

Abstract
Background
Triple-negative breast cancers (TNBCs), especially those non-immune-inflamed tumors, have a poor prognosis and limited therapies. Human leukocyte antigen (HLA)-I not only contributes to antitumor immune response and the phenotype of the tumor microenvironment, but also is a negative predictor of outcomes after immunotherapy. However, the importance of HLA functional status in TNBCs remains poorly understood.
Methods
Using the largest original multiomics datasets on TNBCs, we systematically characterized the HLA-Ⅰ status of TNBCs from the perspective of HLA-Ⅰ homogeneity and loss of heterozygosity (LOH). The prognostic significance of HLA-I status was measured. To explain the potential mechanism of prognostic value in HLA-Ⅰ status, the mutational signature, copy number alteration, neoantigen and intratumoral heterogeneity were measured. Furthermore, the correlation between HLA-Ⅰ functional status and the tumor immune microenvironment was analyzed.
Results
LOH and homogeneity in HLA-I accounted for 18% and 21% of TNBCs, respectively. HLA-I LOH instead of HLA-I homogeneity was an independent prognostic biomarker in TNBCs. In particular, for patients with non-immune-inflamed tumors, HLA-I LOH indicated a worse prognosis than HLA-I non-LOH. Furthermore, integrated genomic and transcriptomic analysis showed that HLA-I LOH was accompanied by upregulated scores of mutational signature 3 and homologous recombination deficiency scores, which implied the failure of DNA double-strand break repair. Moreover, HLA-I LOH had higher mutation and neoantigen loads and more subclones than HLA-I non-LOH. These results indicated that although HLA-I LOH tumors with failure of DNA double-strand break repair were prone to produce neoantigens, their limited capacity for antigen presentation finally contributed to poor immune selection pressure.
Conclusion
Our study illustrates the genomic landscape of HLA-I functional status and stresses the prognostic significance of HLA-I LOH in TNBCs. For “cold” tumors in TNBCs, HLA-I LOH indicated a worse prognosis than HLA-I non-LOH.
Keywords: breast neoplasms, antigen presentation
Introduction
Triple-negative breast cancer (TNBC), which does not express estrogen receptor or progesterone receptor and lacks human epidermal growth factor receptor 2 amplification or overexpression, accounts for 15%–20% of all breast cancers but has the worst prognosis. 1 2 A lack of recognized molecular targets has become the main challenge for patients with TNBC.3 Previous research has classified TNBCs into four subtypes, namely, the luminal androgen receptor (LAR) subtype, basal-like and immune-suppressed (BLIS) subtype, mesenchymal-like (MES) subtype, and immunomodulatory (IM) subtype.4 The IM subtype (so-called “hot” tumor or “immune-inflamed” tumor) is characterized by a high prevalence of both stromal and intratumoral tumor-infiltrating lymphocytes and good prognoses.4 5 In contrast, “cold” tumors have a poor prognosis and limited therapies.5 6
The human leukocyte antigen (HLA) class-I gene group (HLA-A, HLA-B, and HLA-C) is located on chromosome 6p21 and has the highest polymorphism in the human genome.7 8 HLA-I gene products are expressed on all nucleated cells (including tumor cells) and play a crucial role in the presentation of tumor neoantigens with other antigen presentation genes (such as B2M and TAP1/2) to activate the antitumor immune response.9–13 According to previous studies, the higher germline homogeneity in HLA-I molecules contributes to less variation in the peptide-binding region, which leads to this region binding to only a selected repertoire of peptide ligands.11 14–16 Consequently, fewer types of tumor antigens can be presented, and tumor cells are more difficult to recognize by T-cells that can trigger a subsequent antitumor immune response.11 Moreover, a loss of the HLA haplotype may also reduce antigen presentation.17 Therefore, the dysfunction of HLA-Ⅰ might be reflected at the germline and somatic levels. Previous studies have found that homozygosity or loss of heterozygosity (LOH) in HLA-Ⅰ is related to patient prognosis.11 12 18–20 However, few studies systemically discuss germline HLA-Ⅰ mutation or somatic HLA-Ⅰ LOH based on multiomics data, nor do they discuss the correlation between HLA-Ⅰ functional status and the tumor microenvironment, especially in TNBCs.
Here, we questioned the capacity of HLA-Ⅰ to influence the molecular characteristics and prognosis of patients with TNBC. Moreover, we wondered whether HLA-Ⅰ status could improve TNBC immune subtypes from the view of prognosis. With multiomics data for the largest single-center TNBC cohort, we successfully classified 303 TNBC samples into different HLA-I statuses at the germline and somatic levels and explored their biological implications.
Methods
Tumor and normal samples and datasets
Patients diagnosed with malignant breast cancer who were willing to participate in this study were retrospectively selected. Detailed sample selection was described in our previous study.4 In this study, we selected samples qualified to estimate HLA-I germline homogeneity and LOH. In all, 303 patients were retrospectively enrolled in this study.
The follow-up within this cohort ended on June 30, 2017, and the median length of follow-up was 44.7 months (IQR, 34.6–57.4 months). Relapse-free survival (RFS) was defined as the time from primary treatment to first recurrence (local or regional recurrence or distant metastasis) or death due to any cause. Patients without events during follow-up were censored.
