

Abstract
Elastin-like polypeptides (ELP), an increasingly popular tag for protein purification, commonly rely upon inverse transition cycling (ITC) to exploit their lower critical solution temperature characteristics for purification. While considerably faster than chromatography, ITC is still time consuming and often fails to remove host cell contaminants to an acceptable level for in vivo experiments. Here, we present a rapid purification workflow for ELP of broadly varying molecular weight and sequence using a polar organic solvent extraction and precipitation strategy. Four different ELP purification methods were directly compared for their ability to remove host cell protein, nucleic acids, and lipopolysaccharide (LPS) contaminants using a model ELP. On the basis of these findings, an optimized extraction–precipitation method was developed that gave highly pure ELP from bacterial pellets in approximately 2.5 h while removing major host cell contaminants, including LPS to levels below 1 EU/mL, to produce highly pure material that is suitable for in vivo applications. Application of this method to the rapid purification of an ELP–epidermal growth factor fusion gave an isolate that retained its capacity to bind to epidermal growth factor receptor positive cells, thereby demonstrating that this method is capable of producing a functional construct after purification by organic extraction–precipitation.
Graphical Abstract

INTRODUCTION
Approval of the first recombinant peptide therapeutic in 1982—Humulin (human insulin)—by the U.S. Food and Drug Administration (FDA) marked a turning point that led to a new generation of biotherapeutic agents.2 There are now over 239 FDA-approved recombinant proteins in clinical use that account for approximately 10% of the entire pharmaceutical market.3,4 Much of this progress can be attributed to the development of various heterologous expression systems, each with its own unique set of advantages.2,5 Bacterial systems, specifically Escherichia coli (E. coli), are very attractive from the standpoint of industrial scalability, cost effectiveness, rapid cell growth, and ease of genetic and biochemical manipulations.2,6,7 In the field of protein/peptide biopharmaceutics, purification places a significant financial burden on developmental efforts, accounting for 45–92% of the total cost of manufacturing.2 Purification of recombinant proteins from E. coli generally entails the removal of host cell proteins (HCP), nucleic acids, lipopolysaccharides (LPS), and various other cellular metabolites. Of particular concern are LPS, major components of the E. coli cell membrane that have proven difficult to remove.8 Incomplete removal of LPS can elicit potent inflammatory responses in vivo that can be fatal.9 LPS can also alter cell-signaling pathways in vitro, leading to spurious assay results.10 Consequently, most recombinant proteins purified from E. coli require stringent LPS removal step(s) to concentrations below 1 EU/mL (0.1 ng/mL) for biological applications.11
Affinity tags such as polyhistidine (His-tag), glutathione-S-transferase (GST-tag), maltose-binding protein (MBP-tag), or FLAG-tag are commonly used in an affinity chromatography format for protein purification.6,12 They are designed for single-step, bind–wash–elute protocols; however, they generally suffer from high cost, demanding operations, low overall yield, coelution of HCP, and scaling difficulties.7,13 Due to these issues, low-cost nonchromatographic purification tags with high selectivity are still sought. Elastin-like polypeptides (ELP), composed of repeating pentapeptide VPGXG sequences (where X is a “guest residue” that can be any amino acid except proline), have emerged as a viable solution to this challenge. ELP can phase separate and coacervate above their inverse transition temperature (Tt) but resolubilize at temperatures below Tt.14,15 This reversible solubility property is retained even after ELP are fused to other proteins, thus aiding in their purification by the inverse transition cycling (ITC) process.16,17 ITC involves alternate cycles of heating above Tt to precipitate the ELP construct, removal of the contaminants in the supernatant (hot spin), solubilizing in cold buffer, and then centrifuging below Tt to precipitate remaining contaminants.18
In addition to this unique purification approach, the biocompatibility, low immunogenicity, and biodegradable properties of ELP have led to their use in drug delivery19–22 and tissue engineering23 applications. The ELP tag can be removed after purification by incorporating self-cleaving intein tags or retained, often with minimal effect on protein function.24,25 ITC has emerged as an alternative to traditional purification by chromatography; however, it is still time consuming because it often requires at least 3–5 cycles to bring HCP levels below the threshold needed for in vivo applications. Each cycle is accompanied by target protein loss and is frequently unable to remove other macromolecular contaminants such as nucleic acids and LPS without additional purification steps.26–28 Although an additional precipitation of nucleic acids with polyethylenimine is frequently used,14 limitations have been reported when working with acidic ELP constructs.18,29–31 The slowest and most labor-intensive aspect of ITC is the preparation of clarified lysate, particularly on a large scale where resuspension, complete lysis, and clarification can take hours. In addition, quick screening for expression is difficult across multiple conditions unless the fusion protein is easily and reliably detectable in the crude lysate.
Previous work overcame these issues by using an organic solvent extraction method to selectively solubilize ELP based on their intrinsic hydrophobic nature. Complete lysis along with removal of >95% of HCP and nucleic acids was accomplished within 30 min directly from whole cell E. coli.1 Further optimization showed lysis could be achieved through a vortex mixing of the cell pellet with organic solvent to afford near instantaneous isolation of ELP. This approach proved invaluable for optimizing protein expression conditions for a series of related ELP as well as an ELP fusion protein. Beyond expression analysis applications, however, removal of the organic solvent could be problematic. In addition, SDS-PAGE analysis of the extracts revealed the presence of low molecular weight contaminants (LMWC; ≤5 kDa) that were not effectively removed without further purification efforts.
To circumvent these limitations, we report a newly developed purification method by coupling organic solvent extraction to acetonitrile (ACN) precipitation. Compared to the previously reported method, this approach provides three key benefits: (1) faster removal of polar organic solvent, (2) greatly reduced LMWC levels, and (3) enhanced LPS removal. We found that LPS could be further lowered below 1 EU/mL along with removal of LMWC by performing a final polishing round of ITC after organic solvent extraction–precipitation, whereas samples processed by ITC alone typically had approximately 75-fold higher LPS content.
