

Summary
ATAC-seq is a versatile, adaptable, and widely adopted technique for mapping open chromatin regions. However, some biological systems, such as primary neurons, present unique challenges to its application. Conventional ATAC-seq would require the dissociation of the primary neurons after plating but dissociating them leads to rapid cell death and major changes in cell state, affecting ATAC-seq results. We have developed this modified ATAC-seq protocol to address this challenge for primary neurons, providing a high-quality and high-resolution accessible chromatin profile.
For complete details on the use and execution of this protocol, please refer to Maor-Nof et al. (2021).
Subject areas: Genetics, Genomics, Sequencing, Molecular Biology, Neuroscience
Graphical abstract
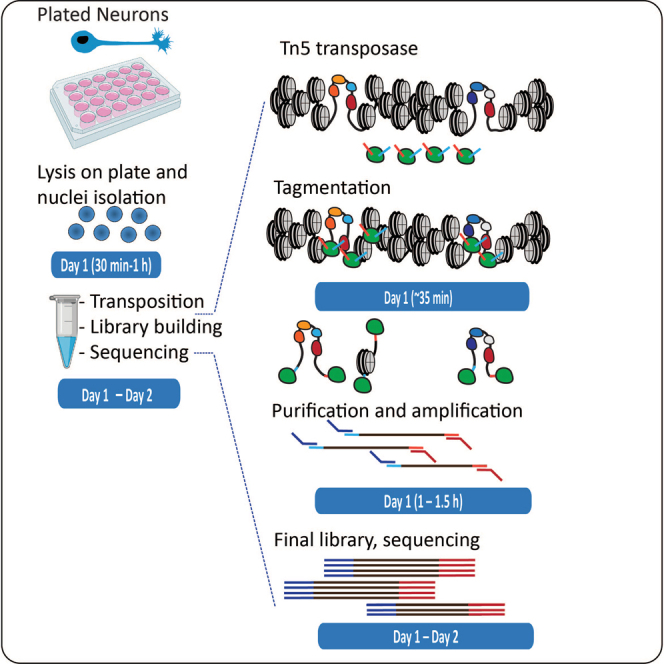
Highlights
-
•
Neuronal ATAC maps chromatin accessibility in primary neurons.
-
•
Neuronal ATAC avoids damage to neurons during cell handling and nuclei preparation.
-
•
Neuronal ATAC provides high-quality and high resolution accessible chromatin profiles.
ATAC-seq is a versatile, adaptable, and widely adopted technique for mapping open chromatin regions. However, some biological systems, such as primary neurons, present unique challenges to its application. Conventional ATAC-seq would require the dissociation of the primary neurons after plating but dissociating them leads to rapid cell death and major changes in cell state, affecting ATAC-seq results. We have developed this modified ATAC-seq protocol to address this challenge for primary neurons, providing a high-quality and high-resolution accessible chromatin profile.
Before you begin
This protocol is used to interrogate the chromatin accessibility landscape of primary neurons cultured on plates. Before you start assays open chromatin in such a setting, it is advisable that:
-
1.
Institutional permission and oversight information for the animal study should be obtained. In this study, animals were bred and used as approved by the Administrative Panel of Laboratory Animal Care (APLAC) of Stanford University, an institution accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC).
-
2.
Cell culture conditions are optimized, and you have made certain that plated neurons are not exhibiting elevated levels of cell death or other undesirable behavior, as this could interfere with the results of the ATAC-seq (or other functional genomic assays carried out).
-
3.
