

Abstract
The angiosperm genus Silene is a model system for several traits of ecological and evolutionary significance in plants, including breeding system and sex chromosome evolution, host-pathogen interactions, invasive species biology, heavy metal tolerance, and cytonuclear interactions. Despite its importance, genomic resources for this large genus of approximately 850 species are scarce, with only one published whole-genome sequence (from the dioecious species Silene latifolia). Here, we provide genomic and transcriptomic resources for a hermaphroditic representative of this genus (S. noctiflora), including a PacBio Iso-Seq transcriptome, which uses long-read, single-molecule sequencing technology to analyze full-length mRNA transcripts. Using these data, we have assembled and annotated high-quality full-length cDNA sequences for approximately 14,126 S. noctiflora genes and 25,317 isoforms. We demonstrated the utility of these data to distinguish between recent and highly similar gene duplicates by identifying novel paralogous genes in an essential protease complex. Furthermore, we provide a draft assembly for the approximately 2.7-Gb genome of this species, which is near the upper range of genome-size values reported for diploids in this genus and threefold larger than the 0.9-Gb genome of Silene conica, another species in the same subgenus. Karyotyping confirmed that S. noctiflora is a diploid, indicating that its large genome size is not due to polyploidization. These resources should facilitate further study and development of this genus as a model in plant ecology and evolution.
Keywords: Silene, PacBio, Iso-Seq, transcriptome, Silene noctiflora, karyotype, genome assembly
Introduction
Silene is the largest genus in the angiosperm family Caryophyllaceae and serves as a model system in many fields of ecology and evolutionary biology (Bernasconi et al. 2009; Jafari et al. 2020). For instance, Silene is used to study breeding system evolution, as the genus includes hermaphroditic, gynodioecious, gynomonoecious, monoecious, and dioecious species (Desfeux et al. 1996; Charlesworth 2006). Despite the diversity of Silene sexual systems, there is only one available whole-genome sequence for the entire genus—from the dioecious species Silene latifolia, which has heteromorphic XY sex chromosomes (Papadopulos et al. 2015; Krasovec et al. 2018). Whole-genome resources are not available for any of the hermaphroditic species, which has limited comparative genomic studies into the evolution of dioecy within this genus.
Silene is also used as a model system for investigating organelle genome evolution and the coevolution between nuclear and cytoplasmic genomes (i.e., cytonuclear interactions) (Olson and Mccauley 2002; Städler and Delph 2002; Klaas and Olson 2006; Garraud et al. 2011). Silene conica and S. noctiflora have two of the largest known plant mitochondrial genomes at 11 and 7 Mb, respectively (Sloan et al. 2012a). In contrast, the mitochondrial genome of S. latifolia is only 0.25 Mb, about 45 times smaller than that of S. conica (Sloan et al. 2012a). Interestingly, the Silene species with expanded mitogenomes also display unusually high evolutionary rates and structural changes in mitochondrial and plastid DNA (Mower et al. 2007; Sloan et al. 2012a). The natural variation in organelle genome evolution found in this genus has been used to study how these differences affect cytonuclear interactions (Havird et al. 2015; Williams et al. 2019).
The ability to use Silene as a model for cytonuclear evolution is still limited by the lack of extensive nuclear genome resources. Previous work has characterized Silene nuclear genome size and chromosome number. Nuclear genome sizes in the genus vary considerably, although not as starkly as mitochondrial genome sizes, ranging roughly 4.5-fold among diploids (haploid sizes of 0.71 to 3.23 Gb) and eightfold when the tetraploid S. stellata (5.77 Gb) is included (Kruckeberg 1960; Siroký et al. 2001; Bai et al. 2012; Dagher-Kharrat et al. 2013; Pellicer and Leitch 2020). Most of the available nuclear sequence data come from short-read RNA sequencing, which has been conducted on multiple Silene species (Blavet et al. 2011; Sloan et al. 2012b; Muyle et al. 2012; Casimiro-Soriguer et al. 2016; Havird et al. 2017; Bertrand et al. 2018; Balounova et al. 2019). These datasets have provided an important resource for molecular studies of Silene, but are limited because of the challenges associated with assembling short-read sequences, especially in distinguishing similar sequences arising from gene duplication, heterozygosity, and/or alternative splicing (Alkan et al. 2011; Schatz et al. 2012; Hahn et al. 2014; Lan et al. 2017).
We have generated genomic resources critical for investigations into S. noctiflora, a species of interest due to its extremely unusual organelle evolution and resultant use as a model for cytonuclear interactions, as well as its status as a hermaphrodite in a genus representing many types of breeding system. We include a high-quality transcriptome using long-read PacBio Iso-Seq technology, genome size estimates, and a draft nuclear genome assembly. These resources will expand opportunities for molecular and ecological studies within the genus.
Materials and methods
Study system
Silene noctiflora (Figure 1) is largely hermaphroditic but can produce a mixture of hermaphroditic and male-sterile flowers on the same plant (gynomonoecy) (Davis and Delph 2005). Also known as the night-flowering catchfly, this annual species is native to Eurasia and introduced throughout much of the world (McNeill 1980; Davis and Delph 2005).
Figure 1.

Silene noctiflora, also known as the night-flowering catchfly.
Plant growth conditions, tissue sampling, and nucleic acid extractions
Plants used for genome sequencing, Iso-Seq, and flow cytometry estimates of genome size were grown under standard greenhouse conditions with 16-hour light/8-hour dark at Colorado State University (Table 1). DNA for short-insert paired-end Illumina libraries was extracted from leaf tissue of a 7-week-old S. noctiflora individual from an Opole, Poland (OPL) population using a Qiagen Plant DNeasy kit. To obtain sufficient DNA quantity for construction of Illumina mate-par libraries, additional DNA was extracted from the same individual 6 weeks later using a modified CTAB protocol (Doyle and Doyle 1987) for construction of Illumina mate-pair libraries. For Iso-Seq library construction, RNA was extracted from a single 12-week-old S. noctiflora OPL individual (grown from seed of the plant used for DNA extraction), using a Qiagen Plant RNeasy kit. RNA extractions were performed for four different tissue samples: (1) a large flower bud with calyx removed, (2) an entire smaller flower bud including calyx, (3) the most recent (top-most) pair of cauline leaves, and (4) one leaf from the second most recent pair of cauline leaves. The four RNA extractions were quantified with Qubit RNA BR kit (Thermo Fisher Scientific). Purity and integrity were assessed with a NanoDrop 2000 (Thermo Fisher Scientific) and TapeStation 2200 (Agilent Technologies). Different tissues and developmental stages were sampled (and eventually pooled; see below) to capture a larger diversity of transcripts and thereby increase the number of genes represented.
