

Abstract
Sepsis reduces the number and function of memory CD8 T-cells within the host, contributing to the long-lasting state of immunoparalysis. Interestingly, the relative susceptibility of memory CD8 T-cell subsets to quantitative/qualitative changes differ after cecal ligation and puncture (CLP)-induced sepsis. Compared to circulatory memory CD8 T-cells (TCIRCM), moderate sepsis (0–10% mortality) does not result in numerical decline of tissue resident memory CD8 T-cells (TRM) which retain their ‘sensing and alarm’ IFNγ-mediated effector function. To interrogate this biologically-important dichotomy, vaccinia virus (VacV)-immune B6 mice containing CD8 TCIRCM and skin TRM underwent moderate or severe (~50% mortality) sepsis. Severe sepsis led to increased morbidity and mortality characterized by increased inflammation compared to moderate CLP or sham controls. Severe CLP mice also displayed increased vascular permeability in the ears. Interestingly, skin CD103+ CD8 TRM, detected by i.v.-exclusion or 2-photon microscopy, underwent apoptosis and subsequent numerical loss following severe sepsis, which was not observed in mice that experienced moderate CLP or sham surgeries. Consequently, severe septic mice showed diminished CD8 T cell-mediated protection to localized skin re-infection. Finally, the relationship between severity of sepsis and demise in circulatory vs tissue-embedded memory CD8 T-cell populations was confirmed by examining tumor-infiltrating and non-specific CD8 T-cells in B16 melanoma tumors. Thus, sepsis can differentially affect the presence and function of Ag-specific CD8 T-cells that reside inside tissues/tumors depending on the severity of the insult, a notion with direct relevance to sepsis survivors and their ability to mount protective memory CD8 T-cell-dependent responses to localized antigen re-encounter.
Introduction
Sepsis presents a tremendous health care challenge throughout the world, accounting for one-third of hospital deaths and affecting almost 2 million people in the United States annually (1–3). The impact of sepsis is more pronounced in the developing world, where it may account for up to 40% of the total deaths in some countries (4, 5). The majority (~75%) of deaths from sepsis usually occur in patients aged 65 and over, but anyone with an uncontrolled local infection can be at risk for developing sepsis (6, 7). Sepsis is classified as the body’s dysregulated immune response to an uncontrolled systemic infection by a bacterial, viral, fungal, or parasitic pathogen. The septic event can occur from systemic infections, with barrier tissues such as the pulmonary, gastrointestinal, and urinary tracts being common sites of original infection. Following the systemic dissemination of a pathogen from the original nidus of infection, the immune system responds with a massive release of pro- and anti-inflammatory cytokines in the blood (8–10). This “cytokine storm” can lead to host tissue damage and is a period of high-risk for acute mortality. Most patients survive the initial septic event, largely through the administration of antibiotics and resuscitation measures, but there are long-lasting lesions in the immune system that compromise the future health of the individual (11, 12). Sepsis survivors are at a significant risk for secondary infection, with 20% of discharged septic survivors being readmitted to the hospital within 30 days (primarily) because of secondary infection complications (13). Also, significant increases in morbidity of septic survivors have been observed due to malignancy and infections that would otherwise be controlled by a healthy, normal immune system (14, 15).
Following the resolution of this acute phase of sepsis, lymphopenia ensues and a state of systemic immune system paralysis follows thereafter. The immunoparalysis phase is characterized by an overall decrease in immune cell number and function, resulting in an increased susceptibility to viral reactivation, secondary infections, and neoplastic malignancy long after the initial septic event (16–18). Using the experimental murine cecal ligation and puncture (CLP) method of sepsis induction, we and others have observed the diminishment of both naïve and memory CD4 and CD8 T cell responses (numbers and Ag-dependent and independent functions) which leave sepsis survivors with increased susceptibility to newly- or previously encountered infections (19–22). These data are consistent with those observed in the human population, where septic survivors have a marked decrease in T cell number and function (23, 24).
Importantly, the sepsis-induced lesions in T cells have mostly been studied in the circulating T cell compartment (25–27). However, moderate sepsis (0–10% mortality) does not evoke numerical decline of tissue-embedded resident memory CD8 T cells (TRM), which also retain their ‘sensing and alarm’ IFNγ-mediated effector functions (28). CD8 TRM are generated at the site of original infection and have been found in many organs such as the skin, lungs, reproductive tract, and gut. They are phenotypically distinct from their circulating counterparts, and they serve to guard and sense local tissue and provide a robust early response upon cognate antigen encounter (29). Recent reports have also showed their egression to secondary lymphoid organs, suggesting a role in priming a recall (or secondary) response (30, 31). Thus, due to their anatomical positioning outside of the vasculature and in previously infected tissue, CD8 TRM are uniquely poised to provide protection and mediate clearance of re-infection. It is also tempting to speculate that TRM are more resistant to the deleterious effects of sepsis due to their seclusion from circulation and sepsis-induced cytokine storm. In this study, we show that depending on the severity of the insult and magnitude and duration of cytokine storm, sepsis has the capacity to also influence tissue- or tumor-embedded bystander memory CD8 T cells, a concept with direct relevance to sepsis survivors and their ability to mount protective memory CD8 T cell-dependent responses to localized/peripheral antigen re-encounter.
Materials and Methods
Mice, pathogens and memory CD8 T cell generation
Experimental procedures using mice were approved by University of Iowa Animal Care and Use Committee under protocol numbers 6121915 and 9101915. The experiments performed followed Office of Laboratory Animal Welfare guidelines and PHS Policy on Humane Care and Use of Laboratory Animals. Inbred C57Bl/6 (B6; Thy1.2/1.2) mice were purchased from the National Cancer Institute (Frederick, MD) and maintained in the animal facilities at the University of Iowa at the appropriate biosafety level. P14 T cell receptor-transgenic (TCR-Tg) mice (Thy1.1/1.1) were bred and maintained at the University of Iowa (Iowa City, IA). B6.CAG.MRFP1-mice were obtained from Jackson labs and crossed with B6.P14-Thy1.1 mice at the University of Iowa to yield P14-RFP Thy1.1 B6 mice. P14 mice were crossed with B6 eGFP (Jackson Laboratory) at the University of Iowa to yield P14-eGFP Thy 1.1 B6 mice.
LCMV-Armstrong (2×105 PFU) was injected i.p.. VacV-GP33 infection of the skin was performed by applying 5×106 PFU in 5 μl saline to the center of the ear pinna and then poking it 30 times with a 27-gauge needle (32). VacV-GP33 viral titers were quantified using plaque assay on BSC-40 cells, as described previously (33). Naïve P14 CD8 T cells obtained from peripheral blood of naïve P14 mice (Thy1.1) were adoptively transferred into B6 recipients (Thy1.2; 5–10×103 Thy1.1+ P14 T cells/recipient) i.v., followed by infection with a pathogen expressing the GP33–41 epitope of LCMV.
Cell isolation
Peripheral blood (PBL) was collected by retro-orbital bleeding. Ears, small intestine, and lungs were treated with collagenase type II (Worthington, 100 U/mL) in RPMI 1640 supplemented with 5% fetal calf serum (FCS) and shaken at 450 RPM for 30–90 min at 37°C. Single cell suspensions were prepared by mashing tissues through a 70 μm cell strainer (Falcon) with the plunger of a 1 mL syringe (BD Biosciences). Samples were centrifuged and re-suspended in RPMI. Single-cell suspensions from spleen, lymph nodes, and tumors were generated after mashing tissue through 70 μm cell strainer without enzymatic digestion.
Flow cytometry, and cytokine detection
Flow cytometry data were acquired on a FACSCanto (BD Biosciences, San Diego, CA) and analyzed with FlowJo software (Tree Star, Ashland, OR). To determine expression of cell surface proteins, mAb were incubated at 4°C for 20–30 min and cells were fixed using Cytofix/Cytoperm Solution (BD Biosciences) and, in some instances followed by mAb incubation to detect intracellular proteins. The following mAb clones were used: CD8 (53–6.7; eBioscience), Thy1.1 (HIS51; eBioscience), B220 (RA3–6B2; eBioscience), CD45.2 (104; eBioscience), IFN-γ (XMG1.2; eBioscience), TNF-α (MP6-XT22, eBioscience), IL-2 (JES6–5H4, eBioscience), CD103 (2E7; eBioscience), CD31 (390, Biolegend), CXCL9 (MIG-2F5.5), VCAM-1 (429; eBioscience), CXCR3 (CXCR3–173, eBioscience), CD62L (MEL-14, eBioscience), CD27 (LG.7F9, eBioscience), KLRG-1 (2F1, eBioscience), CD127 (A7R34, eBioscience), CD122 (5H4, eBioscience), CD69 (H1.2F3, eBioscience), T-bet (eBio410, eBioscience), Eomesodermin (Dan11mag, eBioscience), Granzyme B (MHGB04, Invitrogen), CD107a (1D4B, BD Pharmingen). IFN-γ receptor was detected using CD119-Biotin (2E2; eBioscience) and Streptavidin-PE (eBioscience) after acid stripping, as described previously (34). Plasma concentrations of IFN-γ IL-1α, IL-1β, IL-2, IL-6, TNF-α, CCL5, IL-10, IL-18, CXCL5, G-CSF, LIF, IL-22, IL-27, CCL11, CXCL1, CXCL10, CCL2, CCL7, CCL3, CCL4, CXCL2, IL12p70, GM-CSF, IL-13 were determined using Multiplex Immunoassay (ProcartaPlex; Affymetrix by eBioscience) on a Bio-Rad BioPlex, as performed previously (35). Apoptosis was evaluated using Vybrant FAM Caspase-3/7 Assay Kit (Invitrogen) according to manufacturer’s protocol.
Tissue extraction
At various times after CLP or sham surgery, ears, lungs, and spleens were collected and added to a 1.5-ml centrifuge tube for processing as previously described (36). Each ear was weighed. To each sample was added 0.5 ml of 0.1% Tween 20 in PBS and samples were ground for 3–5 min with a pellet pestle attached to a cordless electric drill. Samples were then quick frozen in liquid N2, thawed in a 37°C water bath, and ground again for 3–5 min. Samples were sonicated for 15 s and then centrifuged for 5 min at 13,000 g. Supernatants were then removed for cytokine measurement by Multiplex Immunoassay. Blood was also collected from these same mice, and then allowed to clot. Serum was collected and run on the same Multiplex Immunoassay.
Intravascular stain protocol to distinguish circulatory from resident cells
Two minutes after APC-conjugated CD45.2 mAb was injected i.v. into mice, peripheral blood was collected by retro-orbital bleeding and used as a positive control with >99% memory P14 CD8 T cells routinely labeled with CD45.2 mAb (37). After an additional minute, mice were euthanized and organs of interest were harvested and processed for flow cytometry.
