

Abstract
The Wnt signaling antagonist, sclerostin, is a potent suppressor of bone acquisition that also mediates endocrine communication between bone and adipose. As a result, Sost−/− mice exhibit dramatic increases in bone formation but marked decreases in visceral and subcutaneous adipose that are secondary to alterations in lipid synthesis and utilization. While interrogating the mechanism by which sclerostin influences adipocyte metabolism, we observed paradoxical increases in the adipogenic potential and numbers of CD45−:Sca1+:PDGFRα+ adipoprogenitors in the stromal vascular compartment of fat pads isolated from male Sost−/− mice. Lineage tracing studies indicated that sclerostin deficiency blocks the differentiation of PDGFRα+ adipoprogenitors to mature adipocytes in association with increased Wnt/β‐catenin signaling. Importantly, osteoblast/osteocyte‐specific Sost gene deletion mirrors the accumulation of PDGFRα+ adipoprogenitors, reduction in fat mass, and improved glucose metabolism evident in Sost−/− mice. These data indicate that bone‐derived sclerostin regulates multiple facets of adipocyte physiology ranging from progenitor cell commitment to anabolic metabolism.
Keywords: adipoprogenitor, adipose, bone, PDGFRα, sclerostin, Wnt, β‐catenin
Abbreviations
- APC
adipoprogenitor cell
- FACS
fluorescent‐activated cell sorting
- gWAT
gonadal white adipose tissue
- HFD
high‐fat diet
- iWAT
inguinal white adipose tissue
- Lrp
low‐density lipoprotein receptor‐related protein
- rScl
recombinant sclerostin
- SVF
stromal vascular fraction
- TM
tamoxifen
1. INTRODUCTION
Lipid‐storing white adipocytes play a key homeostatic role in the regulation of energy reserves in vertebrates. In response to reductions in caloric consumption or heightened energy expenditure, fatty acids are mobilized from white adipose to meet peripheral demands. Conversely, chronic caloric excess or declines in energy utilization results in adipose tissue expansion through the hypertrophy of existing adipocytes and the recruitment and adipogenic differentiation of tissue progenitor cells. 1 , 2 The linkage between prolonged energy excess that leads to obesity and a subsequent increase in the risk for type 2 diabetes, cardiovascular disease, and renal disease 3 , 4 highlights the need to fully understand the genetic and endocrine networks that regulate adipocyte development.
The new adipocytes that replace the ~8% of mature adipocytes that turnover each year and contribute to the increase in adipocyte numbers in obesity 2 , 5 are derived from adipoprogenitor cells (APCs) present within the stromal vascular compartment of adipose depots. Using flow cytometry to isolate specific stromal vascular cell populations, Rodeheffer and colleagues 6 demonstrated that the Lin−:CD29+:CD34+:Sca1+:CD24+ fraction has adipose stem cell properties and can form functional adipose tissue when transplanted in lipodystrophic mice. Subsequent studies revealed that this APC population expresses platelet‐derived growth factor receptor‐α (PDGFRα) and can be traced in vivo, along with more committed CD24− preadipocytes, using the Pdgfrα‐Cre transgene. 7 PDGFRα+ APCs undergo a burst of proliferation in response to obesogenic signals like high‐fat diet feeding 8 and may also give rise to beige adipocytes in perigonadal white adipose tissue after β3‐adrenergic receptor activation. 9 Additional perivascular stem/progenitor cell populations that express the mural cell marker PDGFRβ have also been described and shown to form both white and beige adipocytes. 10 , 11 , 12
The endocrine, cellular, and molecular mechanisms that regulate APC replication and commitment are still poorly understood. The increase in APC proliferation in response to high‐fat diet feeding is mediated by signaling through PI3K‐AKT2, though the ligand:receptor complex that stimulates activation is unknown, and also influenced by sex hormones and adipose depot‐specific factors. 8 , 13 As APCs become committed, the zinc finger protein ZFP423 drives the expression of PPARγ necessary for adipocyte differentiation. 10 , 14 Expression and activity of ZFP423 and APC commitment appears to be under the control of morphogens. For example, BMP signaling through SMADs cooperates with ZFP423, 14 while Wnt signaling suppresses adipogenesis. 15 , 16 , 17
Produced almost exclusively by osteocytes embedded in the bone matrix, the Wnt signaling antagonist sclerostin has primarily been studied for its ability to exert profound control over bone formation. 18 , 19 After binding to its receptor low‐density lipoprotein receptor‐related protein 4 (LRP4), 20 , 21 sclerostin interacts with the first β‐propeller domain of the Wnt co‐receptors LRP5 and LRP6 and thereby suppresses osteo‐anabolic Wnt/β‐catenin signaling. 22 , 23 , 24 Our previous studies indicate that sclerostin also regulates adipose physiology as Sost−/− mice exhibit reductions in white adipose tissue accumulation on both a chow and high‐fat diet, while overproduction of the protein stimulates adipose tissue expansion. 25 , 26 In each case, the alteration in adipose tissue mass was associated with a corresponding change in markers of Wnt/β‐catenin signaling and the ratio of catabolic to anabolic metabolism. These data concur with the association between serum sclerostin levels and fat mass in humans 27 and suggest that circulating sclerostin allows communication between bone and adipose to coordinate the activity of the two tissues.
A paradoxical finding of increased adipogenic potential in stromal vascular cells isolated from Sost−/− mice together with the ability of sclerostin to stimulate adipogenesis of bone marrow‐derived stem cells, 28 led us to question whether sclerostin also influences APC dynamics. Using fluorescence‐activated cell sorting, we report here that sclerostin deficiency due to global or osteoblast/osteocyte‐restricted ablation of the Sost gene leads to an increased rate of stromal vascular cell proliferation and to an expansion of the PDGFRα+ APC pool in white adipose tissue depots. Lineage tracing studies indicated that the maturation of this cell population to lipid‐laden adipocytes is suppressed. These studies point to a regulatory effect of bone‐derived sclerostin on multiple aspects of adipose physiology and further confirm the linkage between bone and adipose.
2. MATERIALS AND METHODS
2.1. Generation of genetic mouse models
All procedures were performed in accordance with the NIH’s Guide for the Care and Use of Laboratory Animals and under the approval of the Johns Hopkins Medical School Animal Care and Use Committee. Sost−/− mice (Sosttm1(KOMP)Vlcg), originally created by Regeneron Pharmaceuticals, Inc using ES cell clone 10069B‐C10, were obtained from the KOMP Repository (www.komp.org/). Experimental control (Sost+/+) and Sost−/− mice were bred in the laboratory from heterozygous breeding pairs. To generate adipocyte‐ or osteoblast/osteocyte‐specific mutants, SostiCOIN mice 29 , 30 were crossed with AdipoQ‐Cre mice 31 or Ocn‐Cre mice, 32 respectively. To generate crossbred strains for lineage tracing, Sost+/− mice were first crossed with Pdgfra‐CreERT2. 33 Progeny were then crossed with ROSA mT/mG mice (Jackson Laboratories; Stock No 007676). For β‐catenin stabilization studies, Ctnnb1 Exon3flox/+ mice, 34 in which exon 3 is flanked by loxP sites, were crossed with Pdgfra‐CreERT2:mT/mG mice. To induce gene recombination in the lineage tracing studies, mice were administered 50 ng/kg BW tamoxifen by intraperitoneal (i.p.) injection on five consecutive days. Mice were euthanized and adipose tissue examined by fluorescent microscopy 2 weeks or 6 weeks (high‐fat diet feeding studies) after the last tamoxifen injection. All mutant mice and littermate controls were maintained on a C57BL/6 background. PCR analysis of tail biopsy specimens was used to confirm genotypes. Mice were housed on ventilated racks on a 14‐h light/10‐h dark cycle and fed ad libitum with a standard chow diet (Extruded Global Rodent Diet, Harlan Laboratories). For the diet‐induced obesity studies, mice were fed a 60% high‐fat diet (D12492, Research Diets) for 6 or 8 weeks.