Table 1.
Genome sizes determined by flow cytometry
| Mean genome size |
|||||
|---|---|---|---|---|---|
| Species | Population | Location | Samples, 2C (pg) | 2C (pg) | 1C (Gb) |
| Silene noctiflora | OPLa | Opole, Poland | 5.65, 5.61, 5.46, 5.44 | 5.54 | 2.71 |
| OSR | Giles County, VA | 5.75, 5.61 | 5.68 | 2.78 | |
| BRP | Nelson County, VA | 5.63, 5.57 | 5.60 | 2.74 | |
| Silene conica | ABR | Abruzzo, Italy | 1.92, 1.92, 1.88 | 1.91 | 0.93 |
| Silene vulgaris | S9L | Giles County, VA | 2.19, 2.16 | 2.18 | 1.07 |
| Silene latifolia | UK2600 | Bedford County, VA | 5.46, 5.45 | 5.46 | 2.67 |
The S. noctiflora OPL population was used for Iso-Seq, genome assembly, and karyotyping.
Units: pg, picogram; Gb, gigabase; 1C, haploid amount; 2C, diploid amount.
PacBio Iso-Seq transcriptome sequencing and analysis
Iso-Seq is an application of Pacific Biosciences (PacBio) long-read sequencing technology that uses cDNA templates to generate high quality reads for full-length transcripts. The high error rate generally associated with PacBio sequencing is drastically reduced using circular consensus sequencing (CCS), which uses hairpin adapters on each end of a double-stranded molecule to create a circular, single-stranded topology (Au et al. 2012; Rhoads and Au 2015; Hestand et al. 2016; Wenger et al. 2019). This topology allows the polymerase to read the same full-length molecule multiple times over, generating an accurate consensus sequence (Ono et al. 2013; Wang et al. 2019). PacBio Iso-Seq has been used to study the transcriptomes of many organisms, often in the context of identifying splice variants, or alternative transcripts (Gordon et al. 2015; Rhoads and Au 2015; Xu et al. 2015; Abdel-Ghany et al. 2016; Guo et al. 2016; Wang et al. 2016; Weirather et al. 2017). Alternative transcripts can be identified using CCS because this technology obtains consensus sequences for full-length single transcripts (Zhao et al. 2019). In the same way, CCS can also be used to distinguish paralogs or gene duplicates.
To create an Iso-Seq library for S. noctiflora, the four RNA extractions (1.5 µg each) were pooled into a single sample and sent to the Arizona Genomics Institute for PacBio Iso-Seq library construction and sequencing. The library was constructed on the pooled RNA sample using Poly(A) selection, following the standard PacBio Iso-Seq protocol (“Procedure & Checklist—Iso-Seq Template Preparation for Sequel Systems,” Pacific Biosciences, PN-101-070-200 Version 06, September 2018), and then was sequenced with a PacBio Sequel (first generation) platform on two SMRT Cells.
Raw movie files of long-read, single-molecule sequences (one per SMRT Cell) were processed using the PacBio Iso-Seq v3.1 pipeline (Anvar et al. 2018; https://www.pacb.com/products-and-services/analytical-software/rna-sequencing/). Circular consensus sequence calling was performed on each movie file separately using the command ccs with the recommended parameters –noPolish and –minPasses 1. Next, primer removal was performed on each dataset by running the command lima with parameters –isoseq and –no-pbi. Poly(A) tails were trimmed and concatemers were removed using the refine command with the parameter –require-polya. Data from the two cells were merged at this point using the commands dataset create –type TranscriptSet and dataset create –type SubreadSet. Finally, the merged data were run through the cluster and polish commands. We also ran the cluster and polish commands on each dataset individually after skipping the merge step.
Trinotate v3.2.0 (Bryant et al. 2017) was used to annotate the final polished sequences produced by the Iso-Seq pipeline after merging the datasets. To complete this process, we used Transdecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder/wiki), SQLite v3 (Kreibich 2010), NCBI BLAST + v2.2.29 (Camacho et al. 2009), HMMER v3.2.1 (including RNAMMER) (Lagesen et al. 2007; Potter et al. 2018), signalP v4 (Petersen et al. 2011), and tmhmm v2 (Krogh et al. 2001). The Pfam (Bateman et al. 2004) and UniProt (UniProt Consortium 2015) databases were included in the Trinotate installation. The transcripts and Transdecoder-predicted peptides were searched against the respective databases, following the standard Trinotate pipeline. All of these results were loaded into a Trinotate SQLite database.
Cogent v4.0.0 (https://github.com/Magdoll/Cogent/wiki) and minimap2 v2.17 (Li 2018) were used to conduct family findings on the final sequences by the Iso-Seq pipeline by partitioning sequences into groups based on similarity. While the Iso-Seq pipeline collapses reads into individual transcripts, it does not collapse alternative transcripts originating from the same gene. Cogent further collapses alternative transcripts into groups, where each group is meant to represent a single gene. Next, coding genome reconstruction was performed on each group from the above step; thus, the Cogent output included both a file containing groups of alternative transcripts (final.partition.txt at https://github.com/alissawilliams/Silene_noctiflora_IsoSeq) and a transcript-based genome. Finally, this transcript-based genome was used to determine total gene and isoform (alternative transcript) counts via cDNA_Cupcake scripts (https://github.com/Magdoll/cDNA_Cupcake/wiki; Jeffries et al. 2020; Wang et al. 2020). A modified form of the script make_file_for_sampling_from_collapsed.py was run with the parameter –include_single_exons in order to include all transcripts in the analysis. Gene and isoform counts were calculated using custom Python and R scripts on the resultant file. These Cogent, minimap2, and cDNA_Cupcake steps were performed on the merged dataset as well as individually on the datasets from each SMRT Cell.