Vascular leakage
Mice were injected i.v. with 200 μl of Evans blue dye (1.0% in PBS) at indicated timepoints. After 30 min, mice were anesthetized, and ears, lungs, and intestines were harvested and treated with formamide. Quantification of the dye was done using optical density reading on a spectrophotometer based on a standard curve with known amounts of Evans blue dye.
Cecal ligation and puncture (CLP) model of sepsis induction
CLP procedure was performed (38) on mice that were anesthetized with ketamine/xylazine (University of Iowa, Office of Animal Resources). Briefly, the abdomen was shaved and disinfected with Betadine (Purdue Products), and a midline incision was made. The distal third of the cecum was ligated with Perma-Hand Silk (Ethicon), punctured once (“moderate”) or twice (“severe”) using a 25-gauge needle, and a small amount of fecal matter extruded. The cecum was returned to abdomen, the peritoneum was closed with 641G Perma-Hand Silk (Ethicon), and skin sealed using surgical Vetbond (3M). Following surgery, 1 mL PBS was administered s.c. to provide post-surgery fluid resuscitation. Bupivacaine (Hospira) was administered at the incision site, and flunixin meglumine (Phoenix) was administered for postoperative analgesia. This procedure created a septic state characterized by loss of appetite and body weight, ruffled hair, shivering, diarrhea, and/or periorbital exudates with 0–10% mortality rate, or 25–50% mortality for moderate and severe sepsis, respectively, similar to our previous reports (39). Sham mice underwent identical surgery excluding cecal ligation and puncture. Removal of punctured cecum as source control is not attempted here due to the short duration of the experiments - most mice were analyzed/sacrificed in the first 2–4 days post CLP surgery.
Bacterial burden
Livers and spleens were obtained from animals 12 hours post-surgery, weighed, and homogenized in antibiotic-free RPMI 1640 (Life Technologies) containing 10% FCS (Life Technologies) with a mechanical disruptor. Serial dilutions of the homogenates were performed, and aliquots were plated on 5-cm petri dishes containing antibiotic-free BHI agar. Plates were incubated at 37°C, 5% CO2 overnight, and colonies were counted by hand the next morning to calculate the bacterial burden per gram of tissue.
Intravital 2-photon microscopy of skin Trms
Fluorescent P14 donor mice were bled and 2–5×103 RFP P14 or 5 × 104 GFP P14 T cells were adoptively transferred to mice by i.v. injection one day prior to infection. VacV-GP33 (5×106 PFU) was administered to the ear (28) and mice underwent CLP or Sham surgery at 30–40 days post-infection. Prior to imaging, mouse ears were treated with Nair (Church and Dwight Co.) for 3–5 minutes to remove hair. Ears were imaged for RFP or GFP P14 T cells with live intravital two-photon microscopy 48 hours post-surgery. The VacV immune ears that contained RFP or GFP P14 TRM cells of live mice were positioned on the microscope base in a continuously heated enclosed chamber (Leica). A custom suction tissue window apparatus (VueBio) was placed on the Ear with 20–25mm Hg of negative pressure to gently immobilize the tissue against a fixed coverslip. All images were acquired on an upright Leica SP8 Microscope (Leica) using a 25x / 0.95 NA water immersion objective with coverslip correction with 1.25x zoom. High resolution (512 × 512) stacks of 15–38 xy sections sampled with a resonant scanning head operating with bi-directional scanning and 5um z spacing were acquired to provide image volumes of 354 × 354 × 75–190um and voxel sizes of 0.693um × 0.693um × 5um. Line averaging of 16 and a kernel-3 median filter were used in LAS X to reduce noise. Images were excited with a tunable Insight laser (Spectra Physics) with excitation at 1040nm and collected by two tunable internal HyD detectors with emission collected for Secondary Harmonic Generation (SHG) at 515–525nm and at 560–630nm for RFP P14s. A tunable Mai Tai HP Sapphire laser (Spectra Physics) at 940 nm was used for detection of eGFP (500–550nm) and SHG (435–485nm) using a quad HyD external detector array equipped with SP565 beam splitter. Tile scans were taken of a large section of the ear (4mm2-12mm2) and individual fields were merged into a single image. Sequences of image stacks were transformed into volume-rendered, 3D images with Imaris Version 9.1 (Bitplane). The Imaris spot function was used to visualize GFP+ P14s. Tile areas with high levels of autofluorescent hair were trimmed from further analysis with the Imaris crop 3D function yielding tiles between 4.00mm2-9.25mm2
Tumor induction and monitoring
The B16 melanoma cell line was provided by Lyse Norian (University of Alabama-Birmingham). B16 cells were grown in DMEM with 4.5 g/l d-glucose, l-glutamine, 10% FCS (HyClone Laboratories) and supplementum complementum (made in-house). Cell lines were passaged every 2–3 d and/or when cell confluency was >80% in 75 cm2-tissue culture flask. Cells were not sequentially passaged longer than 3 weeks. In vitro and in vivo tumor growth did not vary considerably throughout the study. For implantation, 2×104 B16 cells were injected s.c. at 100 μl volume with equal parts B16 medium and Matrigel Matrix (356234; Corning) into the left hind flank of LCMV-infected memory B6 mice that contained P14 memory CD8 T cells (40, 41). Tumor progression was determined by measuring tumor length multiplied by width using an electronic digital caliper. Mice were sacrificed upon reaching any animal protocol threshold including tumor length of >15 mm or tumor ulceration.
Statistical analysis
Unless stated otherwise data were analyzed using Prism6 software (GraphPad) using two-tailed Student t-test, one-way ANOVA, and two-way ANOVA with a confidence interval of >95% to determine significance (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 and N.S. as not significant). Data are presented as standard error of the mean.
Results
The magnitude and kinetics of the cytokine/chemokine storm depend on sepsis severity
Sepsis is an exaggerated host inflammatory response characterized by increased pro- and anti-inflammatory cytokine and chemokine production. This unbridled inflammatory response represents a crucial event in the pathogenesis of sepsis with important implications on the ability of the host to properly respond to ongoing and/or future infections. To firmly establish a correlation between severity of sepsis and cytokine/chemokine storm in our hands, experimental CLP model of sepsis induction was employed to induce moderate and severe sepsis (one or two cecal punctures, respectively). Sham surgery that does not include cecal ligation and puncture was included as control (Fig. 1A). Following surgery, mortality and morbidity (weight loss) were measured (Fig. 1B, C) and serum was collected at 6, 12, 24, 48, and 72 hours post-surgery. Serum cytokine and chemokine concentrations were determined by Bioplex ELISA (Fig. 1D, E). Although no changes in morbidity (weight loss) were observed between the severe and moderate CLP groups, severe CLP mice had a significantly higher mortality - 0% mortality in the CLP-Moderate group compared to 50% mortality in the CLP-Severe group (Fig. 1B, C). Of note: moderate CLP procedure in our hands evoke less than 10% mortality (this is historical average mortality for 500+ mice used in the last 10 years), >90% of mice that succumb to moderate CLP surgery die in the first 48 hours, and mice recover their weight loss in the first 7–10 days after surgery (39, 42).
Figure 1. The magnitude and the kinetics of the cytokine storm depends on the sepsis severity.
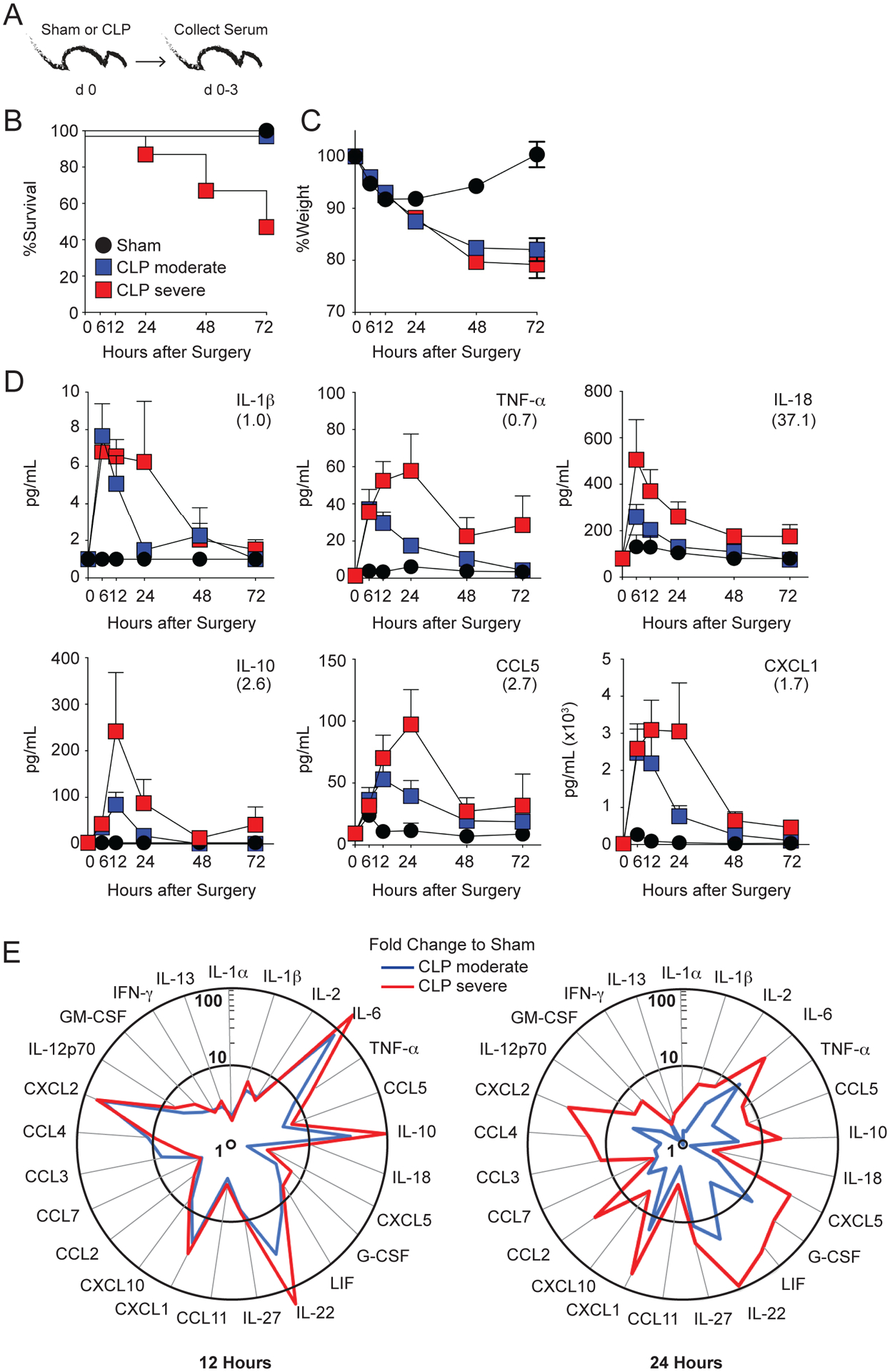
A) Experimental Design. Mice underwent sham or CLP surgery (1 or 2 cecal punctures to induce moderate or severe sepsis, respectively) and serum was collected at the indicated timepoints after surgery. B) Survival and C) weight loss were monitored for 72 h after surgery. D) Detection of indicated analytes in the serum after surgery. Parentheses indicate level of detection for a given cytokine/chemokine. E) Summary data of analyte levels (fold change to sham controls) at 12 (left) and 24 (right) hours after sepsis induction. Data are representative of 3 independent experiments with at least 3 mice per group. Error bars represent the standard error of the mean.