MATERIALS AND METHODS
Elastin-Like Polypeptide Gene Synthesis.
The following ELP constructs (VPGXG)n (where n represents the number of pentapeptide repeats and X represents the guest residue) were used for method development and validation.
Acidic: X = V/I/E [1:3:1]; n = 40 (A40), 80 (A80), 160 (A160).
Neutral: X = V; n = 40 (N40).
Basic: X = V/H/G/A [1:2:1:1]; n = 40 (B40).
Genes that express these protein constructs were assembled in a modified pET28a vector before being transferred to pET32a for expression. Briefly, the modified pET28a vector was generated from pET28a-aECM1 linearized with XbaI and XhoI.32 Purified vector was ligated to a linker sequence formed by annealing complementary oligonucleotides (Table S1) that encoded the Shine–Dalgarno ribosomal binding site [5′-AGGAGG-3′], sites for the type II restriction enzymes BseRI and AcuI, and complementary 3′ overhangs for XbaI and XhoI restriction sites. Complementary oligonucleotides encoding pentameric subunits of the ELP constructs with 2 bp 3′ overhangs (Table S1) were then annealed and phosphorylated before ligation to BseRI-digested and dephosphorylated modified pET28a via the complementary 3′ overhangs. ELP constructs were elongated to the sizes described above through successive rounds of plasmid reconstruction by recursive directional ligation (PRe-RDL), which doubles the ELP length in each step, with AcuI/BglI and BseRI/BglI restriction enzymes.33 The final constructs were then removed with XhoI and NdeI and ligated into pET32a at complementary sites for expression. All complementary oligonucleotides were annealed at 95 °C for 5 min in 100 mM sodium phosphate, 150 mM NaCl, and 1 mM EDTA and allowed to cool to room temperature over 1 h.
N24-EGF was synthesized by inserting a 169 bp sequence encoding for the protein murine epidermal growth factor, NSYPGCPSSYDGYCLNGGVCMHIESLDSYTCNCVIGYSGDRCQTRDLRWWELRGY, into a pET-25b(+) plasmid containing an ELP sequence of (VPGVG)24 as described elsewhere.1 Molecular weights were confirmed by MALDI-MS and isoelectric points, and GRAVY values were determined by ProtParam (Table S2). All molecular biology manipulations were completed according to standard protocols.34
Protein Expression.
All media for bacterial cell culture was autoclaved prior to use and contained an appropriate antibiotic. All protein expressions were performed using BL21(DE3) E. coli as previously reported.1 Briefly, BL21 cells were transformed using Zymo Mix & Go. Colonies were isolated and grown in 5 mL of Terrific Broth (TB) media with 100 μg/mL ampicillin overnight. This culture was used to inoculate a primary culture (1:1000 dilution) of 333 mL of Terrific Broth in a 2 L Erlenmeyer flask, grown at 37 °C for 24 h with shaking at 250 rpm. For the ELP fusion and B40, an induction protocol was used, allowing cells to grow to an optical density of 0.6–0.8 before inducing with 1.2 mM and 0.6 mM of IPTG for N24-EGF and B40, respectively, for 4 h at 37 °C. Cells were collected by centrifugation at 6000g for 15 min and stored at −20 °C until use.
Cell Lysis.
An E. coli cell pellet was thawed at room temperature and then resuspended in 1× phosphate-buffered saline (PBS) using a volume four times the pellet’s weight. The slurry was then incubated with lysozyme at a concentration of 0.1 mg/mL for 1 h at room temperature. It was then probe tip sonicated (Vibra-Cell, Sonics & Materials) for 30 min at a 30% duty cycle on ice. Centrifugation at 13 000g for 1 h was used to clarify the sample.
Inverse Transition Cycling.
Samples were heated to 37 °C, and a saturated (NH4)2SO4 solution was added to a final concentration of 20% v/v. The solution was centrifuged at 15 000g for 20 min at 40 °C (hot spin). The supernatant was removed, and the hot spin pellet (HSS1) was then resuspended in 1× PBS on ice. The resuspended hot spin pellet (HSP1) was cooled to 4 °C and centrifuged at 15 000g for 15 min at 4 °C (cold spin). The cold spin supernatant (CSS1) containing the protein was decanted into a new tube. The number written after “HSP” or “CSS” represents the cycle at which the sample was collected. For A160, 6 M HCl and saturated (NH4)2SO4 solutions were added such that the final HCl concentration was 20 mM.35
Organic Extraction.
An E. coli cell pellet was briefly thawed at room temperature, then extracted with a polar organic solvent four times the bacterial pellet weight (v/w), and vortexed for 1 min. The resulting slurry was centrifuged (8000g, 15 min) to remove the insoluble cell debris, and the supernatant was collected by decanting/pipetting into a new tube. If not followed by back extraction or precipitation, the organic solvent was removed by evaporation under gentle air flow. The protein film was resuspended in 1× PBS (0.4 times the pellet weight, v/w) and centrifuged at 8000g for 15 min to remove insoluble contaminants. Twenty-eight different pure solvents or solvent blends (1:1) (Table S3) were tested for their ability to extract pure protein.
Liquid–Liquid Back Extraction.
Water (4 times pellet weight) and then ethyl acetate (twice the amount of water) were added to the polar organic solvent obtained after extraction. The solution was briefly mixed, vented, and centrifuged at 1000g for 5 min to aid phase separation. The top layer was removed using a serological pipet, while the bottom was evaporated and resuspended in 1× PBS (0.4 times the pellet weight, v/w). This solution was recentrifuged at 8000g for 15 min to remove any insoluble contaminants.
Acetonitrile Precipitation and Clarification Spin.