We used genes from the plastid caseinolytic protease (Clp) as a case study to assess the ability of Iso-Seq dataset to distinguish paralogs (gene duplicates) of various levels of divergence. To identify nuclear-encoded plastid Clp core genes in our dataset, we used blastn in conjunction with the Cogent output. There are eight nuclear-encoded plastid Clp core genes in Arabidopsis thaliana: CLPP3-6 and CLPR1-4 (Nishimura and van Wijk 2015). In addition, the genus Silene shares a duplication of CLPP5, denoted CLPP5A and CLPP5B (Rockenbach et al. 2016). We obtained the sequences of all nine of these genes from a previous study (Rockenbach et al. 2016) and used them as queries in blastn searches against the S. noctiflora Iso-Seq transcriptome. We then identified which groups of collapsed alternative transcripts (from the Cogent output) contained these BLAST hits. BLAST hits for eight of the nine nuclear-encoded Clp core subunits in Silene (including CLPP5A and CLPP5B) were found in a single Cogent group. The sequences within each group were confirmed to represent a single gene via alignment and manual inspection; thus, these eight core subunits are single copy in S. noctiflora. However, in the case of CLPR2, two different Cogent groups contained relevant transcripts, indicating a possible case of gene duplication. Sequence alignment and manual inspection of the transcripts within these two Cogent groups revealed that one group contained two unique sequences. These data, along with sequencing results from a separate project in which we cloned two versions of S. noctiflora CLPR2 using primers designed for S. latifolia CLPR2, suggested that there are actually three distinct CLPR2 sequences in S. noctiflora. In the subsequent phylogenetic analysis of CLPR2, we used the longest sequences from each of the three identified groups.
A phylogenetic tree was constructed using sequences from the three different S. noctiflora CLPR2 genes. In addition to the three S. noctiflora sequences, we also included Agrostemma githago, S. conica, S. latifolia, Silene paradoxa, and Silene vulgaris CLPR2 sequences from a previous study (Rockenbach et al. 2016), as well as three S. undulata CLPR2 sequences identified using blastn against the S. undulata TSA database (accession GEYX00000000). All 11 sequences were aligned using the einsi option in MAFFT v7.222 (Katoh and Standley 2013), and trimmed at the 5′ end based on the trimming conducted in Rockenbach et al. (2016). The resultant sequence file was run through jModelTest v2.1.10 (Darriba et al. 2012) to choose a model of sequence evolution. We chose the top model based on the Bayesian Information Criterion (K80+I) and ran PhyML v3.3 (Guindon et al. 2010) with 1000 bootstrap replicates and 100 random starts.
Genome size estimates by flow cytometry
Leaf or seedling samples were collected from multiple individuals of varying age (between 2 and 14 weeks) for each of our target Silene species and shipped fresh to Plant Cytometry Services (Schijndel, Netherlands). Genome sizes were determined using the CyStain PI Absolute P reagent kit (05-5502). Samples were chopped with a razor blade in 500 μl of ice-cold Extraction Buffer in a plastic petri dish, along with Pachysandra terminalis tissue as an internal standard (3.5 pg/2C). After 30–60 seconds of incubation, 2 ml of Staining Buffer was added. Each sample was then passed through a nylon filter of 50 μm mesh size, and then incubated for 30+ min at room temperature. The filtered solution was then sent through a CyFlow ML flow cytometer (Partec GmbH). The fluorescence of the stained nuclei, which passed through the focus of a light beam with a 50 mW, 532 nm green laser, was measured by a photomultiplier and converted into voltage pulses. The voltage pulses were processed using Flomax version 2.4d (Partec) to yield integral and peak signals. Genome sizes were reported in units of pg/2C. The conversion used to report each size (x) in units of Gb was (x/2)*0.978 (Gregory et al. 2007).
Karyotyping
Silene noctiflora OPL seeds were germinated on wet filter paper and grown for 5 days. Radicles were trimmed off and transferred to ice water for 24 hours. The radicles were then fixed in a 3:1 solution of absolute ethanol and glacial acetic acid and stored at −20°C. Chromosomes were visualized using a squash preparation with Feulgen staining. Fixed radicles were rinsed in distilled water for 5 minutes at 20°C. Radicles were then hydrolyzed in 5 M HCl at 20°C for 60 minutes followed by three rinses in distilled water. The hydrolyzed radicles were transferred to Schiff’s reagent to stain the DNA for 120 minutes at 20°C and were then destained by rinsing in SO2 water at 20°C three times for 2 minutes, two times for 10 minutes, once for 20 minutes, and then transferred to distilled water. Squashes were prepared by placing a piece of tissue in 45% acetic acid for 10 minutes and then minced on glass. A coverslip was placed over the minced tissue and pressed with enough pressure to produce a monolayer of nuclei. Slides were placed on dry ice for 1 minute, and the coverslip was removed. The slides were transferred to 96% ethanol for 2 minutes, air dried, and mounted with a mounting medium. Chromosomes were observed using a compound light microscope at 100× magnification.