Importantly, the amounts of cytokines/chemokines detected and duration of the cytokine storm was dependent on the sepsis severity, with the extended production of pro- and anti-inflammatory cytokines such as IL-1β, TNF-α, IL-10, and IL-2 observed in the severe sepsis groups at 24 hours post-sepsis. (Fig. 1D–E). The concentrations of cytokines/chemokines detected by BioPlex assays used here should only be considered as direct comparison between groups in well controlled experiments and not as accurate measure of their concentrations in the plasma. In addition, bacterial burden detected in the livers and spleens 12 hours post CLP surgery correlated with the severity of sepsis induced (Supp Fig 1A, B). Thus, similar to existing reports (43, 44), a direct correlation between the severity of sepsis, bacteremia, and amount and duration of inflammation was established.
Severe sepsis disrupts the barrier function of vascular endothelium and increases the level of inflammation in situ
Vascular dysfunction is another key hallmark in the host response to sepsis (45). To determine if vascular permeability concordantly increases with severity of sepsis, CLP-Severe, CLP-Moderate, and sham surgeries were performed on naïve B6 mice. Two days later, mice were injected i.v. with Evans Blue dye (Fig. 2A). Thirty minutes after injection, tissues were harvested (ear, intestines, and lungs), processed, and the amount of Evans Blue dye was quantified using optical density reading (46). Morbidity and mortality were assessed (Fig. 2B, C) and an increase in mortality with the mice receiving CLP-Severe surgery compared to CLP-Moderate and sham surgery recipients was observed. Interestingly, the amount of Evans Blue dye (calculation based per gram of tissue) was higher in the tissues of the CLP-Severe mice compared to both the CLP-moderate and Sham mice (Fig 2D, E and Suppl Fig 2A–C), in which the amount of dye did not differ. Importantly, and potentially driven by the changes in the vascular permeability, the level of inflammation (ex. IL-6, IL-10, IL-1β) detected in the ear (skin), lungs, and spleens correlated with the sepsis severity (Suppl Fig 3A–D). Therefore, these data suggest the barrier function of the vascular endothelium in various tissues throughout the body can be disrupted by modulating the severity of sepsis induced, which correlated with the increased inflammatory response(s) detected in situ. Future studies will define the extent to which increase of inflammation inside tissues is driven by the increase in circulatory cytokines/chemokines and/or in situ production by resident cells.
Figure 2. Severe sepsis disrupts the barrier function of skin vascular endothelium.
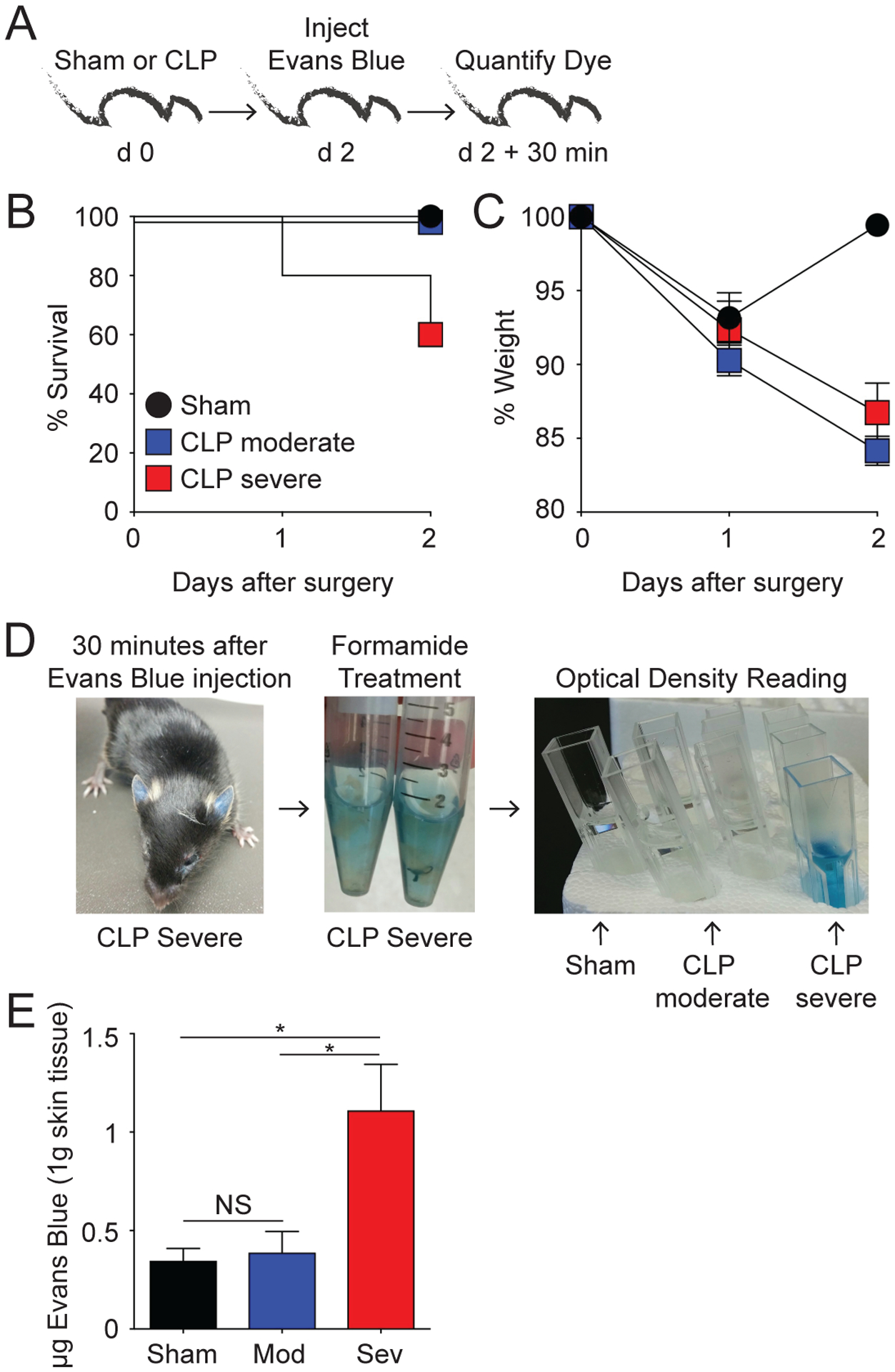
A) Experimental Design. Mice underwent sham, moderate CLP, or severe CLP surgery. B) Survival and C) Weight loss were monitored for 48 h after surgery. D) Representative images of Evans Blue dye in tissues before, during, and after tissue digestion. E) Summary data quantifying the amount of Evans Blue dye in the ear normalized to 1 gram of skin tissue. NS = not significant; * = p<0.05 determined using 1-way ANOVA. Data are representative of 2 independent experiments with at least 3 mice per group. Error bars represent the standard error of the mean.
Severity of sepsis influences the susceptibility of skin CD8 TRM to numerical alterations
Our published data show moderate sepsis (0–10% mortality) does not evoke significant numerical decline of skin CD8 TRM (generated in response to vaccinia virus) when compared to pathogen-specific CD8 TCIRCM. Furthermore, these skin CD8 TRM retained their effector functions (e.g., Ag-dependent IFNγ production) (28). Due to their localization within the tissue parenchyma, we hypothesized the CD8 TRM were “shielded” from the systemic apoptosis-inducing cytokine storm. In moderate sepsis, the barrier function of the vascular endothelium remains intact, which we posit helps facilitate CD8 TRM survival. However, data in Figure 2 show the barrier function of the skin vascular endothelial cells was compromised during severe sepsis. As the endothelial cells lose their barrier integrity, we reasoned that circulating factors (such as cytokines and chemokines) could now leach into the otherwise “shielded” tissue parenchyma and affect the cellular and/or functional integrity of the CD8 TRM. Thus, to define the extent to which disruption of endothelial barrier under more severe sepsis conditions affects the numbers of skin-embedded CD8 TRM, a physiologically relevant number (5×103 cells/mouse) of naïve Thy1.1 TCR-Tg P14 GP33-specific CD8 T cells were adoptively transferred into naïve B6 Thy1.2 mice prior to skin VacV-GP33 infection (Fig 3A) (28). Surgery was performed on VacV immune mice once the CD8 TCIRCM and TRM compartments were firmly established (day 30 p.i.). A fluorescent intravascular label was administered 3 minutes prior to tissue collection on day 2 post-surgery to distinguish circulatory (i.v. positive) and tissue resident (i.v. negative) Thy1.1 P14 memory CD8 T cell subsets (Fig 3A) (37). CD8 TRM are not positively stained using this technique under normal conditions, but the possibility exists that the disruption of the endothelial barrier during severe sepsis could enable the i.v. administered mAb to reach the tissue-embedded Thy1.1 P14 CD8 T cells and mark them as ‘circulatory’ rather than ‘tissue-resident’. To control for this possibility, the frequency of i.v. negative (tissue resident), i.v. positive (circulatory) and CD103+ i.v. positive cells in the skin were determined in all groups of mice. Importantly, no changes in the percentage of i.v. negative and i.v. positive P14 CD8 T cells in the ear skin was observed after severe sepsis compared to moderate sepsis or sham controls (Fig 3B–C). In addition, the frequency of CD103+ i.v. positive P14 CD8 T cells remained low and indistinguishable between groups suggesting the i.v. administered mAb does not label bona-fide TRM even in the severe sepsis group (Fig 3D).
Figure 3. Severity of sepsis influences the susceptibility of skin CD8 TRM to numerical alterations.