Procedures involving patients were performed in accordance with the Declaration of Helsinki. All tissue samples were collected according to the protocols approved by the independent Ethics Committee/Institutional Review Board of the Fudan University Shanghai Cancer Center (FUSCC) Ethical Committee. Each patient provided written informed consent.
Detailed information on biospecimen collection, generation of expression profiles, whole exome sequencing (WES) data and somatic copy number variation (SCNV) data were described in a previous study.4
Gene set enrichment analysis (GSEA) and single sample gene set enrichment analysis (ssGSEA)
GSEA was used to explore enriched pathways and interpret RNA-seq data using predefined gene sets from the Molecular Signatures Database V.7.1.21 All basic and advanced fields were set to default. Gene sets with a false discovery rate (FDR) <0.25 and nominal (NOM) p-value <0.05 were classified as significant and visualized using the Enrichment Map plugin of Cytoscape V.3.7.0. Detailed information for the top 10 gene sets ranked by normalized enrichment score (NES) was visualized using Cleveland plots created with the R package ggplot2. The compendium of microenvironment genes related to microenvironment cell subsets was constructed based on two gene signatures, CIBERSORT22 and MCP-Counter.23 ssGSEA (“GSVA” function in R) was used to estimate the abundance of each microenvironment cell subset for each patient.24
Calculation of TNBC immune subtype
The calculation of TNBC immune subtype was based on the constituent pattern of each microenvironment cell type. To explore the optimal number of stable TNBC immune subtypes, we performed Nbclust testing (“NbClust” function in R, index 1⁄4 “all”). Afterward, k-means (“kmeans” function in R) clustering was used to separate each TNBC immune subtype according to putative optimal number of microenvironment cluster provided by Nbclust testing. The detailed calculation of TNBC immune subtype was described in previous study.5
Determination of HLA-I status
HLA-I status consisted of two aspects of information, HLA-I germline homogeneity and HLA-I LOH. To evaluate HLA-I germline homogeneity, the POLYSOLVER tool25 was used to identify the four-digit HLA genotype for each sample by the exome data of paired normal samples from patients with TNBC (arguments: Asian 1 hg19 STDFQ 0). Patients whose two alleles had the same genotype at any one of the HLA-A, HLA-B, and HLA-C loci were considered to have HLA-I germline homogeneity; otherwise, HLA-I germline heterogeneity was considered. To estimate HLA-I LOH at the somatic level, we used copy number values (nMajor and nMinor) at the segment level adjusted by the ASCAT algorithm.26 Tumor purity has been adjusted when evaluating copy number values in ASCAT. Patients, whose copy number values of one of two alleles at any main HLA-I loci (HLA-A, HLA-B, and HLA-C) equaled zero, were considered HLA-I LOH; otherwise, HLA-I non-LOH.
Survival analysis
Survival curves were constructed using the Kaplan-Meier method and compared with the log-rank test. Univariate and multivariate Cox proportional hazards models were used to explore independent prognostic variables. Age, the number of positive lymph nodes, tumor size, homologous recombination deficiency (HRD) score, PAM50 subtypes, and TNBC immune subtypes were first analyzed in a univariate Cox proportional hazards model. Then, a multivariate analysis of all significant variables was performed using the Cox proportional hazard regression model.
Calculation of neoantigens
NetMHCpan (V.4.0)27 was used to predict neoantigens based on the somatic mutation data (.maf) and HLA genotype data generated by POLYSOVER tools. According to variant classification and variant type in the MAF file, two kinds of neoantigens were predicted separately—neoantigens derived from small insertions and deletions (Indel) (Variant_Classification = “Frame_Shift_Ins”, “Frame_Shift_Del”, “In_Frame_Ins”, “In_Frame_Del”, and Variant_Type = “INS”, “DEL”) and protein-coding single nucleotide variants (SNVs) (Variant_Classification = “Missense_Mutation”, and Variant_Type = “SNP”). Neoantigens were considered to be mutations possessing predicted peptides with a presumptive binding affinity <500 nM. The corresponding gene of those mutations should be expressed as greater than combat value 1 (based on median expression rather than the specific sample). The construction of this algorithm was based on pVAC-seq28 and modified according to the characteristics of our data.
Calculation of HRD scores
The HRD score was calculated as the unweighted sum of the LOH score, telomeric allelic imbalance (TAI) score, and modified large-scale state transition (LSTm) score. The LOH score measures the number of subchromosomal LOH regions longer than 15 Mb. The LST score counts the number of chromosomal breaks of at least 10 Mb between adjacent regions after filtering out regions shorter than 3 Mb. To adjust the effect of ploidy, the LST score was modified using the following formula: LSTm=LST–15.5P (P refers to ploidy). The TAI score was defined as the number of regions of allelic imbalance that extended to one of the subtelomeres but did not cross the centromere. The detailed calculation of these scores was described in a previous study.29
Estimation of subclonal cancer cells
We used PyClone 0.13.0 to infer the clonal composition of each tumor sample.30 The input data included the number of reads overlapping the locus matching the reference allele and variant allele and the copy number of the major allele and minor allele in the malignant cells. Each patient’s subclone number was summarized from the “cluster_id” column in PyClone output tables.