Genome sequencing and assembly
Extracted S. noctiflora OPL DNA samples were used for Illumina library construction and sequencing. A paired-end library with a target insert size of 275-bp was constructed at the Yale Center for Genome Analysis and sequenced on a 2 × 150-bp HiSeq 2500 run (three lanes). Two mate-pair libraries (with target insert sizes of 3–5 and 8–11 kb) were generated at GeneWiz and sequenced on a 2 × 150-bp HiSeq 2500 run (one lane each). Approximately 480, 250, and 230 M read pairs were generated for the 275, 3–5 kb, and 8–11 kb libraries, respectively. These reads are available via the NCBI SRA (accessions SRR9591157-SRR9591159). Reads were trimmed for quality and to remove 3′ adapters, using cutadapt v1.3 (Martin 2011) under the following parameters: -n 3 -O 6 -q 20 -m 30 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC –paired-output. The trimmed reads were assembled with ALLPATHS-LG release 44837 (Gnerre et al. 2011). Estimates of mean insert size and standard deviation for each library were provided as input for the assembly by first mapping a sample of reads to the published S. noctiflora plastid genome (GenBank accession JF715056.1). These estimates were as follows: 274 bp (±22 bp), 3752 bp (±419 bp), and 9873 bp (±1283 bp).
BUSCO analyses
Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis (Seppey et al. 2019) compares an assembly (transcriptomic or genomic) to a set of highly conserved orthologs from a particular clade in order to assess the completeness of the assembly. BUSCO (v4.1.4) analysis was performed on the Iso-Seq transcriptome and the genome assembly, as well as the output of the individual SMRT Cells. In each case, fasta files containing all genomic or transcriptomic sequences were run through BUSCO using the lineage eudicots_odb10 (2020-09-10) and default parameters. The graphical summary of results was produced using the script generate_plot.py included in the BUSCO installation.
Data availability
The original subread bam files and final transcript sequences longer than 199 bp from the PacBio Iso-Seq transcriptome are available at NCBI Sequence Read Archive (SRA accession SRR11784995) and NCBI Transcriptome Shotgun Assembly Sequence Database (TSA accession GIOF01000000), respectively. The genome assembly has been deposited in GenBank (accession VHZZ00000000.1). Additional data have been provided at GitHub (https://github.com/alissawilliams/Silene_noctiflora_IsoSeq): (1) the full transcriptome as outputted by the PacBio Iso-Seq pipeline, (2) the annotation report for the transcriptome, (3) a custom script used to create a gene_trans_map file for our data in order to use Trinotate on non-Trinity-derived data (i.e., transcripts derived from sources other than a Trinity assembly, in this case, Iso-Seq transcripts), (4) the Cogent output containing collapsed groups of transcripts, and (5) the set of trimmed, aligned sequences used in the CLPR2 phylogenetic analysis.
Results and discussion
Silene noctiflora Iso-Seq transcriptome: gene content and duplication
Sequencing of the Iso-Seq library on two Sequel SMRT Cells produced 711,625 and 686,576 reads for the first and second cells, respectively, where each read was derived from a single molecule. The two SMRT Cells differed substantially in data yield, with totals of 12,765,109 and 21,844,543 subreads, corresponding to subread counts of 17.9 and 31.8 per read, respectively. These reads were merged into 65,642 distinct high-quality transcripts according to the thresholds of the Iso-Seq 3.1 merge and polish commands. Of these transcripts, only 14 were found to be nonplant sequences, all of which were derived from Frankliniella occidentalis (the western flower thrip), a common greenhouse pest that likely contaminated our tissue samples. We annotated these transcripts using Trinotate (Bryant et al. 2017); our dataset contains 69,846 total entries for the 65,642 transcripts (transcripts with multiple predicted proteins are represented by multiple entries). Of the 69,846 entries, 48,742 (74.3%) have an annotated PFAM domain, 47,504 (68.0%) have a KEGG annotation, and 55,993 (80.2%) have at least one predicted Gene Ontology term.
Each high-quality transcript represents collapsed reads, meaning that identical or nearly identical sequences are represented by the same final sequence. However, the Iso-Seq pipeline does not collapse alternatively spliced transcripts, or isoforms; thus, this final dataset includes multiple transcripts derived from the same genes. In addition to separately representing isoforms, the transcriptome data could also contain alleles of the same gene and transcripts from paralogs (gene duplicates). Given sufficiently divergent alleles or paralogs, pairs of these types of sequences will also be represented by separate final transcripts in this dataset. Due to the low levels of polymorphism and heterozygosity in S. noctiflora (Sloan et al. 2012a), we did not expect different alleles to comprise a major portion of this dataset.
Based on a BUSCO analysis (Seppey et al. 2019), the Iso-Seq transcriptome had a completeness of 74.9%. This estimate included a large number of duplicated BUSCOs (47.7%), but these do not necessarily represent true gene duplications for the reasons stated above (Figure 2). The merged dataset had a higher completeness percentage than either of the individual SMRT Cells, where the second SMRT Cell was more complete than the first, consistent with the differential data yield between the two cells (Figure 2). The estimated BUSCO completeness of the transcriptome was lower than that of the assembled nuclear genome (see below), which suggests that some genes with low or tissue-specific expression were not captured. Future efforts to generate deeper sequencing across a wider sample of tissues and environments may be beneficial in this respect.
Figure 2.
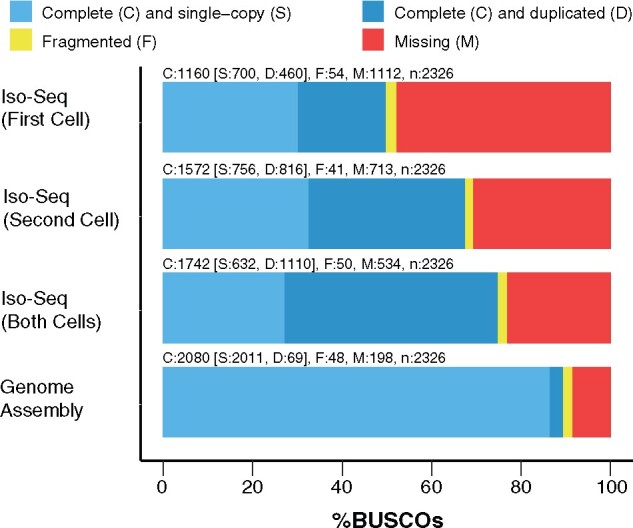
BUSCO analysis of the S. noctiflora genome assembly, Iso-Seq transcriptome (full dataset), and the individual SMRT Cells that were merged to create the Iso-Seq transcriptome.
