bloom JS, Gerke JP, Kruglyak L.. 2014. A variant in the neuropeptide receptor npr-1 is a major determinant of Caenorhabditis elegans growth and physiology. PLoS Genet. 10:e1004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Gerke JP, Shapiro JA, Crissman JR, Ghosh R, et al. 2012. Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat Genet. 44:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen EC, Shimko TC, Crissman JR, Ghosh R, Bloom JS, et al. 2015. A powerful new quantitative genetics platform, combining Caenorhabditis elegans high-throughput fitness assays with a large collection of recombinant strains. G3 (Bethesda). 5:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Albergotti L, Ellebracht B, Huey RB, Phillips PC.. 2011. Does thermoregulatory behavior maximize reproductive fitness of natural isolates of Caenorhabditis elegans? BMC Evol Biol. 11:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Albergotti L, Proulx S, Peden C, Huey RB, et al. 2007. Thermal preference of Caenorhabditis elegans: a null model and empirical tests. J Exp Biol. 210:3107–3116. [DOI] [PubMed] [Google Scholar]
- Barrière A, Félix M-A.. 2005. High local genetic diversity and low outcrossing rate in Caenorhabditis elegans natural populations. Curr Biol. 15:1176–1184. [DOI] [PubMed] [Google Scholar]
- Barrière A, Félix M-A.. 2007. Temporal dynamics and linkage disequilibrium in natural Caenorhabditis elegans populations. Genetics. 176:999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, Walker S.. 2015. Fitting linear mixed-effects models using lme4. J Stat Soft. 67:48. [Google Scholar]
- Berry AJ, Ajioka JW, Kreitman M.. 1991. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics. 129:1111–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd WA, Smith MV, Freedman JH.. 2012. Caenorhabditis elegans as a model in developmental toxicology. Methods Mol Biol. 889:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman JM, Hudson RR, Kaplan NL, Langley CH, Stephan W.. 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics. 140:783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, et al. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Singh RS, Uyenoyama MK.. 2003. The population genetics of life-history evolution In: Singh R. S., Uyenoyama M. K., editors. The Evolution of Population Biology. Cambridge University Press, Cambridge. pp. 216–232. [Google Scholar]
- Cook DE, Zdraljevic S, Roberts JP, Andersen EC.. 2017. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 45:D650–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombie TA, Zdraljevic S, Cook DE, Tanny RE, Brady SC, et al. 2019. Deep sampling of Hawaiian Caenorhabditis elegans reveals high genetic diversity and admixture with global populations. eLife. 8:e50465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD. 2006. Nucleotide polymorphism and linkage disequilibrium in wild populations of the partial selfer Caenorhabditis elegans. Genetics. 172:171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter AD. 2004. Sperm-limited fecundity in nematodes: how many sperm are enough? Evolution. 58:651–655. [PubMed] [Google Scholar]
- Endelman JB. 2011. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome. 4:250–255. [Google Scholar]
- Evans KS, Andersen EC.. 2020. The gene scb-1 underlies variation in Caenorhabditis elegans chemotherapeutic responses. G3 (Bethesda). 10:2353–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KS, Zhao Y, Brady SC, Long L, McGrath PT, et al. 2017. Correlations of genotype with climate parameters suggest Caenorhabditis elegans Niche adaptations. G3 (Bethesda). 7:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu CI.. 2000. Hitchhiking under positive Darwinian selection. Genetics. 155:1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix M-A, Ashe A, Piffaretti J, Wu G, Nuez I, et al. 2011. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 9:e1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix M-A, Braendle C.. 2010. The natural history of Caenorhabditis elegans. Curr Biol. 20:R965–R969. [DOI] [PubMed] [Google Scholar]