Determination of TNBC subtype
Briefly, we performed consensus clustering (“ConsensusClusterPlus” package in R31) and k-means clustering (“kmeans” function in R) to determine the optimal number of TNBC subtypes in RNA sequencing data. Finally, four subtypes, BLIS, IM, LAR, and MES were identified. Detailed methods for subtype determination have been reported previously.4
Mutational signature and SCNV analysis
The package “deconstructSigs”32 was used to identify mutational signatures present in TNBC samples with SNVs. This approach organized sample information in the form of the fraction of mutations in each of 96 trinucleotides and determined the weighted combination of published signatures33 (https://cancer.sanger.ac.uk/cosmic/signatures) that most closely reconstructed the mutational profile. Only signatures that have been observed in human breast cancers were considered (cosmic signatures 1, 2, 3, 5, 6, 8, 13, 17, 18, 20, 26, and 30).34 35
Pearson’s χ2 test and Fisher’s exact test were used to evaluate differences in copy number alteration (CNA) frequency. The FDR method was used to correct p values.
Statistical analysis
In this study, data distributions were characterized by frequency tabulation and summary statistics. Student’s t-test, analysis of variance, and Mann-Whitney Wilcoxon test were used to compare continuous variables, and Pearson’s χ2 test and Fisher’s exact test were employed to compare categorical variables. Before comparison, the Shapiro-Wilk test was conducted in the normality test of distribution. All tests were two-sided. A p-value <0.05 was considered statistically significant, unless otherwise indicated. p-values for multiple comparisons were adjusted to the false discovery rate using FDR correction. All analyses were performed using R package V.4.0.2 (https://cran.r-project.org/).
Data availability
All data can be viewed in The National Omics Data Encyclopedia (http://www.biosino.org/node) by pasting the accession (OEP000155) into the text search box or through online (http://wwwbiosinoorg/node/project/detail/OEP000155). The microarray data and sequence data are also available in the NCBI Gene Expression Omnibus (OncoScan: GSE118527, HTA 2.0: GSE76250) and Sequence Read Archive (WES and RNA-seq: SRP157974).
Results
The landscape of HLA-I status in TNBCs
HLA-I LOH is defined as the monoallelic loss of at least one HLA-I gene (HLA-A, HLA-B, and HLA-C). Individuals whose HLA genotypes showed two identical alleles in at least one HLA-I gene (HLA-A, HLA-B, and HLA-C) were classified as having HLA-I germline homogeneity. Patients with HLA-I germline homogeneity or HLA-I LOH were considered to have HLA-Ⅰ dysfunction and consequently a limited capacity to present antigens.11 17 Using POLYSOLVER and ASCAT algorithms on a cohort of 303 patients with TNBC, we evaluated HLA-I germline homogeneity and HLA-I LOH for each patient.25 26 Only rare patients had >2 homogenous HLA-Ⅰ genes (online supplemental figure 1A). Overall, 18% of patients had HLA-Ⅰ LOH (figure 1A, B), and 21% of patients had HLA-Ⅰ homogeneity (online supplemental figure 1A, B). HLA-Ⅰ statuses were relatively evenly distributed in TNBC intrinsic subtypes and the TNBC immune subtypes (figure 1C and online supplemental figure 1C).
Figure 1.

The landscape of HLA-I LOH statuses in TNBCs. (A) Summary of characteristics of HLA-I LOH and HLA-I non-LOH. (B) Distribution of HLA-I LOH and HLA-I non-LOH. (C) Distribution of the TNBC immune subtype and TNBC intrinsic subtype (FUSCC TNBC subtype). (D) Comparison of Kaplan-Meier curves of RFS between HLA-I non-LOH and LOH patients. (E) Kaplan-Meier curves of RFS between the HLA-I non-LOH and LOH groups in immune-inflamed patients and non-immune-inflamed patients. (F) Kaplan-Meier curves of RFS between immune-inflamed patients and non-immune-inflamed patients with or without HLA-I LOH. BLIS, basal-like and immune-suppression; CNA, copy number alteration; HLA, human leukocyte antigen; IM, immunomodulatory; LAR, luminal androgen receptor; LOH, loss of heterozygosity; MES, mesenchymal-like; ns, no significance; RFS, relapse-free survival; TNBCs, triple-negative breast cancers; ***p<0.001.
jitc-2021-003371supp001.pdf (2MB, pdf)
We further investigated the clinical relevance of HLA-I status in TNBCs. Patients with HLA-Ⅰ LOH showed significantly worse RFS than the HLA-Ⅰ non-LOH group (figure 1D, p=0.013). Regardless of whether patients had germline HLA homogeneity, their prognoses were not significantly different (online supplemental figure 1D, p = 0.36). We conducted further analysis to show the association between specific HLA-I locus homozygosity (HLA-A, HLA-B, and HLA-C) and survival. Specific HLA-I locus homozygosity still showed limited prognostic value (online supplemental figure 1E).
Owing to the close connection between HLA-Ⅰ function and immune response,11 12 20 we subsequently investigated the prognostic value of HLA-Ⅰ function in “cold” and “hot” TNBCs. First, we divided patients with TNBC into an immune-inflamed group and a non-immune-inflamed group.5 In general, non-immune-inflamed tumors had worse RFS than immune-inflamed tumors. When taking HLA-Ⅰ LOH into consideration as well, HLA-Ⅰ LOH identified differences in prognoses among non-immune-inflamed patients rather than immune-inflamed patients (figure 1E). Additionally, although immune-inflamed patients had better prognoses than non-immune-inflamed patients, the prognostic difference between non-immune-inflamed patients without HLA-Ⅰ LOH and immune-inflamed patients was not statistically significant (figure 1F). However, germline HLA homogeneity might not significantly influence the prognosis of the non-immune-inflamed population (online supplemental figure 1F). There was limited prognostic difference between immune-inflamed patients and non-immune-inflamed patients with HLA-Ⅰ homogeneity (online supplemental figure 1G). We reasoned that HLA-Ⅰ LOH was an important factor for the poor prognosis of non-immune-inflamed patients with TNBC.
