

Abstract
Therapies that bind with immune cells and redirect their cytotoxic activity toward diseased cells represent a promising and versatile approach to immunotherapy with applications in cancer, lupus, and other diseases; traditional methods for discovering these therapies, however, are often time-intensive and lack the throughput of related target-based discovery approaches. Inspired by the observation that the cytokine, IL-12, can enhance antileukemic activity of the clinically approved T cell redirecting therapy, blinatumomab, here we describe the structure and assembly of a chimeric immune cell-redirecting agent which redirects the lytic activity of primary human T cells toward leukemic B cells and simultaneously cotargets the delivery of T cell-stimulating IL-12. We further describe a novel method for the parallel assembly of compositionally diverse libraries of these bispecific T cell engaging cytokines (BiTEokines) and their high-throughput phenotypic screening, requiring just days for hit identification and the analysis of composition-function relationships. Using this approach, we identified CD19 × CD3 × IL12 compounds that exhibit ex vivo lytic activity comparable to current FDA-approved therapies for leukemia and correlated drug treatment with specific cell–cell contact, cytokine delivery, and leukemia cell lysis. Given the modular nature of these multivalent compounds and their rapid assembly/screening, we anticipate facile extension of this therapeutic approach to a wide range of immune cells, diseased cells, and soluble protein combinations in the future.
Keywords: nanotechnology, multivalency, leukemia, drug screening
Graphical Abstract
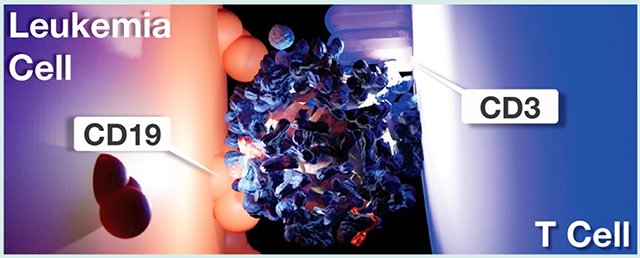
INTRODUCTION
Immune cell redirection (ICR) is a powerful and versatile therapeutic approach in which the cytotoxic activity of endogenous immune cells is redirected toward diseased cells via simultaneous, drug-induced cell binding. This strategy has demonstrated therapeutic benefit in preclinical models of cancer,12 HIV,3,4 lupus,5 and other diseases; however, only one such drug with an Fc-independent mechanism-of-action is currently approved for clinical use in the U.S.: the bispecific antibody, blinatumomab, which redirects T cell killing toward leukemic B cells. Given the ability of ICR therapies to co-opt a wide range of cell types (e.g., T cells, NK cells,6 and macrophages7) against both cell-surface and intracellular targets,8 enthusiasm for future drug development is high with dozens of drug candidates at or in clinical-stage development.2
In addition to their diversity of application, ICR immunotherapies can also vary widely in their composition and mode of delivery. They encompass nanoparticle,9-11 bispecific IgG,12,13 scFv fusion,14 and mRNA15 constructs, as well as vectors based on oncolytic viruses16 and engineered cells.17 While a majority of ICR therapies in clinical testing are produced using traditional genetic engineering techniques, one challenge to their discovery and development is the relatively low-throughput manner in which drug candidates can be investigated and the relatively high dependency of drug action on the affinity of individual cell-binding domains. Fusion protein engineering methods that rely on conventional plasmid vectors18 or de novo protein design19 often require months for expression and purification prior to screening, and the effects of associated modifications on subsequent protein affinity can be challenging to predict.20 Moreover, while response rates to blinatumomab are often impressive, remissions are not always durable.21 Methods to both accelerate the discovery and improve the potency of ICR therapies are therefore urgently needed.