We used the Cogent (https://github.com/Magdoll/Cogent/wiki) family finding algorithm to further collapse the transcripts into groups of isoforms (alternative transcripts) originating from the same gene. Notably, if paralogs (gene duplicates) have high enough sequence similarity, this binning could include them in the same group. We then used the Cogent data along with Cupcake (https://github.com/Magdoll/cDNA_Cupcake/wiki) to calculate the number of genes and isoforms represented in the transcriptome. Based on this analysis, the Iso-Seq transcriptome contains 14,126 S. noctiflora genes and 25,317 isoforms. Of the 14,126 genes, 7,027 had a single isoform (49.7%). We also calculated gene and isoform counts for each individual SMRT Cell; the first SMRT Cell produced 6,790 genes and 10,568 isoforms, while the second SMRT Cell produced 10,283 genes and 17,000 isoforms.
We wanted to test the ability of Iso-Seq to detect and distinguish known paralogs of varying levels of divergence using the Cogent family finding output. To this end, we used a sample gene family—the core subunit genes of the plastid Clp complex, as they have a rich history of paralogy. In Escherichia coli and most other bacteria, the core of the Clp complex, which is responsible for proteolysis, contains 14 identical subunits (Yu and Houry 2007). In cyanobacteria, gene duplication has led to four different core subunit-encoding genes (Stanne et al. 2007). Continued gene duplication in the land plant lineage has further reshaped this complex in plastids; the 14 core subunits are encoded by nine different genes in A. thaliana, eight of which are nuclear encoded (CLPP3-6 and CLPR1-4), and one of which is plastid encoded (clpP1) (Nishimura and van Wijk 2015). Further, we had previously identified a more recent duplication of CLPP5 in Silene, as well as duplications of the plastid-encoded clpP1 in a small number of angiosperm species (Erixon and Oxelman 2008; Rockenbach et al. 2016; Williams et al. 2019). The Clp complex is one of the most highly expressed stromal proteases (Nishimura and van Wijk 2015). It is expressed in most tissues throughout the life stages of the plant, including the tissues from which we extracted RNA (Zheng et al. 2002). Thus, we would expect a transcriptome generated from the tissues we used to yield sequences of the various components of the Clp complex.
We used the Cogent output to examine the nine nuclear-encoded Clp core genes in S. noctiflora. The core genes CLPP3, CLPP4, CLPP5A, CLPP5B, CLPP6, CLPR1, CLPR3, and CLPR4 were each represented by a single group in the Cogent output, whereas CLPR2 was represented by two groups. Upon further examination, one of these groups actually represented two different genes, yielding a total of three CLPR2 genes in S. noctiflora. Thus, CLPR2 was duplicated in this lineage, and then one paralog underwent a second gene duplication. Based on a phylogenetic analysis (Figure 3), these two duplications are shared with Silene undulata but none of the other sampled Silene species. Thus, these duplications likely occurred after Silene section Elisanthe (including S. noctiflora, S. undulata, and Silene turkestanica) diverged from the other members of the genus (Jafari et al. 2020; Moiloa et al. 2021).
Figure 3.

Phylogenetic analysis of CLPR2 genes in S. noctiflora and related species. Branch lengths represent nucleotide sequence divergence. This tree was rooted on the Agrostemma githago sequence. The placement of S. paradoxa is in conflict with the species tree (Jafari et al. 2020), likely due to long branch attraction and the multiple independent evolutionary rate accelerations in this protein across Silene (Rockenbach et al. 2016).
The Iso-Seq data allowed us to identify transcripts from every known nuclear-encoded Clp core gene in S. noctiflora, including the closely related CLPP5A and CLPP5B subunits, as well as an additional, previously unreported triplication of CLPR2. To corroborate the triplication of CLPR2 in S. noctiflora that was identified using the Iso-Seq transcriptome, we used the CLPR2 sequence from Rockenbach et al (2016) as a query in a blastn search against the S. noctiflora genome assembly. This search returned four scaffold hits. Upon examination, each CLPR2 gene identified in the Iso-Seq transcriptome was represented by one scaffold. The fourth scaffold represented all three gene copies in a short region of high sequence identity between them, suggesting collapsing of similar sequence content within the genome assembly. Thus, each CLPR2 gene was fully represented by sequences on two scaffolds—there was one unique scaffold per gene containing most of the sequence and one scaffold containing sequence shared by all three genes.
Silene genome size estimates and chromosome number
Genome sizes of S. noctiflora, S. conica, S. vulgaris, and S. latifolia were determined using flow cytometry. Our estimates for S. vulgaris and S. latifolia (1.07 and 2.67 Gb, respectively; Table 1) were concordant with previously published estimates for these two species of 1.11 and 2.64 Gb (Costich et al. 1991; Siroký et al. 2001). Interestingly, despite their similar and extreme patterns of organelle evolution (Sloan et al. 2012a, 2014), including large mitochondrial genomes, S. noctiflora and S. conica have very different nuclear genome sizes. We found their respective genome sizes to be approximately 2.74 and 0.93 Gb, respectively (Table 1), which are on opposite ends of the spectrum for Silene diploids (Pellicer and Leitch 2020). The S. noctiflora nuclear genome is almost threefold larger than that of S. conica, suggesting that mitochondrial genome size is not necessarily correlated with nuclear genome size.
Most diploids in the genus, including S. noctiflora, have a chromosome number of 2n = 24, which is likely the ancestral number (Bari 1973; McNeill 1980; Yildiz et al. 2008; Kemal et al. 2009; Gholipour and Sheidai 2010; Ghasemi et al. 2015; Mirzadeh Vaghefi and Jalili 2019). There are also numerous polyploid Silene species, including tetraploid, hexaploid, and octaploid forms (Kruckeberg 1960; Popp and Oxelman 2001, 2007; Popp et al. 2005; Bai et al. 2012). Silene noctiflora has been previously reported as a diploid (McNeill 1980; Yildiz et al. 2008; Ghasemi et al. 2015). Given its relatively large genome size, we sought to confirm this result in our sampled population with a karyotype analysis (Figure 4), which indeed supported the conclusion that S. noctiflora OPL is diploid.
