

Abstract
Stearoyl-CoA desaturases (SCD) are endoplasmic reticulum (ER)-associated enzymes that catalyze the synthesis of the monounsaturated fatty acids (MUFAs). As such, SCD play important roles in maintaining the intracellular balance between saturated fatty acid (SFAs) and MUFAs. The roles of SCD in CD4+ T-helper cell responses are currently unexplored. Here, we have found that murine and human follicular helper T (TFH) cells express higher levels of SCD compared to non-TFH cells. Further, the expression of SCD in TFH cells is dependent on the TFH lineage-specification transcription factor BCL6. We found that the inhibition of SCD impaired TFH cell maintenance and shifted the balance between TFH and follicular regulatory T (TFR) cells in the spleen. Consequently, SCD inhibition dampened germinal center B-cell responses following influenza immunization. Mechanistically, we found that SCD inhibition led to increased ER stress and enhanced TFH cell apoptosis in vitro and in vivo. These results reveal a possible link between fatty acid metabolism and cellular and humoral responses induced by immunization or potentially, autoimmunity.
Keywords: Endoplasmic reticulum stress, Follicular help T cells, Germinal center B cells, Lipid metabolism, Stearoyl-CoA desaturase 1
Introduction
Follicular helper T (TFH) cells are specialized effector CD4+ T helper cells that promote the formation of germinal centers (GCs) and the production of class-switched high-affinity immunoglobulins [1]. TFH cells are required for the generation of antibody-mediated protection against microbes, but aberrant TFH responses may cause autoimmunity [2]. Therefore, it is important to understand the positive and/or negative regulators of TFH-cell responses for the proper induction during vaccination or the inhibition of TFH responses during autoimmunity.
TFH differentiation in vivo is a multifactorial multistep process. The transcription factor BCL6 is considered the master regulator for the differentiation of TFH cells and subsequent GC responses [3, 4]. Interestingly, recent evidence suggests that cell metabolic processes may play important roles in modulating TFH development in vivo. BCL6 was shown to repress glycolysis in activated CD4+ T cells [5, 6]. Consistently, it has been demonstrated that TFH cells exhibit diminished glycolysis and mitochondrial respiration compared to TH1 cells [7]. However, a recent study has also suggested mTOR kinase complex 1 (mTORC1)-related glucose metabolism is required for the TFH generation in vivo [8]. Therefore, the roles of glucose metabolism in negative and positive regulation of TFH differentiation and/or maintenance remain to be elucidated.
Besides glucose metabolism, emerging evidence has suggested that lipid metabolism also plays important roles in regulating T-helper cell responses. The inhibition of acetyl-CoA carboxylase 1 (ACC1), a key enzyme for fatty acid synthesis, reduces human and mouse TH17 cell development in favor of regulatory T (Treg) cell differentiation [9]. Simvastatin, the inhibitor for 3-hydroxy-3-methyglutaryl (HMG)-CoA reductase, has been reported to promote Treg differentiation whereas it suppresses TH17 development [10]. Furthermore, the suppression of fatty acid synthase (FASN) during T-cell activation reduces the development of CD4+ T cell memory [11]. Currently, the roles of lipid metabolism in TFH differentiation have not been explored. Stearoyl-CoA desaturases (SCD) are endoplasmic reticulum (ER) proteins that catalyze the synthesis of monounsaturated fatty acids (MUFAs) from saturated fatty acids (SFAs). Besides the needs of MUFAs for the synthesis of complex lipids, such as diacyglycerols (DG), triglycerides (TGs), and cholesterol esters, MUFAs also have important functions in cell signaling and membrane fluidity [12]. Therefore, SCD are highly regulated and conserved enzymes that play important roles in regulating obesity, insulin resistance, inflammation, and cancer progression [13, 14]. Humans have two SCD isoforms (SCD1 and SCD5), while mouse has four (SCD1–4) that share homology with human SCD1, but not human SCD5 [15, 16]. The roles of SCD in regulating adaptive lymphocyte responses have not been explored.
Given the importance of lipid synthesis and metabolism in adaptive immunity, we examined the roles of SCD in TFH responses. We found that SCD1 was highly expressed in both human and murine CD4+CD44+PD1hiCXCR5hi TFH cells compared to non-TFH cells. BCL6-deficient CD4+ T cells exhibited diminished Scd1 gene expression. We showed that in vivo inhibition of SCD activity impaired the maintenance of TFH and GC B cells. Furthermore, SCD inhibitor increased apoptosis in TFH cells in vivo and in vitro. Finally, the inhibition of SCD promoted the expression of ER stress genes whereas SCD1 overexpression showed opposite results in TFH cells. Our results suggest that BCL6-mediated SCD expression promotes TFH-cell maintenance and efficient GC B-cell responses in vivo.
Results
SCD inhibitor suppresses TFH and GC B-cell responses
To explore the function of lipid metabolism in regulating TFH and GC B-cell responses, we immunized influenza X31 virus and then treated mice with Statin (an inhibitor of HMG-CoA reductase that is required cholesterol synthesis) [17], C75 (an inhibitor of FASN that is required for the generation of long-chain fatty acids) [18], or A939572 (an inhibitor of SCD1) [19]. Since lipid metabolism can potentially regulate APC function and early T-cell activation [20, 21], we administered these inhibitors at day 4 postimmunization to minimize the effects of these inhibitors on early T-cell priming. We then checked TFH (CD4+CD44+PD1hiCXCR5hi), non-TFH (CD4+CD44+PD1low/−CXCR5low/−), and GC B- (B220+FAS+ GL-7+) cell responses in the spleen at 14 days postimmunization. We found that both Statin and A939572 markedly diminished TFH magnitude, but only moderately decreased non-TFH responses (Fig. 1A). A939572, but not Statin, also diminished GC B-cell responses following influenza immunization (Fig. 1A). In contrast, C75 treatment had no effects on TFH or GC B-cell responses (Fig. 1A). SCD inhibitor did not alter the total number of CD4+ T and B cells (Fig. 1B), nor the production of TH1 cytokine IFN-gamma in CD4+ and CD8+ T cells following antigen-specific stimulation (Fig. 1C). These results suggest that SCD inhibition selectively impairs TFH but not TH1-cell formation following influenza immunization.
Figure 1.