2.2. Cell culture
Primary adipocyte precursors were harvested from the inguinal fat pads by collagenase digestion 35 and cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, Sigma‐Aldrich) and 1% Pen/Strep (Invitrogen) at 37℃ in a humidified incubator at 5% CO2. For colony‐forming studies, 5 × 103 were seeded to 100 mm tissue culture plates. For proliferation studies, stromal vascular cells cultured in six well plates were labelled for 2 h with 10 μM 5‐ethynyl‐2′‐deoxyuridine (EdU) in media containing 1% FBS. EdU staining prior to flow cytometric analysis was performed using a Base‐Click 488 EdU Flow Cytometry Kit (Sigma‐Aldrich). For differentiation studies using stromal vascular cells or flow‐sorted adipoprogenitor cells, cells were grown to confluence and then induced to differentiate by treatment with 0.5 mM 3‐isobutyl‐1‐methylxanthin (IBMX), 1 μM dexamethasone, and 167 nM insulin for 2 days, followed by continued culture in 167 nM insulin, which was changed every 2 days thereafter until analysis. Recombinant mouse sclerostin (R&D System) and recombinant mouse Wnt‐3a (R&D System) were replaced with each media change. Oil red O staining was carried out according to standard technique.
2.3. Stromal vascular cell fractionation and flow cytometry
For fluorescence‐activated cell sorting (FACS) analysis, SVFs were resuspended in red blood cell lysis buffer (BD Biosciences) for 15 min at room temperature and followed by cell surface marker staining using anti‐PDGFRa (CD140a)‐PE (rat, 1:200; BioLegend, San Diego, CA, USA), anti‐Sca1‐PE/Cy7 (rat, 1:300; BioLegend), and anti‐CD45‐APC/Cy7 (rat, 1:300; BioLegend). Cell sorting and analytic cytometry were performed using FASCAria (Becon Dickinson) and BD LSR II (BD Biosciences, San Jose, CA, USA) flow cytometers, respectively. All compensation was performed using single‐color controls in BD FACSDiVa (BD Biosciences) software at the time of acquisition. 100,000 stromal vascular cells were examined for each sample. Raw data were processed using FlowJo software (Tree Star, Ashland, OR, USA). Results of analytic cytometry are presented as the fraction of CD45−:Sca1+:PDGFRα+ per total cells analyzed.
2.4. Gene expression studies
Total RNA was extracted using TRIzol (Life Technologies). For adipose tissue, samples were centrifuged prior to RNA purification to remove excess lipid. Reverse transcriptase reactions were carried out using 1 μg of RNA and the iScript cDNA Synthesis system (Bio‐Rad). Real‐time qPCR was carried out using iQ Sybr Green Supermix (Bio‐Rad). Reactions were normalized to endogenous 18S reference transcripts. Primer sequences are shown in Table 1. Protein lysates were obtained from flow sorted PDGFRα+ APCs after pooling cells from 3 to 4 mice and used for Western blot analysis according to standard technique. Antibodies specific for active, non‐phosphorylated β‐catenin (#19807) and actin (#3700) were obtained from Cell Signal Technologies.
TABLE 1.
qPCR primers
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Acaca | CTCCCGATTCATAATTGGGTCTG | TCGACCTTGTTTTACTAGGTGC |
| Axin2 | TGACTCTCCTTCCAGATCCCA | TGCCCACACTAGGCTGACA |
| Cebpa | GCGGGAACGCAACAACATC | GTCACTGGTCAACTCCAGCAC |
| Bmp2 | GGGACCCGCTGTCTTCTAGT | TCAACTCAAATTCGCTGAGGAC |
| Bmp4 | ATTCCTGGTAACCGAATGCTG | CCGGTCTCAGGTATCAAACTAGC |
| Bmpr1 | TGGCACTGGTATGAAATCAGAC | CAAGGTATCCTCTGGTGCTAAAG |
| Bmpr2 | CCTCGGCCCAAGATCCTA | CCTAGACATCCAGAGGTGACA |
| Ctnnb1 | ATGGAGCCGGACAGAAAAGC | TGGGAGGTGTCAACATCTTCTT |
| Dkk1 | CAGTGCCACCTTGAACTCAGT | CCGCCCTCATAGAGAACTCC |
| Fabp4 | AAGGTGAAGAGCATCATAACCCT | TCACGCCTTTCATAACACATTCC |
| Fasn | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| Lpl | TTGCCCTAAGGACCCCTGAA | TTGAAGTGGCAGTTAGACACAG |
| Lrp5 | GGACAGATGTGAGCGAGGAG | GGTCCTGCCAGAAGA GAA CC |
| Lrp6 | CGGGACTTGAGATTGGTTGA | ATC CGGGGACAATAATCC AG |
| Nkd2 | GAGCGGAAGAAACGGACCG | GAACCCTTGTCGTCCCAGA |
| Pparg | GGAAGACCACTCGCATTCCTT | GTAATCAGCAACCATTGGGTCA |
| Plin1 | CTGTGTGCAATGCCTATGAGA | CTGGAGGGTATTGAAGAGCCG |
| Sost | AGCCTTCAGGAATGATGCCAC | CTTTGGCGTCATAGGGATGGT |
2.5. Metabolic phenotyping and bioassays
Serum sclerostin and insulin were measured in plasma by ELISA (Alpco). Glucose levels were measured using a Bayer Contour hand‐held glucose monitor. For glucose tolerance testing, glucose (2 g/kg BW) was injected IP after a 6‐h fast. Tissues for histological analysis were collected at necropsy, weighed, and then fixed in 4% paraformaldehyde before embedding and sectioning. Adipocyte size was assessed using ImageJ. DNA was isolated from tissues using the Quick‐DNA Universal kit (Zymo Research) following proteinase K digestion.
2.6. Histology and fluorescence microscopy
For histology, mouse adipose tissue samples were fixed in 4% formaldehyde for 24 h at 4℃ and washed with PBS. Paraffin‐embedded tissue sections were stained with H&E using standard methods. For immunofluorescence, the snap‐frozen fat tissues were sectioned at 15 μm thickness and fixed with acetone at −20℃ for 5 min and directly imaged under a fluorescent microscope (Keyence BZ‐X700).
2.7. MicroCT imaging
High‐resolution images of the mouse femur were acquired using a desktop micro‐tomographic imaging system (Skyscan 1275, Bruker) in accordance with the recommendations of the American Society for Bone and Mineral Research (ASBMR). 36 Bones were scanned at 65 keV and 153 μA using a 1.0 mm aluminum filter with an isotropic voxel size of 10 μm. Trabecular bone parameters were assessed in a region of interest 500 μm proximal to the growth plate and extending for 2 mm (200 CT slices).