If using homemade Tn5 transposase rather than one obtained from commercial sources, it is good to check its quality by generating test libraries on any cell culture suspensions that might be available in the lab, sequencing 1–2M reads, and examining the enrichment for open chromatin (see below for more details).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Papain Dissociation System | Worthington Biochemical Corporation | LK003150 |
| B-27 serum-free supplement Poly-L-lysine-solution (0.01% (wt/vol)) |
Life Technologies Sigma-Aldrich |
17504-044 P4832 |
| Neurobasal Medium | Life Technologies | 21103-049 |
| GlutaMAX | Invitrogen | 35050-061 |
| Tn5a | Illumina | FC-131-1024 |
| IGEPAL CA-630 detergentb | Sigma | 11332465001 |
| Tween-20 detergentc Digitonin detergentd |
Sigma Promega |
11332465001 G9441 |
| 1M Tris-HCl pH 7.5 | Thermo Fisher | 15567027 |
| 5M NaCl | Thermo Fisher | AM9759 |
| 1M MgCl2 | Thermo Fisher | AM9530G |
| Dimethyl formamide | Sigma | D4551 |
| Deoxyribonuclease I (DNAse I) | Worthington | LS006331 |
| 10mM dNTP Mix | Thermo Fisher | 18427013 |
| SYBR Green I Nucleic Acid Gel Stain | Thermo Fisher | S7563 |
| PhiX Control v3 | Illumina | FC-110-3001 |
| Critical commercial assays | ||
| Sequencing library primers/adapterse,f | Illumina | FC-131-1096 |
| NEBNext High-Fidelity 2× PCR Master Mix | NEB | M0541S |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530L |
| MinElute PCR Purification Kit | QIAGEN | 28004/28006 |
| Zymo DNA Clean and Concentrator Kit | Zymo | D4013/D4014 |
| QuBit dsDNA HS Assay Kit | Thermo Fisher | Q32854 |
| Deposited data | ||
| Raw and analyzed data | Maor-Nof et al. (2021) | GEO: GSE162048 |
| Experimental models: organisms/strains | ||
| C57BL/6J | The Jackson Laboratory | JAX:100012 |
| Experimental models: cell lines | ||
| Primary mouse cortical neurons | This paper | N/A |
| Other | ||
| Nuclease-free H2O | Thermo Fisher | AM9916 |
| 1× PBS buffer solution | Thermo Fisher | 10010023 |
| qPCR machine (StepOne or equivalent) | Thermo Fisher | 4376357 |
| 200-μL PCR tubes | Thermo Fisher | AB0620 |
| 1.5-mL Microcentrifuge tubesg | Eppendorf | 022431021 |
| Thermomixer | Eppendorf | 5382000023 |
| Tabletop centrifuge | Eppendorf | 5427R |
| Qubit fluorometer | Thermo Fisher | Q33238 |
| QuBit tubes | Thermo Fisher | Q32856 |
| TapeStation | Agilent | G2991BA |
| TapeStation D1000 tape | Agilent | 5067-5582 |
| TapeStation D1000 reagents | Agilent | 5067-5583 |
| Thermocycler | Eppendorf | 2231000813 |
a Tn5 is the key reagent in the ATAC-seq protocol; it can be obtained from Illumina as listed here, but it can also be prepared in-house, following the protocol described previously by Picelli et al. (2014). The Tn5 enzyme is called “Amplicon Tagment Mix” in the Illumina kit
b Supplied as a 10% solution
c Supplied as a 10% solution; store at 4°C
d Supplied as a 2% solution in DMSO; (store at -20°C)
e PCR primers for amplifying ATAC-seq libraries can also be ordered directly from other sources; the i7 primer sequence is 5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3′, the i5 sequence is 5′AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3′, where [i7] and [i5] are the index sequences (typically 8-bp long). Dissolve and dilute to 25μM.
f -bp long
g Tubes should be preferably low protein- and DNA-binding
Materials and equipment
ATAC-RSB buffer (50 mL)
| Reagent | Final concentration | Amount per sample |
|---|---|---|
| 1M Tris-HCl pH 7.4 | 10 mM | 500 μL |
| 5M NaCl | 10 mM | 100 μL |
| 1M MgCl2 | 3 mM | 150 μL |
| H2O | 49.25 mL |
ATAC-RSB lysis buffer (1 mL)
| Reagent | Final concentration | Amount per sample |
|---|---|---|
| 10% IGEPAL CA-630 | 0.1% | 10 μL |
| 10% Tween-20 | 0.1% | 10 μL |
| 2% Digitonin | 0.01% | 5 μL |
| ATAC-RSB | 970 μL |
ATAC-RSB wash buffer (10 mL)
| Reagent | Final concentration | Amount per sample |
|---|---|---|
| 10% Tween-20 | 0.1% | 100 μL |
| ATAC-RSB | 9.9 mL |
2× TD buffer (10 mL)
| Reagent | Final concentration | Amount per sample |
|---|---|---|
| Tris-HCl pH 7.5 | 20 mM | 200 μL |
| 1M MgCl2 | 10 mM | 100 μL |
| Dimethyl Formamide | 20% | 2 mL |
| H2O | 9.78 mL |
Note: store the ATAC-RSB master buffer at 4°C (no specific time limit), and only prepare the ATAC-RSB-Lysis and ATAC-RSB-Wash buffers prior to use. Stored the 2× TD buffer (up to 1 year) and the Tn5 enzyme (according to the manufacture expiry date) at −20°C.