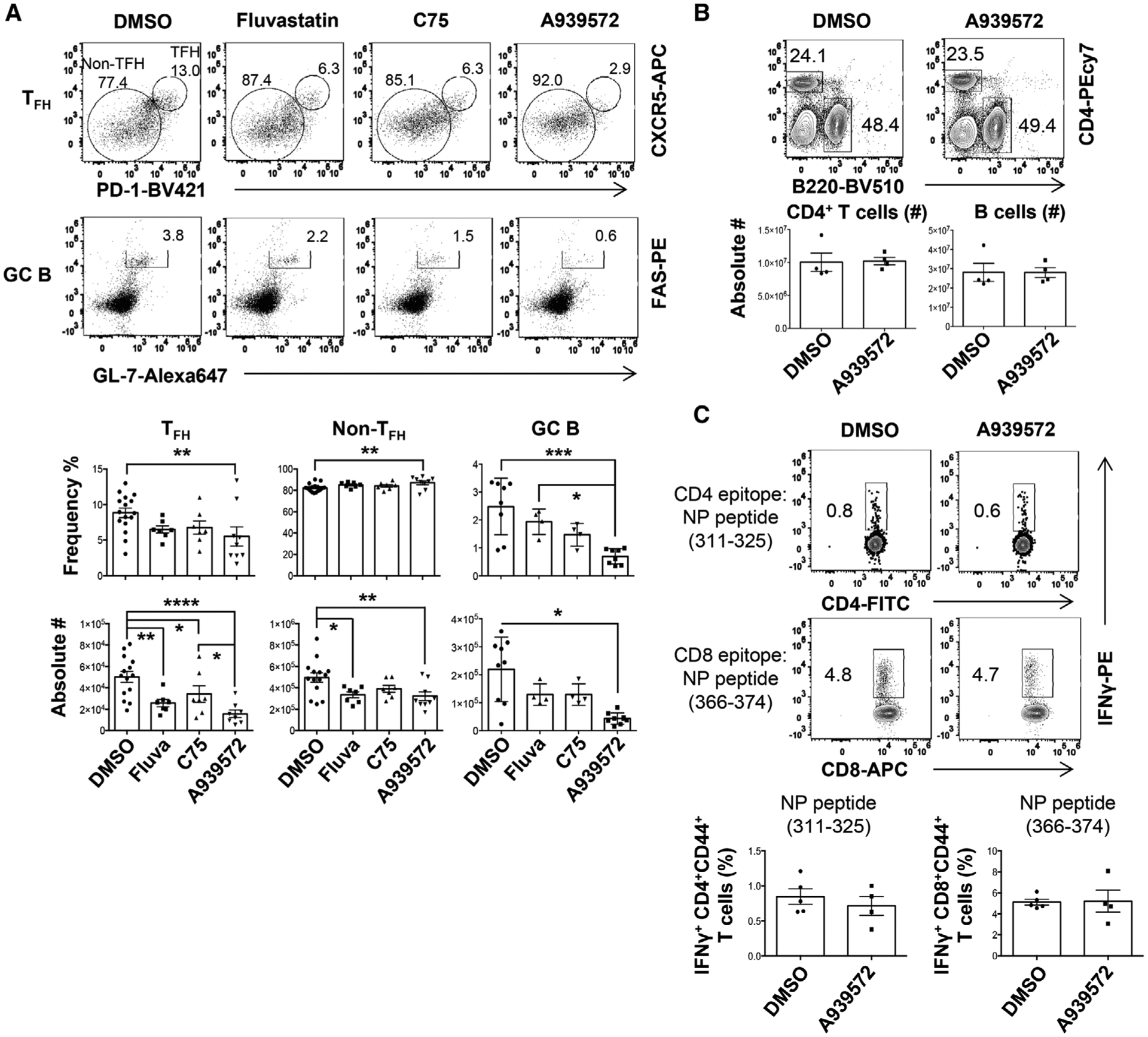
SCD inhibition suppresses TFH but not TH1 responses. WT B6 mice were immunized with X31 and treated with indicated inhibitors starting at 4 days postimmunization (d.p.i.). (A) Frequencies and absolute cell numbers of splenic TFH, non-TFH, GC B cells were measured by flow cytometry at 14 d.p.i. (B) Representative dot plot and absolute cell number of splenic CD4+ T or B cells at 14 d.p.i. (C) IFN-γ production by CD4+ or CD8+ T cells were measured through intracellular staining (ICS) following restimulation with NP311–325 (CD4+ T-cell epitope) or NP366–374 (CD8+ T-cell epitope) peptides in vitro at 14 d.p.i. Combined results in A are obtained from three independent experiments (two to five mice per group). Representative data in B and C are obtained from at least two independent experiments (four to five mice per group). Results are given as mean ± SE with one-way ANOVA (A) or mean ± SEM unpaired t-test (B, C). *indicates significant differences (p < 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001.
SCD are highly expressed by human and murine TFH cells
To identify the expression of SCD in the TFH and non-TFH cells, we first analyzed a published microarray data set (GEO# GSE50391) of CD45RO+CXCR5high, CD45RO+CXCR5int, and CD45RO+CXCR5− from human tonsil [22]. We found that CD45RO+CXCR5high GC-TFH cells exhibit significantly higher SCD1 expression compared to non-TFH cells (Fig. 2A). Mouse TFH cells isolated from KLH/CFA immunized animals also exhibited higher levels of Scd1 and Scd2 relative to non-TFH cells [23] (GEO# GSE40068) (Fig. 2B). In another published RNAseq dataset, TFH cells, particularly LN and splenic TFH cells, had enhanced Scd1 and Scd2 genes expressed relative to conventional non-TFH cells (TConv) [24] (GEO #: GSE124883) (Fig. 2C). However, the expression of Fasn was comparable between non-TFH and TFH cells (Fig. 2B and C). We then sorted murine TFH and non-TFH (CD4+CD44+PD1−CXCR5−) from spleens of day 14 postinfluenza (X31) immunized mice. We examined Scd1, Scd2, and Fasn expression in TFH and non-TFH by quantitative RT-PCR. We found that TFH cells exhibited drastic enhanced levels of Scd1 and modest increased Scd2 expression compared with non-TFH cells (Fig. 2D). Together, these results reveal that both murine and human TFH cells have enhanced Scd gene expression.
Figure 2.
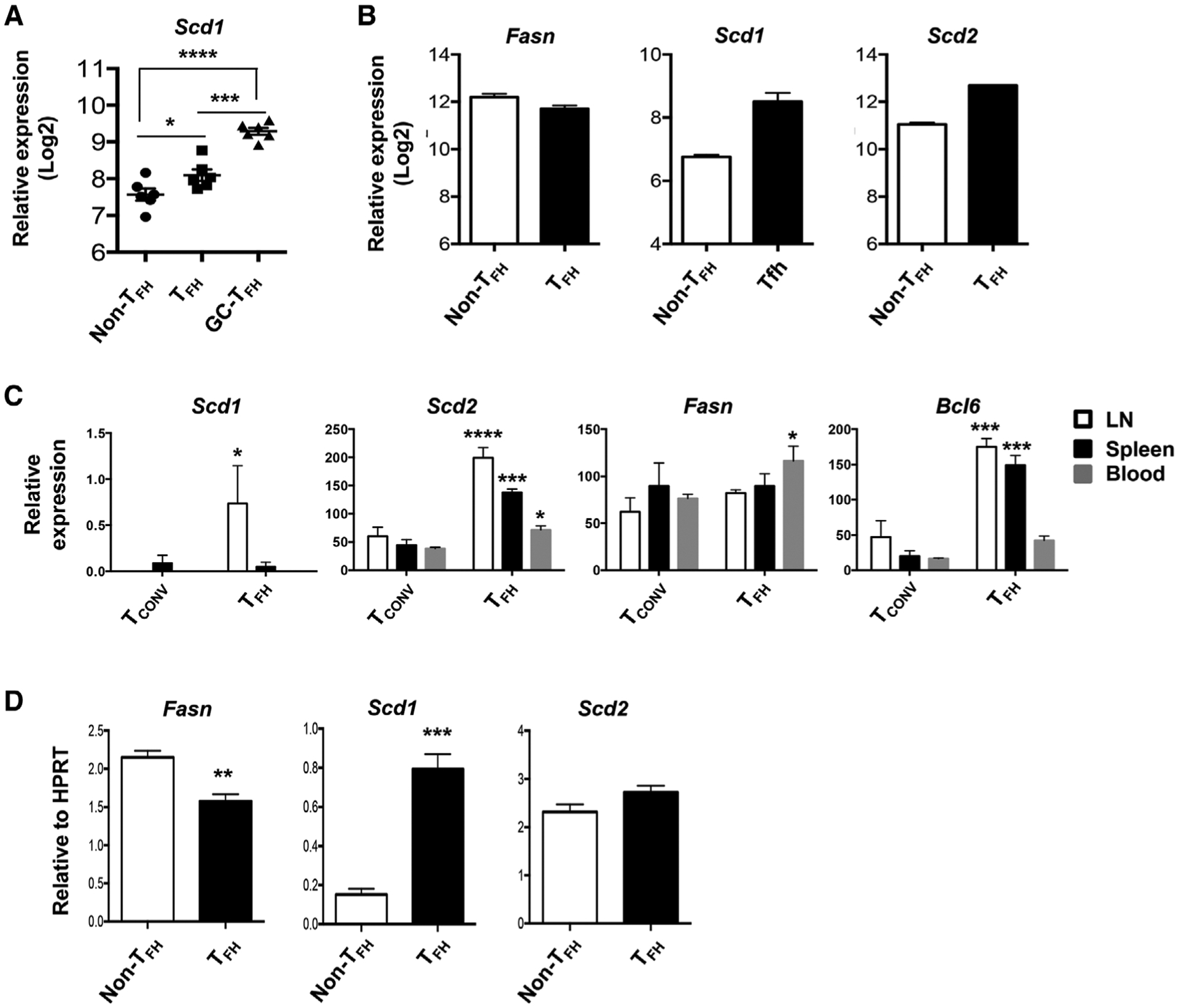
Human and mouse TFH express higher levels of SCD mRNA. (A) Relative SCD1 expression in published microarray data (GEO# GSE50391) CD45RO+CXCR5− (non-TFH), CD45RO+CXCR5int (TFH), and CD45RO+CXCR5hi (GC-TFH) cells from human tonsils (five donors per group). (B) Relative expression of Fasn, Scd1, and Scd2 in published microarray data (GEO# GSE40068) from BCL6hiCXCR5+ (TFH) and BCL6−CXCR5− (non-TFH) cells of the mice immunized with KLH in CFA (two samples each pooled from three mice per group). (C) Relative expression of Fasn, Scd1, Scd2, and Bcl6 in published RNAseq data (GEO# GSE124883) from LN, spleen, or blood TFH and conventional T cells (Tconv) following NP-OVA immunization (three samples each pooled form ten mice per group). (D) Relative expression of Fasn, Scd1, and Scd2 in sorted splenic TFH (PD-1+CXCR5+) and non-TFH (PD1−CXCR5−) cells isolated from X31 immunized mice at 14 d.p.i. Representative results of (D) obtained from two independent experiments (five mice per groups). Results are given as mean ± SEM with one- (A) or two- (C) way ANOVA or unpaired t-test (D). *indicates significant differences (p < 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001.