To eliminate confounding factors, we subsequently performed univariate and multivariate Cox regression. Age, the number of positive lymph nodes, tumor size, HRD score, PAM50 subtype, tumor HLA-Ⅰ LOH, and TNBC immune subtype were included. After univariate and multivariate Cox regression, the number of positive lymph nodes, tumor size, tumor HLA-Ⅰ LOH, and TNBC immune subtype were considered independent prognostic factors. Notably, among these factors, tumor HLA-Ⅰ LOH was one of the strongest independent prognostic factors with statistical significance (table 1, non-LOH: HR=0.307, p<0.001). Moreover, we validated that HLA-I LOH was still an independent poor prognostic indicator in non-immune-inflamed patients with TNBC (online supplemental table S1, non-LOH: HR=0.255, p<0.001).
Table 1.
Univariate and multivariate Cox proportional hazard models for RFS
| Variable | Univariate | Multivariate | ||
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Age, years | 0.997 (0.971 to 1.024) | 0.837 | Not Included | |
| Positive lymph node, number | 1.089 (1.049 to 1.131) | <0.001 | 1.093 (1.048 to 1.140) | <0.001 |
| Tumor size, cm | 1.462 (1.117 to 1.816) | 0.001 | 1.368 (1.085 to 1.724) | 0.008 |
| HRD score | 0.997 (0.989 to 1.006) | 0.525 | Not Included | |
| PAM50 | ||||
| Basal | Ref | Not Included | ||
| Others | 1.375 (0.749 to 2.524) | 0.304 | ||
| Tumor HLA-I LOH | ||||
| LOH | Ref | Ref | ||
| Non-LOH | 0.459 (0.245 to 0.860) | 0.015 | 0.307 (0.158 to 0.596) | <0.001 |
| TNBC immune subtype | ||||
| Immune-inflamed | Ref | Ref | ||
| Non-immune-inflamed | 2.519 (1.067 to 5.946) | 0.035 | 2.784 (1.169 to 6.629) | 0.033 |
HRs and p-values of the covariates in the univariate and multivariate Cox proportional hazard models for RFS.
CI, confidence interval; HLA, human leukocyte antigen; HRD, homologous recombination deficiency; LOH, loss of heterozygosity; RFS, relapse-free survival; TNBCs, triple-negative breast cancers.
In summary, both of HLA-Ⅰ LOH and HLA-I germline homogeneity accounted for approximately 20% of patients with TNBC, and HLA-I LOH instead of HLA-I homogeneity was a potential prognostic biomarker. HLA-I LOH indicated worse prognoses than HLA-I non-LOH, especially for patients with non-immune-inflamed tumors.
Genomic alterations among HLA-I statuses
As HLA-I LOH had strong prognostic significance, we further investigated whether this was driven by specific oncogenic mutations and CNAs. Initially, we discussed the differences in CNAs between the HLA-I non-LOH group and the LOH group. Most of the segments had no significant differences in copy number amplification or deletion, except for the short arm of chromosome 6, where HLA-Ⅰ genes were located (figure 2A). This result suggested that HLA-I LOH was potentially a driver event rather than a passage event in CNAs. Moreover, the mutation landscape between the HLA-I non-LOH group and LOH group was compared. Among the top 20 most frequent mutations, none of them showed statistically significant differences between the HLA-Ⅰ LOH group and the HLA-I non-LOH group (figure 2B). Interestingly, among all breast cancer-related mutational signatures, mutational signature 3, which was associated with failure of DNA double-strand break repair, showed a significant difference between the HLA-Ⅰ LOH and non-LOH groups (figure 2C).32 33
Figure 2.

Genomic alterations between different HLA-I LOH statuses in TNBCs. (A) Comparison of the somatic copy number variations between HLA-I non-LOH groups and HLA-I LOH groups. The top two plots illustrate the frequency of the amplification (dark red), gain (light red), loss (light blue), and deletion (dark blue) of each gene in each cluster, and the bottom plot illustrates the –log10 FDR value of each gene when compared among all four clusters in the amplification-centric (light yellow) or deletion-centric (light green) calculations. (B) Somatic mutation profile in HLA-I LOH and HLA-I non-LOH patients. Top 20 most frequent genes are shown. (C) Heatmap showing the contribution of breast cancer-related mutational signatures to HLA-I non-LOH and HLA-I LOH. HLA, human leukocyte antigen; LOH, loss of heterozygosity; TNBCs, triple-negative breast cancers; *p<0.05.
We also explored the difference in oncogenic mutations and CNAs between the HLA-Ⅰ heterogeneity group and germline HLA-Ⅰ homogeneity group. There were no statistically significant copy number alterations between the germline HLA-Ⅰ heterogeneity group and germline HLA-Ⅰ homogeneity group (online supplemental figure S2A). PIK3CA mutation was enriched in the homogeneity group (29% vs 13%, p=0.009; online supplemental figure S2B). There was no difference in mutational signatures between the homogeneity and heterogeneity groups (online supplemental figure S2C).