Recently, we identified IL-12 as a key mediator of the immune response to leukemia cells in mouse models of B cell acute lymphoblastic leukemia (ALL), including that recombinant IL-12 therapy alone could improve T cell activation, immunologic memory, and overall survival in mouse models of the disease.22 Based on these findings, we hypothesized that the activity of ICR therapies targeting T cells and leukemic B cells may be improved by concurrent delivery of IL-12, particularly if the two agents were tethered to one another in order to improve the typically poor circulation of IL-12 that limits its therapeutic potential.23
To examine this hypothesis, here we describe a method for the rapid assembly and screening of multivalent ICR drug candidates that redirect the lytic activity of T cells toward leukemic B cells and simultaneously codeliver T cell-stimulating IL-12 to yield multifunctional therapies which we term, bispecific T cell-engaging cytokines (BiTEokines). Using this discovery platform, we show that cytokine codelivery can dramatically alter the antileukemic activity of ICR immunotherapies and that the generation and screening of diverse libraries of BiTEokine candidates can be achieved in a matter of days, rather than weeks or months, thereby greatly accelerating the process of hit identification. Future extensions of this approach could enable the rapid identification of drug compounds with activity against cancer, autoimmune diseases, or pathogen infections and, given its modular nature, could be extended to a wide range of immune cells, diseased cells, and soluble protein combinations in the future.
RESULTS AND DISCUSSION
IL-12 Enhances Bispecific T Cell Engager Activity.
We22 and others previously found that IL-12 can improve cancer immune elimination via enhanced CD8+ T cell proliferation,24 cytotoxicity,17,25 survival,17 and T cell receptor (TCR) signaling.26,27 As clinically approved T cell engager therapies are thought to act primarily on a subset of CD8+ T cells,28,29 we posited that IL-12 may improve the lytic activity of blinatumomab which bispecifically targets T cell CD3 and leukemic B cell CD19. Following prolonged coculture of primary CD8+ T cells with CD19+ NALM-6 leukemia cells, we observed significant improvement in target leukemia cell lysis in the presence of IL-12, as measured by flow cytometry, (Figure 1a-c) as well as an associated increase in T cell proliferation (Figure 1d, Figure S1), and T cell activation (Figure 1e), as measured by dye dilution and interferon gamma (IFNγ) secretion, respectively. Together, these data demonstrate that IL-12 can improve the performance of T cell redirecting therapies in an ex vivo assay that is heavily relied upon to prioritize ICR drug candidates30,31 and that these effects are attributable, in part, to cytokine-enhanced T cell proliferation and activation.
Figure 1.

Recombinant IL-12 enhances activity of the bispecific T cell engager therapy, blinatumomab. (a) Schematic of assay conditions for the coculture of primary human CD8+ T cells with CD19+ NALM-6 leukemia cells. Blinatumomab-induced (b,c) lysis of NALM-6 leukemia cells and (d) T cell proliferation enhanced by coincubation with IL-12 as measured by flow cytometry. (e) Blinatumomab and IL-12 synergize to enhance T cell activation as measured by ELISA of IFNγ secretion into coculture supernatants. Data in (c,d) report representative dot plots and dye-dilution histograms, respectively. Cocultures in (c–e) were treated by blina (7 ng/mL) with or without IL-12 (3.5 ng/mL) in comparison to PBS vehicle over (c,d) 72 h or (e) 48 h. Values report (b) mean ± SEM (n = 3 donors) as analyzed by the mixed-effects model with correction for multiple comparisons and (e) mean ± SEM (n = 3 donors) as analyzed by one-way ANOVA with Tukey’s correction for multiple comparisons. See Supporting Information for associate gating strategies. *p < 0.05, ****p < 0.0001.
This marked effect of IL-12 on blinatumomab activity is significant in that other T cell mitogens such as IL-2 have been previously combined with blinatumomab with relatively little impact on lytic activity.14,30 While the synergy observed here may be unique to IL-12, such differential effects may arise due to the fact that typical lysis assays are performed over much shorter durations (e.g., 4 h) and that the effects of IL-12 result, in part, due to cytokine-induced T cell proliferation (P = 0.020, Figure 1d, Figure S1). Interestingly, however, T cell expansion observed in the presence of both drugs was, alone, insufficient to fully account for the large change in target cell lysis (P = 0.027), thus future studies focusing on the role of IL-12 in modulating the selective expansion, differentiation,32 or activation CD8+ T cells in the presence of blinatumomab are warranted.
Also, while not investigated further in this work, the observation of synergy between blinatumomab and recombinant IL-12 is significant in that the former is currently approved to treat relapsed/refractory and minimal residual disease positive (MRD+) B-ALL in both adults in children. And while IL-12 therapies have not advanced to phase III trials due to poor circulation and toxicity, several novel IL-12 drug candidates currently under investigation may benefit from combination with blinatumomab including adenoviral,33 plasmid,34 mRNA,35 and affinity-targeted24,25 IL-12.