BCL6 promotes SCD expression in TFH cells
To determine whether TFH master transcription factor BCL6 is involved in Scd gene expression, we measured Scd1 and Scd2 expression in vitro cultured WT or BCL6-deficient (BCL6 KO) CD4+ T cells under the TFH-like polarization conditions. Both Scd1 and Scd2 were diminished in BCL6 KO CD4+ T cells (Fig. 3A). Western blot analysis confirmed that BCL6 deficiency decreased SCD1 protein levels in CD4+ T cells under TFH polarization conditions (Fig. 3B). We next preactivated naive CD4+ T cells for 2 days and then transduced the cells with BCL6-expressing retroviruses. Scd1 and Scd2 were measured in the isolated transduced cells. The BCL6-overexpressed cells exhibited significantly enhanced Scd1, and modestly upregulated Scd2 expression (Fig. 3C). To probe the relationship between BCL6 and SCD1 in vivo, we adoptively transferred WT OTII TCR-transgenic T cells or BCL6 KO-OTII cells into CD45.1 congenic mice. Following adoptive transfer, mice were immunized with X31-OTII virus. At day 14 postimmunization, OTII cells were sorted for measuring Scd1 and Scd2 expression. Similar to in vitro culture results, the expression of Scd1 was impaired in BCL6 KO-OTII cells (Fig. 3D). Together, these results demonstrated that BCL6 promotes the expression of SCD expression in TFH cells.
Figure 3.
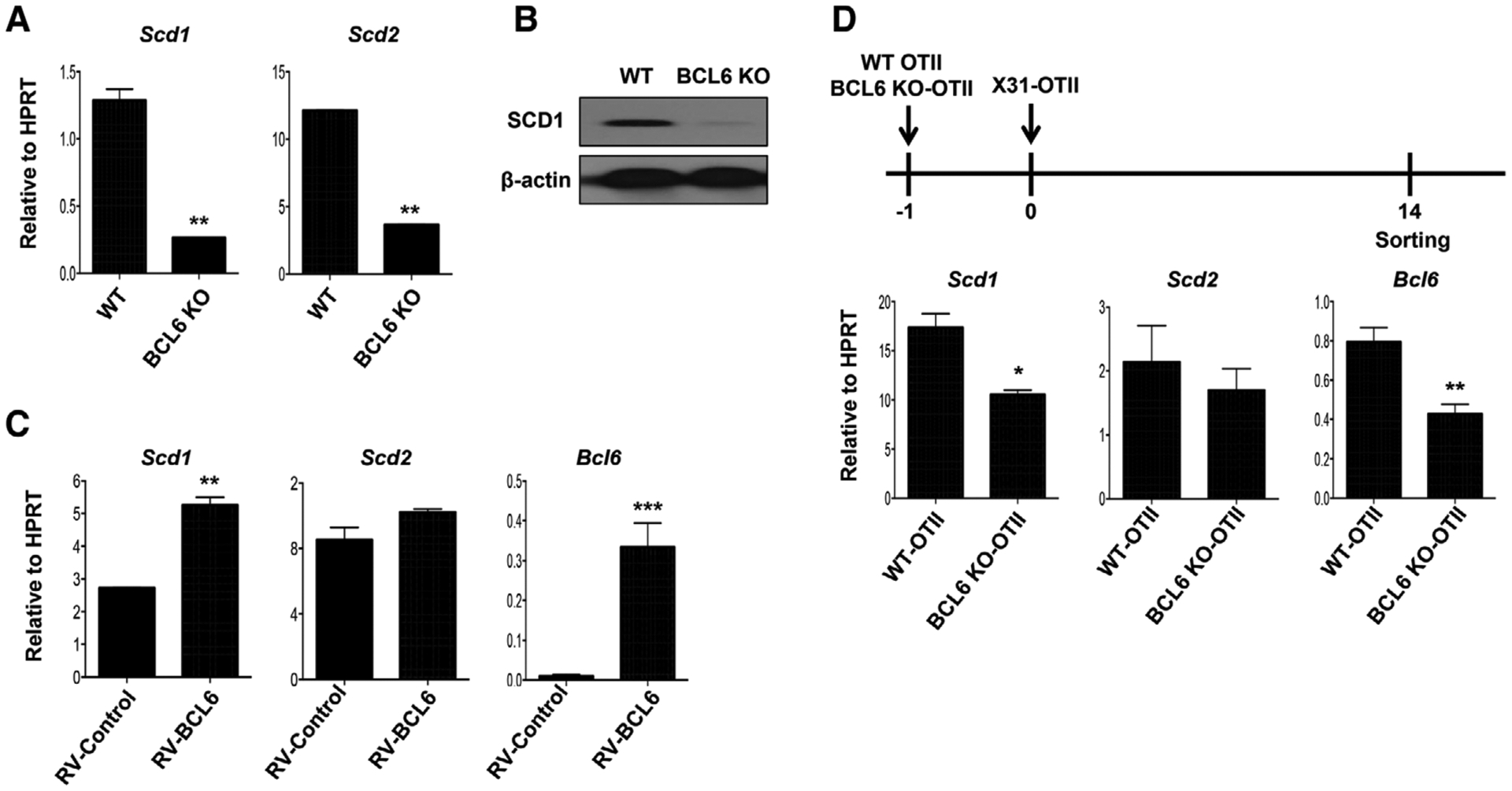
BCL6 promotes SCD1 expression. (A) Relative expression of Scd1 and Scd2 in WT or BCL6 KO CD4+ T cells under the TFH cell polarizing condition for 4 days. (B) Western blot analysis of SCD1 protein levels in WT or BCL6 KO CD4+ T cells under the TFH cell polarizing condition for 5 days. (C) Scd1, Scd2, and Bcl6 expression in the BCL6-retroviral transduced CD4+ T cells under the TFH cell polarizing condition. (D) WT or BCL6 KO-OTII cells were transfer into CD45.1 congenic mice at one day before X31-OTII virus immunization. After day 14 postimmunization, the OTII cells were sorted and then Scd1 and Scd2 were measured by quantitative RT-PCR (three mice per group in two independent experiments). Representative graphs are obtained from at least two (A, B, and C) independent experiments (two mice per group). Results are given as mean ± SEM with unpaired t-test: *p < 0.05; **p < 0.01; and ***p < 0.001.