After organic extraction, protein in the polar organic solvent was precipitated by adding 100% acetonitrile (or acetone) until its final composition was 70% v/v. After gentle mixing, the sample was centrifuged at 15 000g for 20 min. The supernatant was removed, and the protein was resuspended in 1× PBS (0.4 times the pellet weight, v/w). After resuspension, a clarification spin was performed to remove any insoluble debris (8000g, 15 min).
SDS-PAGE.
Proteins were electrophoresed through a 15% resolving gel with a 5% stacking gel in a Tris-glycine buffer system. BLUEstain 2 was used as the molecular weight ladder. Samples were prepared using a 4× loading solution consisting of Laemmli sample buffer with heating for 5 min at 95 °C. Electrophoresis was performed at 180 V for 45–60 min. Following electrophoresis, proteins were stained with Colloidal Coomassie Brilliant Blue G-25036 and then imaged using a Bio-Rad Chemidoc Touch imaging system. Within each SDS-PAGE, all of the conditions were derived from the same culture split in different tubes to keep the protein expression level consistent. The screen gels shown in the Supporting Information were analyzed using ImageJ densitometry analysis. The following assumptions were made for gel analysis: (1) Coomassie stains all of the protein bands equally well within a gel, (2) empty lanes have no protein bands and any signal from it corresponds to background noise, and (3) distortion or noise caused by LMWC did not significantly affect ELP band intensity. The relative amount of protein was determined by comparing the intensity of the protein band to the intensity of the molecular weight closest to it in the ladder. All of the relative amounts were deduced using the reference intensity of the ladder; conditions that were lower than 70% were marked in blue letters (Figures S1 and S2).
Agarose Gel Electrophoresis.
Agarose gel electrophoresis was used to reveal the presence of contaminating nucleic acids. In brief, 0.5% agarose gels were cast using TBE buffer (pH 8.7) with a 1:10 000 dilution of the nucleic acid detection reagent GelRed. Samples were prepared with a 6× sample loading buffer and compared with a 1 kb DNA ladder. The gel was run at 80 V for 90 min and then imaged with a Bio-Rad Chemidoc Touch imaging system.
Lipopolysaccharide Quantitation by Limulus Amoebocyte Lysate (LAL) Test.
Endotoxin analysis was performed on A160 purified by various methods from two separate experiments. The solvent used for extraction was 1:1 butanol:ethanol (BD). LPS content was normalized against ACN precipitation within each experiment. To help lower LPS contamination, the second experiment was performed away from the primary lab area where bacterial cultures were handled. Both LAL tests were performed by an independent testing lab (Nelson Laboratories, Salt Lake City, UT).
Fluorescein Labeling of ELP.
Purified ELP in 0.1 M Na2CO3 buffer (pH 9) was stirred at 4 °C with 10 molar excess equivalents of fluorescein isothiocyanate (FITC) for 12 h. Excess FITC was removed using LH-20 Sephadex as recommended by the manufacturer.
Flow Cytometry.
MB49 cells, generously provided by Dr. Timothy Ratliff (Purdue University), were seeded at a density of 20 000 cells in 96-well plates overnight in DMEM with serum. Wells were then rinsed 3 times with PBS before incubating under test conditions with DMEM serum-free media. After incubation, the cells were again rinsed 3 times with PBS and trypsinized (20 μL per well) for 5 min at 37 °C. PBS (100 μL) was used to lift the cells from each well, and 4 wells per condition were combined and filtered through a round-bottom tube with a cell strainer cap to minimize clumping. Samples were run on a BD LSRFortessa cell analyzer using a FITC channel, and data was further processed using FCS Express 7 and FlowJo.
RESULTS AND DISCUSSION
Extraction Efficiency Is Independent of Sequence Identity and Polarity.
We performed a 28 organic solvents extraction screen on ELP of the same sequence length but with different guest residues (X): A40 (acidic, X = V/I/E [1:3:1]), N40 (neutral, X = V), and B40 (basic, X = V/H/G/A [1:2:1:1]). Samples of these different ELP were extracted, evaporated, and analyzed by SDS-PAGE (Figures 1 and S1). Regardless of the ELP composition, alcohol-based solvent mixtures performed the best, with isopropyl alcohol and its combinations typically giving the highest yield and purity as assessed by SDS-PAGE. This finding is consistent with our previous experience with a lysine-containing triblock ELP.1,6 Although the protein expression levels varied across these constructs, guest residue played little or no role in the extractability of the ELP (Figure S1).
Figure 1.

SDS-PAGE analysis of isopropanol-containing solvent extraction screens of ELP with similar MW but varying charges. (A) Acidic short chain: A40. (B) Neutral short chain: N40. (C) Basic short chain: B40. All solvent combinations refer to a 1:1 v/v mixture, e.g., combination AB refers to a 1:1 (v/v) mixture of A (isopropanol) and B (1-butanol). Outcomes of the other organic solvent extractions appear in Figure S1.
Extraction Efficiency Is Independent of ELP Molecular Weight.
To probe the effect of ELP sequence length on its extractability, we then performed the 28 organic solvents extraction screen on constructs of the same chemical nature (acidic: X = V/I/E [1:3:1]) but with varying lengths: A40 (17.19 kDa), A80 (34.15 kDa), A160 (68.06 kDa). Once again, we consistently found that low molecular weight alcohols gave the best yields and purities regardless of molecular weights (Figures 2 and S2). However, some combinations of 1-butanol and ethyl acetate gave suboptimal results due to their lower water miscibility.
Figure 2.

SDS-PAGE analysis of isopropanol-containing solvent extraction screens of ELP with different MW but identical charge distribution. (A) A40, MW 17.19 kDa. (B) A80, MW 34.15 kDa. (C) A160, MW 68.06 kDa (due to the size of the protein a 12% SDS-PAGE was used in this case). All solvent combinations refer to a 1:1 v/v mixture, e.g., AB refers to a 1:1 v/v mixture of A (isopropanol) and B (1-butanol). Outcomes of the other organic solvent extractions appear in Figure S2.
Extraction Is Also Effective for an ELP Fusion.