Design and Rapid Screening of CD19 × CD3 × IL12 BiTEokines.
Having shown that IL-12 potentiates the antileukemic activity of T cell-redirecting immunotherapy, we next devised a drug architecture that (i) directs the lytic activity of T cells toward leukemic B cells, (ii) simultaneously codelivers T cell-stimulating IL-12, and (iii) features a modular design amenable to combinatorial assembly of test compound libraries (Figure 2). We based the core scaffold of these structures on magnetic iron oxide nanoparticles due to their track-record of clinical use,36,37 ability to accommodate a wide range of IgG antibodies via Fc-protein G affinity, and rapid purification via magnetic field sedimentation. Antibody clones were selected due to their prior clinical testing as CAR-T cell constructs (CD19, SJ25-C1)38 or antibody-drug conjugates (CD3, UCHT1),39 and their comparable IgG1-protein G affinity. Using this modular design, we surmised that varying BiTEokine protein abundance would result in differential capacity for drug-induced leukemia cell lysis through altered cytokine concentration or affinity/avidity toward cell-surface epitopes.
Figure 2.

Structure and assembly of bispecific T cell engaging cytokines (BiTEokines). Schematic of drug-induced synapse formation between T cells and leukemic B cells, as well as synapse-targeted delivery of the cytokine, IL-12. Inset illustrates the modular and rapid self-assembly of CD19 × CD3 × IL12 BiTEokines via addition of human IgG to protein G-conjugated iron oxide nanoparticles and subsequent cytokine complexation. The solid beige arrow denotes cytokine release or trans-presentation.
To assemble BiTEokine test compound libraries, we dispensed equivalent numbers of magnetic nanoparticles into individual wells of a standard 96 well plate, each containing varying cocktails of fluorochrome-labeled antibodies directed against human CD19, CD3ε, (non-neutralizing) IL-12, or isotype control (Figure 3a, Figure S2). After incubation, magnetic field-induced sedimentation, and analysis of antibody abundance using a standard fluorescence plate reader, we then sterile-filtered the beads and added varying volumes of a novel single-chain variant of IL-12 (scIL-12), followed by incubation and further purification. Test compounds prepared using this method were highly reproducible batch-to-batch, exhibiting stable hydrodynamic size and consistent antibody composition (Figure S3). Using this approach, BiTEokine libraries were prepared and characterized over the course of just 8 to 9 h.
Figure 3.

High-throughput assembly and screening enables rapid identification of BiTEokines that induce efficient leukemia cell lysis. (a) Schematic of test compound library assembly. (b) Structural diversity of the BiTEokine library and (c,d) representative test compound size/morphology as measured by antibody fluorescence intensity, dynamic light scattering, and transmission electron microscopy, respectively. (e) Schematic of coculture assay conditions and (f) parallel screening results rank-ordered by drug-induced lysis of CD19+ NALM-6 leukemia cells by primary human CD8+ T cells. (g) Heatmaps illustrating concordance between target cell lysis the ratio of αCD3 to αCD19 per BiTEokine and (h) significance of antibody components to measured lysis values as modeled by multivariate least-squares regression. Univariate composition-function relationships reporting NALM-6 leukemia cell lysis versus (i–k) antibody abundance per particles and (l) T cell division. Values in (f) represent mean ± SEM of 2–3 T cell donors, each analyzed in duplicate. Lines in (i–l) report linear regressions with 95% confidence intervals. E:T, effector-to-target cell ratio. *p < 0.05, **p < 0.01.