SCD inhibitor promotes accelerated contraction of TFH and GC B cells
Next, we examined whether SCD inhibition is required for development and/or maintenance of TFH and/or GC B cells. To this end, the frequencies and cell numbers of TFH and GC B cells were measured 0, 8, 10, and 14 days post-X31 immunization following SCD inhibition. SCD inhibitor significantly diminished both TFH and GC B-cell responses at day 14, but not at day 8 or 10 postimmunization (Fig. 4A and B). Notably, TFH-cell numbers begun to diminish from day 10 postimmunization and SCD inhibitor accelerated the contraction of TFH cells in vivo. The expression of BCL6 and ICOS in the TFH cells between vehicle (DMSO)- and SCD inhibitor-treated group were comparable (Supporting Information Fig. 1). These data suggested that SCD inhibition may not suppress the formation of TFH and GC B cells, but promotes accelerated contraction of TFH cells (i.e. inhibition of TFH maintenance). Recently, T-follicular regulatory (TFR) (CD4+CD44+PD1+CXCR5+Foxp3+) cells have been reported as a major regulator repressing excessive TFH and GC B-cell responses [25]. We found that SCD inhibition impaired TFH but not TFR-cell magnitude (Fig. 4C and D). As such, the frequencies of TFR cells were inflated relative to TFH cells following SCD inhibition (Fig. 4C). This indicates that SCD inhibition alters the balance of TFH and TFR responses. Consequently, influenza-specific IgG antibody production was impaired in the serum following SCD inhibition (Fig. 4E). Together, these results suggest that SCD is required for the maintenance of TFH and GC B cells through the regulation of the balance between TFH and TFR cells in the spleen.
Figure 4.
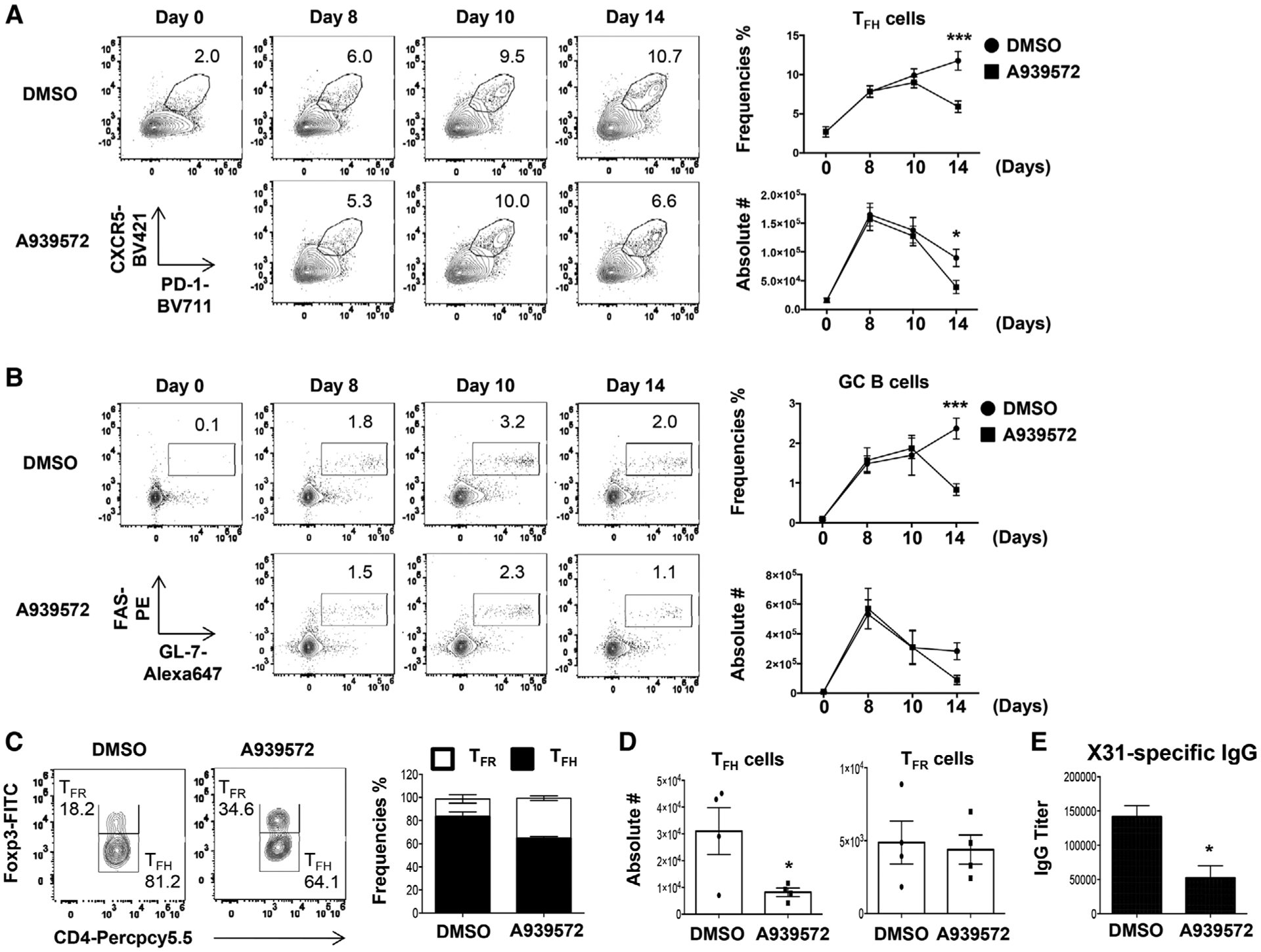
Inhibition of SCD suppresses TFH and GC B maintenance, but not TFR responses. (A-B) Frequencies and absolute cell number of splenic TFH and GC B cells in the mice immunized with X31 virus and treated with SCD inhibitor at indicated d.p.i. (C) Frequencies and (D) cell numbers of splenic TFR and TFH cells were measured in the mice immunized with X31 virus and treated with SCD inhibitor at 14 d.p.i. (E) The titer of influenza-specific IgG was measured by ELISA from blood serum at 14 d.p.i. Representative data are obtained from at least two independent experiments (two to four mice per group). Results are given as mean ± SE with two-way ANOVA (A and B) or unpaired t-test (C and E): *p < 0.05; and ***p < 0.001.
Inhibition of SCD promotes apoptosis and ER stress gene expression
To probe the underlying mechanism, we examined T-cell apoptosis and proliferation following SCD inhibitor treatment. At day 8 post-X31 immunization, active caspase 3/7 activities in TFH cells following SCD inhibitor treatment were similar to vehicle (DMSO)-treated group. However, at day 14 postimmunization, caspase 3/7 activities were significantly higher in TFH cells following SCD inhibition compared to DMSO-treated group (Fig. 5A). In contrast to apoptosis, SCD inhibition had no effects on the proliferation of TFH cells at both day 8 and 14 post-X31 immunization (Fig. 5B). In vitro cell apoptosis was significantly enhanced following SCD inhibitor treatment in a dose-dependent manner under TFH-polarizing conditions (Fig. 5C). Taken together, these data demonstrate that SCD activity promotes TFH-cell viability. A recent study demonstrated excessive accumulation of SFAs following SCD inhibition promotes ER stress-related cell apoptosis [26]. Thus, we examined whether SCD inhibition promoted the expression of ER stress-related genes. The expression of activating transcription factor (Atf) 4 and Atf3 were increased in CD4+ T cells following SCD inhibition (Fig. 5D). ATF4 protein levels were also markedly increased following SCD inhibition (Fig. 5E). Furthermore, the expression of multiple ER stress-related genes including Atf4, Atf3, Perk, and sXbp1, were downregulated in CD4+ T cells transduced with SCD1-expressing retroviruses (Fig. 5F). These results suggest that SCD promote TFH-cell survival likely due to their regulation of ER stress genes.