- Félix M-A, Duveau F.. 2012. Population dynamics and habitat sharing of natural populations of Caenorhabditis elegans and C. briggsae. BMC Biol. 10:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filina O, Haagmans R, van Zon JS.. 2020. Temporal scaling in C. elegans larval development. bioRxiv. 2020.09.21.306423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatt T. 2020. Life-history evolution and the genetics of fitness components in Drosophila melanogaster. Genetics. 214:3–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatt T, Heyland A.. 2011. Mechanisms of Life History Evolution: The Genetics and Physiology of Life History Traits and Trade-Offs. Oxford: OUP. [Google Scholar]
- Frézal L, Félix M-A.. 2015. The natural history of model organisms: C. elegans outside the Petri dish. eLife. 4:e05849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González AP, Ritter AD, Shrestha S, Andersen EC, Yilmaz LS, et al. 2017. Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell. 169:431–441.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert KJ, Zdraljevic S, Cook DE, Cutter AD, Andersen EC, et al. 2020. The distribution of mutational effects on fitness in Caenorhabditis elegans inferred from standing genetic variation. Cold Spring Harbor Lab. bioRxiv 2020.10.26.355446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JWM, Snoek LB, Kammenga JE, Harvey SC.. 2013. Genetic mapping of variation in dauer larvae development in growing populations of Caenorhabditis elegans. Heredity. 111:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JWM, Stastna JJ, Orbidans HE, Harvey SC.. 2014. Highly polygenic variation in environmental perception determines dauer larvae formation in growing populations of Caenorhabditis elegans. PLoS One. 9:e112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahnel SR, Zdraljevic S, Rodriguez BC, Zhao Y, McGrath PT, et al. 2018. Extreme allelic heterogeneity at a Caenorhabditis elegans beta-tubulin locus explains natural resistance to benzimidazoles. PLoS Pathog. 14:e1007226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J, Doniach T.. 1997. Natural variation and copulatory plug formation in Caenorhabditis elegans. Genetics. 146:149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D. 1992. Comparing evolvability and variability of quantitative traits. Genetics. 130:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, et al. 2008. Efficient control of population structure in model organism association mapping. Genetics. 178:1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH.. 1989. The “hitchhiking effect” revisited. Genetics. 123:887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Nielsen R.. 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics. 167:1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiontke KC, Félix M-A, Ailion M, Rockman MV, Braendle C, et al. 2011. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol. Biol. 11:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight GR, Robertson A.. 1957. Fitness as a measurable character in Drosophila. Genetics. 42:524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Zdraljevic S, Cook DE, Frézal L, Hsu J-C, et al. 2019. Selection and gene flow shape niche-associated variation in pheromone response. Nat Ecol Evol. 3:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Zdraljevic S, Stevens L, Wang Y, Tanny RE, et al. 2021. Balancing selection maintains hyper-divergent haplotypes in Caenorhabditis elegans. Nat Ecol Evol. 2021 Apr 5:1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall KE, Sinclair BJ.. 2010. Repeated stress exposure results in a survival-reproduction trade-off in Drosophila melanogaster. Proc Biol Sci. 277:963–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath PT, Rockman MV, Zimmer M, Jang H, Macosko EZ, et al. 2009. Quantitative mapping of a digenic behavioral trait implicates globin variation in C. elegans sensory behaviors. Neuron. 61:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H, Zdraljevic S, Tanny RE, Walhout AJM, Andersen EC.. 2020. Natural variation in a glucuronosyltransferase modulates propionate sensitivity in a C. elegans propionic acidemia model. PLoS Genet. 16:e1008984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz EM. 2019. vcf2phylip v2.0: convert a VCF matrix into several matrix formats for phylogenetic analysis. URL https://doi org/105281/zenodo, 2540861.