To test the broader applicability of this extraction method, we fused a short ELP to an epidermal growth factor sequence (N24-EGF). We chose N24 for this evaluation due to the simplicity of the ELP containing valine as its guest residue. To confirm expression of the N24-EGF fusion, we visualized the extracts by Western blot using an anti-EGF antibody (Figure S3). Once expression was verified, a broad solvent extraction screen was performed using the same 28 solvents as well as several more nuanced ratios besides the typical 1:1 ratio used in our standard screen. Interestingly, we found that a 1:5 ethanol:methanol solution gave the highest yield by dot blot analysis (Figure S4).
Development of a Method to Reduce Sample Preparation Time and Enhance ELP Purification Efficacy.
Organic extraction provides a near instantaneous cell lysis and selective isolation of ELP compared to traditional lysis methods (first step in Figure 3A and 3B). We sought a method to rapidly execute both a solvent switch and removal of LMWC after extraction to enable direct use of ELP for biological experiments. Four different organic solvent removal methods were tested for their impact on ELP recovery and purity: (i) evaporation, (ii) evaporation followed by ITC, (iii) liquid–liquid back extraction, and (iv) ACN precipitation (Figure 3).
Figure 3.

Summary of ELP purification procedures evaluated in this work. Typical macromolecular contaminants observed during each method: host cell proteins (HCP), low molecular weight contaminants (LMWC), nucleic acid, and lipopolysaccharide (LPS). Times mentioned for methods B and C include the average time for evaporation, a parameter that varies based on the solvent.
These methods were applied to the purification of A160 and compared using SDS-PAGE analysis (Figures 4, S5, and S10). A160 was chosen as the test case because of its high expression and low transition temperature, making it favorable for ITC. The solvents used for extraction were based on hits from the previous organic solvent screen (Figures 2c and S2b). Extraction with evaporation gave a clean protein band; however, it was accompanied by significant LMWC (Figure 4). Although these can be removed with one round of ITC, this workflow still requires several hours of processing time due to the slow evaporation step. Traditional ITC removes LMWC completely, however, it requires considerable labor-intensive processing compared to simple evaporation (Figure 4). To bring together the advantages of these techniques, we developed ACN precipitation as an intermediate step. ACN precipitation led to reductions in LMWC content and removed host cell proteins after extraction in a fraction of the time needed for evaporation (Figures S6 and S7). It can easily be coupled to ITC to remove LMWC below detectable levels by PAGE analysis and is considerably faster than performing multiple ITC rounds. Similar trends were observed when a different solvent was used for extraction (Figure S5). Direct comparison against back extraction is shown in Figure S10.
Figure 4.

SDS-PAGE analysis comparing protein yield and purity obtained from various methods of A160 (68.06 kDa) purification. Lanes: 1 = ladder; 2 = empty; 3 = cell lysate; 4 = cold spin supernatant from the first round of ITC (CSS1) after lysis; 5 = CSS3 after lysis; 6 = empty; 7 = extraction (BD); 8 = CSS1 after extraction (BD); 9 = ACN precipitation after extraction (BD); 10 = CSS1 after ACN precipitation (BD). Sample volumes were normalized across lanes 5–10, allowing for direct comparisons of ELP yield and purity. Estimated time needed is displayed above each lane. LMWC: smear around 5 kDa can be seen in lanes 7 and 9.
Parameters Controlling Efficiency of Precipitation after Extraction.
Optimization of the extraction–ACN precipitation method revealed three parameters that could be fine tuned: temperature, precipitation solvent, and percentage of precipitation solvent.
Temperature.
The temperature for precipitation may be varied. For A160 extracted with BD as shown in Figure 4, ACN precipitations were directly compared between 4 and 22 °C (Figure S7). Since the differences were modest, these steps could be conducted at 22 °C unless the stability of the protein of interest requires a colder temperature.
Solvent for Precipitation.
ACN was typically used in our direct comparisons at both 4 and 22 °C for A160 (Figure S7); however, acetone is a viable alternative. We found that acetone performed similarly to ACN at 22 °C with respect to ELP purity and yield. Depending on the ELP and solvent used for extraction, only minor differences in the protein yield and purity were observed after precipitation; thus, both acetone and acetonitrile are viable precipitation solvents.
Percentage of Solvent for Precipitation.
Extensive screening with different solvent mixtures showed that mixtures starting from 70% ACN or acetone yielded similar amounts of protein. On the basis of these results, 70% ACN or acetone minimizes the amount of solvent needed as well as the LMWC content (Figure S8).
Applicability of Precipitation to ELPs of Varying Charge and Size.
Once the key parameters for precipitating a long acidic ELP (A160) were determined, we tested its applicability with short, basic, and neutral ELP (B40 and N40). Viable extraction solvents that were previously identified in our solvent screen were tested with ACN or acetone for their ability to precipitate. The optimal combinations of extraction–precipitation for B40 and N40 are shown in Figure 5. Direct comparison of the extraction–precipitation method to extraction alone revealed little to no loss in protein yield and a significant reduction in LMWC.
Figure 5.

SDS-PAGE analysis of purification efficacy for ELP with varying guest residues. Results shown are for optimized precipitation conditions for each construct compared to extraction. Lanes: 1 = ladder; 2 = empty; 3 = N40 (16.62 kDa) extraction (CD); 4 = empty; 5 = N40 ACN precipitation after extraction (CD); 6 = empty; 7 = B40 (16.66 kDa) extraction (AD); 8 = empty, 9 = B40 acetone precipitation after extraction (AD); 10 = empty. The faint higher MW band in Lane 9 is attributed to a dimeric form; ELP dimerization has been previously reported.1 Extr. = extraction, Ppt. = ACN precipitation.
Contaminant Profiling.
Challenges associated with protein purification from E. coli expression systems involve tedious removal of host cell proteins, nucleic acids, lipopolysaccharides (LPS), and small molecule metabolites. Using a variety of analytical tools, we probed changes in ELP yield and contaminant content as a function of purification method.