In conclusion, HLA-I LOH was potentially a driver CNA event and was accompanied by the failure of DNA double-strand break repair.
HLA-I status correlates with selection pressure
Selection pressure from the immune system influences tumor evolution. Weak immune selection reflects immune escape and expands the tumor population.36 Previous research indicated that HLA-I LOH might facilitate genome evolution by decreasing selection pressure and result in tumor progression in non-small-cell lung cancer (NSCLC).17 We further compared the differences in selection pressure between each HLA-I status in TNBCs. Mutation load, neoantigen load and the number of subclones were chosen to reflect the selection pressure.36 We divided all patients into a HLA-I LOH group and a HLA-I non-LOH group. A violin plot showed that the HLA-I LOH group had higher neoantigen and tumor mutation loads (figure 3A, B). Moreover, we found that the HLA-I LOH group had more subclones than the HLA-I non-LOH group (figure 3C). Similar results were also observed when we only included non-immune-inflamed TNBCs (figure 3D, E, F). However, for patients with immune-inflamed tumors, there was no significant difference in neoantigen load, mutation load or subclone number (figure 3G, H, I).
Figure 3.
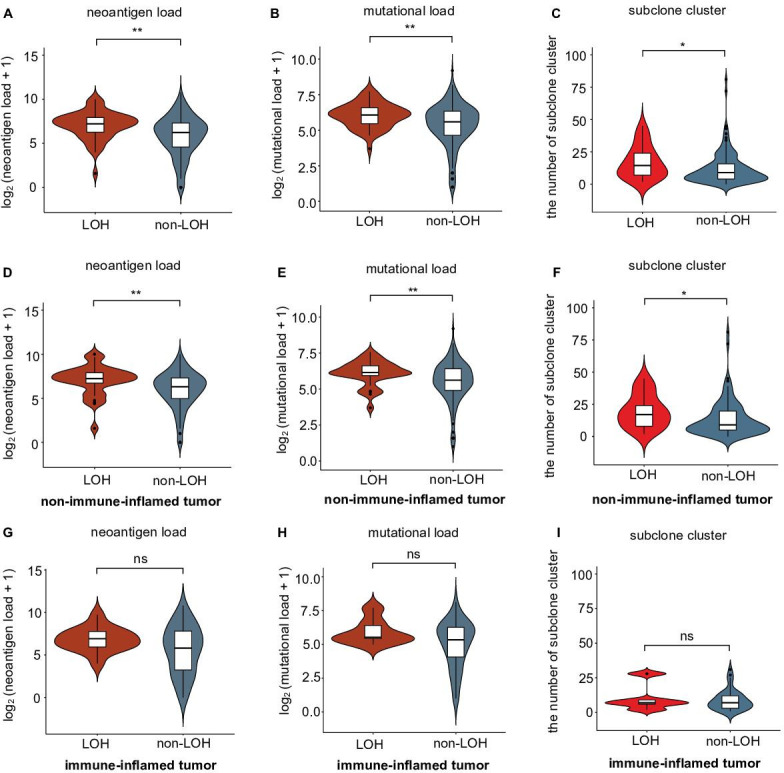
Correlation of selective pressure and HLA-I LOH statuses in TNBCs. (A–B) Comparison of (A) neoantigen and (B) mutational loads between the HLA-I LOH and non-LOH groups. (C) Comparison of subclone clusters between the HLA-I LOH and non-LOH groups. (D–E) Comparison of (D) neoantigen and (E) mutational loads between the HLA-I LOH and non-LOH groups in the non-immune-inflamed tumor population. (F) Comparison of subclone clusters between the HLA-I LOH and non-LOH groups in the non-immune-inflamed tumor population. (G–H) Comparison of (G) neoantigen and (H) mutational loads between the HLA-I LOH and non-LOH groups in the inflamed tumor population. (I) Comparison of subclone clusters between the HLA-I LOH and non-LOH groups in the inflamed tumor population. **p<0.01; *p<0.05; ns, no significance; LOH, HLA-I LOH group; non-LOH, non-HLA-I LOH group. HLA, human leukocyte antigen; LOH, loss of heterozygosity; TNBCs, triple-negative breast cancers.
In contrast, regardless of TNBC immune subtype, mutation load, neoantigen load, and the number of subclones were not significantly different between the germline HLA-I homogeneity group and germline HLA-I heterogeneity group (online supplemental figure S3).
Taken together, these data suggested that TNBCs with HLA-I LOH had higher mutation and neoantigen loads and more subclones than HLA-I non-LOH TNBCs, indicating that HLA-I LOH tumors were subjected to poor selection pressure due to antigen presentation dysfunction.