Figure 4.

Micrograph verifying the diploidy of S. noctiflora at 100× magnification. Although an exact chromosome count is difficult to determine, this image suggests that S. noctiflora is a diploid with the typical number of 24 chromosomes previously documented in this species and the genus in general, rather than polyploid with 48 or more chromosomes (Bari 1973; McNeill 1980; Yildiz et al. 2008; Kemal et al. 2009; Gholipour and Sheidai 2010; Ghasemi et al. 2015; Mirzadeh Vaghefi and Jalili 2019).
The Silene noctiflora nuclear genome
Illumina sequencing produced ∼50× coverage of the S. noctiflora genome for a 275-bp paired-end library and ∼15–20× for each of two mate-pair libraries. By performing a de novo assembly of these reads, we obtained a total assembly length (including estimated scaffold gaps) of 2.58 Gb, which is generally consistent with our estimate based on flow cytometry for S. noctiflora OPL (2.71 Gb). Given that we relied entirely on short-read sequencing technology, it was not surprising that the resulting assembly of this large genome was highly fragmented (79,768 scaffolds with a scaffold N50 of 59 kb; 222,040 contigs [minimum length of 1 kb for reporting contigs] with a contig N50 of 4.8 kb). Moreover, assembly gaps made up 73% of the total scaffold length, presumably representing the highly repetitive content that is typical of plant nuclear genomes. As such, the assembled gap-free sequences amount to only about a quarter of the genome (702 Mb). Given the expected low levels of polymorphism and heterozygosity in S. noctiflora (Sloan et al. 2012a), the assembly was interpreted as a single haplotype and no attempt was made to phase the two distinct haplotypes within the diploid.
BUSCO analysis (Seppey et al. 2019) provided an estimate of 89.5% completeness for the S. noctiflora genome assembly (Figure 2). Only 3.0% of BUSCOs were reported to be duplicated, in great contrast to the transcriptome, where 47.7% of BUSCOs were duplicated. Given that the final Iso-Seq dataset includes alternatively spliced transcripts as separate entries, it is not surprising that the transcriptome had a higher percentage of duplicated BUSCOs than the genome assembly.
As a complement to the Iso-Seq transcriptome, this S. noctiflora genome assembly should provide a useful resource to query for sequences of interest, especially in genic regions, and to compare against S. latifolia and other members of this genus. However, a more complete assembly that includes repetitive regions of the genome will require additional data from long-read technologies such as PacBio or nanopore sequencing. The Iso-Seq data generated in this study may be helpful in combination with improved genomic sequencing data in the future, as a means to improve scaffolding (Zhu et al. 2018), resolve paralogs (e.g., the collapsed regions of the CLPR2 paralogs in the genome assembly), and annotate gene models.
Acknowledgments
The authors thank Jocelyn Cuthbert and Zhiqiang Wu for assistance with plant growth and DNA extraction, Suzanne Royer for preliminary investigations into Silene karyotyping, and Joel Sharbrough for assistance with PacBio data analysis. They also thank three anonymous reviewers for their valuable comments on an earlier version of this manuscript.
Funding
This work was supported by a National Science Foundation (NSF) grant (MCB-1733227), start-up funds from Colorado State University, and graduate fellowships from NSF (DGE-1321845) and the National Institutes of Health (T32-GM132057).
Conflicts of interest
None declared.
Literature cited
- Abdel-Ghany SE, Hamilton M, Jacobi JL, Ngam P, Devitt N, et al. 2016. A survey of the sorghum transcriptome using single-molecule long reads. Nat Commun. 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE.. 2011. Limitations of next-generation genome sequence assembly. Nat Methods. 8:61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anvar SY, Allard G, Tseng E, Sheynkman GM, de Klerk E, et al. 2018. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol. 19:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KF, Underwood JG, Lee L, Wong WH.. 2012. Improving PacBio long read accuracy by short read alignment. PLoS One. 7:e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C, Alverson WS, Follansbee A, Waller DM.. 2012. New reports of nuclear DNA content for 407 vascular plant taxa from the United States. Ann Bot. 110:1623–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balounova V, Gogela R, Cegan R, Cangren P, Zluvova J, et al. 2019. Evolution of sex determination and heterogamety changes in section Otites of the genus Silene. Sci Rep. 9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bari EA. 1973. Cytological studies in the Genus Silene L. New Phytol. 72:833–838. [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32:D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi G, Antonovics J, Biere A, Charlesworth D, Delph LF, et al. 2009. Silene as a model system in ecology and evolution. Heredity (Edinb). 103:5–14. [DOI] [PubMed] [Google Scholar]
- Bertrand YJK, Petri A, Scheen A-C, Töpel M, Oxelman B.. 2018. De novo transcriptome assembly, annotation, and identification of low-copy number genes in the flowering plant genus Silene (Caryophyllaceae). bioRxiv 290510.
- Blavet N, Charif D, Oger-Desfeux C, Marais GA, Widmer A.. 2011. Comparative high-throughput transcriptome sequencing and development of SiESTa, the Silene EST annotation database. BMC Genomics. 12:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Johnson K, DiTommaso T, Tickle T, Couger MB, et al. 2017. A tissue-mapped Axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 18:762–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, et al. 2009. BLAST+: architecture and applications. BMC Bioinformatics. 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro-Soriguer I, Narbona E, Buide ML, del Valle JC, Whittall JB.. 2016. Transcriptome and biochemical analysis of a flower color polymorphism in Silene littorea (Caryophyllaceae). Front Plant Sci. 7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D. 2006. Evolution of plant breeding systems. Curr Biol. 16:R726–R735. [DOI] [PubMed] [Google Scholar]
- Costich DE, Meagher TR, Yurkow EJ.. 1991. A rapid means of sex identification inSilene latifolia by use of flow cytometry. Plant Mol Biol Rep. 9:359–370. [Google Scholar]
- Dagher-Kharrat MB, Abdel-Samad N, Douaihy B, Bourge M, Fridlender A, et al. 2013. Nuclear DNA C-values for biodiversity screening: case of the Lebanese flora. Plant Biosystems. 147:1228–1237. [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D.. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9:772–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SL, Delph LF.. 2005. Prior selfing and gynomonoecy in Silene noctiflora L. (Caryophyllaceae): opportunities for enhanced outcrossing and reproductive assurance. Int J Plant Sci. 166:475–480. [Google Scholar]
- Desfeux C, Maurice S, Henry JP, Lejeune B, Gouyon PH.. 1996. Evolution of reproductive systems in the genus Silene. Proc Biol Sci. 263:409–414. [DOI] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL.. 1987. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochemical Bulletin. 19:11–15.