HCP and LMWC.
We found that all of the optimized purification methods were effective in removing host cell proteins and LMWC based on SDS-PAGE analysis (Figures 4, S5, and S10). To determine whether this extraction–precipitation method led to protein degradation, the LMWC band near 5 kDa was digested and analyzed by LC-ESI-MS/MS. For protein identification, LC-ESI-MS/MS data was searched using MaxQuant, applying a 1% false discovery rate criterion in both protein and peptide.37 Proteins identified with at least two unique peptides and at least 4 MS/MS counts were considered as true/confident identification.38 Applying these criteria, there were no apparent ELP breakdown products or any host cell proteins detectable above the noise levels (Table S4). We inferred from these findings that the observed LMWC near 5 kDa in the SDS-PAGE are likely cellular metabolites.
Nucleic Acids.
Elimination of detectable nucleic acids by polar organic solvent extraction was observed by agarose gel analysis in the case of A160; however, it takes multiple rounds of ITC to achieve similar reductions (Figure S9). The removal of nucleic acids below the limit of detection during extraction is especially noteworthy in the case of basic ELP, where electrostatic interactions prevent its efficient removal via ITC as previously reported.1
LPS.
Table 1 shows LPS amounts obtained using different methods of purification as detected by Limulus Amoebocyte Lysate assay. The assays were performed from two separate experiments (Figures S10 and S11); both included samples from ACN precipitation for comparison against other methods. Precipitation leads to a 6-fold reduction in LPS content when compared to extraction alone. ITC had a higher LPS concentrations when compared to ACN precipitation (2.4-fold). Interestingly, when ITC is coupled to ACN precipitation, LPS levels are reduced below the critical 1 EU/mL limit.
Table 1.
LPS Content Analyzed from Two Independent Experiments by Limulus Amoebocyte Lysate Test
| EU/mL | ratio [condition/ACN] | |
|---|---|---|
| experiment 1 conditions | ||
| extraction | 268 | 6.23 |
| back extraction | 151 | 3.51 |
| ACN ppt. | 43.0 | 1 |
| experiment 2 conditions | ||
| 3 rounds ITC | 46.8 | 2.40 |
| ACN ppt. | 19.5 | 1 |
| ACN ppt. + ITC | 0.607 | 0.031 |
N24-EGF Binding in MB49 Cells.
To determine whether the function of EGF was maintained after purification by organic extraction and precipitation, a binding experiment was performed with an epidermal growth factor receptor (EGFR) positive cell line. MB49 cells, a urothelial carcinoma cell line used widely for in vitro and in vivo models of bladder cancer, were used to test for EGFR binding using N24-EGF. N40, an ELP of similar MW to the N24-EGF fusion, was used as an ELP control to probe for nonspecific binding to MB49 cells. Figure 6 shows a robust increase in MB49 cell associated fluorescence with N24-EGF relative to N40 or PBS controls.
Figure 6.
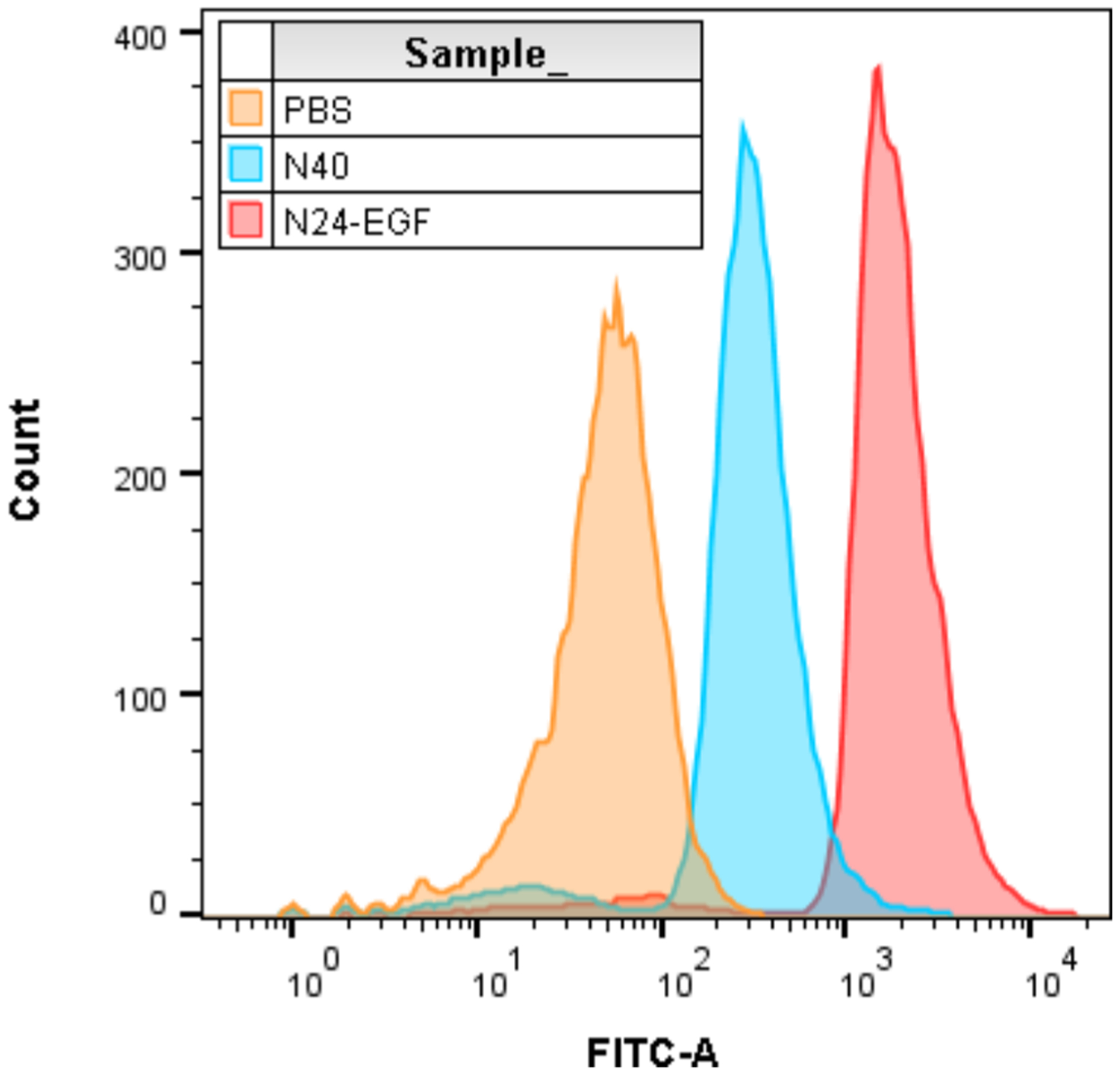
Flow cytometry data representing binding of ELP constructs chemically labeled with fluorescein and incubated with MB49 cells for 45 min before rinsing with PBS and FACS analysis. Analysis was performed using the FITC channel and 10 000 events.