Figure 5.
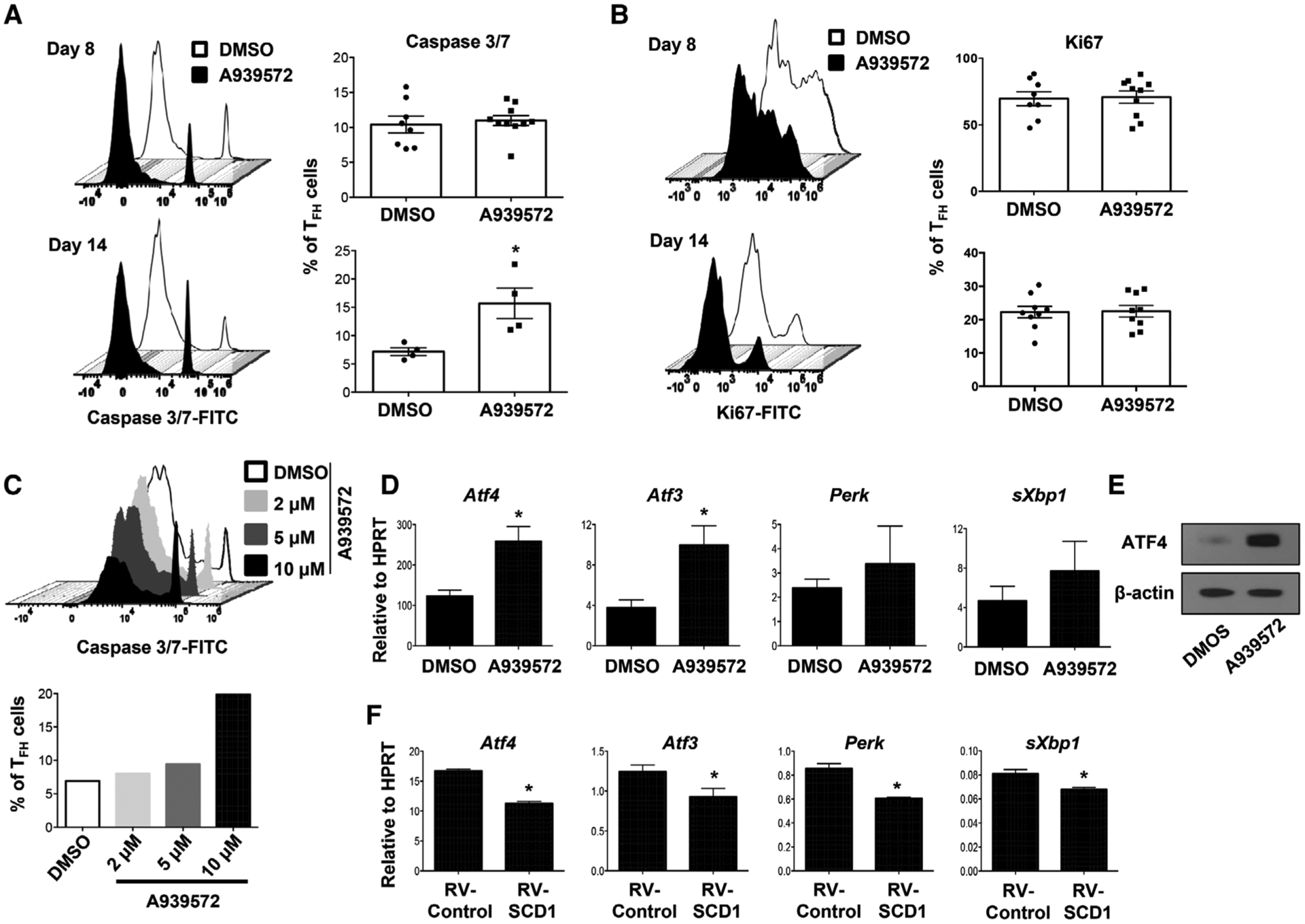
SCD inhibition promotes TFH apoptosis and ER stress gene expression. (A) Caspase 3/7 activity and (B) proliferation (indicated by Ki67 staining) of splenic TFH cells from the mice immunized with X31 virus and treated with SCD inhibitor at 8 and 14 d.p.i. (C) Naïve CD4+ T cells were stimulated under TFH polarization condition and treated with increased doses of SCD inhibitor, active caspase 3/7+ cells were measured at day 4 postactivation. (D) The expression of ER stress-related genes in CD4+ T cells under TFH polarization with or without SCD inhibitor (10 μM) was determined by qRT-PCR. (E) ATF4 protein level in CD4+ T cells under TFH polarization with or without SCD inhibitor. (F) The expression of ER stress-related genes in CD4+ T cells under TFH polarization with or without SCD1 transduction was determined by qRT-PCR. Combine data (A and B) are obtained from two independent experiments (two to five mice per group). Representative results (C and E) are shown from two independent experiments (two mice per group). Results are given as mean ± SEM with unpaired t-test: *p < 0.05.
Enhanced Scd1 expression promotes TFH responses in vivo
To identify whether SCD expression contributes cell-intrinsic maintenance of TFH cells, we transduced WT OTII cells with control or SCD1-overexpressing retroviruses. We then adoptively transferred the OT-II cells into CD45.1 congenic mice and immunized the mice with X31-OTII virus (Fig. 6A). We found that SCD1 overexpression did not significantly increase TFH-cell numbers, possibly due to the slightly diminished overall OTII T-cell magnitude following SCD1 overexpression (Fig. 6B). However, the frequency of TFH cells within OTII cells was elevated following SCD1 overexpression, suggesting that ectopic SCD1 expression promotes TFH differentiation and/or maintenance relative to non-TFH cells (Fig. 6B). Ectopic SCD1 expression in OTII cells did not affect overall GC B-cell responses (Fig. 6C), which is likely due to the relatively low numbers of the transferred TFH cells compared to endogenous TFH cells. Together, our data suggest that ectopic SCD1 expression facilitates TFH-cell responses relative to non-TFH cells.
Figure 6.