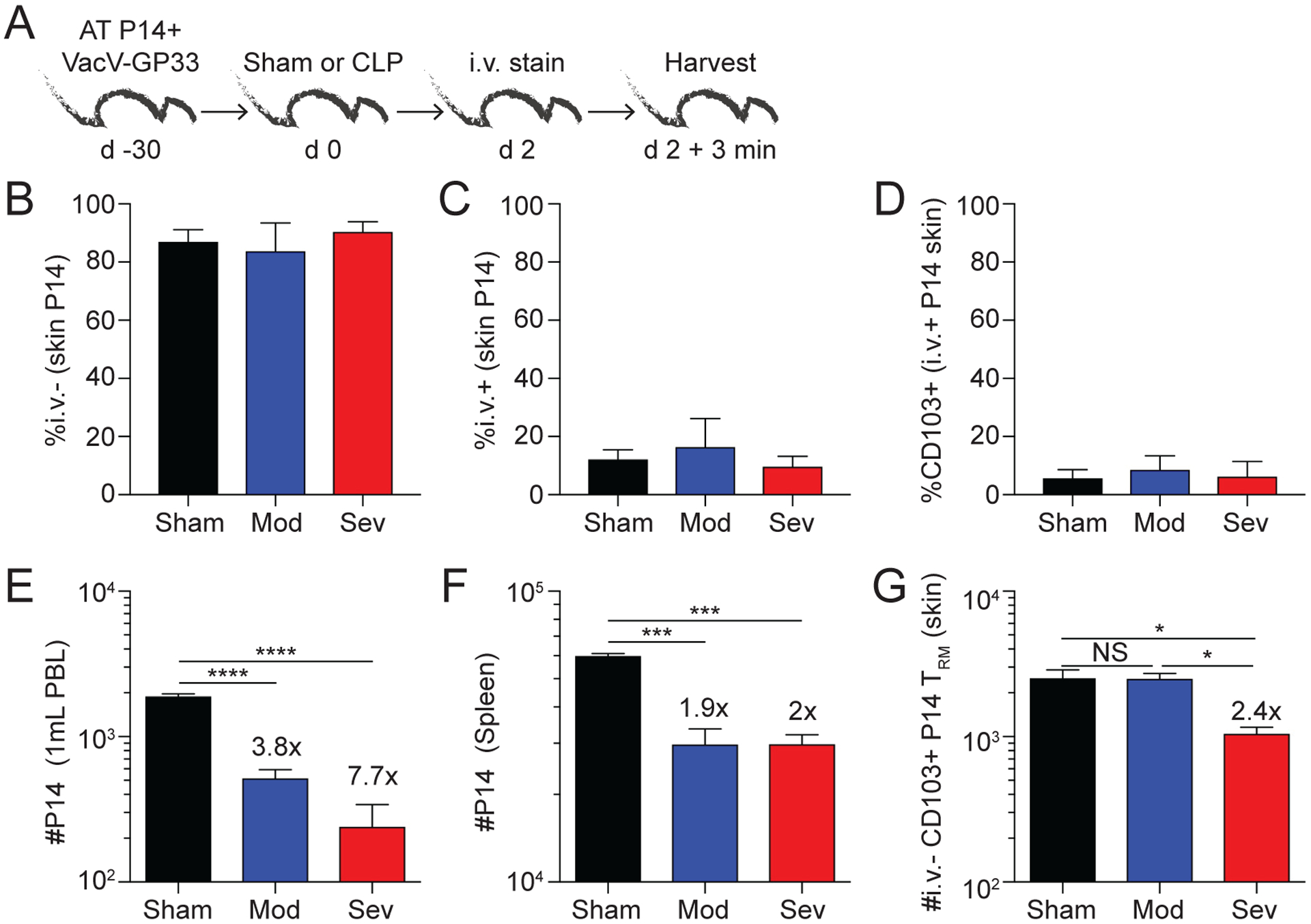
A) Experimental Design. VacV-GP33 immunized mice containing memory P14 CD8 T cells underwent sham or CLP (moderate or severe) surgery. After 2 days, the mice received an i.v. injection of anit-CD45.2 mAb, tissues were collected 3 minutes later, and cells were analyzed by flow cytometry. B) Frequency of CD45.2− (i.v.−) P14 CD8 T cells in the skin 2 days after surgery. C) Frequency of CD45.2+ (i.v.+) P14 CD8 T cells in the skin 2 days after surgery. D) Frequency of CD103+ P14 CD8 T cells of CD45.2+ (i.v.+) P14 CD8 T cells in the skin 2 days after surgery. Number of P14 CD8 TCIRCM cells in E) blood and F) spleen 2 days after surgery. G) Number of CD103+ skin P14 CD8 TRM 2 days after surgery. Fold change is the difference between sham and CLP groups. NS = not significant; * = p<0.05 determined using 1-way ANOVA, * = p<0.05; *** = p<0.001; **** = p<0.0001 using 1-way ANOVA corrected for multiple comparisons using Dunn’s test. Data are representative of 3 independent experiments with at least 3 mice per group. Error bars represent the standard error of the mean.
As expected, both sepsis modalities led to significant reductions in number of P14 TCIRCM in the blood and spleen (Fig 3E, F). In line with previous data (28), moderate sepsis did not influence the number of CD103+ i.v. negative P14 TRM in the skin (Fig 3G). In contrast, severe sepsis led to a 2.4-fold decrease in the total number of P14 TRM (Fig 3G). Thus, the data in Figures 2 and 3 collectively show CD8 TRM, which are protected in nonlymphoid tissues during a moderate septic event, become potentially susceptible to the detrimental effects of the cytokine storm following a severe septic event as a result of breeches in the vascular endothelium.
In vivo imaging of skin CD8 TRM confirms numerical decline after severe sepsis
To formally prove CD8 TRM in the skin undergo numerical decline during a severe septic event, two-photon microscopy – an approach that does not rely on labeling cells with i.v.-injected mAb – was employed to visualize and quantitate bona-fide CD8 TRM. To achieve this, naïve Thy1.1+ P14-RFP T cells (104 cells/mouse) were adoptively transferred into naïve B6 recipients, followed by VacV-GP33 infection the next day. Sham, moderate CLP, and severe CLP surgery was performed 30 days later, and the total number of RFP+ cells was determined in infected ears 2 days post-surgery using in vivo two-photon microscopy (28, 47). We observed a marked decrease in the number of RFP+ P14 CD8 T cells in CLP-Severe mice compared to CLP-moderate mice (Fig 4A, B). Moreover, we saw no difference in the number of RFP+ P14 CD8 T cells when comparing the sham and CLP-moderate groups (Fig 4C). To increase the resolution of the in vivo imaging, the experiment was repeated using GFP+ P14 CD8 T cells transferred at 5-fold higher numbers (5×104 cells/mouse) prior to VacV-GP33 infection (Fig 4F). Importantly, severe sepsis significantly diminished the number of ear-resident GFP+ P14 CD8 T cells compared to Sham and/or moderate sepsis groups (Fig 4G and Suppl Fig 4). Thus, these data confirm the number of tissue-embedded memory CD8 T cells can be differentially affected depending on the severity of the septic insult.
Figure 4. Visualizing the numerical decline skin CD8 TRM by in vivo imaging.
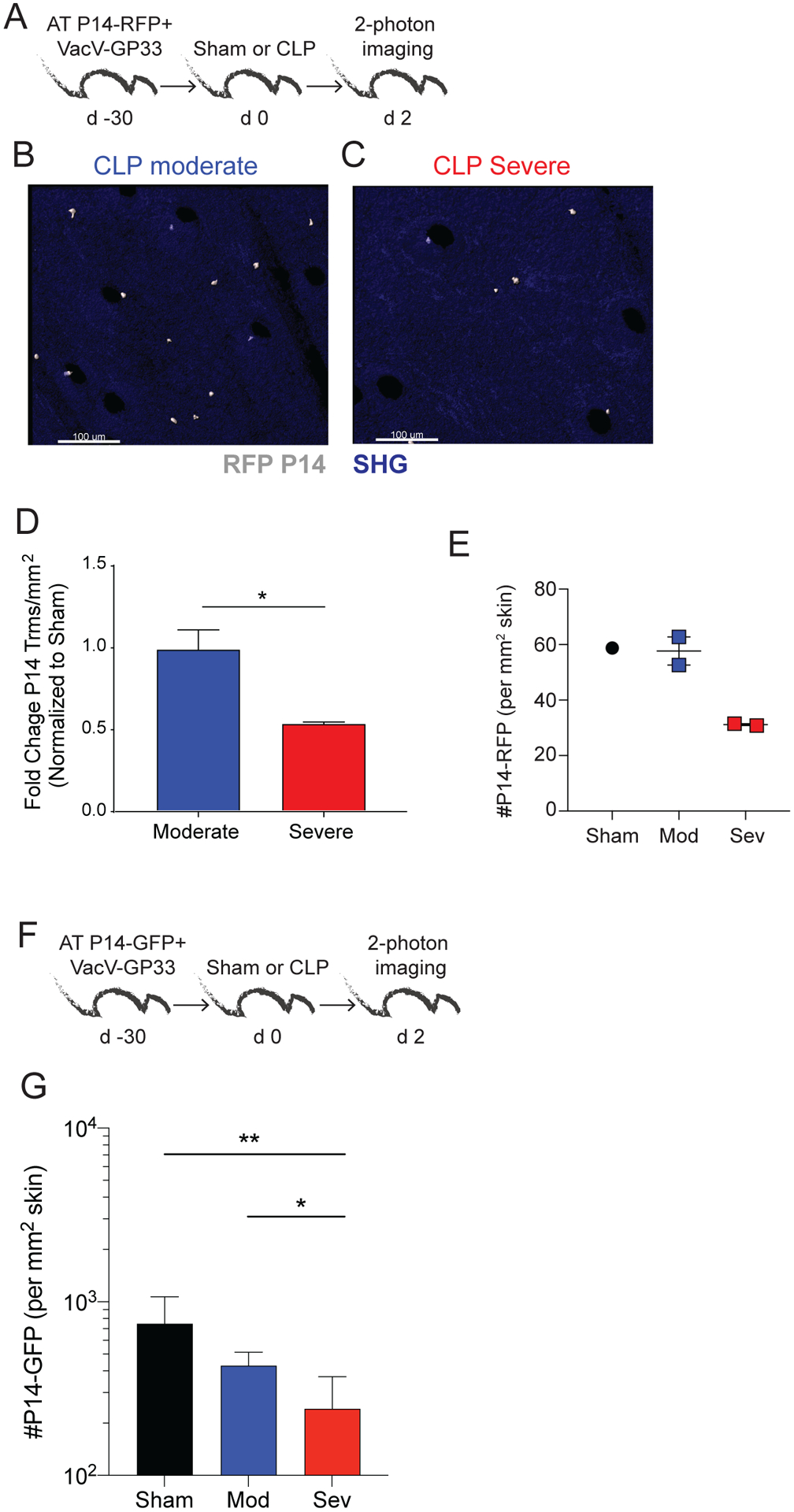
A) Experimental Design. Mice received 104 naïve P14-RFP CD8 T cells followed by VacV-GP33 infection in the ear. Immunized mice received sham or CLP surgery 30 days later. The number of P14-RFP CD8 T cells in the skin was quantified 2 days later using two-photon microscopy. Representative images of P14-RFP CD8 T cells (denoted with asterisk) in the skin of mice after B) CLP moderate and C) CLP severe surgery. D) Fold change of P14 CD8 TRM/mm2 in CLP moderate and severe mice normalized to sham mice 2 days after surgery. E) Number of P14-RFP CD8 T cells/mm2 of skin 2 days after surgery. F) Experimental Design. Mice received 5 × 104 naïve P14-eGFP CD8 T cells followed by VacV-GP33 infection in the ear. Immunized mice received sham or CLP surgery 30 days later. The number of P14-eGFP CD8 T cells in the skin was quantified 2 days later using two-photon microscopy. G) Number of P14-eGFP CD8 T cells/mm2 of skin 2 days after surgery. Data in F-G are from a single experiment with 3–5 mice per group. * = p<0.05; ** = p<0.01; **** = p<0.0001 using two tailed students T-test or 1-way ANOVA corrected for multiple comparisons using Dunn’s test. Error bars represent the standard error of the mean.