High homologous recombination deficiency in HLA-I LOH and non-immune-inflamed TNBCs
Given that the occurrence of HLA-I LOH might be a driver event and strongly contribute to the poor prognosis of patients with non-immune-inflamed tumors in TNBCs, we further explored potential therapeutic strategies for these TNBCs. We compared the expression profiles between the non-immune-inflamed population with HLA-I LOH and without HLA-I LOH. In GSEA, among all statistically significant pathways, cell cycle-related pathways, DNA repair pathways and ribosome-related pathways were enriched in the non-immune-inflamed population with HLA-I LOH compared with the non-immune-inflamed population without HLA-I LOH (figure 4A). Specifically, among the top 20 pathways with the highest NES, most pathways reflected an elevated level of ribosome activity and cell cycle-related activities (figure 4B). Furthermore, in non-immune-inflamed tumors, the mean HRD score of patients with HLA-I LOH was 43, which was significantly higher than that of patients with HLA-I non-LOH, whose average HRD score was 14 (figure 4C, p<0.001). The HRD score is the sum of TAI, LSTm and LOH score. All these scores were higher in the HLA-I LOH group than in the HLA-I non-LOH group among non-immune-inflamed tumors (online supplemental figure S4A-C, TAI: p=0.027, LSTm: p<0.001; LOH: p<0.001). For the non-immune-inflamed population without HLA-I LOH, the top 20 statistically significant pathways with the highest NES value were mostly related to metabolism, including carbohydrate metabolism (online supplemental figure S5).
Figure 4.

High homologous recombination deficiency in non-immune-inflamed TNBCs with HLA-I LOH. (A) Enrichment map shows pathways enriched in non-immune-inflamed TNBCs with HLA-I LOH when compared with non-immune-inflamed TNBCs with HLA-I non-LOH. Nodes in the network represent pathways, and similar pathways with many common genes are connected. Groups of similar pathways are indicated. The size and color of each node represent the p-value and NES value of each pathway, respectively. All pathways included were statistically significant (p-value <0.05 and q-value <0.25). (B) Cleveland plot shows the top 15 statistically significant pathways (p-value <0.05 and q-value <0.25) with the highest NES value. All pathways included are statistically significant. (C) Comparison of homologous recombination deficiency scores between the HLA-I LOH and non-LOH groups in non-immune-inflamed tumors. (D) Potential clinical translations of HLA-I LOH status. ***, p<0.001. HLA, human leukocyte antigen; LOH, loss of heterozygosity; NES, normalized enrichment score; TNBCs, triple-negative breast cancers.
Overall, non-inflamed-tumors with HLA-Ⅰ LOH had a worse prognosis and the upregulation of cell cycle-related and DNA repair pathways (figure 4D).
Discussion
Using multiomics data from the largest single-center TNBC cohort (FUSCC TNBC cohort), our study investigated the clinical significance of HLA-Ⅰ function from the view of germline HLA-Ⅰ status and somatic tumor HLA-Ⅰ status. We found that HLA-Ⅰ LOH played a more important role than germline homogeneity in clinical outcomes of patients with TNBC. We further combined HLA-Ⅰ statuses with the TNBC immune subtype and indicated that so-called “cold” tumors with HLA-Ⅰ LOH had the worst prognosis.
Our study delves deeply into the prognostic value of HLA-I status. Survival analysis indicating LOH rather than germline homogeneity of HLA-Ⅰ suggested a poor prognosis in the largest Chinese TNBC cohort, which was consistent with previous studies based on Western populations and pan-cancer cohort.20 Furthermore, we explained the potential mechanism of poor prognosis in patients who had HLA-I LOH. The analysis of subclones and neoantigens implied lower negative selection but higher intratumor heterogeneity in HLA-I LOH tumors. We assumed that LOH of HLA-I largely inhibited tumor antigen presentation, blocking the initiation of the immune response, weakening the elimination of the immune system, and ultimately promoting immune escape.17 20 37 In this process, due to weak negative selection, tumors were able to accumulate relatively massive mutations and subclones. The increasing intratumor heterogeneity of HLA-I LOH tumor led to drug resistance and cancer development.38 39 Therefore, dysfunction of antigen presentation and concomitant intra-tumor heterogeneity might partially explain the different outcomes between HLA-I LOH tumors and HLA-I non-LOH tumors. Interestingly, it seemed that germline HLA homogeneity might be not as important as HLA-I LOH in predicting prognosis. One potential explanation was the relatively lower prognostic impact of germline-level mutations on HLA-I function in TNBCs, which should be strictly proven in future studies. We noticed that previous studies showed that the prognostic value of single germline HLA homogeneity was controversial.11 20 40 41 However, it merely focused on NSCLC or advanced melanoma with immunotherapy. We attributed the different results to the distinct histological types of cancer (TNBC vs NSCLC or melanoma) and cohort (with or without immunotherapy).
This study provides a unique insight for TNBC immune subtype. First, based on previous research, TNBCs can be divided into “hot” tumors (immune-inflamed) and “cold” tumors (non-immune-inflamed), and the former immune subtype always has a better prognosis.5 When applying HLA statuses in immune-inflamed and non-immune-inflamed populations, we found that so-called “cold” tumors were actually heterogeneous in prognosis from the view of somatic HLA-I mutations rather than germline HLA-I mutations. Specifically, even for “cold“ tumors, as long as there was no HLA-Ⅰ LOH, their prognoses were the same as those of “hot” tumors. The analysis of subclones and neoantigens in immune-inflamed and non-immune-inflamed tumors further indicated weak negative selection in non-immune-inflamed tumors with HLA-I LOH. However, HLA-I status might not influence selective pressure in immune-inflamed tumors. These corresponded to the result of survival analysis. We assumed that the poor prognosis of non-immune-inflamed tumors might be mainly ascribed to both HLA-I LOH and lower tumor-infiltrating lymphocytes. This result indicated the limitation of previous immune microenvironment subtype. Second, after analysis based on the expression profile, we found that within non-immune-inflamed tumors, HLA-Ⅰ LOH might result in upregulation of cell cycle-related and DNA repair pathways and a higher HRD score. Moreover, mutational signature 3, a biomarker indicating failure of DNA double-strand break repair, was enriched in patients with HLA-Ⅰ LOH, which probably strengthened our finding.