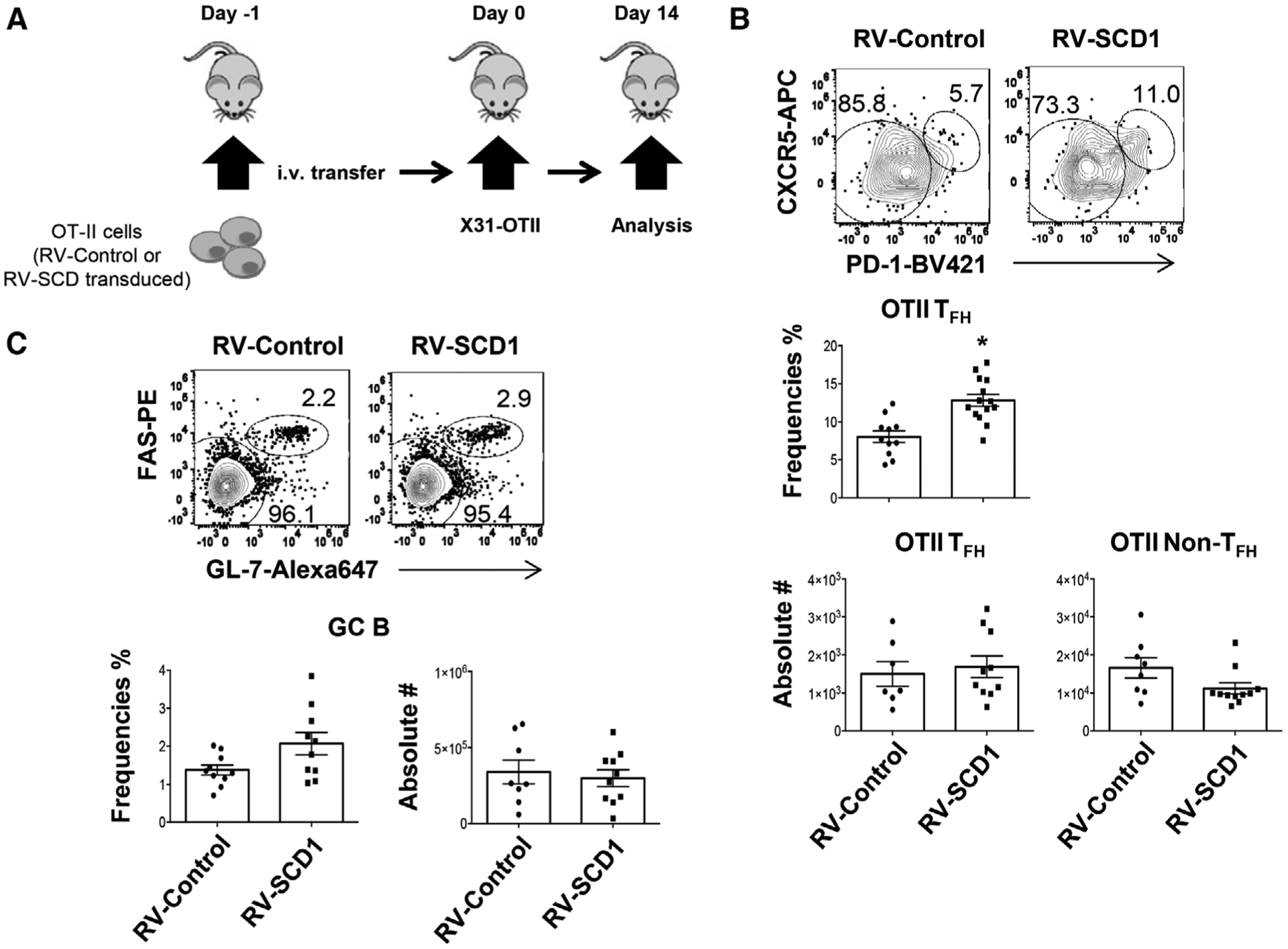
Enforced SCD1 expression in CD4+ T cells promotes TFH responses in vivo. Mock or SCD1-retroviral transduced OTII cells were adoptively transferred into CD45.1 congenic mice. Then the mice were immunized with X31-OTII. (A) Schematics of experimental design. (B) Frequency of TFH cells in OTII cells and cell numbers of TFH and non-TFH cells in CD45.2+ donor cells at 14 d.p.i. (C) Frequency and cell numbers of GC B cells in spleen at 14 d.p.i. Results were pooled from three independent experiments (three to four mice per group). Results are given as mean ± SEM with unpaired t-test: *p < 0.05.
Discussion
In this report, we show that human and murine TFH cells exhibit higher levels of the lipogenic gene expression in the form of SCD. The expression of Scd1 is regulated in a BCL6-dependent manner both in vivo and in vitro condition. Importantly, a SCD inhibitor, A939572, diminished the magnitude of TFH and GC B-cell responses whereas it promoted the frequency of TFR cells in vivo following immunization with X31-influenza virus. Therefore, our results have revealed a nonredundant role of SCD and lipid desaturation in regulating TFH maintenance and GC B-cell responses.
Excessive or prolonged ER stress, also known as unfolded protein response (UPR), suppresses normal cell growth through inhibition of translation and ultimately apoptosis. Lipotoxicity, induced by overload of lipids and their metabolites, disturbed ER homeostasis and caused metabolic abnormalities and cell death [27]. Knockdown of SCD1 enhanced the accumulation of SFAs, resulted in increased expression of ER stress-related genes such as C/EBP homologous protein (CHOP), glucose-regulated protein 78 (GRP78), and splicing of Xbox-binding protein 1 (sXBP1) [26]. However, these enhanced ER stress-induced genes and cell death were rescued by treatment of unsaturated fatty acid in Hela cells [28]. Interestingly, oleate and palmitoleate, the major products of SCD1 activity, have different metabolic functions. The oleate, but not palmitoleate, normalized the excess ER stress in SCD1 liver-specific deficient mice [12]. Thus, SCD expression in TFH cells may contribute to cellular homeostasis through the maintenance of balance between SFAs and MUFAs. It must be noted that even though SCD inhibition induces the upregulation of ER stress genes in T cells, and excessive ER stress has been associated with cellular apoptosis. Evidence presented in this study linking ER stress and TFH apoptosis is correlative in nature. Future mechanistic investigations are needed to test whether excessive ER stress is the actual cause of enhanced TFH apoptosis following SCD inhibition in vitro and in vivo.
Nevertheless, SCD1 have been reported to be highly expressed in several human cancer cells including breast, prostate, lung and ovary [29]. SCD inhibitor decreased the proliferation and increased apoptosis in clear cell renal cell carcinoma (ccRCC) in vivo and in vitro [30]. SCD1 inhibition promoted autophagy-induced apoptosis accompanied with autophagy formation in the human hepatocellular carcinoma (HCC) cells via AMPK activation [31]. Thus, SCD inhibition might be utilized to targeted cell therapy against SCD-expressing cells including malignant cells and TFH cells.
Enhanced TFH cells have been reported to be associated with the development and progression of many autoimmune diseases [32]. Recently, as the potential target of autoimmune disease therapy, inhibition of lipid metabolism gene has been reported in human patients and animal models. Inhibition of FASN reduced the development of TH17 cells, resulting in diminished EAE disease progression [33]. Simvastatin, inhibitor of HMG-CoA reductase, diminished the serum TNF-α and CRP levels in RA patients [34]. In our model, compared to C75 (fasn inhibitor) and fluvastatin, SCD inhibitor (A939572) exhibited superior activities in terms of the inhibition of TFH and GC B-cell responses. Interestingly, it has been reported that obese humans have higher levels of SCD1 expression and MUFA contents compared to lean objects [35]. SCD1 has been associated with obesity-related abnormal metabolic diseases such as hyperlipidemia, nonalcoholic liver disease, and type 2 diabetes in animal model and human [12]. These reports suggest that T-cell lipid metabolism, particularly SCD pathway, may represent a promising target to dampen autoimmunity, especially in those obesity-associated autoimmunity [36].
In this study, we show that inhibition of SCD reduces the number of TFH and GC B-cells postinfluenza virus immunization. It is possible that SCD inhibitor may have off-target effects in vivo to attenuate TFH and/or GC B-cell responses. Furthermore, SCD inhibitor may directly inhibit B cells or other cell types, such as DCs, to diminish TFH maintenance. Particularly, B cells and DCs have been shown to be important in TFH-cell generation and/or maintenance [1, 37, 38]. Therefore, future studies employing cell-type specific deletion of SCD in different cell types (such as T cells, DCs, and/or B cells) are warranted to address these potential limitations of our studies. However, as shown in Figure 6, Scd1 overexpression promoted TFH responses in vivo, indicating that the promotion of the T cell-intrinsic SCD1 expression facilitates TFH responses in vivo.
In conclusion, we found SCD activity promotes TFH-cell maintenance by inhibiting their apoptosis and sustaining GC B-cell responses. Therefore, SCD may be considered as a novel target to modulate excessive TFH responses during vaccination and/or autoimmunity.