Taken together, these findings indicate that low molecular weight alcohols provide the best capacity for extracting ELP regardless of molecular weight and guest residue charge. While we primarily tested pure or 1:1 ratios of various organic solvents, our results with an ELP fusion construct demonstrated that additional optimization of solvent ratios may further enhance purified ELP yields. Organic extraction provides the advantages of near instantaneous cell lysis and removal of >95% HCP and nucleic acids. When combined with a sensitive, antibody-based detection technique, this method becomes a robust platform for rapid optimization of protein expression and purification. Recent work by Mackay and co-workers made ELP detection possible by developing the first monoclonal antibody that holds broad specificity to ELP of various aliphatic guest residues.39 Once suitable organic solvent extraction conditions are identified, this method can be combined with different approaches to enhance the overall purity needed for biological applications (Figure 3).
The first step toward this goal is removal of the organic extraction solvent. This was previously performed by back extraction and (or) evaporation. Unfortunately, this approach requires long processing times and still is compromised by LPS and low molecular weight compound contamination. These limitations compelled the development of an extraction–precipitation sequence that enables both a rapid solvent switch and a reduction in LMWC and LPS content. The precipitation step was seamlessly integrated with hits found from our extraction screens for all ELP tested. For further reductions in LMWC content, a size-based purification technique such as dialysis or size-exclusion chromatography is effective. In our case, ITC was the preferred polishing step due to its scalability and speed relative to dialysis. We found that this extraction–precipitation–ITC method was able to reduce LPS levels below 1 EU/mL, a threshold that was not possible with ITC alone. This agrees with previous reports showing the necessity of alternative techniques to remove LPS when ITC is used to purify ELP from E. coli.27 We hypothesize that small amounts of organic solvent present in the extraction–precipitation samples help to solvate LPS in the aqueous phase and minimize its interaction with the aggregated ELP during ITC. It is important to note that these experiments were performed on a standard benchtop and not in a biological hood; thus, an even greater reduction in LPS levels may be realized in a more controlled environment.
On the basis of our previous demonstration of ELP fusion protein extraction,1 we sought to expand this work by producing a novel ELP fusion, N24-EGF, for functional testing of the material purified by organic solvent extraction–precipitation. A lower molecular weight ELP was selected for testing our extraction–precipitation method since sequences of this length have a limited ability to transition effectively via ITC. Furthermore, optimized purifications of EGF from E. coli frequently include urea to assist in protein solubilization;40,41 however, the use of organic solvent extraction obviates this step to enable direct extraction from E. coli. A binding experiment was performed using MB49 murine bladder cancer cells to test for EGF binding activity after purification by extraction–precipitation. Flow cytometry revealed a robust binding signal for N24-EGF compared to N40 and PBS controls, indicating that EGF function was maintained even after purification by the extraction–precipitation process. Even though the use of organic solvents in protein purifications is typically avoided due to concerns about their role in denaturing proteins, there are reports showing that some enzymes are highly compatible with organic solvents.42,43 Furthermore, purification protocols employing denaturants for purification followed by refolding methods to produce active protein isolates may also be great candidates for replacement by this method.44 In fact, EGF isolations frequently require denaturants when purified; however, the EGF fusion purification by extraction–precipitation reported here did not require this time-consuming step.
CONCLUSIONS
This work shows the broad feasibility of organic extraction–precipitation over a wide range of ELP constructs and for an ELP fusion that retains its functionality after purification in this manner. A nearly universal extractability was found with several polar organic solvents and combinations, most notably isopropanol. Organic solvent extraction is an operationally simple and inexpensive method to burst E. coli cells and selectively solubilize ELP. Subsequent treatment of the ELP extracts with an acetonitrile or acetone precipitation step (≥70% organic solvent), followed by an ITC final polish, provides the most efficient removal of LMWC and LPS. Since ELP are gaining increasingly important roles in biomedical research, this facile nonchromatographic method for the purification of ELP could greatly enhance their scope and impact since it rapidly removes immunostimulatory contaminants in a simple and scalable operation. Future efforts will be focused on investigating the mechanistic basis for this facile extraction–precipitation method for ELP purification.
Supplementary Material
ACKNOWLEDGMENTS
This publication is dedicated to the groundbreaking work on elastin biophysics by Dan W. Urry. The authors gratefully acknowledge the support of NIH Grant P30 CA023168 Phase I Small Grants Program of the Purdue University Center for Cancer Research and the resources of the Bindley Bioscience Center, a core facility of the NIH-funded Indiana Clinical and Translational Sciences Institute. Special thanks are also due to the technical expertise and proteomics support of Dr. Uma Aryal, Director of the Purdue Proteomics Facility.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.1c00096.