- Petersen C, Dirksen P, Prahl S, Strathmann EA, Schulenburg H.. 2014. The prevalence of Caenorhabditis elegans across 1.5 years in selected North German locations: the importance of substrate type, abiotic parameters, and Caenorhabditis competitors. BMC Ecol. 14:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poullet N, Vielle A, Gimond C, Ferrari C, Braendle C.. 2015. Evolutionarily divergent thermal sensitivity of germline development and fertility in hermaphroditic Caenorhabditis nematodes. Evol Dev. 17:380–397. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richaud A, Zhang G, Lee D, Lee J, Félix M-A.. 2018. The local coexistence pattern of selfing genotypes in Caenorhabditis elegans natural metapopulations. Genetics. 208:807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Kruglyak L.. 2009. Recombinational landscape and population genomics of Caenorhabditis elegans. PLoS Genet. 5:e1000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV, Skrovanek SS, Kruglyak L.. 2010. Selection at linked sites shapes heritable phenotypic variation in C. elegans. Science. 330:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel BS, Rowedder H, Braendle C, Félix M-A, Ruvkun G.. 2016. Caenorhabditis elegans responses to bacteria from its natural habitats. Proc Natl Acad Sci USA. 113:E3941–E3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep KP. 2011. phangorn: phylogenetic analysis in R. Bioinformatics. 27:592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW.. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimko TC, Andersen EC.. 2014. COPASutils: an R package for reading, processing, and visualizing data from COPAS large-particle flow cytometers. PLoS One. 9:e111090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J.. 1974. The hitch-hiking effect of a favourable gene. Genet Res. 23:23–35. [PubMed] [Google Scholar]
- Stearns SC. 1976. Life-history tactics: a review of the ideas. Q Rev Biol. 51:3–47. [DOI] [PubMed] [Google Scholar]
- Stearns SC. 1989. Trade-offs in life-history evolution. Funct Ecol. 3:259–268. [Google Scholar]
- Stephan W. 2019. Selective sweeps. Genetics. 211:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W, Song YS, Langley CH.. 2006. The hitchhiking effect on linkage disequilibrium between linked neutral loci. Genetics. 172:2647–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. 2006. Maintenance of C. elegans. WormBook 11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilieva LL, Lynch M.. 1999. The rate of spontaneous mutation for life-history traits in Caenorhabditis elegans. Genetics. 151:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkers RJM, Snoek LB, Hubar C. J V H, Coopman R, Chen W, et al. 2013. Gene-environment and protein-degradation signatures characterize genomic and phenotypic diversity in wild Caenorhabditis elegans populations. BMC Biol. 11:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward S, Carrel JS.. 1979. Fertilization and sperm competition in the nematode Caenorhabditis elegans. Dev Biol. 73:304–321. [DOI] [PubMed] [Google Scholar]
- Wegewitz V, Schulenburg H, Streit A.. 2008. Experimental insight into the proximate causes of male persistence variation among two strains of the androdioecious Caenorhabditis elegans (Nematoda). BMC Ecol. 8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Smith DK, Zhu H, Guan Y, Lam TT.. 2017. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 8:28–36. [Google Scholar]
- Zdraljevic S, Fox BW, Strand C, Panda O, Tenjo FJ, et al. 2019. Natural variation in C. elegans arsenic toxicity is explained by differences in branched chain amino acid metabolism. Elife. 8:e40260. Communicating editor: K. Gunsalus [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets, code for generating all figures, and Supplementary figures can be found at https://github.com/AndersenLab/swept_broods. Supplementary File S1 contains the haplotype data of 403 C. elegans isotypes from CeNDR release 20200815. Supplementary File S2 contains genetic relatedness of 403 C. elegans isotypes. Supplementary File S3 contains lifetime fecundity of 121 C. elegans strains, their classification of swept strains and divergent strains, and the assay blocks of these strains. Supplementary File S4 contains daily fecundity and daily intrinsic growth rate of 121 C. elegans strains. Supplementary File S5 contains GWA results on lifetime fecundity of 121 C. elegans strains. Supplementary File S6 contains genotype and phenotype data of 121 C. elegans strains at the peak markers of GWA mapping. Supplementary File S7 contains the sampling locations of 121 C. elegans strains. Supplementary File S8 contains the GPS coordinates of sampling locations of 121 C. elegans strains. Supplementary File S9 contains lifetime fecundity and swept and divergent classifications of each of the four swept chromosomes for each of the 121 C. elegans strains. Supplementary File S10 contains LD results among the three QTL of GWA using 121 C. elegans strains. Supplementary File S11 contains the shared haplotypes of the 121 strains within the QTL of GWA mapping. Supplementary File S12 contains GWA results on fecundity data of 236 strains from a previous study (Hahnel et al. 2018). Supplementary File S13 contains genotype and phenotype data of 236 strains at the peak marker of GWA mapping. Supplementary File S14 contains the shared haplotypes of the 236 strains within the QTL of GWA mapping. Supplementary File S15 contains the linkage mapping results for the 402 RIAILs in 1% water condition. Supplementary File S16 contains genotype and phenotype data of the 402 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results. Supplementary File S17 contains the linkage mapping results for the 417 RIAILs in 1% DMSO condition. Supplementary File S18 contains genotype and phenotype data of the 417 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results. Supplementary File S19 contains the linkage mapping results for the 432 RIAILs in 0.5% DMSO condition. Supplementary File S20 contains genotype and phenotype data of the 432 RIAILs at the peak markers and phenotype data of the parents in linkage mapping results.