2.8. Statistical analysis
All results are expressed as mean ± SEM. Statistical analyses were performed using unpaired, two‐tailed Student's t‐tests or ANOVA tests followed by post‐hoc tests using Prism Graphpad software. A p‐value <.05 was considered significant.
3. RESULTS
3.1. Adipogenic potential is increased in stromal vascular cells isolated from Sost−/− mice
In our previous work, 26 we demonstrated that the secreted glycoprotein sclerostin exerts endocrine control over the accumulation of white adipose tissue in addition to its well‐established, paracrine effects on bone formation. 18 , 19 In line with this function, male Sost−/− mice exhibited normal body weight (Figure 1A), but reductions in both the mass of white adipose depots (Figure 1B) and the expression of adipocyte markers in inguinal adipose when compared to control littermates (Figure 1C). While attempting to confirm that this phenotype was due to cell non‐autonomous effects of bone‐derived sclerostin on adipose function, we isolated stromal vascular cells via collagenase digestion 35 of the inguinal fat pads of male Sost+/+ and Sost−/− mice for in vitro adipogenesis assays. Since the Sost gene is not expressed in this tissue (Figure 1C), we expected that differentiation potential under adipogenic conditions would be similar in cells isolated from control and mutant mice. Like inguinal adipose, freshly isolated stromal vascular cells from Sost−/− mice exhibited a decrease in the mRNA levels of the adipogenic transcription factors CEBPα and PPARγ relative to controls (Figure 1D). However, stromal vascular cells isolated from male Sost−/− mice and cultured in vitro exhibited a surprising increase in differentiation relative to those from Sost+/+ mice when indexed by Oil Red O staining for lipid accumulation and qPCR analyses of adipocytic genes (Figure 1E,F). Since the stromal vascular fraction contains putative stem/progenitor cell populations, these data led us to hypothesize that the paradoxical enhancement in adipogenic potential is due to an increase in the abundance of APCs within the white adipose depots of Sost−/− mice. Indeed, stromal vascular cells from Sost−/− mice exhibited an approximately fourfold increase in colony‐forming efficiency when compared to cells from Sost+/+ mice (Figure 1G,H). Colony size (Figure 1I) was similar in Sost+/+ and Sost−/− cultures, but a more direct assessment of proliferation, using EdU incorporation, indicated a higher rate of proliferation in stromal vascular cell isolated from Sost−/− mice (Figure 1J). Thus, the increase in adipogenic differentiation potential of the stromal vascular fraction isolated Sost−/− mice appears to be secondary to an increase in APC numbers.
FIGURE 1.
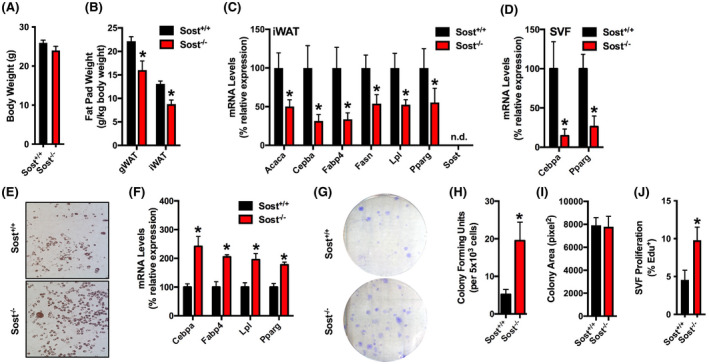
The stromal vascular fraction isolated from Sost−/− mice exhibits a paradoxical increase in adipocyte differentiation in vitro. (A) Body weight of 8‐week‐old male Sost+/+ and Sost−/− mice (n = 7 mice/genotype). (B) Gonadal (gWAT) and inguinal (iWAT) fat pad weights (n = 7 mice/genotype). (C) qPCR analysis of mRNA samples isolated from the inguinal fat pad of 8‐week‐old mice (n = 6 mice/genotype). (D) qPCR analysis of mRNA samples from stromal vascular fraction (SVF) cells freshly isolated from the inguinal fat pad (n = 4–6 mice/genotype). (E and F) In vitro differentiation of SVF cells isolated from the inguinal fat pad of 8‐week‐old male Sost+/+ and Sost−/− mice was assessed by Oil Red O staining (E) and qPCR analysis (F, n = 4–6 mice/genotype). (G–I) Colony‐forming capacity and colony size were assessed by seeding 5 × 103 SVF cells per 100 mm plate (n = 10–11 mice/genotype). (J) EdU incorporation was assessed as a marker of proliferation (n = 6–7 mice/genotype). All data are represented as mean ± SEM. *p < .05
3.2. The abundance of PDGFRα+APCs is increased in the fat pads of male Sost−/− mice
To more carefully characterize changes in the APC pool in the white adipose depots of Sost−/− mice, we isolated stromal vascular cells from the inguinal and gonadal fat pads of male Sost+/+ and Sost−/− mice and quantified APC numbers by FACS. We focused our attention on the CD45−:Sca1+:PDGFRα+ population as the descendants of PDGFRα+ stromal vascular cells give rise to nearly all mature adipocytes and also contribute to the increase in adipocyte numbers in response to an obesogenic diet. 7 , 9 Consistent with the increased colony‐forming efficiency, the percentage of CD45−:Sca1+:PDGFRα+ cells in 100,000 stromal vascular cells analyzed per mouse was markedly increased in both the inguinal and gonadal fat pads of 8‐week‐old Sost−/− mice relative to controls (Figure 2A,B). Age‐related declines in the percentage of CD45−:Sca1+:PDGFRα+ APCs were evident in both Sost+/+ and Sost−/− mice (16weeks) but the PDGFRα+ APC pool remained larger in knockouts.
FIGURE 2.