In total, we synthesized 47 unique BiTEokine test compounds which varied widely in antibody composition, achieving a consistent, and near theoretical maximum, total coverage of 134 ± 15 IgG per particle (αCD19:1.5 ± 0.8 to 42 ± 4; αCD3:2.5 ± 0.5 to 59 ± 3; αIL12 0.12 ± 0.08 to 15 ± 0.7; Figure 3b, Figure S4). Dynamic light scattering measurements indicated high stability of the subsequent test compounds in buffer, with hydrodynamic size increasing from 71.5 to 108.1 nm upon antibody surface-assembly (Figure 3c) with no appreciable change in particle morphology as measured by transmission electron microscopy (Figure 3d). We note that such size increases (ca. 36.5 nm) correspond closely to what one would expect following addition of a single monolayer of IgG1 (10–12 nm hydrodynamic diameter)40 about these particles, augmenting their size to well-above the size threshold for renal clearance in humans, thus potentially improving the circulation and associated therapeutic benefit of IL-12 therapy both alone and in combination with ICR immunotherapy. Together these data demonstrate that BiTEokine test compound libraries can be assembled rapidly, in parallel with a wide range of structural diversity.
With a test compound library in-hand, we next screened the lytic activity of BiTEokines following incubation with cocultures of primary human CD8+ T cells and CD19+ NALM-6 leukemia cells and analysis by flow cytometry (Figure 3e,f). These screens were performed over 72 h using a low31 effector-to-target (E:T) ratio of 1:1 to allow for observable T cell proliferation and to closely reflect cell counts present in the peripheral blood of patients with MRD+ B-ALL41 and post-transplant relapsed B-ALL,42 populations for whom blinatumomab therapy is currently FDA-approved. As anticipated, BiTEokine test compounds varied widely in their corresponding lytic potential with median activity just 11% (n = 3) that of blinatumomab’s. The top eight screening hits, in contrast, exhibited lytic activity closely comparable to, and statistically indistinguishable (P > 0.98) from, blinatumomab (e.g., 035:62 ± 26%). Interestingly, these top performing BiTEokines displayed only a small number of B cell-targeting antibodies per particle (2 ± 1 to 3.2 ± 0.3) and an abundance for CD3 antibodies (24 ± 1 to 59 ± 3), thus largely limiting the potential for interaction with multiple B cells. Based on our prior studies in leukemia-bearing mice,22 we anticipate additional therapeutic benefits from the delivery and prolonged circulation of IL-12 in vivo, due to its actions alone and when cross-exposed to antigens from lysed target cells.
Another attractive feature of this combinatorial screening approach is its ability to rapidly shed light on composition-function relationships unique to these novel multivalent drug architectures. For example, the bispecific antibody, blinatumomab, is well-known to target T and leukemic B cells with low and high affinity, respectively.43 Multivariate least-squares regression modeling of screening data (R2 = 0.82) indicated significant contributions from all BiTEokine antibody components; however surprisingly, here we observed that high αCD3/αCD19 ratio was, in fact, more closely associated with favorable lytic activity (Figure 3g,h). We were unable to rationalize this effect based on disparate antigen ratio (approx.44 1.2:1.0 CD19:CD3) or density given that this cell line was used extensively in the preclinical development of blinatumomab.30,31 The nonobvious finding that optimally lytic structures displayed effector cell antibodies 10- to 25-fold in excess of those toward target cells is significant in that prior studies of other multivalent ICR immunotherapies have previously focused on either single (i.e., approximately equimolar)9,11 or narrow ranges of antibody composition (e.g., 0.33- to 3-fold).10 Antibody ratios in these ranges induced only low levels of leukemia cell lysis in this work (Figure 3g); thus, the already impressive performance of prior multivalent ICR immunotherapies may be further improved by the systematic discovery approach described here. We further found that αCD19 abundance was a negative correlate of lytic activity and, conversely, that αCD3, αIL12, and T cell division (% divided) were positive correlates of leukemia cell lysis. Target cell lysis was also nominally improved by the tethering of IL-12 to BiTEokines in comparison to coadministration of cytokine with αIL12-deficient analogs (Figure S5, P = 0.26). Interestingly, the impact of IL-12 on drug activity appeared to bifurcate depending on its relative abundance on BiTEokines with low amounts of cytokine (approximately 2.3 ng/mL) inducing high T cell division but low leukemia cell lysis, and high IL-12 (approximately 270 ng/mL) inducing less rapid T cell division and high target cell lysis (Figure 3i-l). While future studies will be required to attribute the origin of these differential effects from IL-12, we speculate that (i) the novel ability of multivalent BiTEokines to induce TCR clustering45 may allow them to redirect the activity of CD8+ T cell subsets outside of those typically acted upon by bispecific antibodies and (ii) that IL-12 concentration-dependent CD8+ T cell differentiation32 may enrich for T cell subsets with differential dependency on costimulation for drug-induced lysis (e.g., memory precursor or short-lived effector cells).