- Erixon P, Oxelman B.. 2008. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the Chloroplast clpP1 Gene. PLoS One. 3:e1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraud C, Brachi B, Dufay M, Touzet P, Shykoff JA.. 2011. Genetic determination of male sterility in gynodioecious Silene nutans. Heredity (Edinb). 106:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemi FS, Jalili A, Mirzadeh Vaghefi SS.. 2015. Chromosome report of three species of flora of Iran. The Iranian Journal of Botany. 21:165–168. [Google Scholar]
- Gholipour A, Sheidai M.. 2010. Karyotype analysis and new chromosome number reports in Silene species (sect. Auriculatae, Caryophyllaceae). Biologia. 65:23–27. [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci USA. 108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, et al. 2015. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One. 10:e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR, Nicol JA, Tamm H, Kullman B, Kullman K, et al. 2007. Eukaryotic genome size databases. Nucleic Acids Res. 35:D332–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59:307–321. [DOI] [PubMed] [Google Scholar]
- Guo W, Grewe F, Fan W, Young GJ, Knoop V, et al. 2016. Ginkgo and Welwitschia mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol Biol Evol. 33:1448–1460. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Zhang SV, Moyle LC.. 2014. Sequencing, assembling, and correcting draft genomes using recombinant populations. G3 (Bethesda). 4:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird JC, Trapp P, Miller CM, Bazos I, Sloan DB.. 2017. Causes and consequences of rapidly evolving mtDNA in a plant lineage. Genome Biol Evol. 9:323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird JC, Whitehill Nicholas S, Snow Christopher D, Sloan Daniel B.. 2015. Conservative and compensatory evolution in oxidative phosphorylation complexes of angiosperms with highly divergent rates of mitochondrial genome evolution. Evolution. 69:3069–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestand MS, Houdt JV, Cristofoli F, Vermeesch JR.. 2016. Polymerase specific error rates and profiles identified by single molecule sequencing. Mutat Res. 784–785:39–45. [DOI] [PubMed] [Google Scholar]
- Jafari F, Zarre S, Gholipour A, Eggens F, Rabeler RK, et al. 2020. A new taxonomic backbone for the infrageneric classification of the species-rich genus Silene (Caryophyllaceae). Taxon. 69:337–368. [Google Scholar]
- Jeffries AR, Leung S, Castanho I, Moore K, Davies JP, et al. 2020. 2020 Full-length transcript sequencing of human and mouse identifies widespread isoform diversity and alternative splicing in the cerebral cortex. bioRxiv 10.14.339200. [DOI] [PMC free article] [PubMed]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemal Y, Minareci E, Çirpici A.. 2009. Karyotypic study on Silene, section Lasiostemones species from Turkey. Caryologia. 62:134–141. [Google Scholar]
- Klaas AL, Olson MS.. 2006. Spatial distributions of cytoplasmic types and sex expression in Alaskan populations of Silene acaulis. Int J Plant Sci. 167:179–189. [Google Scholar]
- Krasovec M, Chester M, Ridout K, Filatov DA.. 2018. The mutation rate and the age of the sex chromosomes in Silene latifolia. Curr Biol. 28:1832–1838.e4. [DOI] [PubMed] [Google Scholar]
- Kreibich JA. 2010. Using SQLite. Sebastopol, CA: O’Reilly Media, Inc. [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL.. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 305:567–580. [DOI] [PubMed] [Google Scholar]
- Kruckeberg AR. 1960. Chromosome numbers in Silene (Caryophyllaceae). II. Madroño. 15:205–215. [Google Scholar]
- Lagesen K, Hallin P, Rødland EA, Staerfeldt H-H, Rognes T, et al. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, Renner T, Ibarra-Laclette E, Farr KM, Chang T-H, et al. 2017. Long-read sequencing uncovers the adaptive topography of a carnivorous plant genome. Proc Natl Acad Sci USA. 114:E4435–E4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 17:10. [Google Scholar]
- McNeill J. 1980. The biology of Canadian weeds.: 46. Silene noctiflora L. Can J Plant Sci. 60:1243–1253. [Google Scholar]
- Mirzadeh Vaghefi SS, Jalili A.. 2019. Chromosome numbers of some vascular plant species from Iran. Iranian J Bot. 25:140–144. [Google Scholar]
- Moiloa NA, Mesbah M, Nylinder S, Manning J, Forest F, et al. 2021. Biogeographic origins of southern African Silene (Caryophyllaceae). Mol Phylogenet Evol. 107199. [DOI] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD.. 2007. Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol. 7:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyle A, Zemp N, Deschamps C, Mousset S, Widmer A, et al. 2012. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol. 10:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, van Wijk KJ.. 2015. Organization, function and substrates of the essential Clp protease system in plastids. Biochimica Biophysica Acta. 1847:915–930. [DOI] [PubMed] [Google Scholar]
- Olson MS, Mccauley DE.. 2002. Mitochondrial DNA diversity, population structure, and gender association in the gynodioecious plant Silene vulgaris. Evolution. 56:253–262. [DOI] [PubMed] [Google Scholar]
- Ono Y, Asai K, Hamada M.. 2013. PBSIM: PacBio reads simulator—toward accurate genome assembly. Bioinformatics. 29:119–121. [DOI] [PubMed] [Google Scholar]
- Papadopulos AST, Chester M, Ridout K, Filatov DA.. 2015. Rapid Y degeneration and dosage compensation in plant sex chromosomes. Proc Natl Acad Sci USA. 112:13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicer J, Leitch IJ.. 2020. The Plant DNA C-values database (release 7.1): an updated online repository of plant genome size data for comparative studies. New Phytol. 226:301–305. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H.. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8:785–786. [DOI] [PubMed] [Google Scholar]