Materials and methods
Mouse and immunization
Mouse studies were approved by the Institutional Animal Care and Use Committee at the Mayo Clinic or Indiana University School of Medicine (IUSM). All methods were performed in accordance with the relevant guidelines and regulations. WT C57/BL6, CD45.1 congenic, OTII and CD4-cre mice were originally purchased from the Jackson Laboratory and bred in house. BCL6 KO mice (CD4-cre Bcl6 fl/fl) were generated by crossing CD4-cre transgenic mice with Bcl6 fl/fl mice [39]. BCL6 KO OTII mice were generated by crossing OTII transgenic mice to BCL6 KO mice. Influenza immunization was achieved i.p. with Influenza H3N2 A/X31 or X31-OTII (~1.2 × 105 pfu/mouse). A939572 (6 mg/kg, BioFine International, WA, USA), C75 (15 mg/kg, Cayman chemical, MI, USA), or Fluvastatin (20 mg/kg, Cayman chemical) was injected to the X31 immunized mice from day 4 to day 13 for daily via i.p. route.
Flow cytometry
Splenic TFH and TFR cells were stained with antibodies against mouse CD4 (Clone: RM4–5, Biolegend, San Diego, CA, USA), CD44 (Clone:IM7, Biolegend), PD-1 (Clone: 29F.1A12, Biolegend), CXCR5 conjugated with biotin (Clone: 2G8, BD Biosciences, San Jose, CA, USA), streptavidin conjugated with APC (Biolegend), Ki67 (Clone: SolA15, Thermo Fisher Scientific, Waltham, MN, USA), Foxp3 (Clone: FJK-16s, Thermo Fisher Scientific), BCL6 (Clone: K112–91, BD Biosciences), ICOS (Clone: C398.4A, Biolegend), and GC B cells were stained with antibodies against mouse B220 (Clone: RA3–6B2, Biolegend), GL7 (Clone: GL7, Biolegend), and CD95 (FAS, Clone: SA367H8, Biolegend). For BCL6-retroviral transduced cell gating, anti-mouse H2K antibody (Clone: 36–7-5, BD Biosciences) was used and for SCD1-retroviral transduced cell gating anti-human CD4 antibody (Clone: OKT4, Biolegend) was employed. For intracellular staining, cells were restimulated with NP 311–325 peptide or NP 366–374 peptide (AnaSpecs, CA, USA) in the presence of 1 μg/mL of monensin (BD Biosciences) and brefeldin A (Biolegend) for 4 h. Then, cells were stained with surface antibodies and fixed/permeabilization buffers (Biolegend) for staining with anti-IFN-γ antibody as previously described [40]. To detect cell apoptosis, the CellEvent Caspase 3/7 Green Detection Reagent (Thermo Fisher Scientific) was added to the cells with complete media for 30 min in 37°C and the caspase 3/7 positive cells were measured on an Attune NXT flow cytometer (Life Technologies, Carlsbad, CA, USA). For TFH-cell sorting, CD4+ T cells were enriched from spleen with CD4 microbeads (Clone: L3-T4, Miltenyi Biotec, San Diego, CA, USA). The enriched CD4+ T cells were stained with antibodies to define TFH cells as described in flow methods below. And the cells were sorted on a FACSAria (BD Biosciences,). All the flow cytometry analysis was conducted accordingly to the “Guidelines for the use of flow cytometry and cell sorting in immunological studies” [41].
In vitro T-cell culture
Naïve CD4+ T cells were isolated from WT (C57BL/6) or Bcl6 fl/fl CD4-Cre (BCL6 KO). Cells were cultured with TFH-polarizing condition (precoated with anti-CD3 [1 μg/mL], soluble anti-CD28 [2 μg/mL], anti-IFN-γ [20 μg/mL], anti-IL-2 [10 μg/mL] antibodies, rmIL-21 [20 ng/mL], and rmIL-6 [20 ng/mL]) for 4–5 days.
Quantitative RT-PCR
RNA was extracted by RNA miniprep kit (Sigma-Aldrich) according to the manufacturer’s instructions. RT was performed using random primers (Invitrogen, Grand Island, NY, USA) and MMLV (Invitrogen). cDNAs were amplified using SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY, USA) using quantistudio 3 PCR system (Applied Biosystems). Data were generated with the comparative threshold cycle method by normalizing to hypoxanthine phosphoribosyltransferase (HPRT), as we previously reported [42].
Retroviral transduction
Naïve polyclonal CD4+ T or OTII cells were preactivated with precoated anti-CD3 (5 μg/mL) and soluble anti-CD28 (2 μg/mL) antibodies for 2 days. Then cells were transduced with bicistronic retroviruses through spin transduction (2,500 rpm, 90 min) as reported [43]. After transduction, cells were cultured with TFH polarized condition for an additional 2 days.
Adoptive cell transfer
OTII, BCL6 KO-OTII cells, control retroviral-transduced or SCD1-retroviral transduced OTII cells were adoptively transferred into CD45.1 congenic mice. One day post-transfer, A/X31-OVA (X31-OTII) (~5.0 × 105 pfu/mouse) was immunized for 14 days via i.p. route.
ELISA
Levels of X31-specific IgG antibody in the blood serum were measured by ELISA. In brief, 96-well plates (Biolegend) were precoated with 100 μL/well of 7 × 104 PFU of X31 virus in PBS overnight at 4°C. After blocking with PBS containing 3% of BSA (Sigma) for 1 h at room temperature (RT), each serum sample was serially diluted in blocking buffer to each well and incubated for 2 h at RT. After three times wash with wash buffer (0.05% of Tween 20 in PBS), HRP-conjugated goat anti-mouse IgG (Promega, Madison, WI, USA) was added into each well. After incubation for 2 h at RT, tetramethylbenzidine (TMB, Thermo Fisher Scientific) was added to develop the color and then the reaction was stopped by adding 2 M H2SO4. The absorbance at wavelength 450 nm was measured by an ELISA reader (Molecular Devices, Sunnyvale, CA, USA). Endpoint titers were calculated as the titer of dilution that gave an absorbance value (O.D.) of 0.2.
Western blot analysis
Naïve CD4+ T cells were purified from WT or BCL6 KO mice with naïve CD4+ T-cell isolation kit (Miltenyi Biotec). Then, the cells were activated and cultured under the TFH polarizing condition as above. The cultured cells were lysed in sample lysis buffer (Cell Signaling Technology) with a protease inhibitor cocktail (Thermo Scientific) and incubated on ice for 30 min. Proteins in the supernatants were heat denatured, separated by 4% to 12% gradient Bis-Tris gel (Invitrogen) and transferred into a nitrocellulose membrane (Bio Rad). Membrane was blocked with 5% of nonfat milk in TBST for 1 h at RT. After washing with TBST (Tris-buffered saline with 0.1% Tween 20), membrane was incubated with following primary antibodies for overnight: Rabbit-anti SCD1 (Cell signaling), rabbit-anti ATF4 (Cell signaling) or mouse-anti actin (Santa Cruz). Membrane was then rinsed with TBST and incubated with the HRP-conjugated secondary antibodies (Promega) for 1 h at RT. ECL kit (Thermo) was used for film (Fisher Scientific) development.
Statistical analysis
Graphs were generated by Graph Pad Prism software. Statistical significance was evaluated by calculating ρ-values using one/two-way ANOVA or Student’s t-test. Significance between the groups was judged based on p-value< 0.05 (two tailed).
Supplementary Material
Acknowledgments:
We thank Mayo flow cytometry core for help cell sorting. This work was supported by the US National Institutes of Health grants (RO1 HL126647, AG047156, and AI147394 J.S.; RO1 AI132771 to A.D.; T32 AG049672 to N.P.G.) and Kogod Aging Center High Risk Pilot Grant to J.S.