Activity and Specificity of BiTEokines.
After identifying hits from the BiTEokine activity screen, we sought to confirm that these compounds specifically targeted T and leukemic B cells via flow cytometry. As anticipated, we observed particle abundance-dependent increases in the labeling of T cells with CD3 antibodies and B cells with CD19 antibodies, but no apparent αIL12-dependent cell specificity from BiTEokine test compounds (Figure 4a). In addition, we further confirmed that BiTEokine lytic activity arose from precise combinations of antibodies, rather than nonspecific antibody interactions, using compounds either fully or partially conjugated with isotype control antibody to approximately equivalent total amounts of IgG. While isotype control BiTEokines elicited only basal levels of activity (1.9% lysis), we observed >17-fold increases in lytic responses from lead compounds 35 and 37 (Figure 4b). Together, these data correlate cell-specific binding by BiTEokines with the lysis of leukemic B cells.
Figure 4.

CD19 × CD3 × IL12 BiTEokines bind specifically and induce efficient leukemia cell lysis. (a) Cell fluorescence from various BiTEokine antibodies observed in cocultures gated on (top) primary human T cells or (bottom) NALM-6 leukemia cells as measured by flow cytometry. Top left: CD8+ T cells exhibit CD3 antibody fluorescence that increases in intensity with relative abundance on BiTEokines. Bottom middle: CD19+ leukemia cells exhibit CD19 antibody fluorescence that increases in intensity with relative abundance on BiTEokines. (b) Representative dot plots of BiTEokine-induced NALM-6 leukemia cell killing in comparison to isotype control-conjugated particles. Experimental conditions in (a), (b) are noted in Figure 3.
To further characterize the role of BiTEokines in inducing leukemia cell lysis, we performed imaging flow cytometry on cocultures of primary human CD8+ T cells and CD19+ NALM-6 leukemia cells treated with fluorescently labeled hit compound 35 from the prior activity screen, as well as blinatumomab. Gating on doublets of T and B cells, we observed similar patterns of LAMP-1 (CD107a) positive vesicle accumulation, indicative of lytic granules and lysosomes, in both blinatumomab- and BiTEokine-treated cocultures (Figure 5). Strikingly, we observed distinct accumulation of BiTEokines at the interface between T and leukemic B cells, further associating BiTEokine treatment with cell–cell contact with leukemia cell lysis.
Figure 5.

BiTEokines localize at the interface between primary human T cells and NALM-6 leukemic B cells. Imaging flow cytometry of CD8+ T cells cocultured with NALM-6 leukemia cells at a 1:1 E:T ratio and treated with (a) blinatumomab or (b) CD19 × CD3 × IL12 BiTEokines (35) for 24 h at equimolar concentrations (130 pM). (a,b) report images from two different T cell donors. Arrowheads indicate localization of BiTEokines at the T-B cell interface. E:T, effector-to-target cell ratio. Scale bar is 7 μm.
CONCLUSIONS
Here, we describe a methodology for the rapid discovery of immune cell-redirecting therapies which are combinatorially self-assembled from recombinant proteins and magnetic nanoparticles. Motivated by the antileukemic activity of IL-12, both alone and in combination with the T cell engager therapy, blinatumomab, we devised a modular and convergently assembled drug structure that redirects the lytic activity of T cells toward leukemic B cells and simultaneously cotargets the delivery of T cell-stimulating IL-12. We show that compositionally diverse libraries of these CD19 × CD3 × IL12 BiTEokines can be assembled and screened over the course of just days—rather than months that are typically required using traditional recombinant techniques—enabling rapid hit identification and the delineation of important composition-function relationships. Using this approach, we identified BiTEokine hit compounds which exhibit ex vivo lytic activity comparable to current FDA-approved therapies for leukemia. Detailed analysis of BiTEokine activity strongly correlated drug treatment with specific cell–cell contact, IL-12 delivery, and leukemia cell lysis. These results are particularly promising given that we anticipate additional in vivo therapeutic benefit from IL-12, due to immune memory resulting from crossexposure of cytokine with antigens from lysed target cells. Although future lead optimization may be required in order to maximize in vivo activity from these compounds, these studies demonstrate that optimal antibody composition and density can be rapidly determined using this approach; such information could be used to inform the synthesis of structurally analogous antibody-conjugated liposomes, viruslike particles (VLPs),46,47 and self-assembled protein cages48 for subsequent translation. Future studies investigating the impact of core scaffold size, spatial ordering antibodies, targeted cytokine neutralization, rather than delivery, may also lead to further improvements in BiTEokine activity or an expansion of disease targets, respectively. While a limited number of promising synthetic ICR agents have been previously described,9-11 these studies are the first—to our knowledge—to present a method for the discovery and screening-based optimization of this promising class of immunotherapy. Given the rapid and modular nature of the approach presented here, we also anticipate facile extension to a wide range of immune cells, diseased cells, and soluble protein combinations in the future.