Severe sepsis leads to increase in apoptosis of CD8 TRM
We next aimed to determine potential mechanisms responsible for the numerical decline in CD8 TRM during severe sepsis. To determine if increased apoptotic death was occurring, mice were seeded with P14 CD8 T cells, infected with VacV-GP33 to generate CD8 TRM, and then underwent sham, moderate CLP, or severe CLP surgery 30 days later (Fig 5A). On day 2 post-surgery, mice were injected with anti-CD45.2 mAb i.v. and tissues were collected and processed to determine the extent of Caspase3/7 activity (Fig 5A). When examining PBL and P14 CD8 TCIRCM cells from the spleen, we noted a stepwise increase in Caspase3/7+ cells from CLP-moderate and CLP-severe mice compared to Sham controls (Fig 5B–C). Consistent with our previous data (28, 47), moderate sepsis did not result in an increase in Caspase3/7+ CD8 TRM in the skin (Fig 5D), which is supported by the observation of no measurable decline in numbers of these cells (as shown in Fig 3G and 4E). There was, however, a significant increase in frequency of Caspase3/7+ CD8 TRM in the severe sepsis group (Fig 5D). Thus, these data show severe sepsis leads to an increase in apoptotic CD8 TRM.
Figure 5. Severe sepsis leads to increased apoptosis of CD8 TRM.
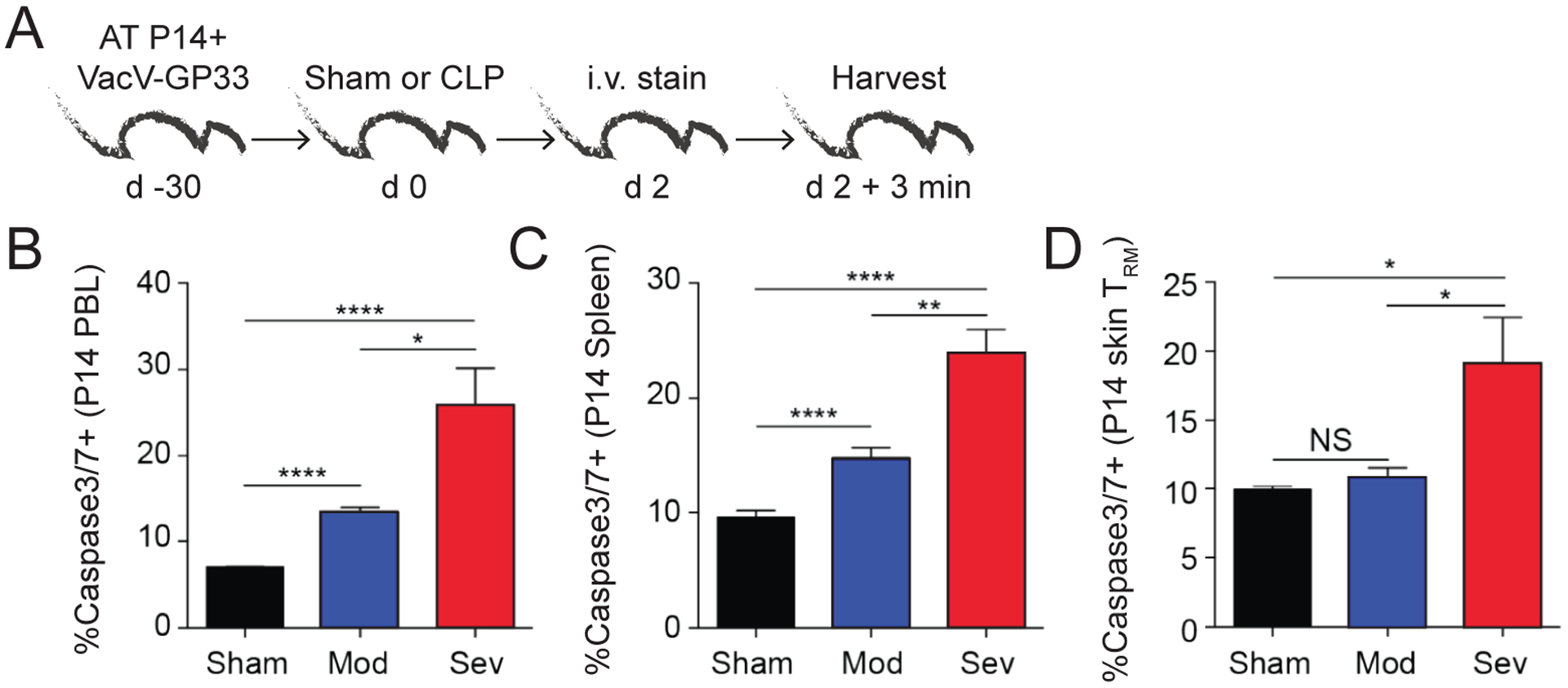
A) Experimental Design. VacV-GP33 immunized mice containing memory P14 CD8 T cells underwent sham or CLP (moderate or severe) surgery. After 2 days, the mice received an i.v. injection of anti-CD45.2 mAb, tissues were collected 3 minutes later, and cells analyzed by flow cytometry. Frequency of activated caspase3/7+ P14 CD8 T cells in the B) PBL, C) spleen, and D) CD103+ skin P14 CD8 TRM 2 days after surgery. Data representative from a single experiment with 3 mice per group. NS = not significant. * = p<0.05; ** = p<0.01; **** = p<0.0001 using 1-way ANOVA corrected for multiple comparisons using Dunn’s test. Error bars represent the standard error of the mean.
The severity of sepsis influences the capacity of skin CD8 TRM to protect against homologous skin infection
Following the observation that skin CD8 TRM decrease in number during severe sepsis, we next examined the functional consequences of this phenomenon. We generated P14 CD8 TRM and performed sham or CLP surgery 30 days later. Groups of naïve, non-immunized B6 mice were included at the time of surgery as infection controls. Mice were then infected with VacV-GP33 in the ear 2 days after surgery. Ears were excised 2 days later and viral load in the skin was measured (Fig 6A). It is important to note that while sepsis does increase the susceptibility of naïve mice to localized VacV skin infection, the initial (up to day 2 post-infection) viral titers were similar in the naïve sham or CLP-moderate groups after primary infection (28). All of the groups of non-vaccinated mice had similar viral titer, indicating the existence of memory CD8 T cells could mediate local protection against viral re-infection (Fig 6B). However, within the vaccinated hosts CLP surgery reduced the level of protection, as both the CLP-moderate and CLP-severe groups had significantly higher viral titers than sham mice (Fig 6B). Furthermore, CLP-severe surgery resulted in even greater loss of protection than CLP-moderate surgery (Fig 6B), confirming the relationship between the severity of the septic event, number of CD8 TRM, and protection they mediate.
Figure 6. The severity of sepsis influences the capacity of skin CD8 TRM to protect against homologous skin infection.
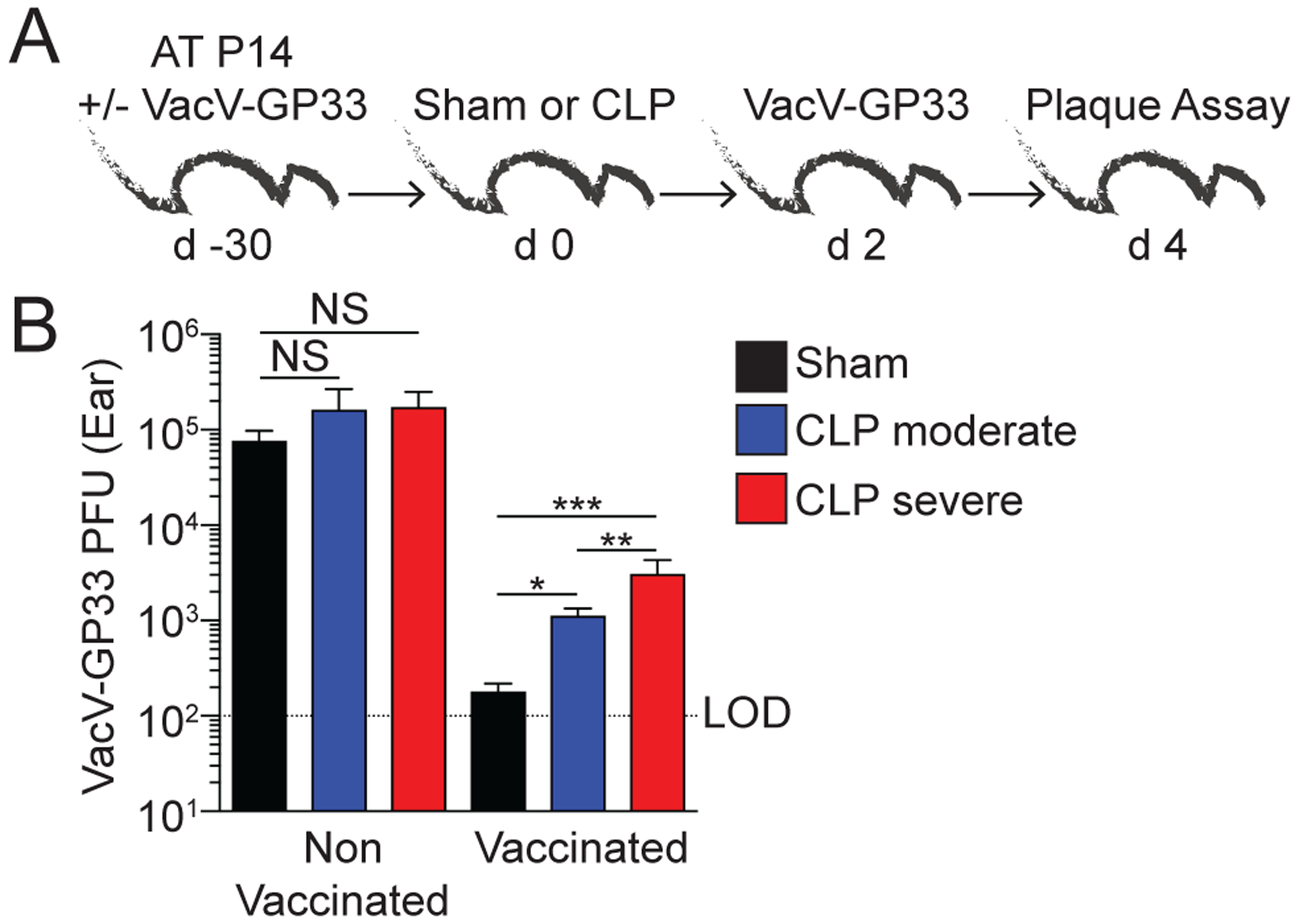
A) Experimental Design. Naïve and VacV-GP33 immunized P14 memory CD8 T cell mice underwent sham or CLP (moderate or severe) surgery. After 2 days, the mice received a homologous skin infection with VacV-GP33. Viral titers in the skin were determined 2 days later. B) VacV-GP33 PFU in the skin 2 days after re-infection. Data are pooled from 3 independent experiments with at least 6 mice per group. NS = not significant. * = p<0.05; ** = p<0.01; *** = p<0.001 using 1-way ANOVA corrected for multiple comparisons using Dunn’s test. Error bars represent the standard error of the mean.