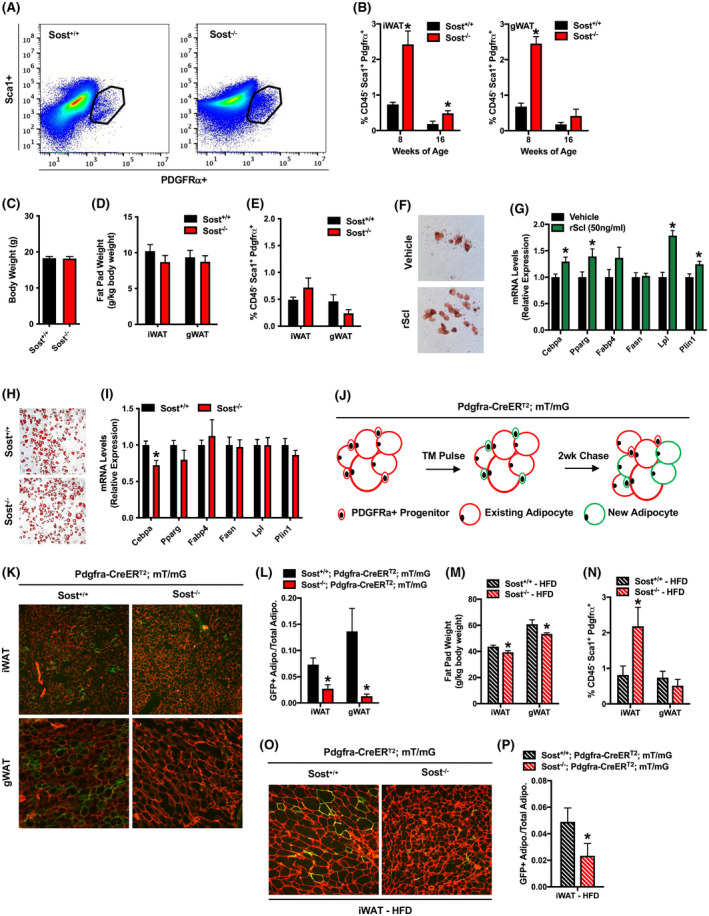
Sclerostin deficiency inhibits the differentiation of adipoprogenitor cells. (A) Representative flow cytometry plot of CD45−:Sca1+:PDGFRα+ APCs isolated from the inguinal fat pad of Sost+/+ and Sost−/− mice. (B) Quantification of CD45−:Sca1+:PDGFRα+ APCs in the inguinal (iWAT, left panel) and gonadal (gWAT, right panel) fat pads of 8‐ and 16‐week‐old male Sost+/+ and Sost−/− mice (n = 4–6 mice/genotype). (C–E) Assessment of body weight (C), fat pad weight (D) and the abundance of CD45−:Sca1+:PDGFRα+ APCs in the iWAT and gWAT of 8‐week‐old female Sost+/+ and Sost−/− mice (n = 7–9 mice/genotype). (F and G) In vitro differentiation of CD45−:Sca1+:PDGFRα+ APCs isolated from the inguinal fat pad and treated with vehicle or recombinant mouse sclerostin (rScl, 50 ng/ml) was assessed by Oil Red O staining (F) and qPCR analysis (G, n = 4 cell isolations). (H and I) In vitro differentiation of identical numbers (80,000 cells/well) of CD45−:Sca1+:PDGFRα+ APCs isolate from the inguinal fat pads of male Sost+/+ and Sost−/− mice was assessed by Oil Red O staining (H) and qPCR analysis (I, n = 3–4 cell isolations). (J) Experimental design for lineage tracing experiments. 7‐week‐old Pdgfra‐CreERT2:mT/mG mice were treated with tamoxifen on five consecutive days (TM Pulse). Tissues were harvested 14 days (2 wk Chase) after the last tamoxifen injection. (K) Fluorescent micrographs of mRFP and mGFP expression in the iWAT and gWAT of Pdgfra‐CreERT2:mT/mG mice expressing or lacking sclerostin. (L) Quantification of the ratio of mGFP+ adipocytes to total adipocytes from lineage tracing studies (n = 4–6 mice/genotype). (M and N) Fat pad weight (M) and quantification of the CD45−:Sca1+:PDGFRα+ APC pool (N) in male Sost+/+ and Sost−/− mice fed a high‐fat diet for 6 weeks (n = 8–9 mice/genotype). (O) Fluorescent micrographs of mRFP and mGFP expression in the iWAT of high‐fat diet fed mice. (P) Quantification of the ratio of mGFP+ adipocytes to total adipocytes from HFD lineage tracing studies (n = 4–6 mice/genotype). All data are represented as mean ± SEM. *p < .05
As we previously reported 26 that female Sost−/− mice exhibit the increases in bone volume and insulin sensitivity evident in male Sost mutants, but do not exhibit alterations in body composition, we also examined PDGFRα+ APC abundance in females. Consistent with our previous data, body weight (Figure 2C) and fat pad weights (Figure 2D) were comparable in female Sost+/+ and Sost−/− mice at 8 weeks of age. Likewise, the percentages of CD45−:Sca1+:PDGFRα+ APCs that were evident in iWAT and gWAT were similar in female control and knockout mice (Figure 2E). These data suggest the influence of sclerostin deficiency on PDGFRα+ APC abundance is specific to male mice.
Since recombinant sclerostin enhanced the adipogenic differentiation of CD45−:Sca1+:PDGFRα+ cells cultured under adipogenic conditions in vitro (Figure 2F,G), we predicted that the increased abundance of PDGFRα+ APCs in male Sost−/− mice was also due to an inhibition of their differentiation to mature adipocytes in vivo. To test this hypothesis, we first isolated CD45−:Sca1+:PDGFRα+ cells from 8‐week‐old Sost+/+ and Sost−/− mice and cultured identical numbers of PDGFRα+ APCs under adipogenic conditions in vitro. When PDGFRα+ APC numbers were normalized (80,000 per well) in cell cultures isolated from Sost+/+ and Sost−/− mice Oil Red O staining (Figure 2H) and the expression of adipocytic marker genes (Figure 2I) was identical (except for a small decrease in Cebpa levels in Sost−/− cultures). These data indicated that PDGFRα+ APCs from the control and knockout mice have similar differentiation potential and that the accumulation of these cells in Sost−/− mice is due to cell non‐autonomous effects on proliferation and differentiation.
We next crossed Sost+/− mice with Pdgfra‐CreERT2:mT/mG mice 33 and performed lineage tracing studies (Figure 2J). Male mice were dosed with tamoxifen on five consecutive days and the abundance of mGFP+ adipocytes derived from PDGFRα+ APCs were assessed 2 weeks later. mGFP was not detected in the absence of tamoxifen treatment or in tamoxifen‐treated mT/mG mice that lacked the Pdgfra‐CreERT2 transgene (data not shown). In Sost+/+; Pdgfra‐CreERT2:mT/mG mice, 7.3% and 13.6% of adipocytes were labeled with mGFP in the inguinal and gonadal fat pads, respectively (Figure 2K,L). Sost−/−; Pdgfra‐CreERT2:mT/mG mice exhibited a paucity of newly formed adipocytes as only 2.7% and 1.2% of adipocytes were mGFP+ in the inguinal and gonadal depots.
We performed identical analyses in mice that were fed a high‐fat diet since our previous studies suggested a protective effect of sclerostin deficiency. 26 After 8 weeks of high‐fat diet feeding (60% of kcal from fat), Sost−/− mice retained a decrease in white adipose tissue mass (Figure 2M) and an increase in the percentage of CD45−:Sca1+:PDGFRα+ APCs in the stromal vascular fraction of inguinal fat (Figure 2N). Likewise, lineage tracing indicated a suppression of PDGFRα+ APC differentiation in this depot (Figure 2O,P) after high‐fat diet feeding. The smaller increase in the pool of CD45−:Sca1+:PDGFRα+ APCs in the gonadal fat pad of chow‐fed Sost−/− mice was normalized by high‐fat diet feeding (Figure 2I) and likely reflects the differential responses of gonadal and inguinal fat depots to obesogenic signals. 8 , 13 When taken together, these data indicate that sclerostin deficiency stimulates proliferation within the stromal vascular fraction and inhibits the differentiation of PDGFRα+ APCs to mature adipocytes which leads at least in part to the reduction in white adipose mass in Sost−/− mice.