Abbreviations:
- ACC1
acetyl-CoA carboxylase 1
- Atf
activating transcription factor
- DG
diacyglycerols
- FASN
fatty acid synthase
- mTORC1
mTOR kinase complex 1
- MUFAs
monounsaturated fatty acids
- RT
room temperature
- SCD
Stearoyl-CoA desaturases
- SFAs
saturated fatty acids
- TConv
conventional non-TFH cells
- TFH
follicular helper T
- TFR
follicular regulatory T
- TG
triglycerides
Footnotes
Conflict of interest: The authors declare no commercial or financial conflict of interest.
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Crotty S, T follicular helper cell differentiation, function, and roles in disease. Immunity 2014. 41: 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet ME, Lazaro E, Duffau P et al. , T follicular helper cells in autoimmune disorders. Front. Immunol 2018. 9: 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH et al. , Bcl6 mediates the development of T follicular helper cells. Science 2009. 325: 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C et al. , ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011. 34: 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V and Weinmann AS, BCL-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol 2014. 15: 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie MM, Amet T, Liu H, Yu Q and Dent AL, AMP kinase promotes BCL6 expression in both mouse and human T cells. Mol. Immunol 2017. 81: 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ray JP, Staron MM, Shyer JA, Ho P-C, Marshall HD, Gray SM, Laidlaw BJ et al. , The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity 2015. 43: 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, Cloer C et al. , mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 2016. 45: 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A et al. , De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med 2014. 20: 1327–1333. [DOI] [PubMed] [Google Scholar]
- 10.Kagami S, Owada T, Kanari H, Saito Y, Suto A, Ikeda K, Hirose K et al. , Protein geranylgeranylation regulates the balance between Th17 cells and Foxp3+ regulatory T cells. Int. Immunol 2009. 21: 679–689. [DOI] [PubMed] [Google Scholar]
- 11.Ibitokou SA, Dillon BE, Sinha M, Szczesny B, Delgadillo A, Reda Abdelrahman D, Szabo C et al. , Early inhibition of fatty acid synthesis reduces generation of memory precursor effector T cells in chronic infection. J. Immunol 2018. 200: 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.AM AL, Syed DN and Ntambi JM, Insights into stearoyl-CoA desaturase-1 regulation of systemic metabolism. Trends Endocrinol. Metab 2017. 28: 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sampath H and Ntambi JM, The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann. NY Acad. Sci 2011. 1243: 47–53. [DOI] [PubMed] [Google Scholar]
- 14.Mancini R, Noto A, Pisanu ME, De Vitis C, Maugeri-Sacca M and Ciliberto G, Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene 2018. 37: 2367–2378. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Ge L, Parimoo S, Stenn K and Prouty SM, Human stearoyl-CoA desaturase: alternative transcripts generated from a single gene by usage of tandem polyadenylation sites. Biochem. J 1999. 340 (Pt 1): 255–264. [PMC free article] [PubMed] [Google Scholar]
- 16.Paton CM and Ntambi JM, Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab 2009. 297: E28–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azuma RW, Suzuki J, Ogawa M, Futamatsu H, Koga N, Onai Y, Kosuge H et al. , HMG-CoA reductase inhibitor attenuates experimental autoimmune myocarditis through inhibition of T-cell activation. Cardiovasc. Res 2004. 64: 412–420. [DOI] [PubMed] [Google Scholar]
- 18.Wong A, Chen S, Yang LK, Kanagasundaram Y and Crasta K, Lipid accumulation facilitates mitotic slippage-induced adaptation to anti-mitotic drug treatment. Cell Death Discov 2018. 4: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L, Ren J, Yang L, Li Y, Fu J, Li Y, Tian Y et al. , Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep 2016. 6: 19665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wculek SK, Khouili SC, Priego E, Heras-Murillo I and Sancho D, Metabolic control of dendritic cell functions: digesting information. Front. Immunol 2019. 10: 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, Marbois BN et al. , Sterol regulatory element–binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol 2013. 14: 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, Su LF et al. , Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 2013. 39: 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, Martin-Orozco N et al. , Bcl6 expression specifies the T follicular helper cell program in vivo. J. Exp. Med 2012. 209: 1841–1852, S1841–S1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou S, Clement RL, Diallo A, Blazar BR, Rudensky AY, Sharpe AH and Sage PT, FoxP3 and Ezh2 regulate Tfr cell suppressive function and transcriptional program. J. Experiment. Med 2019. 216: 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sage PT and Sharpe AH, T follicular regulatory cells. Immunol. Rev 2016. 271: 246–259. [DOI] [PubMed] [Google Scholar]
- 26.Masuda M, Miyazaki-Anzai S, Keenan AL, Okamura K, Kendrick J, Chonchol M, Offermanns S et al. , Saturated phosphatidic acids mediate saturated fatty acid-induced vascular calcification and lipotoxicity. J. Clin. Invest 2015. 125: 4544–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han J and Kaufman RJ, The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res 2016. 57: 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ariyama H, Kono N, Matsuda S, Inoue T and Arai H, Decrease in membrane phospholipid unsaturation induces unfolded protein response. J. Biol. Chem 2010. 285: 22027–22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley BJ, Yang WP et al. , Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res 2011. 9: 1551–1561. [DOI] [PubMed] [Google Scholar]
- 30.von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW et al. , Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res 2013. 19: 2368–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang GM, Jiang QH, Cai C, Qu M and Shen W, SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett 2015. 358: 180–190. [DOI] [PubMed] [Google Scholar]
- 32.Tangye SG, Ma CS, Brink R and Deenick EK, The good, the bad and the ugly—TFH cells in human health and disease. Nat. Rev. Immunol 2013. 13: 412–426. [DOI] [PubMed] [Google Scholar]
- 33.Young KE, Flaherty S, Woodman KM, Sharma-Walia N and Reynolds JM, Fatty acid synthase regulates the pathogenicity of Th17 cells. J. Leukoc. Biol 2017. 102: 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tikiz C, Utuk O, Pirildar T, Bayturan O, Bayindir P, Taneli F, Tikiz H et al. , Effects of angiotensin-converting enzyme inhibition and statin treatment on inflammatory markers and endothelial functions in patients with long-term rheumatoid arthritis. J. Rheumatol 2005. 32: 2095–2101. [PubMed] [Google Scholar]
- 35.Hulver MW, Berggren JR, Carper MJ, Miyazaki M, Ntambi JM, Hoffman EP, Thyfault JP et al. , Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab 2005. 2: 251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Versini M, Jeandel PY, Rosenthal E and Shoenfeld Y, Obesity in autoimmune diseases: not a passive bystander. Autoimmun. Rev 2014. 13: 981–1000. [DOI] [PubMed] [Google Scholar]
- 37.Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, Wu R et al. , Migratory CD11b+ conventional dendritic cells induce T follicular helper cell–dependent antibody responses. Science Immunol 2017. 2: eaam9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krishnaswamy JK, Alsén S, Yrlid U, Eisenbarth SC and Williams A, Determination of T follicular helper cell fate by dendritic cells. Front. Immunol 2018. 9: 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amet T, Son YM, Jiang L, Cheon IS, Huang S, Gupta SK, Dent AL et al. , BCL6 represses antiviral resistance in follicular T helper cells. J. Leukoc. Biol 2017. 102: 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao S, Jiang L, Moser EK, Jewett LB, Wright J, Du J, Zhou B et al. , Control of pathogenic effector T-cell activities in situ by PD-L1 expression on respiratory inflammatory dendritic cells during respiratory syncytial virus infection. Mucosal Immunol 2015. 8: 746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cossarizza A, Chang H-D, Radbruch A, Acs A, Adam D, Adam-Klages S, Agace WW et al. , Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z et al. , The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 2019. 51: 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH and Sun J, Interferon regulatory factor 4 sustains CD8+ T cell expansion and effector differentiation. Immunity 2013. 39: 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.