The severity of sepsis controls the numerical loss of tumor-infiltrating and non-specific memory CD8 T cells
The experiments described so far have focused on understanding the impact of sepsis severity on skin CD8 TRM within the context of virus infection. To expand the relevance of our data showing how sepsis, depending on the severity, has a capacity to influence the number and function of CD8 T cells outside of the vasculature, we used the B16 melanoma model to interrogate role of sepsis on tumor-infiltrating virus-specific memory CD8 T cells (TILs). Specifically, naive Thy1.1+ P14 CD8 T cells (104 cells/mouse) were transferred into naïve Thy1.2+ B6 mice prior to LCMV-Armstrong challenge. Once memory had been firmly established (30 d post-infection), all immune mice were subcutaneously challenged with B16 melanoma in the hind flank (40, 41) and then underwent sham or CLP surgery 15 d later (Fig 7A). Of note, tumor-infiltrating virus-specific memory CD8 T cells residing inside tumor mass are considered to be bystander, tumor non-specific, memory CD8 T cells (48) enabling an additional model in which demise in memory CD8 T cell subsets can be interrogated by modulating severity of septic insult.
Figure 7. The severity of sepsis governs the extent of numerical loss of tumor-infiltrating CD8 T cells.
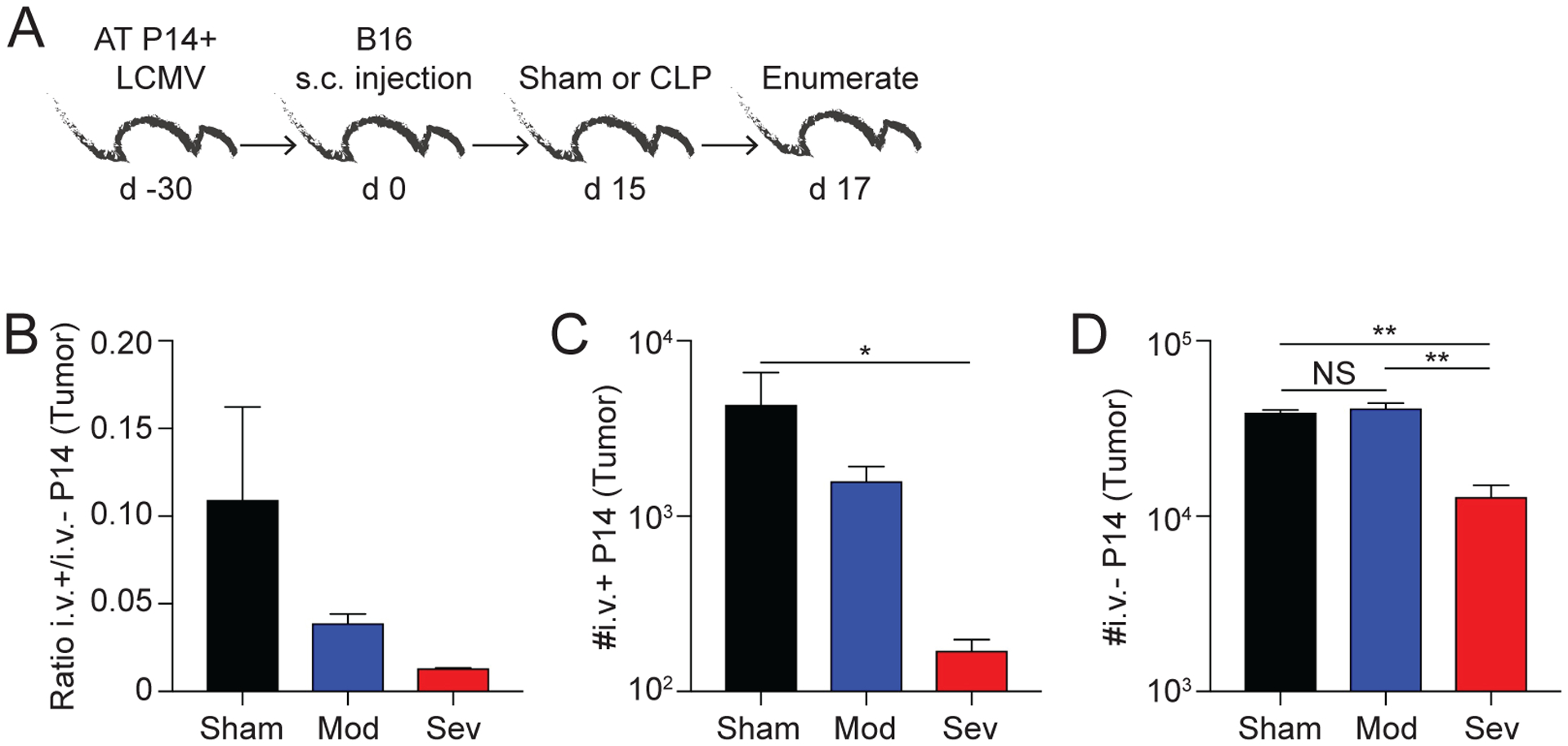
A) Experimental Design. LCMV-immune P14 memory CD8 T cell mice received an s.c. injection of B16 melanoma cells in the hind flank. Mice underwent sham or CLP surgery (moderate or severe) 15 days later. After 2 days, mice received an i.v. injection of anti-CD45.2 mAb and tumors were collected 3 minutes later. B) Ratio of CD45.2+ (i.v.+) to CD45.2- (i.v.−) P14 CD8 T cells in the tumor. Number of C) i.v.+ and D) i.v.− P14 CD8 T cells in the tumor 2 days after surgery. Data are representative from 2 experiments with 3 mice per group. NS = not significant. * = p<0.05; ** = p<0.01 using 1-way ANOVA corrected for multiple comparisons using Dunn’s test. Error bars represent the standard error of the mean.
Tumors were collected after i.v. injection of anti-CD45.2 mAb, and processed to enumerate the virus-specific memory P14 CD8 TILs by flow cytometry (Fig 7A). To determine how the severity of the septic event influences the ratio of memory P14 CD8 T cells in the circulation and within the tumor, the ratio of i.v.+/i.v.− P14 CD8 T cells was analyzed across the groups. We observed a trending decrease in the ratio of i.v.+/i.v.− P14 CD8 T cells in mice receiving CLP surgery compared to mice who received sham surgery, highlighting the correlative loss of TCIRCM with the increased severity of the septic event (Fig 7B). Next, we determined the number of i.v.+ and i.v.− P14 CD8 T cells in the tumor. In both cases, mice that experienced CLP-severe surgery had significantly fewer i.v.+ and i.v.− P14 CD8 T cells compared to sham surgery recipients. By comparison, moderate CLP surgery did not lead to a significant decline in i.v.− P14 CD8 TIL numbers (Fig 7C, D). Collectively, these data show the number of tumor-infiltrating virus-specific bystander memory CD8 T cells present after sepsis is dependent on the severity of the septic insult, which was similar to what was seen for pathogen-induced CD8 TRM present at the site of the infection.
Discussion
Sepsis is a serious medical condition that accounts for ~20% of deaths around the world. Sepsis is characterized by an overreactive immune response to a systemic infection and leads to uncontrolled inflammation, vascular dysfunction, acute lymphopenia, and long-term immunoparalysis. It is important to note that a spectrum of disease severity exists for septic patients: some patients are able to overcome the septic event, while others succumb and die. Our lab and others have employed the CLP model of sepsis because of its similarities in the clinical manifestation and presentation of the disease (49–52). In this report, we used CLP to model the spectrum of disease severity seen in patients. Specifically, we modulated the severity of the septic event by altering the “puncture” portion of the CLP protocol – a single puncture of the cecum was used to induce a moderate septic event, whereas two punctures induced a more severe sepsis. The increased duration and magnitude of the cytokine storm observed in CLP-severe mice models the disease pathogenesis seen in human populations, with more septic patients having increased amounts of cytokine present in the serum (53). Furthermore, CLP-severe resulted in increased vascular dysfunction and more pronounced lymphopenia when compared to CLP-moderate, again better modeling the spectrum of disease severity.
Of the patients that survive a septic event, they display increased susceptibility to secondary infections and viral reactivation due to long-term immunoparalysis (16). Studies have detailed the impact of sepsis on naïve and antigen-experienced CD4 and CD8 T cells in the circulation (25, 39, 54), but the influence of sepsis specifically on CD8 TRM has been neglected until recently (28). Previous data from our lab have demonstrated CD8 TRM are numerically stable and maintain their function in the context of a moderate septic event (0–10% mortality). The maintenance of CD8 TRM number and retention of their function during moderate sepsis is most likely due to their localization in tissue parenchyma, where they are likely shielded from the apoptosis-inducing cytokines circulating in the vasculature. However, vascular junction integrity is lost during a more severe septic event (i.e., one resulting in ~50% mortality), and cytokines (and likely other factors) are now able to leach into the tissue. In this setting, we were now able to detect increased CD8 TRM apoptosis and subsequent numerical decline (which is intent of our experimental design), but we expect other pathophysiological effects were being experienced in the tissue parenchyma in the CLP-severe mice. Our utilization of Evan’s Blue dye to quantify vascular dysfunction supports this hypothesis, but this technique does not give us any indication which anatomical locations (e.g., blood vessel vs capillary bed) are experiencing endothelial barrier disruption. Supernatants from tissues sections may be examined for cytokine levels to confirm elevated levels present in the parenchyma.