3.3. Increased Wnt/β‐catenin signaling inhibits APC differentiation in Sost−/− mice
The abundance of genetic and molecular evidence indicates that sclerostin functions as a Wnt/β‐catenin signaling antagonist in the bone microenvironment. 22 , 23 , 24 To determine whether sclerostin modulates APC differentiation via an identical mechanism, we examined Wnt‐ and BMP‐related gene expression in freshly isolated APCs. Consistent with the idea that sclerostin also acts to inhibit Wnt/β‐catenin signaling in adipose tissue, PDGFRα+ APCs isolated from Sost−/− mice exhibited significantly higher mRNA levels of Axin2 and Nkd2 (Figure 3A), two genes that are directly activated by Wnt signaling, 37 , 38 when compared to APCs isolated from Sost+/+ mice. Western blot analysis revealed that the levels of active, non‐phosphorylated β‐catenin protein were also consistently increased in APCs isolated from Sost−/− mice (Figure 3B). mRNA levels for β‐catenin, the Wnt antagonist Dkk1, and the Wnt signaling co‐receptors Lrp5 and Lrp6 were comparable in APCs isolated from Sost+/+ and Sost−/− mice (Figure 3A), suggesting that the increase in Wnt signaling activation is unlikely to be due to changes in the expression of the pathway's components. By contrast, the expressions of Bmp2, Bmp4, Bmpr1, and Bmpr2 were comparable in APCs isolated from Sost+/+ and Sost−/− mice (Figure 3C), indicating that the activators of the BMP pathway in APCs are not influenced by sclerostin deficiency. To examine the effect on Wnt signaling activation on the differentiation of PDGFRα+ APCs, cell cultures were next treated with Wnt‐3a in vitro. Wnt‐3a dose dependently inhibited the adipogenic differentiation of PDGFRα+ APCs (Figure 3D). Relative to untreated APC cultures, Wnt‐3a at a concentration of 1ng/ml was sufficient to significantly reduce the expression of Cebpa and Lpl. At higher concentrations, Wnt‐3a stimulation significantly reduced the expression of Fabp4, Fasn, and Pparg.
FIGURE 3.
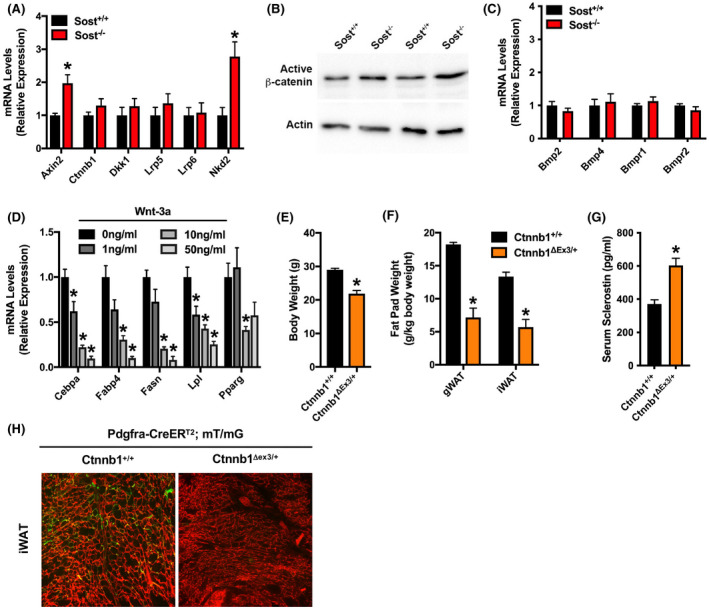
Wnt signaling inhibits APC differentiation. (A) qPCR analysis of Wnt‐related gene expression in CD45−:Sca1+:PDGFRα+ APCs (n = 6 mice/genotype). (B) Western blot analysis of active, non‐phosphorylated β‐catenin in APCs. Analysis for two lysates is shown for each genotype. (C) qPCR analysis of BMP‐related gene expression in CD45−:Sca1+:PDGFRα+ APCs (n = 6 mice/genotype). (D) Adipogenic gene expression in CD45−:Sca1+:PDGFRα+ APCs treated with 0–50 ng/ml recombinant mouse Wnt‐3a (n = 6/treatment group). (E and F) Body weight (E) and fat pad weight (F) in Pdgfra‐CreERT2:mT/mG mice that contain wildtype Ctnnb1 alleles (Ctnnb1+/+) or a Cre‐inducible, constitutively active mutant allele (Ctnnb1Δex3/+) (n = 5–7 mice/genotype). (G) Serum sclerostin levels measured by ELISA in Ctnnb1+/+ and Ctnnb1Δex3/+ mice (n = 5–7 mice/genotype). (H) Representative fluorescent micrographs of mRFP and mGFP expression in the iWAT from lineage tracing studies with Ctnnb1Δex3/+; Pdgfra‐CreERT2:mT/mG and control littermates (n = 5–7 mice/genotype). All data are represented as mean ± SEM. *p < .05
To ensure that β‐catenin activation is sufficient to inhibit APC differentiation and determine whether it phenocopies the effect of sclerostin deficiency, we next examined fat mass and performed lineage tracing studies with Pdgfra‐CreERT2:mT/mG mice that also contain one allele of Ctnnb1 with loxP sites flanking exon 3. 34 Exon 3 of Ctnnb1 encodes the serine and threonine residues phosphorylated by GSK‐3β and Cre‐mediated deletion (Ctnnb1Δex3/+) in this model results in the expression of a constitutively active mutant. Two weeks after tamoxifen administration, Ctnnb1Δex3/+ mice exhibited significant reductions in both body weight (Figure 3E) and white adipose depot mass (Figure 3F) but significantly increased serum sclerostin levels (Figure 3G) when compared to littermates with wildtype Ctnnb1 alleles. Lineage tracing revealed that these rapid changes in body composition in Ctnnb1Δex3/+ mice were accompanied by a near complete inhibition of new adipogenesis (Figure 3H). Thus, the activation of Wnt/β‐catenin signaling is sufficient to alter PDGFRα+ APCs dynamics and adipose accumulation and is likely to contribute to the inhibition of APC differentiation in the context of sclerostin deficiency.
3.4. Bone‐derived sclerostin regulates fat mass and APC differentiation
While sclerostin expression is not detectable in white adipose tissue (Figure 1B), 26 its expression has been detected outside of bone during development. 39 , 40 Therefore, to ensure that bone‐derived sclerostin contributes to the regulation of fat mass and APC differentiation, we ablated the expression of the Sost gene in mature osteoblasts and osteocytes. Sost conditional loss of function mice (SostiCOIN/iCOIN) 29 , 30 were crossed with Ocn‐Cre mice 32 to generate control and osteoblast/osteocyte‐specific knockouts (referred to hereafter as ObΔSost), which was confirmed by allele‐specific PCR (Figure 4A). At 12 weeks of age, male ObΔSost mice exhibited a 76% reduction in serum sclerostin levels (Figure 4B) and the expected increase in bone volume (Figure 4C,D) when compared to controls. The mutant mice also phenocopied many of the metabolic changes evident in Sost−/− mice. 26 Despite normal body weight when fed either a chow or high‐fat diet (Figure 4E), white adipose tissue mass was significantly reduced in ObΔSost mice (Figure 4F). Inguinal adipocytes were noticeably smaller than those in control littermates (Figure 4G) and exhibited an increase in the abundance of multi‐locular adipocytes characteristic of white adipose tissue beiging. Likewise, ObΔSost mice exhibited improvements in glucose homeostasis (Figure 4H–K) evident by reductions in serum insulin levels and increased glucose tolerance relative to control. Most importantly, ObΔSost mice exhibited the expansion of the CD45−:Sca1+:PDGRFα+ APC pool observed in global knockouts (Figure 4L,M). By contrast, the disruption of the Sost gene in adipose tissue (AdΔSost, Figure 4N) by crossing SostiCOIN/iCOIN mice with AdipoQ‐Cre 31 had no effect on serum sclerostin levels, body weight, fat pad weight, or the percentage of CD45−:Sca1+:PDGRFα+ APCs in inguinal fat (Figure 4O–R). Thus, sclerostin released by mature osteoblasts and osteocytes contributes to the regulation of APC numbers and likely differentiation.