Step-by-step method details
Note: This protocol has been adapted from the omniATAC version of the ATAC-seq assay, previously described in Corces et al., 2017
Note: We advise to perform at least two independent replicates for each condition assayed.
Preparation of primary mouse cortical neurons
Timing: ∼3 h plus subsequent culturing.
In this step, neurons are harvested from mouse cortices and then transferred onto plates, where they can be subjected to various treatments and manipulations.
-
1.
Dissociate primary mouse cortical neurons into single-cell suspensions from E16.5 mouse cortices with a papain dissociation system following manufacturer’s recommended protocols (papain dissociation protocol) with 40 min incubation time in papain solution at 37C.
-
2.
Plate neurons onto poly-L-lysine- coated –(add the solution to the plates and incubate for 1 h, then remove it, and rinse three times with H2O) 12-well plates for DNA extraction (450,000 cells/well).
-
3.
Grow neurons in Neurobasal medium supplemented with B-27 serum-free supplement, GlutaMAX, and penicillin–streptomycin in a humidified incubator at 370C, with 5% CO2.
-
4.
Perform half media changes every 4 or 5 days, or as required.
DNAse treatment of cells
Note: DNAse treatment helps improve signal to noise by removing free-floating DNA and digesting DNA from dead cells.
-
5.
Add DNAse at a concentration of 200 U/mL to the media in the plate with the neurons.
-
6.
Incubate at 37°C for 30 min.
-
7.
Remove the media with the DNAse from the plate.
-
8.
Add cold 1× PBS to cover the plate and remove the PBS.
-
9.
Repeat step 8 three additional times.
Preparation of nuclei
In this step, nuclei are isolated from the rest of cells (thus removing mitochondria and various other debris) and prepared for transposition.
Note: To avoid cell death caused by trypsinization, neurons are directly lysed on the plate, and the nuclei are prepared from the lysate. Carry out all steps on ice and centrifugations at 4°C.
-
10.
Add 1 mL cold ATAC-RSB-Lysis Buffer directly to the neurons on plate.
-
11.
Incubate on ice for 10 min.
-
12.
Use cell scraper to collect neurons from the plate and count them on a hemocytometer after staining nucleus with trypan blue.
-
13.
Centrifuge ∼50,000 nuclei at 500 g for 5 min in a pre-chilled 4°C fixed-angle centrifuge.
Note: Nuclei can be counted directly using a cell counter or they can be estimated based on the input number of cells from the previous step.
-
14.
Carefully aspirate the supernatant in two steps, by first removing most of it, then using the P200 pipette to remove the last ∼100 μL.
-
15.
Resuspend the pellet in 1 mL of ATAC-RSB-Wash Buffer.
-
16.
Centrifuge for 10 min at 500 g at 4°C.
-
17.
Carefully aspirate the supernatant in two steps as described above.
Transposition
In this step nuclei are incubated with the transposase enzyme, which will insert itself into accessible chromatin regions.
Carry out transposition as follows:
-
18.
Immediately resuspend the pellet in the transposase reaction mix (prepare a master mix for multiple samples in the same proportions):
| 25 μL TD buffer |
| 2.5 μL Tn5 |
| 5 μL nuclease-free H20 |
| 16.75 μL 1× PBS |
| 0.25 μL 2% digitonin |
| 0.5 μL 10% Tween-20 |
-
19.
Incubate at 37°C for 30 min in a Thermomixer with shaking at 1000 RPM.