A number of laboratories, including ours, have begun to include two-photon microscopy to visualize CD8 TRM in situ (28, 55). We used this methodology to confirm the loss of CD8 TRM in non-lymphoid tissue after a septic event, and our data show significant reduction in the overall TRM cellularity of a tissue. Interestingly, some areas appeared to become completely devoid of CD8 TRM during sepsis, while other areas were spared of this loss. This difference in CD8 TRM susceptibility may be due to their location with respect to blood vessels and capillary beds, allowing parts of the tissues to be bathed in apoptosis-inducing cytokines and other factors, while the localized concentrations in other parts of the tissues did not reach critical levels to trigger apoptosis in those CD8 TRM.
The molecular driver(s) of T cell apoptosis during sepsis has been difficult to ascertain, whether this is for circulating T cells or those resident in non-lymphoid tissues (such as the TRM specifically investigated in this study). Data generated from a number of papers have suggested sepsis-induced T cell apoptosis is regulated by a complex network of pro- and anti-apoptotic proteins within the T cells, which can be further influenced by a number of extracellular factors. Similarly, administration of mAb specific for TIGIT, 2B4, OX40, and PD-1 (among others) can modulate the number of T cells in the circulation or primary lymphoid organs by changing the frequency of apoptotic cells(56–60), but it remains to be determined if these same proteins contribute to the apoptosis of TRM during severe sepsis. Interestingly, recent data by Nedeva et al (61) identified the importance of TREML4 receptor in regulating the inflammatory cytokine response and innate immune cell (i.e., neutrophils) after cecal slurry injection. It would be interesting to use the same CRISPR-screening approach to confirm the importance of the various proteins that have been suggested to control T cell (both circulating and tissue-resident) apoptosis during sepsis, as well as identify other new regulatory proteins
To examine the functional consequences of local CD8 TRM decline after a septic event, mice were given a homologous vaccinia virus challenge. The extent of viral clearance was measured two days after infection via plaque assay. We observed higher viral load in the CLP-severe mice compared to CLP-moderate mice, suggesting the local loss of CD8 TRM resulted in reduced protection from homologous infection. This observation also has important translational implications, where patients who survived a septic event may have lost the memory immune cells responsible for protection against common pathogens at barrier tissues – a primary function of CD8 TRM. While others have documented the effects of sepsis on B cells and antibody production (62, 63), our focus was primarily on the CD8 T cell arm of adaptive immunity. We cannot rule out the contribution of B cells and/or vaccinia-specific antibodies in this homologous re-infection model, and further interrogation may elucidate additional mechanisms that contribute to the loss of protection. Furthermore, these data raise the question of whether septic survivors would benefit from re-vaccination. Our data makes it tempting to speculate secondary (booster) immunizations would help to restore CD8 TRM in barrier tissues of survivors of a severe septic event, as their prior immunity may have waned as a result of sepsis-induced CD8 TRM attrition. To extend our findings to other relevant models, we examined how the severity of sepsis influenced the maintenance of tumor-infiltrating lymphocytes (TILs). CD8 TILs control tumor growth, both in a non-specific (bystander) and antigen-specific manner (41, 64, 65). Since these CD8 TILs are thought to be embedded in the tumors and outside of the circulation, we interrogated how a severe septic event may influence the numbers of this important population of cells. We observed a significant decrease in the number of tissue-infiltrating (i.v.−) P14 CD8 TILs present in the tumor. Since these CD8 T cells are specific for GP33, an antigen not expressed by the B16 melanoma tumor cells, these cells model bystander CD8 TILs. It would be interesting to observe the effect of a severe septic event on tumor-specific CD8 TILs, and the consequence of the loss of these CD8 TILs on tumor growth, in future studies.
In summary, our data collectively show that increasing the severity of the septic event increases the length and magnitude of cytokine production, impairs the barrier function of vascular endothelium, and results in the correlational numerical decline of both the CD8 TCIRCM and TRM compartments. Following a severe septic event, the amount of a number of cytokines (such as IL-10 and TNFα) more than doubled at their peaks when compared to a moderate septic event. Moreover, other cytokines (such as IL-1β and IL-2) were present at 24 hours post sepsis in CLP-severe mice, while they were almost undetectable in CLP-moderate mice. This finding indicates a prolonged and more intense cytokine storm is present in CLP-severe mice, which in turn, may lead to a more profound numerical decline in a number of CD8 T cell memory populations. Furthermore, we showed increased caspase activity indicates increased levels of CD8 TRM apoptosis, likely driving the numerical decline of these cells. As a result of the loss of the CD8 TRM, CLP-severe mice were less protected from viral rechallenge.
Finally, we extended our findings to cancer immunology to show severe sepsis can also lead a numerical decline of tumor-infiltrating bystander (i.e., virus-specific) memory CD8 T cells. These data, together with previously published findings, further elucidate how CD8 TRM can be affected during a septic event and the functional consequences of their numerical decline. These data also give us insight into how blood vessel physiology during sepsis can shape immunological landscapes within tissues. Highly vascularized tissues may experience increased loss of CD8 TRM compared to tissues with lower vascularization due to the amount of apoptotic factors that flow into these tissues during sepsis.
Overall, this report adds important information to the current body of literature surrounding the impact of sepsis on memory CD8 T cells subpopulations, and specifically dissects how the severity of sepsis concordantly influences the numerical decline of CD8 TRM. The translational implication of this study relates directly to the spectrum of disease severity observed in septic humans. It is integral to determine how variation in sepsis severity dictates the extent of impairment in memory CD8 T cell subpopulations as lesions in this population render the host susceptible to secondary, repeated and/or nosocomial infections. Furthermore, our study sheds light on some of the potential mechanisms behind the increase in susceptibility to re-infections among sepsis survivors and informs future studies designed to boost the recovery of protective circulatory and resident memory CD8 T cells after sepsis. Future human studies that analyze the recovery of these memory T cell populations will benefit from the newfound understanding into how the severity of the initial septic insult influences the numbers and function of pre-existing infection and/or vaccine induced memory CD8 T cells.
Supplementary Material
Key Points:
The severity of sepsis influences the cytokine storm and vascular permeability
Skin CD103+ CD8 TRM undergo apoptosis and numerical loss following severe sepsis
Severe sepsis leads to loss of Trm-mediated protection to local viral re-challenge
Acknowledgements
We thank members of our laboratories for technical assistance and helpful discussions.
Supported by NIH Grants GM134880 (V.P.B.), AI114543, AI151183 (V.P.B. and J.T.H.), GM115462 (T.S.G.), R35GM140881 (T.S.G.), T32AI007485 (D.B.D. and I.J.J.), AI42767, AI085515, AI100527, (J.T.H), T32 5T32HL007 (S.M.A). The Holden Comprehensive Cancer Center at The University of Iowa and its National Cancer Institute Award P30CA086862 (V.P.B) and a Veterans Administration Merit Review Award I01BX001324 (T.S.G.).
Footnotes
The authors declare no competing interest
References
- 1.Hotchkiss RS, and Karl IE. 2003. The pathophysiology and treatment of sepsis. N Engl J Med 348: 138–150. [DOI] [PubMed] [Google Scholar]
- 2.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, Klompas M, and Program CDCPE. 2017. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA 318: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin GS, Mannino DM, Eaton S, and Moss M. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348: 1546–1554. [DOI] [PubMed] [Google Scholar]
- 4.Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, Fleischmann-Struzek C, Machado FR, Reinhart KK, Rowan K, Seymour CW, Watson RS, West TE, Marinho F, Hay SI, Lozano R, Lopez AD, Angus DC, Murray CJL, and Naghavi M. 2020. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395: 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultz MJ, Dunser MW, Dondorp AM, Adhikari NK, Iyer S, Kwizera A, Lubell Y, Papali A, Pisani L, Riviello BD, Angus DC, Azevedo LC, Baker T, Diaz JV, Festic E, Haniffa R, Jawa R, Jacob ST, Kissoon N, Lodha R, Martin-Loeches I, Lundeg G, Misango D, Mer M, Mohanty S, Murthy S, Musa N, Nakibuuka J, Serpa Neto A, Nguyen Thi Hoang M, Nguyen Thien B, Pattnaik R, Phua J, Preller J, Povoa P, Ranjit S, Talmor D, Thevanayagam J, and Thwaites CL. 2017. Current challenges in the management of sepsis in ICUs in resource-poor settings and suggestions for the future. Intensive Care Med 43: 612–624. [DOI] [PubMed] [Google Scholar]
- 6.Thompson K, Venkatesh B, and Finfer S. 2019. Sepsis and septic shock: current approaches to management. Intern Med J 49: 160–170. [DOI] [PubMed] [Google Scholar]
- 7.Martin GS, Mannino DM, and Moss M. 2006. The effect of age on the development and outcome of adult sepsis. Crit Care Med 34: 15–21. [DOI] [PubMed] [Google Scholar]
- 8.Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, Goncalves L, Gómez-Herreras JI, de Lejarazu RO, and Bermejo-Martin JF. 2011. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw 22: 82–87. [DOI] [PubMed] [Google Scholar]
- 9.Cohen J 2002. The immunopathogenesis of sepsis. Nature 420: 885–891. [DOI] [PubMed] [Google Scholar]
- 10.Rittirsch D, Flierl MA, and Ward PA. 2008. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 8: 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruse N, Leijte GP, Pickkers P, and Kox M. 2019. New frontiers in precision medicine for sepsis-induced immunoparalysis. Expert Rev Clin Immunol 15: 251–263. [DOI] [PubMed] [Google Scholar]