FIGURE 4.
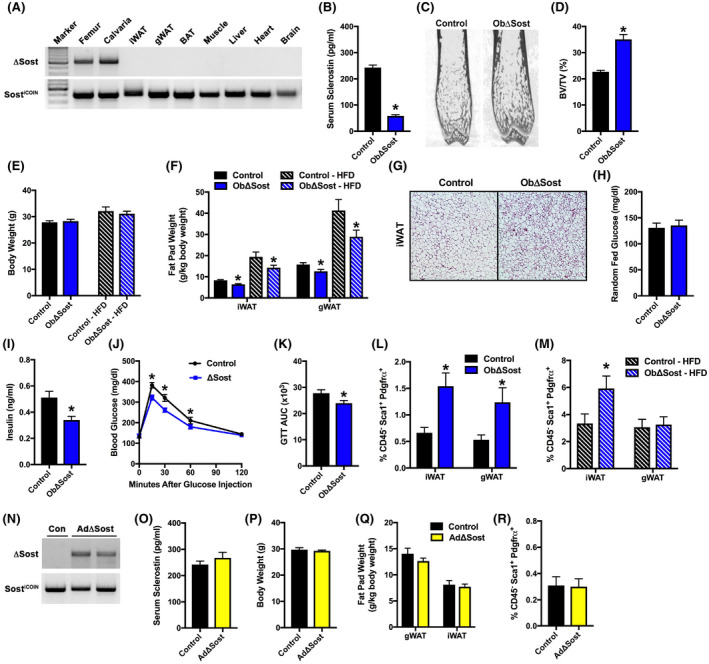
Osteoblast/osteocyte‐derived sclerostin regulates PDGFRα+ adipoprogenitor cell behavior. (A) PCR analysis of Sost gene recombination in tissues of male, 12‐week‐old osteoblast‐specific Sost knockout (ObΔSost) mice. (B) Serum sclerostin levels in male, 12‐week‐old control and osteoblast/osteocyte‐specific Sost knockout (ObΔSost) mice (n = 6–8 mice/genotype). (C and D) Representative microCT images and quantification of bone volume per tissue volume in the distal femur of control and ObΔSost mice (n = 8–9 mice/genotype). (E) Body weight of control and ObΔSost mice fed a chow or high‐fat diet (HFD, 8 weeks on diet) (n = 9–13 mice/genotype). (F) Gonadal (gWAT) and inguinal (iWAT) fat pad weight (n = 9–13 mice/genotype). (G) Representative iWAT histological sections. 10× magnification. (H and I) Random fed blood glucose and serum insulin levels (n = 6–9 mice/genotype). (J and K) Glucose tolerance (GTT) and area under the curve (AUC) analysis (n = 8–10 mice/genotype). (L and M) Quantification of CD45−:Sca1+:PDGFRα+ adipoprogenitors in iWAT of chow‐fed (L) and HFD‐fed (M) mice (n = 9–13 mice/genotype). (N) PCR analysis of Sost gene recombination in the iWAT of male, 12‐week‐old control (Con) and adipocyte‐specific Sost knockout (AdΔSost) mice. (O) Serum sclerostin levels (n = 7–8 mice/genotype). (P) Body weight (n = 7–8 mice/genotype). (Q) Gonadal and inguinal fat pad weight (n = 7–8 mice/genotype). (R) Quantification of CD45−:Sca1+:PDGFRα+ adipoprogenitors in iWAT (n = 7–8 mice/genotype). All data are represented as mean ± SEM. *p < .05
4. DISCUSSION
Sclerostin‐mediated inhibition of Wnt/β‐catenin signaling is most closely associated with the regulation of bone acquisition. Hormonal or genetic suppression of Sost gene expression leads to rapid bone growth, while increased Sost expression represses it. 18 , 19 , 41 , 42 In our previous work, 25 , 26 we demonstrated that sclerostin also exerts endocrine actions that influence adipose tissue accumulation. Relative to wildtype littermates, Sost−/− mice, and those lacking the Sost gene specifically in mature osteoblasts and osteocytes as shown herein, exhibit a reduction in fat mass with small adipocyte. We reasoned that this phenotype was the result of a change in the ratio of anabolic to catabolic metabolism as de novo lipid synthesis was reduced and fatty acid oxidation in white adipose was increased. 26
In this work, we identify a second mechanism by which sclerostin influences the accumulation of white adipose tissue in male mice. Despite a lack of sclerostin expression in adipose, we found that the stromal vascular fraction isolated from male Sost−/− mice exhibited an increase in differentiation potential relative to that isolated from Sost‐expressing mice when cultured in vitro under conditions in which sclerostin is present in the serum. This paradoxical finding, given the reduction in fat mass in Sost−/− mice and reduced expression of adipocytic genes in these mice in vivo, led us to hypothesize that sclerostin influences the commitment of a stem/progenitor cell population present in the stromal vascular compartment of white adipose tissue. Consistent with this idea, flow cytometric studies revealed an accumulation of PDGFRα+ APCs, cells that represent a major source for new adipocytes, 7 , 9 in the inguinal and gonadal fat pads of Sost−/− mice. We observed a similar increase in the abundance of PDGFRα+ APCs in the fat pads of ObΔSost mice, but not when the Sost gene was ablated in mature adipocytes. These genetic studies indicate that bone‐derived sclerostin regulates PDGFRα+ APCs abundance via an endocrine mechanism. It remains possible that other adipoprogenitor cell pools, like PDGFRβ+ progenitors that give rise to white and beige adipocytes, 10 , 11 , 12 are also influenced by serum sclerostin levels. Such studies would require the development of genetic models to track PDGFRβ+ cell fate.
Interestingly, we found that the effect of sclerostin deficiency on PDGFRα+ APCs behavior is sexually dimorphic. While male Sost−/− mice exhibited an increase in APC abundance, this was not the case in female knockouts. These data likely explain the difference in the body composition phenotypes of male and female Sost−/− mice. 26 Both male and female knockouts exhibit increased bone mass, improve insulin sensitivity, and reduce adipocyte hypertrophy, but female Sost−/− mice do not exhibit the overall reduction in visceral and subcutaneous fat mass that is evident in males. To maintain normal fat mass in the face of reduced adipocyte hypertrophy, female Sost−/− mice must have a greater number of adipocytes in white adipose tissue depots than wildtype littermates. Identifying the underlying mechanism for this sexual dimorphism will require additional work, but estrogen signaling has been shown to strongly regulate adipocyte commitment 43 and it is possible that the sex hormone has more potent effects on differentiation than sclerostin deficiency.