DNA purification
Transposed DNA needs to be purified before it can be amplified.
Note: Reactions can be cleaned up either with the Zymo DNA Clean and Concentrator or the Qiagen MinElute Cleanup kits, with equivalent results.
-
20.
Immediately stop the reaction using 250 μL (i.e., 5×) of PB buffer (if using MinElute) or DNA Binding Buffer (if using Zymo).
-
21.
Purify samples following the kit instructions.
-
22.
Elute with 10 μL of Elution Buffer.
PCR amplification and library generation
Transposed DNA is minuscule in its amount given the low number of input cells, and needs to be amplified sufficiently to be sequenced.
Note: When amplifying transposed DNA, the initial extension is needed to fill in the gap left from the transposition itself and allow PCR primers to land in subsequent amplification cycles. Hot-start polymerase mixes, in which the polymerase is only activated by exposing it to denaturation temperatures, are therefore not recommended for amplifying ATAC-seq libraries.
-
23.
Set up the following PCR reaction:
| 10 μL transposition eluate |
| 10 μL Nuclease-free H2O |
| 2.5 μL of Adapter 1 |
| 2.5 μL of Adapter 2 |
| 25 μL NEBNext High-Fidelity 2× PCR Master Mix |
-
24.
Optimize PCR conditions, pre-amplification. Amplify DNA for 5 cycles as follows:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial | 72°C | 3 min | 1 |
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 5 |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 30 s | |
| Hold | 4°C | forever | |
-
25.
Determine additional cycles using qPCR. Use 5 μL of the pre-amplified reaction in a total qPCR reaction of 15 μL as follows:
| 3.76 μL nuclease-free H2O |
| 0.5 μL of Adapter 1 |
| 0.5 μL of Adapter 2 |
| 0.24 μL 25× SYBR Green (in DMSO) |
| 5 μL NEBNext High-Fidelity 2× PCR Master Mix |
| 5 μL pre-amplified sample |
-
26.
Determine additional cycles using qPCR. Run the qPCR reaction with the following settings in a qPCR machine:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 20 |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 30 s | |
| Hold | 4°C | forever | |
-
27.
Assess the amplification profiles and determine the required number of additional cycles to amplify.
-
28.
Carry out final amplification by placing the remaining 45 μL in a thermocycler and running the following program:
Nadd cycles of:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Denaturation | 98°C | 10 s | Nadd |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 30 s | |
| Hold | 4°C | forever | |
Where Nadd is the number of additional cycles.
In practice, 8–10 total cycles are usually sufficient to amplify a standard ATAC library thus if a large number of samples are being processed at the same time, the following reaction can be run:
-
29.
Single-step PCR.
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial | 72°C | 3 min | 1 |
| Initial Denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 8–10 |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 30 s | |
| Hold | 4°C | forever | |
-
30.
Purify the amplified library as described above for the purified ATAC reaction7. Elute in 20 μL Elution Buffer.
Note: In our experience, primer dimers are efficiently removed from ATAC libraries with simple column purification and size selection with beads is not necessary.
Library size distribution profiling
A highly informative quality control step is to evaluate the distribution of final libraries. i.e., to check whether it shows the typical nucleosomal pattern, or it deviates from it in potentially important ways.
-
31.
There are multiple options for carrying out this step, e.g., the TapeStation and BioAnalyzer instruments. We prefer to use a TapeStation with the D1000 or HS D1000 kits due to its ease of use, flexibility and rapid turnaround time. Follow the manufacturer’s instructions depending on the exact instrument and kit used (TapeStation online protocol).
Library quantification
Before they are sequenced, libraries need to be quantified, and this has to be done in terms of molarity, not just mass, as molarity is what is important for accurate loading on the sequencer.
Note: This step is typically carried out using a Qubit fluorometer for most high-throughput sequencing libraries that exhibit a unimodal fragment length distribution. However, ATAC-seq fragment distribution is usually not unimodal and ATAC-seq libraries often include fragments longer than what can be sequenced on standard Illumina instruments. Effective library concentrations therefore often differ from apparent library concentrations measured using Qubit. The best way to estimate effective library concentration is thus qPCR. Commercial kits such as the NEBNext Library Quant Kit for Illumina or KAPA Library Quantification Kits can also be used, in a similar manner.