Our study has important implications for clinical translations. First, we found that HLA-Ⅰ status, especially HLA-Ⅰ LOH, had prognostic value for patients with “cold” tumors, which, to the best of our knowledge, had not been reported. Previous studies merely focused on the prognostic value of the immune microenvironment or discussed the role of HLA-I without differentiating between “cold” and “hot” tumors.5 11 12 17–20 The emergence of Nanostring (nCounter Breast Cancer 360 Panel) makes it possible to relatively quickly type the immune microenvironment and transform our discovery into clinical practice. Second, genomic characterization, including a higher HRD score and mutation signature 3 of non-immune-inflamed tumors with HLA-I LOH, was associated with PARPi or platinum sensitivity.33 42–44 Further studies could explore the feasibility of PARPi or platinum therapy for these patients who have the worst prognoses but are not sensitive to immune therapy. Notably, unlike this study, the majority of studies mainly discuss the relationship between HLA-I and response to immunotherapy rather than chemotherapy or targeted therapy.11 12 20 40 41
To the best of our knowledge, this research is the first study to systemically discuss germline HLA-Ⅰ mutation and somatic HLA-Ⅰ LOH in TNBCs based on multiomics data and to combine it with mature immune microenvironment subtypes. However, our research had limitations. First, due to the lack of HLA-Ⅰ-related genes in our targeted NGS panel, we could not validate our conclusion in a large-scale treatment cohort retrospectively or prospectively. Second, considering that our data originated from bulk sequences, our tumor subclonal analysis needs to be further expanded by single-cell sequencing. Third, detecting HLA status required raw data from whole exome sequencing, which were inaccessible. Thus, we were unable to perform external validation in a Western population from TCGA.
In conclusion, our study revealed that a patient’s HLA-Ⅰ status had unique prognostic value. For so-called “cold” tumors, HLA-I LOH indicated a worse prognosis than HLA-I non-LOH.
Acknowledgments
Some parts of Figure 4D were created with BioRender.com.
Footnotes
Y-FZ, YX and XJ contributed equally.
Contributors: Conceptualization: Y-FZ, YX, Y-ZJ, and Z-MS. Methodology: Y-FZ and YX. Formal analysis: Y-FZ and YX. Writing-original draft: Y-FZ. Writing-review and editing: Y-FZ, YX, XJ, G-HD, Y-ZJ, and Z-MS. Supervision: G-HD, Y-ZJ and Z-MS.
Funding: This study was supported by grants from the National Key Research and Development Project of China (2020YFA0112304), the National Natural Science Foundation of China (82002792, 81902684, and 82072922), Shanghai Rising-Star Program (20YF1408600), Shanghai three-year action plan for Traditional Chinese Medicine (ZY(2018-2020)-CCCX-2005-04) and the Shanghai Key Laboratory of Breast Cancer (12DZ2260100).
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available in a public, open access repository. All data can be viewed in The National Omics Data Encyclopedia (http://www.biosino.org/node) by pasting the accession (OEP000155) into the text search box or through the URL: http://www.biosino.org/node/project/detail/OEP000155. The microarray data and sequence data are also available in the NCBI Gene Expression Omnibus (OncoScan: GSE118527, HTA 2.0: GSE76250) and Sequence Read Archive (WES and RNA-seq: SRP157974).
Ethics statements
Patient consent for publication
Not required.
References
- 1.Garrido-Castro AC, Lin NU, Polyak K. Insights into molecular classifications of triple-negative breast cancer: improving patient selection for treatment. Cancer Discov 2019;9:176–98. 10.1158/2159-8290.CD-18-1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harbeck N, Penault-Llorca F, Cortes J, et al. Breast cancer. Nat Rev Dis Primers 2019;5:66. 10.1038/s41572-019-0111-2 [DOI] [PubMed] [Google Scholar]
- 3.Waks AG, Winer EP. Breast cancer treatment: a review. JAMA 2019;321:288–300. 10.1001/jama.2018.19323 [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y-Z, Ma D, Suo C, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell 2019;35:428–40. 10.1016/j.ccell.2019.02.001 [DOI] [PubMed] [Google Scholar]
- 5.Xiao Y, Ma D, Zhao S, et al. Multi-Omics profiling reveals distinct microenvironment characterization and suggests immune escape mechanisms of triple-negative breast cancer. Clin Cancer Res 2019;25:5002–14. 10.1158/1078-0432.CCR-18-3524 [DOI] [PubMed] [Google Scholar]
- 6.Tekpli X, Lien T, Røssevold AH, et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat Commun 2019;10:5499. 10.1038/s41467-019-13329-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes AL, Yeager M. Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 1998;32:415–35. 10.1146/annurev.genet.32.1.415 [DOI] [PubMed] [Google Scholar]
- 8.Robinson J, Guethlein LA, Cereb N, et al. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet 2017;13:e1006862. 10.1371/journal.pgen.1006862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGranahan N, Furness AJS, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 11.Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018;359:582–7. 10.1126/science.aao4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowell D, Krishna C, Pierini F, et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med 2019;25:1715–20. 10.1038/s41591-019-0639-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samstein RM, Lee C-H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. 10.1038/s41588-018-0312-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrington M, Nelson GW, Martin MP, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 1999;283:1748–52. 10.1126/science.283.5408.1748 [DOI] [PubMed] [Google Scholar]