Before differentiating to mature lipid‐laden adipocytes, adipoprogenitors exhibit a transient period of proliferation. 8 In the mixed population of stromal vascular cells isolated from Sost−/− mice, the rate of EdU incorporation indicative of cellular proliferation could account for the increase in APC numbers and increased colony‐forming capacity relative to cells isolated from Sost+/+ mice. Indeed, studies examining sclerostin's effects on bone acquisition have noted that its suppression increases osteoprogenitor proliferation. 44 However, our data suggest that the accumulation of PDGFRα+ APCs in the absence of endocrine sclerostin is also due to a block on their differentiation. Lineage tracing studies revealed that the development of new adipocytes in the inguinal and gonadal fat pads after the activation of the CreERT2 enzyme in PDGFRα+ cells was greatly reduced in mice deficient for sclerostin. A similar inhibition of PDGFRα+ APC differentiation and increased abundance of PDGFRα+ APC was evident in the inguinal fat pad of Sost−/− mice fed an obesogenic high‐fat diet. Still further support for this interpretation can be drawn from the similar differentiation potential of PDGFRα+ APC when similar numbers of flow‐sorted cells are differentiated in vitro and the ability of recombinant sclerostin to enhance PDGFRα+ APCs adipogenesis in vitro as well as the protein's ability to promote the differentiation of 3T3‐L1 adipocytes as described by others. 45
While interpreting our lineage tracing studies, it is important to note that tamoxifen has been reported to induce transient adipocyte necrosis that is followed by de novo adipogenesis. 46 As a result, the levels of adipogenesis observed in our lineage tracing studies are likely to be higher than the basal level of turnover. We have no reason to suspect that sclerostin deficiency or the expression of a constitutively active β‐catenin in adipose should influence the metabolism of tamoxifen or the susceptibility of adipocytes to tamoxifen‐induced necrosis.
As indicated above, sclerostin primarily affects bone formation by antagonizing the osteo‐anabolic Wnt/β‐catenin signaling pathway. 23 Likewise, we demonstrated previously that markers of Wnt signaling are elevated in the white adipose tissue of Sost−/− mice and mice that lack the Lrp4 receptor, which facilitates sclerostin's inhibition of Wnt signaling, in adipocytes. 25 , 26 Here, we found that indicators of activated Wnt signaling, including increases in expression of Axin2 and Nkd2 and the abundance of active, non‐phosphorylated β‐catenin protein, are also elevated in PDGFRα+ APCs isolated from Sost−/− mice and that Wnt‐3a inhibits the differentiation of this cell population to adipocytes. These data are consistent with the effect of expressing a constitutively active β‐catenin mutant in PDGFRα+ cells on adipogenesis in vivo in our lineage tracing studies, as well as the known inhibitory effects of Wnt signaling on adipogenesis. 47 , 48 Thus, sclerostin deficiency likely regulates the behavior of mature adipocytes and APCs via a similar mechanism. We also examined the expression of mediators of BMP signaling in APCs because we reported 26 that the levels of SMAD1/5/9 phosphorylation are reduced in the white adipose tissue of Sost−/− mice and that rScl increased the expression of BMP4 and the activation of its signaling pathway in primary adipocytes. The expression of these genes was not altered in APCs isolated from Sost−/− mice, but we cannot rule out the possibility that reductions in BMP production by mature adipocytes influence the differentiation of APCs in vivo.
In our examination of APCs by FACs, the fraction of PDGFRα+ APCs among SVF cells decreased with age in both Sost+/+ and Sost−/− mice. Age‐related declines in stem/progenitor cell abundance and differentiation potential are common features in many tissues and occur via a variety of mechanisms, including changes in asymmetrical division, alterations in the niche, and the development of cellular senescence. 49 , 50 However, it is notable that the abundance of PDGFRα+ APCs remained higher in the inguinal adipose of Sost−/− mice than those expressing sclerostin. This phenotype may explain why the body composition phenotype in these mutants is more pronounced with age 26 and have implications for the improved metabolic phenotype of Sost−/− mice. Gao and colleagues 51 recently demonstrated that the acceleration of APC aging via the manipulation of telomerase predisposes mice to the development of metabolic dysfunction. The maintenance of APCs numbers in Sost−/− mice as they age may therefore provide protection against metabolic insults like high‐fat diet feeding. The increase abundance of beige adipocytes observed previously in the white adipose depots of Sost−/− mice 26 and in the inguinal adipose of ObΔSost mice also likely contributes to this protective effect and is currently being examined in our laboratory.
Recent studies have indicated that sclerostin also contributes to the regulation of adipogenesis in the bone marrow. Fairfield and colleagues 28 demonstrated that recombinant sclerostin protein enhanced adipogenesis of marrow stromal cells, while marrow adiposity is decreased in Sost−/− mice and can be reduced by the administration of a sclerostin neutralizing antibody in wildtype mice. Likewise, Balani 52 reported that sclerostin neutralization increases the abundance of Sox9CreER+ skeletal progenitors and prevents their differentiation to marrow adipocytes. We attempted to use identical strategies to determine whether PDGFRα+ cells form bone marrow adipocyte and whether impairments in their differentiation contribute to the reduction in bone marrow adipocytes evident in Sost−/− mice, but the broad expression of the Pdgfra‐CreERT2 transgene in bone marrow prevented these analyses.
In summary, these studies extend the effects of bone‐derived sclerostin from the mature adipocyte to the adipoprogenitor cells at least in male mice. Thus, sclerostin regulates the accumulation of white adipose tissue by influencing lipid accumulation as well as the differentiation of the progenitor cells responsible for adipose tissue maintenance. These data help to explain the associations between serum sclerostin levels and fat mass in humans and will likely have implications for the use of sclerostin neutralizing therapeutics. It is possible that this strategy may have benefits in the treatment of metabolic disorders as well as the intended effects on low bone mass.
DISCLOSURES
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization: Ryan C. Riddle; Methodology, Soohyun P. Kim, Lei Wang, and Mei Wan; Investigation: Soohyun P. Kim, Hao Da, Lei Wang, and Ryan C. Riddle; Critical Resources: Makoto M. Taketo; Writing, Soohyun P. Kim and Ryan C. Riddle; Funding Acquisition, Ryan C. Riddle; Supervision, Ryan C. Riddle.
ACKNOWLEDGMENTS
We are grateful for the assistance of staff from the Johns Hopkins University School of Medicine, Division of Hematology, Flow Cytometry Core Facility. We thank Dr Dwight Bergles of The Solomon H. Snyder Department of Neuroscience at the Johns Hopkins University School of Medicine for providing Pdgfra‐CreERT2 mice and Dr Aris Economides of Regeneron for providing the SostiCOIN mice. This work was supported by a Merit Review Award from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development (BX003724, RCR) and a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (DK099134, RCR).