-
32.
Generate a standard curve using Illumina PhiX standard (10nM) by first making a 50× dilution to 200 pM, then making additional serial 2× dilutions to 100 pM, 50 pM, 25 pM, 12.5 pM, 6.25 pM, 3.125 pM, and 1.56 pM.
-
33.
Set up a 20 μL qPCR reactions as follows:
| 7.9 μL nuclease-free H2O |
| 5 μL ATAC-seq 400× diluted library or PhiX standards |
| 4 μL Phusion HF Buffer |
| 1μL Adapter 1 |
| 1μL Adapter 2 |
| 0.4 μL 10mM dNTP mix |
| 0.5 μL 25× SYBR Green (in DMSO) |
| 0.2 μL NEB Phusion HF |
-
34.
Run the qPCR reaction with the following settings in a qPCR machine:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 98°C | 30 s | 1 |
| Denaturation | 98°C | 10 s | 20 |
| Annealing | 63°C | 30 s | |
| Extension | 72°C | 30 s | |
| Hold | 4°C | forever | |
-
35.
Create a standard curve based on the PhiX dilutions and estimate the library’s true molarity based on it.
Sequencing
This protocol generates libraries intended to be sequenced on Illumina sequencers.
Note: Make sure to use optimally compatible index pairs when pooling libraries prior to sequencing.
Note: Once libraries have been made, the optimal sequencing format needs to be decided on, as there are multiple different Illumina kits, which differ in their output, read length, and cost.
Note: ATAC-seq libraries should not be sequenced in a single-end format, as the analysis of fragment lengths is important for the quality evaluation of ATAC-seq datasets and for a number of downstream analyses, which are only possible in a paired-end format. In addition, some analytical tasks, (such as transcription factor foot printing) focus on Tn5 insertions rather than read coverage, and paired-end reads produce twice as many such data points for the same cost.
Note: We also note that the post-sequencing ATAC-seq insert length distribution peaks between 50 and 100 bp (Figure 2A). It is accordingly most cost-effective to sequence ATAC libraries in 2×36 bp or 2 × 50 bp formats (depending on whether using a NextSeq or some of the higher-throughput Illumina instruments). However, some applications (e.g. analyzing the effects of sequence variation on chromatin accessibility) can benefit from longer reads, and this should be kept in mind depending on the goals of the particular study.
Figure 2.

Typical ATAC-seq results after processing of sequencing data
(A) Length distribution for mapped fragments (shown is the dataset corresponding to SRA accession SRR13120289).
(B) TSS profile (for the same sample).
(C) TSS scores for the library from Maor-Nof et al. (2021).
(D) ATAC-seq profiles around protein coding TSSs in the genome.
(E) ATAC-seq shows activation of one of the promoters of the Cdkn1a gene in (PR)50 cells relative to TDP-43 and control GFP cells.
(F) Example of transcription factor footprint detection with ATAC-seq (a metaplot of ATAC-seq cleavage around occupied CTCF motif instances is shown).
Expected outcomes
Figure 1 shows examples of typical ATAC-seq library size profiles. A clear nucleosomal pattern is expected, with peaks corresponding to subnucleosomal, mononucleosomal, dinucleosomal, and so on fragments, due to the inhibitory effect of nucleosomes on Tn5 insertion. Occasionally, it is possible to see a “flattened” profile, in which the nucleosomal peaks stand out less than usual. This is often due to the presence of a large amount of mitochondrial genome-derived fragments and does not necessarily affect enrichment for open chromatin in the nuclear genome.
Figure 1.

Typical TapeStation profile (D1000 TapeStation in this case) of an ATAC-seq library
ATAC-seq libraries tend to display a nucleosomal pattern with dominant peaks corresponding to subnucleosomal fragments (the ∼180-∼250 range; note that the length of adapters is included in these values), mononucleosomes, dinucleosomes, and so on. The relative height of peaks can occasionally vary between different libraries (A and B).
Quantification and statistical analysis
Quality characterization and evaluation after sequencing is based on the following criteria:
-
1.