Additional information on methods and suppliers; list of primers; MALDI-TOF data; organic solvents used for screens; SDS-PAGE with organic solvent screens of A40, N40, B40, A80, and A160; Western blot of N24-EGF; dot blot for organic solvent screen of N24-EGF; A160 purification methods comparison; A160 stepwise comparison between ITC and ACN precipitation; percent acetone or ACN for precipitation; effect of temperature on precipitation; nucleic acid contamination analysis; SDS-PAGE for experiments 1 and 2 of LPS analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biomac.1c00096
The authors declare no competing financial interest.
Contributor Information
Logan Readnour, Department of Agricultural and Biological Engineering, Purdue University, West Lafayette, Indiana 47907, United States; Bindley Biosciences Center, Purdue University, West Lafayette, Indiana 47907, United States.
Kevin V. Solomon, Department of Agricultural and Biological Engineering, Laboratory of Renewable Resources Engineering (LORRE) and Bindley Biosciences Center, Purdue University, West Lafayette, Indiana 47907, United States;.
David H. Thompson, Department of Chemistry, Purdue Center for Cancer Research, Multi-disciplinary Cancer Research Facility and Bindley Biosciences Center, Purdue University, West Lafayette, Indiana 47907, United States;.
REFERENCES
- (1).VerHeul R; Sweet C; Thompson DH Rapid and Simple Purification of Elastin-Like Polypeptides Directly from Whole Cells and Cell Lysates by Organic Solvent Extraction. Biomater. Sci 2018, 6, 863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Owczarek B; Gerszberg A; Hnatuszko-Konka K A Brief Reminder of Systems of Production and Chromatography-Based Recovery of Recombinant Protein Biopharmaceuticals. Biomed. Res. Int 2019, 2019, 4216060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Usmani SS; Bedi G; Samuel JS; Singh S; Kalra S; Kumar P; Ahuja AA; Sharma M; Gautam A; Raghava GPS THPdb: Database of FDA-approved Peptide and Protein Therapeutics. PLoS One 2017, 12, No. e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fosgerau K; Hoffmann T Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- (5).Trono D Recombinant Enzymes in the Food and Pharmaceutical Industries; Elsevier, 2019; pp 349–387. [Google Scholar]
- (6).Wingfield PT Overview of the Purification of Recombinant Proteins. Curr. Protoc. Protein Sci 2015, 80, 1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yadav DK; Yadav N; Yadav S; Haque S; Tuteja N An Insight Into Fusion Technology Aiding Efficient Recombinant Protein Production for Functional Proteomics. Arch. Biochem. Biophys 2016, 612, 57–77. [DOI] [PubMed] [Google Scholar]
- (8).Saraswat M; Musante L; Ravidá A; Shortt B; Byrne B; Holthofer H Preparative Purification of Recombinant Proteins: Current Status and Future Trends. Biomed. Res. Int 2013, 2013, 312709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Heumann D; Roger T Initial Responses to Endotoxins and Gram-Negative Bacteria. Clin. Chim. Acta 2002, 323, 59–72. [DOI] [PubMed] [Google Scholar]
- (10).Shi H; Guo Y; Liu Y; Shi B; Guo X; Jin L; Yan S The In Vitro Effect of Lipopolysaccharide on Proliferation, Inflammatory Factors and Antioxidant Enzyme Activity in Bovine Mammary Epithelial Cells. Animal Nutrit. 2016, 2, 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schwarz H; Gornicec J; Neuper T; Parigiani MA; Wallner M; Duschl A; Horejs-Hoeck J Biological Activity of Masked Endotoxin. Sci. Rep 2017, 7, 44750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Young CL; Britton ZT; Robinson AS Recombinant Protein Expression and Purification: A Comprehensive Review of Affinity Tags and Microbial Applications. Biotechnol. J 2012, 7, 620–634. [DOI] [PubMed] [Google Scholar]
- (13).Li Y; Stern D; Lock LL; Mills J; Ou S-H; Morrow M; Xu X; Ghose S; Li ZJ; Cui H Emerging Biomaterials for Downstream Manufacturing of Therapeutic Proteins. Acta Biomater. 2019, 95, 73–90. [DOI] [PubMed] [Google Scholar]
- (14).Meyer DE; Chilkoti A Purification of Recombinant Proteins by Fusion With Thermally-Responsive Polypeptides. Nat. Biotechnol 1999, 17, 1112–1115. [DOI] [PubMed] [Google Scholar]
- (15).Urry DW Physical Chemistry of Biological Free Energy Transduction As Demonstrated by Elastic Protein-Based Polymers. J. Phys. Chem. B 1997, 101, 11007–11028. [Google Scholar]
- (16).Christensen T; Hassouneh W; Trabbic-Carlson K; Chilkoti A Predicting Transition Temperatures of Elastin-Like Polypeptide Fusion Proteins. Biomacromolecules 2013, 14, 1514–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).McPherson DT; Xu J; Urry DW Product Purification by Reversible Phase Transition Following Escherichia Coli Expression of Genes Encoding Up To 251 Repeats of the Elastomeric Pentapeptide GVGVP. Protein Expression Purif. 1996, 7, 51–7. [DOI] [PubMed] [Google Scholar]