METHODS
Primary Cells and Cell Lines.
Deidentified, normal donor blood samples were obtained from ZenBio (Durham, NC). PBMCs were isolated from buffy coats by ficoll density gradient centrifugation. CD8+ T cells were isolated from PBMCs by negative selection using EasySep (Human CD8+ T cell Isolation Kit, Stemcell), assessed for ≥90% purity by flow cytometry, and cryopreserved. Primary cells and the NALM-6 cell line (gifted from Dr. Lia Gore, University of Colorado) were cultured in RPMI (10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin). Hek-Blue IL-12 reporter cells were obtained from Invivogen and cultured in DMEM (10% FBS, 50 U/mL penicillin, 50 μg/mL streptomycin, 100 μg/mL Normocin, 1× HEK-Blue selection). All cells were cultured at 37 °C in a 5% CO2 humidified atmosphere and tested regularly for mycoplasma.
Therapeutic Antibodies and Recombinant Proteins.
Fluorochrome-conjugated IgG antibodies were purchased from Biolegend: anti-CD3 (UCHT1), anti-CD19 (SJ25-C1), anti-IL-12 (C11.5), and IgG1 isotype control (MOPC21C). Single chain human IL-12 was purchased from Invivogen. Blinatumomab (anti-hCD19-CD3) was obtained from Invivogen. Protein concentrations were measured via UV optical absorption (Nanodrop, Thermo).
BiTEokine Synthesis and Characterization.
Magnetic nanoparticles (50–80 nm) functionalized with protein G were obtained from Ocean Nanotech. Particles were validated for lot-specific sizing via DLS (DynaPro III, Wyatt). BiTEokine test compounds were prepared via addition of 1.65 × 1011 particles to antibody mixtures (PBS) for 10 min at room temperature with agitation at 200 rpm. Antibody amounts per well ranged from 0.11 μg to 2.28 μg (αCD19), 1.25 μg to 24.31 μg (αCD3), 0.91 μg to 8.38 μg (αIL12), 1.76 μg to 11.27 μg (Isotype). Unbound antibodies were removed via magnetic field-induced sedimentation (≥8 min) and washing twice with PBS. Previously obtained calibration curves for antibody-particle binding were used to establish conditions for compound library preparation. Intermediate compounds were then passed through 0.45 μm sterile PVDF filters and 2 eq. of recombinant human single chain IL-12 (relative to αIL12 binding sites) was added to a subset of particles for 30 min at room temperature with agitation at 200 rpm. Purified test compounds were obtained after magnetic field-induced sedimentation (≥8 min) and washing twice with PBS. Antibody abundance on test compounds was determined from spillover-corrected fluorescence intensity and comparison to standard curves for each fluorochrome-conjugated antibody. Particle-bound antibody fluorescence was linearly related to input antibody fluorescence over the ranges tested here.
BiTEokine test compounds were imaged via transmission electron microscopy at 80 kV using a Hitachi HT-7700 instrument following sample application to Formvar/carbon coated copper grids (400 mesh, Electron Microscopy Sciences) for 15 min and washing for 2 s with ultrapure water. Hydrodynamic size was measured via dynamic light scattering using a DynaPro III plate reader (Wyatt).
Standard Flow Cytometry.