Evaluation of the fragment length distribution (Figure 2A). It is possible to have a very prominent subnucleosomal peak without a strong mononucleosomal one and still have quite high enrichment for open chromatin, but high-quality ATAC libraries in eukaryotes typically display the characteristic nucleosomal signature in their fragment length distribution.
-
2.
Evaluation of open chromatin enrichment (Figures 2B and 2C). To this end, the average profile around the transcription start sites (TSS) of protein coding genes is a very useful, intuitive and independent of ad hoc parameters such as peak calling thresholds measure. It can also be distilled to a single “TSS score” number, which is calculated as the ratio of signal (in read-per-million-mapped reads units or read counts) over the region immediately (e.g., ±100 bp) around the TSS versus the average signal over the two regions of equal size located ±2 kb on the flanks either side of the TSS (Marinov and Shipony 2021). High-quality ATAC libraries tend to have TSS scores ≥10 for mammalian genomes. TSS scores for the libraries from Maor-Nof et al. (2021) are shown in Figure 2C.
-
3.
Evaluation of the extent of mitochondrial contamination. As mitochondria do not have nucleosomes and their DNA is highly accessible, they are preferentially transposed by Tn5. High levels of mitochondrial reads are not necessarily associated with poor open chromatin enrichment in the nuclear genome, but they result in having to sequence much deeper to obtain the same effective sequencing coverage and are a sign of a need to optimize the protocol. While in early versions of the ATAC-seq protocol (Buenrostro et al., 2013), most of ATAC libraries consisted of mitochondria-derived fragments, since then the omniATAC protocol (Corces et al., 2017), on which the plated neuronal ATAC protocol is based, has greatly reduced the level of mitochondrial contamination. The fraction of chrM reads for the libraries from Maor-Nof et al. (2021) is shown in Figure 2C.
-
4.
The molecular complexity of libraries – high-quality libraries should contain a large number of distinct fragments.
-
5.
The effective sequencing depth – in general we aim for ∼20–30 million reads after deduplicating fragments mapping to the nuclear genome.
Note: ATAC-seq libraries should also display visible enrichment over promoters and other regulatory elements when examined using a genome browser or globally, as shown in Figures 2D and 2E.
Note: ATAC-seq libraries can also be used to evaluate footprinting of transcription factors (although mostly globally rather than at individual sites). An example of global protection footprints around sites occupied by the CTCF is shown in Figure 2F.
Note: It is in practice often highly useful to sequence a small number of reads (a few hundred thousand to a million) to evaluate library quality before committing to a full production run, as most of these metrics, especially TSS enrichment and mitochondria contamination, can be calculated on just a small sampling of the final library.
Note: We refer the reader to previous treatments of the subject (Marinov and Shipony, 2021) regarding the details of ATAC-seq processing and analysis.
Limitations
The usual limitations to the ATAC-seq assay, even if very few, also apply to the version described here. The signal-to-noise ratio is sensitive to the condition of the cells used as input and Tn5’s strong preference for inserting into unprotected genomic regions means many mitochondrial reads (which are of little use for most analyses) can be observed in libraries. Below we list some tips on improving the quality of datasets if such issues are encountered.
Troubleshooting
Problem 1
Primary neurons do not extend their axons (steps 1–4).
Potential solution
The number of cortical neurons you plate will have tremendous impact on the growth of their axons. There are a minimal number of neurons that needs to be present in order for the neurons to survive and grow processes. This number changes according to the area surface size in the plate. Here for the 12 well plate it is critical not to use lower number of neurons than 450,000 cells/well.
Problem 2
Libraries exhibit low TSS enrichment, i.e., TSS scores substantially below 10 (steps 31–35).
Potential solution
ATAC-seq is quite robust and usually works well in terms of producing good enrichment for open chromatin. When issues with poor enrichment are encountered, this is typically due to problems with the input material, such as the presence of many dead and nonviable cells (which contain significant quantities of dechromatinized DNA) or free floating DNA. The DNAse pretreatment step is designed to address these issues.
Problem 3
Libraries contain a high fraction of mitochondrial reads (steps 32–35).