- Popp M, Erixon P, Eggens F, Oxelman B.. 2005. Origin and evolution of a circumpolar polyploid species complex in Silene (Caryophyllaceae) inferred from low copy Nuclear RNA polymerase introns, rDNA, and Chloroplast DNA. Syst Bot. 30:302–313. [Google Scholar]
- Popp M, Oxelman B.. 2001. Inferring the history of the polyploid Silene aegaea (Caryophyllaceae) using plastid and homoeologous Nuclear DNA sequences. Mol Phylogenet Evol. 20:474–481. [DOI] [PubMed] [Google Scholar]
- Popp M, Oxelman B.. 2007. Origin and evolution of North American polyploid Silene (Caryophyllaceae). Am J Bot. 94:330–349. [DOI] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, et al. 2018. HMMER web server: 2018 update. Nucleic Acids Res. 46:W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A, Au KF.. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 13:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenbach K, Havird JC, Monroe JG, Triant DA, Taylor DR, et al. 2016. Positive selection in rapidly evolving plastid–Nuclear enzyme complexes. Genetics. 204:1507–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz MC, Witkowski J, McCombie WR.. 2012. Current challenges in de novo plant genome sequencing and assembly. Genome Biol. 13:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppey M, Manni M, 2019. BUSCO: Assessing Genome Assembly and Annotation Completeness. In: Zdobnov EM, Kollmar M, editors. Gene Prediction: Methods and Protocols. New York, NY: Methods in Molecular Biology, Springer. p. 227–245. [DOI] [PubMed] [Google Scholar]
- Siroký J, Lysák MA, Dolezel J, Kejnovský E, Vyskot B.. 2001. Heterogeneity of rDNA distribution and genome size in Silene spp. Chromosome Res. 9:387–393. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, et al. 2012a. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10:e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Keller SR, Berardi AE, Sanderson BJ, Karpovich JF, et al. 2012b. De novo transcriptome assembly and polymorphism detection in the flowering plant Silene vulgaris (Caryophyllaceae). Mol Ecol Resour. 12:333–343. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Triant DA, Forrester NJ, Bergner LM, Wu M, et al. 2014. A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol Phylogenet Evol. 72:82–89. [DOI] [PubMed] [Google Scholar]
- Städler T, Delph LF.. 2002. Ancient mitochondrial haplotypes and evidence for intragenic recombination in a gynodioecious plant. Proc Natl Acad Sci USA. 99:11730–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanne TM, Pojidaeva E, Andersson FI, Clarke AK.. 2007. Distinctive types of ATP-dependent Clp Proteases in Cyanobacteria. J Biol Chem. 282:14394–14402. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Kumar V, Olson A, Ware D.. 2019. Reviving the transcriptome studies: an insight into the emergence of single-molecule transcriptome sequencing. Front Genet. 10:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Tseng E, Baybayan P, Eng K, Regulski M, et al. 2020. Variant phasing and haplotypic expression from long-read sequencing in maize. Commun Biol. 3:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Tseng E, Regulski M, Clark TA, Hon T, et al. 2016. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat Commun. 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirather JL, de Cesare M, Wang Y, Piazza P, Sebastiano V, et al. 2017. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Res. 6:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, et al. 2019. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 37:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AM, Friso G, van Wijk KJ, Sloan DB.. 2019. Extreme variation in rates of evolution in the plastid Clp protease complex. Plant J. 98:243–259. [DOI] [PubMed] [Google Scholar]
- Xu Z, Peters RJ, Weirather J, Luo H, Liao B, et al. 2015. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 82:951–961. [DOI] [PubMed] [Google Scholar]
- Yildiz K, Minareci E, Çirpici A, Dadandı MY.. 2008. A karyotypic study on Silene, section Siphonomorpha species of Turkey. Nordic J Bot. 26:368–374. [Google Scholar]
- Yu AYH, Houry WA.. 2007. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 581:3749–3757. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang H, Kohnen MV, Prasad KVSK, Gu L, et al. 2019. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and Nanopore-based direct RNA sequencing. Front Genet. 10:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Halperin T, Hruskova-Heidingsfeldova O, Adam Z, Clarke AK.. 2002. Characterization of Chloroplast Clp proteins in Arabidopsis: localization, tissue specificity and stress responses. Physiol Plant. 114:92–101. [DOI] [PubMed] [Google Scholar]
- Zhu B-H, Xiao J, Xue W, Xu G-C, Sun M-Y, et al. 2018. P_RNA_scaffolder: a fast and accurate genome scaffolder using paired-end RNA-sequencing reads. BMC Genomics. 19:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original subread bam files and final transcript sequences longer than 199 bp from the PacBio Iso-Seq transcriptome are available at NCBI Sequence Read Archive (SRA accession SRR11784995) and NCBI Transcriptome Shotgun Assembly Sequence Database (TSA accession GIOF01000000), respectively. The genome assembly has been deposited in GenBank (accession VHZZ00000000.1). Additional data have been provided at GitHub (https://github.com/alissawilliams/Silene_noctiflora_IsoSeq): (1) the full transcriptome as outputted by the PacBio Iso-Seq pipeline, (2) the annotation report for the transcriptome, (3) a custom script used to create a gene_trans_map file for our data in order to use Trinotate on non-Trinity-derived data (i.e., transcripts derived from sources other than a Trinity assembly, in this case, Iso-Seq transcripts), (4) the Cogent output containing collapsed groups of transcripts, and (5) the set of trimmed, aligned sequences used in the CLPR2 phylogenetic analysis.