Kim SP, Da H, Wang L, Taketo MM, Wan M, Riddle RC. Bone‐derived sclerostin and Wnt/β‐catenin signaling regulate PDGFRα+ adipoprogenitor cell differentiation. FASEB J. 2021;35:e21957. doi: 10.1096/fj.202100691R
REFERENCES
- 1. Hirsch J, Batchelor B. Adipose tissue cellularity in human obesity. Clin Endocrinol Metab. 1976;5:299‐311. [DOI] [PubMed] [Google Scholar]
- 2. Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19:1338‐1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nguyen NT, Magno CP, Lane KT, Hinojosa MW, Lane JS. Association of hypertension, diabetes, dyslipidemia, and metabolic syndrome with obesity: findings from the National Health and Nutrition Examination Survey, 1999 to 2004. J Am Coll Surg. 2008;207:928‐934. [DOI] [PubMed] [Google Scholar]
- 4. Wormser D, Kaptoge S, Di Angelantonio E, et al. Separate and combined associations of body‐mass index and abdominal adiposity with cardiovascular disease: collaborative analysis of 58 prospective studies. Lancet. 2011;377:1085‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783‐787. [DOI] [PubMed] [Google Scholar]
- 6. Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240‐249. [DOI] [PubMed] [Google Scholar]
- 7. Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol. 2013;15:302‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot‐specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. 2015;17:376‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee YH, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by beta3‐adrenoceptor activation and high‐fat feeding. Cell Metab. 2012;15:480‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gupta RK, Mepani RJ, Kleiner S, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15:230‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vishvanath L, MacPherson KA, Hepler C, et al. Pdgfrbeta+ mural preadipocytes contribute to adipocyte hyperplasia induced by high‐fat‐diet feeding and prolonged cold exposure in adult mice. Cell Metab. 2016;23:350‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang W, Zeve D, Suh JM, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jeffery E, Wing A, Holtrup B, et al. The adipose tissue microenvironment regulates depot‐specific adipogenesis in obesity. Cell Metab. 2016;24:142‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gupta RK, Arany Z, Seale P, et al. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464:619‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hammarstedt A, Hedjazifar S, Jenndahl L, et al. WISP2 regulates preadipocyte commitment and PPARgamma activation by BMP4. Proc Natl Acad Sci U S A. 2013;110:2563‐2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeve D, Seo J, Suh JM, et al. Wnt signaling activation in adipose progenitors promotes insulin‐independent muscle glucose uptake. Cell Metab. 2012;15:492‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J, Farmer SR. Regulating the balance between peroxisome proliferator‐activated receptor gamma and beta‐catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation‐defective mutant of beta‐catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279:45020‐45027. [DOI] [PubMed] [Google Scholar]
- 18. Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860‐869. [DOI] [PubMed] [Google Scholar]
- 19. van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte‐expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xiong L, Jung JU, Wu H, et al. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc Natl Acad Sci U S A. 2015;112:3487‐3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chang MK, Kramer I, Huber T, et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci U S A. 2014;111:E5187‐E5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bourhis E, Wang W, Tam C, et al. Wnt antagonists bind through a short peptide to the first beta‐propeller domain of LRP5/6. Structure. 2011;19:1433‐1442. [DOI] [PubMed] [Google Scholar]
- 23. Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883‐19887. [DOI] [PubMed] [Google Scholar]
- 24. Ellies DL, Viviano B, McCarthy J, et al. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738‐1749. [DOI] [PubMed] [Google Scholar]
- 25. Kim SP, Da H, Li Z, et al. Lrp4 expression by adipocytes and osteoblasts differentially impacts sclerostin's endocrine effects on body composition and glucose metabolism. J Biol Chem. 2019;294:6899‐6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim SP, Frey JL, Li Z, et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci U S A. 2017;114:E11238‐E11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urano T, Shiraki M, Ouchi Y, Inoue S. Association of circulating sclerostin levels with fat mass and metabolic disease—related markers in Japanese postmenopausal women. J Clin Endocrinol Metab. 2012;97:E1473‐E1477. [DOI] [PubMed] [Google Scholar]
- 28. Fairfield H, Falank C, Harris E, et al. The skeletal cell‐derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol. 2018;233:1156‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Economides AN, Frendewey D, Yang P, et al. Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc Natl Acad Sci U S A. 2013;110:E3179‐E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yee CS, Manilay JO, Chang JC, et al. Conditional deletion of Sost in MSC‐derived lineages identifies specific cell‐type contributions to bone mass and B‐cell development. J Bone Miner Res. 2018;33:1748‐1759. [DOI] [PubMed] [Google Scholar]
- 31. Eguchi J, Wang X, Yu S, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang M, Xuan S, Bouxsein ML, et al. Osteoblast‐specific knockout of the insulin‐like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005‐44012. [DOI] [PubMed] [Google Scholar]
- 33. Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta‐catenin gene. EMBO J. 1999;18:5931‐5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Negrel R, Dani C. Cultures of adipose precursor cells and cells of clonal lines from animal white adipose tissue. Methods Mol Biol. 2001;155:225‐237. [DOI] [PubMed] [Google Scholar]
- 36. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res. 2010;25:1468‐1486. [DOI] [PubMed] [Google Scholar]
- 37. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta‐catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yan D, Wiesmann M, Rohan M, et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta ‐catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98:14973‐14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Bezooijen RL, Deruiter MC, Vilain N, et al. SOST expression is restricted to the great arteries during embryonic and neonatal cardiovascular development. Dev Dyn. 2007;236:606‐612. [DOI] [PubMed] [Google Scholar]
- 40. Weivoda MM, Youssef SJ, Oursler MJ. Sclerostin expression and functions beyond the osteocyte. Bone. 2017;96:45‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866‐5875. [DOI] [PubMed] [Google Scholar]
- 43. Lapid K, Lim A, Clegg DJ, Zeve D, Graff JM. Oestrogen signalling in white adipose progenitor cells inhibits differentiation into brown adipose and smooth muscle cells. Nat Commun. 2014;5:5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boyce RW, Brown D, Felx M, et al. Decreased osteoprogenitor proliferation precedes attenuation of cancellous bone formation in ovariectomized rats treated with sclerostin antibody. Bone Rep. 2018;8:90‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ukita M, Yamaguchi T, Ohata N, Tamura M. Sclerostin enhances adipocyte differentiation in 3T3‐L1 cells. J Cell Biochem. 2016;117:1419‐1428. [DOI] [PubMed] [Google Scholar]
- 46. Ye R, Wang QA, Tao C, et al. Impact of tamoxifen on adipocyte lineage tracing: Inducer of adipogenesis and prolonged nuclear translocation of Cre recombinase. Mol Metab. 2015;4:771‐778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bennett CN, Ross SE, Longo KA, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. 2002;277:30998‐31004. [DOI] [PubMed] [Google Scholar]
- 48. Longo KA, Wright WS, Kang S, et al. Wnt10b inhibits development of white and brown adipose tissues. J Biol Chem. 2004;279:35503‐35509. [DOI] [PubMed] [Google Scholar]
- 49. Shefer G, Van de Mark DP, Richardson JB, Yablonka‐Reuveni Z. Satellite‐cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Dev Biol. 2006;294:50‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pinho S, Frenette PS. Haematopoietic stem cell activity and interactions with the niche. Nat Rev Mol Cell Biol. 2019;20:303‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao Z, Daquinag AC, Fussell C, et al. Age‐associated telomere attrition in adipocyte progenitors predisposes to metabolic disease. Nat Metab. 2020;2:1482‐1497. [DOI] [PubMed] [Google Scholar]
- 52. Balani DH, Trinh S, Xu M, Kronenberg HM. Sclerostin antibody administration increases the numbers of Sox9creER+ skeletal precursors and their progeny. J Bone Miner Res. 2021;36:757‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]