Potential solution
The omniATAC protocol and its derivatives are usually quite successful at minimizing the extent of mitochondrial contamination. A typical reason for very high levels of chrM-mapping reads is failure to aspirate all the supernatant (which contains the mitochondria) during the nuclei preparation procedure prior to transposition. Make sure to remove all of it while being careful not to disturb the pellet.
Problem 4
Final ATAC-seq libraries show low yield (low concentration and a barely visible TapeStation/BioAnalyzer trace), and after sequencing contain few distinct fragments and are of generally low molecular complexity (steps 31–35).
Potential solution
This issue could be due to cell loss during the nuclei preparation procedure. Because ATAC-seq works on relatively small numbers of cells/nuclei – only ∼50,000 – cell and nuclei pellets are often quite small and barely visible in tubes. It can thus be easy to inadvertently disturb them while aspirating supernatants, leading to cell loss. Be careful to avoid pellets by using the usual methods of spinning tubes consistently with one side pointing outwards within the centrifuge then carefully pipetting out liquids on the opposite side of the tube.
Problem 5
Poor sequencing yield, i.e., many fewer raw reads obtained than the expected based on the characteristics of the sequencing kit used (steps 31–35).
Potential solution
ATAC-seq libraries are generally difficult to quantify because the fragment length distribution is not uniform and often fragments longer than the maximum length that can be sequenced on an Illumina instrument are present in the library. Thus the effective concentration is usually lower than the apparent concentration, which can result in underloading of sequencing flowcells. For optimal yields, be very careful when measuring library concentration using qPCR; it can also help to try to “overload” the flowcell by a factor of 20–30%.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aaron D. Gitler (agitler@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by NIH grants R35NS097263(10) (A.D.G.), P50HG007735 (W.J.G.), R01HG008140 (W.J.G.), U19AI057266 (W.J.G.), UM1HG009442 (W.J.G.), and 1UM1HG009436 (W.J.G.) and the Brain Rejuvenation Project of the Wu Tsai Neurosciences Institute (A.D.G.). W.J.G. is a Chan Zuckerberg Biohub investigator. Some of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. This work used the Genome Sequencing Service Center by Stanford Center for Genomics and Personalized Medicine Sequencing Center, supported by NIH grant S10OD020141. Some of the figures were created with BioRender.com.
Author contributions
Conceptualization, M.M.-N., Z.S., and A.D.G.; methodology and writing, M.M.-N., G.K.M., and Z.S.; supervision, W.J.G. and A.D.G.
Declaration of interests
A.D.G. is a scientific founder of Maze Therapeutics. W.J.G. has affiliations with 10× Genomics (consultant), Guardant Health (consultant), and Protillion Biosciences (co-founder and consultant).
Contributor Information
Maya Maor-Nof, Email: maormaya@stanford.edu.
Aaron D. Gitler, Email: agitler@stanford.edu.
Data and code availability
The raw and analyzed sequence data from our original paper carrying out ATAC-seq in primary cultured neurons (Maor-Nof et al., 2021) can be found on NCBI GEO (GSE162048).
References
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.G. Transposition of na-tive chromatin for fast and sensitive epigenomic pro-filing of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017;14:959–962. doi: 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor-Nof M., Shipony Z., Lopez-Gonzalez R., Nakayama L., Zhang Y.J., Couthouis J., Blum J.A., Castruita P.A., Linares G.R., Ruan K. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR) Cell. 2021;184:689–708.e20. doi: 10.1016/j.cell.2020.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinov G.K., Shipony Z. Interrogating the accessible chromatin landscape of eukaryote Ge-nomes using ATAC-seq. Methods Mol. Biol. 2021;2243:183–226. doi: 10.1007/978-1-0716-1103-6_10. [DOI] [PubMed] [Google Scholar]
- Picelli S., Bjorklund A.K., Reinius B., Sagasser S., Winberg G., Sandberg R. Tn5 transposase and tagmentation procedures for massively scaled se-quencing projects. Genome Res. 2014;24:2033–2040. doi: 10.1101/gr.177881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw and analyzed sequence data from our original paper carrying out ATAC-seq in primary cultured neurons (Maor-Nof et al., 2021) can be found on NCBI GEO (GSE162048).