- 12.Cao C, Yu M, and Chai Y. 2019. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis 10: 782–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donnelly JP, Hohmann SF, and Wang HE. 2015. Unplanned Readmissions After Hospitalization for Severe Sepsis at Academic Medical Center-Affiliated Hospitals. Crit Care Med 43: 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yende S, D’Angelo G, Kellum JA, Weissfeld L, Fine J, Welch RD, Kong L, Carter M, and Angus DC. 2008. Inflammatory markers at hospital discharge predict subsequent mortality after pneumonia and sepsis. Am J Respir Crit Care Med 177: 1242–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotchkiss RS, Monneret G, and Payen D. 2013. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13: 260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, Pachot A, Brooks TL, Deych E, Shannon WD, Green JM, Storch GA, and Hotchkiss RS. 2014. Reactivation of Multiple Viruses in Patients with Sepsis. PLOS ONE 9: e98819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, and Hotchkiss RS. 2010. Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun 78: 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, and Hotchkiss RS. 2011. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA 306: 2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurung P, Rai D, Condotta SA, Babcock JC, Badovinac VP, and Griffith TS. 2011. Immune unresponsiveness to secondary heterologous bacterial infection after sepsis induction is TRAIL dependent. J Immunol 187: 2148–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sjaastad FV, Kucaba TA, Dileepan T, Swanson W, Dail C, Cabrera-Perez J, Murphy KA, Badovinac VP, and Griffith TS. 2020. Polymicrobial Sepsis Impairs Antigen-Specific Memory CD4 T Cell-Mediated Immunity. Front Immunol 11: 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serbanescu MA, Ramonell KM, Hadley A, Margoles LM, Mittal R, Lyons JD, Liang Z, Coopersmith CM, Ford ML, and McConnell KW. 2016. Attrition of memory CD8 T cells during sepsis requires LFA-1. J Leukoc Biol 100: 1167–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie J, Crepeau RL, Chen CW, Zhang W, Otani S, Coopersmith CM, and Ford ML. 2019. Sepsis erodes CD8(+) memory T cell-protective immunity against an EBV homolog in a 2B4-dependent manner. J Leukoc Biol 105: 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotchkiss RS, Tinsley KW, Swanson PE, Schmieg RE Jr., Hui JJ, Chang KC, Osborne DF, Freeman BD, Cobb JP, Buchman TG, and Karl IE. 2001. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 166: 6952–6963. [DOI] [PubMed] [Google Scholar]
- 24.Hotchkiss RS, Coopersmith CM, and Karl IE. 2005. Prevention of lymphocyte apoptosis--a potential treatment of sepsis? Clin Infect Dis 41 Suppl 7: S465–469. [DOI] [PubMed] [Google Scholar]
- 25.Jensen IJ, Sjaastad FV, Griffith TS, and Badovinac VP. 2018. Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J Immunol 200: 1543–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Tulzo Y, Pangault C, Gacouin A, Guilloux V, Tribut O, Amiot L, Tattevin P, Thomas R, Fauchet R, and Drénou B. 2002. Early circulating lymphocyte apoptosis in human septic shock is associated with poor outcome. Shock 18: 487–494. [DOI] [PubMed] [Google Scholar]
- 27.Duong S, Condotta SA, Rai D, Martin MD, Griffith TS, and Badovinac VP. 2014. Polymicrobial sepsis alters antigen-dependent and -independent memory CD8 T cell functions. J Immunol 192: 3618–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Danahy DB, Anthony SM, Jensen IJ, Hartwig SM, Shan Q, Xue HH, Harty JT, Griffith TS, and Badovinac VP. 2017. Polymicrobial sepsis impairs bystander recruitment of effector cells to infected skin despite optimal sensing and alarming function of skin resident memory CD8 T cells. PLoS Pathog 13: e1006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masopust D, and Soerens AG. 2019. Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol 37: 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, Wijeyesinghe SP, Langlois RA, Osum KC, Mitchell JS, and Masopust D. 2020. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med 217: e20192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, Borges da Silva H, Vezys V, Youngblood B, and Masopust D. 2020. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol 21: 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan TN, Mooster JL, Kilgore AM, Osborn JF, and Nolz JC. 2016. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med 213: 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudson CJ, Weiss KA, Hartwig SM, and Varga SM. 2014. The pulmonary localization of virus-specific T lymphocytes is governed by the tissue tropism of infection. J Virol 88: 9010–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haring JS, Corbin GA, and Harty JT. 2005. Dynamic regulation of IFN-gamma signaling in antigen-specific CD8+ T cells responding to infection. J Immunol 174: 6791–6802. [DOI] [PubMed] [Google Scholar]
- 35.Khan SH, Hemann EA, Legge KL, Norian LA, and Badovinac VP. 2014. Diet-induced obesity does not impact the generation and maintenance of primary memory CD8 T cells. J Immunol 193: 5873–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferguson TA, Dube P, and Griffith TS. 1994. Regulation of contact hypersensitivity by interleukin 10. J Exp Med 179: 1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D. 2014. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9: 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sjaastad FV, Jensen IJ, Berton RR, Badovinac VP, and Griffith TS. 2020. Inducing Experimental Polymicrobial Sepsis by Cecal Ligation and Puncture. Curr Protoc Immunol 131: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Condotta SA, Rai D, James BR, Griffith TS, and Badovinac VP. 2013. Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J Immunol 190: 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danahy DB, Kurup SP, Winborn CS, Jensen IJ, Harty JT, Griffith TS, and Badovinac VP. 2019. Sepsis-Induced State of Immunoparalysis Is Defined by Diminished CD8 T Cell-Mediated Antitumor Immunity. J Immunol 203: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Danahy DB, Jensen IJ, Griffith TS, and Badovinac VP. 2019. Cutting Edge: Polymicrobial Sepsis Has the Capacity to Reinvigorate Tumor-Infiltrating CD8 T Cells and Prolong Host Survival. J Immunol 202: 2843–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen IJ, McGonagill PW, Butler NS, Harty JT, Griffith TS, and Badovinac VP. 2021. NK Cell-Derived IL-10 Supports Host Survival during Sepsis. J Immunol 206: 1171–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chousterman BG, Swirski FK, and Weber GF. 2017. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 39: 517–528. [DOI] [PubMed] [Google Scholar]
- 44.Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, Weingarden AR, Jensen IJ, Danahy DB, Badovinac VP, Jameson SC, Vezys V, Masopust D, Khoruts A, Griffith TS, and Hamilton SE. 2019. Microbial Exposure Enhances Immunity to Pathogens Recognized by TLR2 but Increases Susceptibility to Cytokine Storm through TLR4 Sensitization. Cell Rep 28: 1729–1743 e1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang JC 2019. Sepsis and septic shock: endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb J 17: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baccala R, Welch MJ, Gonzalez-Quintial R, Walsh KB, Teijaro JR, Nguyen A, Ng CT, Sullivan BM, Zarpellon A, Ruggeri ZM, de la Torre JC, Theofilopoulos AN, and Oldstone MB. 2014. Type I interferon is a therapeutic target for virus-induced lethal vascular damage. Proc Natl Acad Sci U S A 111: 8925–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurup SP, Anthony SM, Hancox LS, Vijay R, Pewe LL, Moioffer SJ, Sompallae R, Janse CJ, Khan SM, and Harty JT. 2019. Monocyte-Derived CD11c(+) Cells Acquire Plasmodium from Hepatocytes to Prime CD8 T Cell Immunity to Liver-Stage Malaria. Cell Host Microbe 25: 565–577.e566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, Pennell CA, Blazar BR, Chen CC, Geller MA, Vezys V, and Masopust D. 2019. Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun 10: 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW 3rd, Bland KI, and Chaudry IH. 2005. Cecal ligation and puncture. Shock 24 Suppl 1: 52–57. [DOI] [PubMed] [Google Scholar]
- 50.Rittirsch D, Huber-Lang MS, Flierl MA, and Ward PA. 2009. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 4: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deitch EA 2005. Rodent models of intra-abdominal infection. Shock 24 Suppl 1: 19–23. [DOI] [PubMed] [Google Scholar]
- 52.Remick DG, Newcomb DE, Bolgos GL, and Call DR. 2000. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock 13: 110–116. [DOI] [PubMed] [Google Scholar]
- 53.Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, and Nagarkatti M. 2013. Role of cytokines as a double-edged sword in sepsis. In Vivo 27: 669–684. [PMC free article] [PubMed] [Google Scholar]
- 54.Cabrera-Perez J, Condotta SA, James BR, Kashem SW, Brincks EL, Rai D, Kucaba TA, Badovinac VP, and Griffith TS. 2015. Alterations in antigen-specific naive CD4 T cell precursors after sepsis impairs their responsiveness to pathogen challenge. J Immunol 194: 1609–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, and Masopust D. 2018. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol 19: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Y, Anyalebechi JC, Sun H, Yumoto T, Xue M, Liu D, Liang Z, Coopersmith CM, and Ford ML. 2021. Anti-TIGIT differentially affects sepsis survival in immunologically experienced versus previously naive hosts. JCI Insight 6: e141245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie J, Chen CW, Sun Y, Laurie SJ, Zhang W, Otani S, Martin GS, Coopersmith CM, and Ford ML. 2019. Increased attrition of memory T cells during sepsis requires 2B4. JCI Insight 4: e126030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherwood ER, and Williams DL. 2021. Reversal of sepsis-induced T cell dysfunction: OX-40 to the rescue? J Leukoc Biol 109: 689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Unsinger J, Walton AH, Blood T, Tenney DJ, Quigley M, Drewry AM, and Hotchkiss RS. 2021. Frontline Science: OX40 agonistic antibody reverses immune suppression and improves survival in sepsis. J Leukoc Biol 109: 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakamori Y, Park EJ, and Shimaoka M. 2020. Immune Deregulation in Sepsis and Septic Shock: Reversing Immune Paralysis by Targeting PD-1/PD-L1 Pathway. Front Immunol 11: 624279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nedeva C, Menassa J, Duan M, Liu C, Doerflinger M, Kueh AJ, Herold MJ, Fonseka P, Phan TK, Faou P, Rajapaksha H, Chen W, Hulett MD, and Puthalakath H. 2020. TREML4 receptor regulates inflammation and innate immune cell death during polymicrobial sepsis. Nat Immunol 21: 1585–1596. [DOI] [PubMed] [Google Scholar]
- 62.Gustave CA, Gossez M, Demaret J, Rimmele T, Lepape A, Malcus C, Poitevin-Later F, Jallades L, Textoris J, Monneret G, and Venet F. 2018. Septic Shock Shapes B Cell Response toward an Exhausted-like/Immunoregulatory Profile in Patients. J Immunol 200: 2418–2425. [DOI] [PubMed] [Google Scholar]
- 63.Sjaastad FV, Condotta SA, Kotov JA, Pape KA, Dail C, Danahy DB, Kucaba TA, Tygrett LT, Murphy KA, Cabrera-Perez J, Waldschmidt TJ, Badovinac VP, and Griffith TS. 2018. Polymicrobial Sepsis Chronic Immunoparalysis Is Defined by Diminished Ag-Specific T Cell-Dependent B Cell Responses. Front Immunol 9: 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smazynski J, and Webb JR. 2018. Resident Memory-Like Tumor-Infiltrating Lymphocytes (TIL(RM)): Latest Players in the Immuno-Oncology Repertoire. Front Immunol 9: 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong B-H, Koh T, Hillmer AM, Takano A, Lim TKH, Tan EH, Zhai W, Tan DSW, Tan IB, and Newell EW. 2018. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557: 575–579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.