- (18).Macewan SR; Hassouneh W; Chilkoti A Non-chromatographic Purification of Recombinant Elastin-like Polypeptides and their Fusions with Peptides and Proteins from Escherichia coli. J. Visualized Exp 2014, 88, No. 51583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Banskota S; Yousefpour P; Kirmani N; Li X; Chilkoti A Long Circulating Genetically Encoded Intrinsically Disordered Zwitterionic Polypeptides for Drug Delivery. Biomaterials 2019, 192, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Luginbuhl KM; Schaal JL; Umstead B; Mastria EM; Li X; Banskota S; Arnold S; Feinglos M; D’Alessio D; Chilkoti A One-Week Glucose Control via Zero-Order Release Kinetics from an Injectable Depot of Glucagon-Like Peptide-1 Fused to a Thermo-sensitive Biopolymer. Nat. Biomed. Eng 2017, 1, 0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sarangthem V; Cho EA; Yi A; Kim SK; Lee B-H; Park R-W Application of Bld-1-Embedded Elastin-Like Polypeptides in Tumor Targeting. Sci. Rep 2018, 8, 3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Le DHT; Sugawara-Narutaki A Elastin-Like Polypeptides as Building Motifs Toward Designing Functional Nanobiomaterials. Mol. Sys. Des. Eng 2019, 4, 545–565. [Google Scholar]
- (23).Fletcher EE; Yan D; Kosiba AA; Zhou Y; Shi H Biotechnological Applications of Elastin-Like Polypeptides and the Inverse Transition Cycle in the Pharmaceutical Industry. Protein Expression Purif. 2019, 153, 114–120. [DOI] [PubMed] [Google Scholar]
- (24).Banki MR; Feng L; Wood DW Simple Bioseparations Using Self-Cleaving Elastin-Like Polypeptide Tags. Nat. Methods 2005, 2, 659–662. [DOI] [PubMed] [Google Scholar]
- (25).Yang C-G; Lang M-F; Fu X; Lin H; Zhang L-C; Ge G-S; Sun J; Hu X-J Application of Short Hydrophobic Elastin-Like Polypeptides for Expression and Purification of Active Proteins. 3 Biotech 2020, 10, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bahniuk MS; Alshememry AK; Unsworth LD High-Yield Recombinant Expression and Purification of Marginally Soluble, Short Elastin-Like Polypeptides. BioTechniques 2016, 61, 297–304. [DOI] [PubMed] [Google Scholar]
- (27).Lin C-Y; Liu JC Incorporation of Short, Charged Peptide Tags Affects the Temperature Responsiveness of Positively-Charged Elastin-Like Polypeptides. J. Mater. Chem. B 2019, 7, 5245–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yeboah A; Cohen RI; Rabolli C; Yarmush ML; Berthiaume F Elastin-Like Polypeptides: A Strategic Fusion Partner for Biologics. Biotechnol. Bioeng 2016, 113, 1617–1627. [DOI] [PubMed] [Google Scholar]
- (29).Hassouneh W; Christensen T; Chilkoti A Elastin-Like Polypeptides as a Purification Tag for Recombinant Proteins. Curr. Protoc. Protein Sci 2010, 61, 6.11.1–6.11.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bahniuk MS; Alshememry AK; Unsworth LD High-Yield Recombinant Expression and Purification of Marginally Soluble, Short Elastin-Like Polypeptides. BioTechniques 2016, 61, 297–304. [DOI] [PubMed] [Google Scholar]
- (31).Wang E; Lee S-H; Lee S-W Elastin-Like Polypeptide Based Hydroxyapatite Bionanocomposites. Biomacromolecules 2011, 12, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu JC; Heilshorn SC; Tirrell DA Comparative Cell Response to Artificial Extracellular Matrix Proteins Containing the RGD and CS5 Cell-Binding Domains. Biomacromolecules 2004, 5, 497–504. [DOI] [PubMed] [Google Scholar]
- (33).McDaniel JR; Mackay JA; Quiroz FGA; Chilkoti A Recursive Directional Ligation by Plasmid Reconstruction Allows Rapid and Seamless Cloning of Oligomeric Genes. Biomacromolecules 2010, 11, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Russell DW; Sambrook J Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, 2001; Vol. 1. [Google Scholar]
- (35).MacKay JA; Callahan DJ; FitzGerald KN; Chilkoti A Quantitative Model of the Phase Behavior of Recombinant pH-Responsive Elastin-Like Polypeptides. Biomacromolecules 2010, 11, 2873–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dyballa N; Metzger S Fast and Sensitive Colloidal Coomassie G-250 Staining for Proteins in Polyacrylamide Gels. J. Visualized Exp 2009, 3, No. 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tyanova S; Temu T; Cox J The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc 2016, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- (38).Old WM; Meyer-Arendt K; Aveline-Wolf L; Pierce KG; Mendoza A; Sevinsky JR; Resing KA; Ahn NG Comparison of Label-free Methods for Quantifying Human Proteins by Shotgun Proteomics. Mol. Cell. Proteomics 2005, 4, 1487–1502. [DOI] [PubMed] [Google Scholar]
- (39).Kouhi A; Yao Z; Zheng L; Li Z; Hu P; Epstein AL; MacKay JA Generation of a Monoclonal Antibody to Detect Elastin-like Polypeptides. Biomacromolecules 2019, 20, 2942–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Pouranvari S; Ebrahimi F; Javadi G; Maddah B Cloning, Expression, and Cost Effective Purification of Authentic Human Epidermal Growth Factor With High Activity. Iranian Red Crescent Med. J 2016, 18, No. e24966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang Y; Zhang K; Wan Y; Zi J; Wang Y; Wang J; Wang L; Xue X A pH-Induced, Intein-Mediated Expression and Purification of Recombinant Human Epidermal Growth Factor in Escherichia coli. Biotechnol. Prog 2015, 31, 758–764. [DOI] [PubMed] [Google Scholar]
- (42).Klibanov AM Improving Enzymes by Using Them in Organic Solvents. Nature 2001, 409, 241–246. [DOI] [PubMed] [Google Scholar]
- (43).Stepankova V; Bidmanova S; Koudelakova T; Prokop Z; Chaloupkova R; Damborsky J Strategies for Stabilization of Enzymes in Organic Solvents. ACS Catal. 2013, 3, 2823–2836. [Google Scholar]
- (44).Yu Y; Wang J; Shao Q; Shi J; Zhu W The Effects of Organic Solvents on the Folding Pathway and Associated Thermodynamics of Proteins: A Microscopic View. Sci. Rep 2016, 6, 19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.