- 15.Penn DJ, Damjanovich K, Potts WK. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc Natl Acad Sci U S A 2002;99:11260–4. 10.1073/pnas.162006499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul S, Weiskopf D, Angelo MA, et al. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol 2013;191:5831–9. 10.4049/jimmunol.1302101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 2017;171:e11:1259–71. 10.1016/j.cell.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung JT, Hamilton RL, Ohnishi K, et al. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clin Cancer Res 2013;19:1816–26. 10.1158/1078-0432.CCR-12-2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenbaum E, Seier K, Bandlamudi C, et al. HLA genotyping in synovial sarcoma: identifying HLA-A*02 and its association with clinical outcome. Clin Cancer Res 2020;26:5448–55. 10.1158/1078-0432.CCR-20-0832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montesion M, Murugesan K, Jin DX, et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov 2021;11:282–92. 10.1158/2159-8290.CD-20-0672 [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. 10.1038/nmeth.3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17:218. 10.1186/s13059-016-1070-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol 2015;33:1152–8. 10.1038/nbt.3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A 2010;107:16910–5. 10.1073/pnas.1009843107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurtz V, Paul S, Andreatta M, et al. NetMHCpan-4.0: improved peptide-MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 2017;199:3360–8. 10.4049/jimmunol.1700893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hundal J, Carreno BM, Petti AA, et al. pVAC-Seq: a genome-guided in silico approach to identifying tumor neoantigens. Genome Med 2016;8:11. 10.1186/s13073-016-0264-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res 2014;16:475. 10.1186/s13058-014-0475-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roth A, Khattra J, Yap D, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014;11:396–8. 10.1038/nmeth.2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–3. 10.1093/bioinformatics/btq170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016;17:31. 10.1186/s13059-016-0893-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov LB, Jones PH, Wedge DC, et al. Clock-like mutational processes in human somatic cells. Nat Genet 2015;47:1402–7. 10.1038/ng.3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nik-Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nik-Zainal S, Morganella S. Mutational signatures in breast cancer: the problem at the DNA level. Clin Cancer Res 2017;23:2617–29. 10.1158/1078-0432.CCR-16-2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakatos E, Williams MJ, Schenck RO, et al. Evolutionary dynamics of neoantigens in growing tumors. Nat Genet 2020;52:1057–66. 10.1038/s41588-020-0687-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer 2021;21:298–312. 10.1038/s41568-021-00339-z [DOI] [PubMed] [Google Scholar]
- 38.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 2017;168:613–28. 10.1016/j.cell.2017.01.018 [DOI] [PubMed] [Google Scholar]
- 39.Dentro SC, Leshchiner I, Haase K, et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021;184:e39:2239–54. 10.1016/j.cell.2021.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abed A, Calapre L, Lo J, et al. Prognostic value of HLA-I homozygosity in patients with non-small cell lung cancer treated with single agent immunotherapy. J Immunother Cancer 2020;8:e001620. 10.1136/jitc-2020-001620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negrao MV, Lam VK, Reuben A, et al. Pd-L1 expression, tumor mutational burden, and cancer gene mutations are stronger predictors of benefit from immune checkpoint blockade than HLA class I genotype in non-small cell lung cancer. J Thorac Oncol 2019;14:1021–31. 10.1016/j.jtho.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 42.Robson ME, Tung N, Conte P, et al. OlympiAD final overall survival and tolerability results: olaparib versus chemotherapy treatment of physician's choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol 2019;30:558–66. 10.1093/annonc/mdz012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loibl S, Weber KE, Timms KM, et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response-final results from GeparSixto. Ann Oncol 2018;29:2341–7. 10.1093/annonc/mdy460 [DOI] [PubMed] [Google Scholar]
- 44.Fasching PA, Jackisch C, Rhiem K, et al. GeparOLA: a randomized phase II trial to assess the efficacy of paclitaxel and olaparib in comparison to paclitaxel/carboplatin followed by epirubicin/cyclophosphamide as neoadjuvant chemotherapy in patients (PTS) with HER2-negative early breast cancer (bc) and homologous recombination deficiency (HRD). Journal of Clinical Oncology 2019;37:506. 10.1200/JCO.2019.37.15_suppl.506 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-003371supp001.pdf (2MB, pdf)
Data Availability Statement
All data can be viewed in The National Omics Data Encyclopedia (http://www.biosino.org/node) by pasting the accession (OEP000155) into the text search box or through online (http://wwwbiosinoorg/node/project/detail/OEP000155). The microarray data and sequence data are also available in the NCBI Gene Expression Omnibus (OncoScan: GSE118527, HTA 2.0: GSE76250) and Sequence Read Archive (WES and RNA-seq: SRP157974).
Data are available in a public, open access repository. All data can be viewed in The National Omics Data Encyclopedia (http://www.biosino.org/node) by pasting the accession (OEP000155) into the text search box or through the URL: http://www.biosino.org/node/project/detail/OEP000155. The microarray data and sequence data are also available in the NCBI Gene Expression Omnibus (OncoScan: GSE118527, HTA 2.0: GSE76250) and Sequence Read Archive (WES and RNA-seq: SRP157974).