Primary cells were stained for purity post-isolation using anti-CD45 (HI30, BD), anti-CD8 (RPA-T8, BD), and Near IR Live/dead stain (Invitrogen), then fixed with 4% formaldehyde (Thermo) and analyzed using a BD LSR II or a Cytek Aurora cytometer. Data were analyzed using FlowJo 10 software.
Imaging Flow Cytometry.
Cell multimers were fixed by gently adding 4% formaldehyde directly to cell cocultures to a final concentration of 2% for 15 min at room temperature. Cells were then permeabilized with saponin (BD Perm/wash), stained with anti-LAMP1 AF488 (eBioH4A3, Thermo), washed 2× and resuspended in PBS, then analyzed using an ImageStreamx Mk II (Amnis) instrument with 60× magnification in extended depth-of-field mode. Side scatter measurements were obtained using the default 785 nm laser line. Data were analyzed using IDEAS software (Amnis).
Cytotoxicity Assay.
Target leukemia cells were stained with CFSE (Tonbo) or CellTrace Violet (Thermo) and T cells were left unstained or stained with CellTrace Yellow prior to coculture. Target cells and T cells were cocultured in 96 well u-bottom plates for up to 72 h. Count beads (Invitrogen) were added to each sample to determine absolute cell counts. After coculture, cells were stained with Near IR Live/Dead for viability and fixed with 4% formaldehyde prior to flow cytometric analysis. Test compounds for which multiple donors or multiple replicates were unobtained, or T cell donors from whom drug-induced lysis was not observed, were excluded from analysis. Specific lysis was calculated using the equation: %specific lysis = 100(%treated sample [violet+NIR LD+] - %isobeads[violet+NIR LD+])/(100–%isobeads[violet +NIR LD+]). Lysis measurements in all cases, including blinatumomab-treated controls, were <100%.
Proliferation Assay.
Proliferation of T cells was measured by dye dilution of CellTrace Yellow (Thermo). Gating was performed using FlowJo software, and analysis of dye dilution data was performed using ModFit software (Verity) to determine proliferation index and percent divided.
ELISA Assay.
Human IFNγ was quantified via sandwich ELISA assay (430107, Biolegend) and per the manufacturer’s recommended conditions.
Statistics and Software.
Analyses were performed in Graphpad Prism, JMP Pro 14, and Modfit. Statistical comparisons were performed via one-way or two-way ANOVA with correction for multiple comparisons using Graphpad Prism. Analysis of BiTEokine variables contributing to optimal lytic activity was performed via standard least-squares modeling in JMP Pro 14.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the U.S. Department of Defense (Idea Award CA180783), the AAI Careers in Immunology Fellowship Program, the National Institutes of Health Research Training Program in Immunoengineering (T32EB021962), the American Cancer Society (IRG-17-181-05), the Coulter Department of Biomedical Engineering, and the Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta. We are also grateful for assistance from the Children’s Healthcare of Atlanta and Emory University’s Pediatric Flow Cytometry Core, the Robert P. Apkarian Integrated Electron Microscopy Core, and the Emory Chemical Biology Discovery Center. The content here is solely the responsibility of the authors and does not necessarily represent the official views of the organizations acknowledged herein.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscombsci.0c00081.
Neutralizing capacity of anti-IL-12 clone C11.5. Batch-to-batch reproducibility and additional test compound characterization. Composition of the BiTEokine test compound library (PDF)
Contributor Information
Priscilla Do, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30322, United States.
Lacey A Perdue, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30322, United States.
Andrew Chyong, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30322, United States.
Rae Hunter, Department of Pediatrics, Emory School of Medicine, Atlanta, Georgia 30322, United States; Winship Cancer Institute of Emory University, Atlanta, Georgia 30322, United States; Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Emory School of Medicine, Atlanta, Georgia 30322, United States.
Jodi Dougan, Department of Pediatrics, Emory School of Medicine, Atlanta, Georgia 30322, United States; Winship Cancer Institute of Emory University, Atlanta, Georgia 30322, United States; Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Emory School of Medicine, Atlanta, Georgia 30322, United States.
Curtis J Henry, Department of Pediatrics, Emory School of Medicine, Atlanta, Georgia 30322, United States; Winship Cancer Institute of Emory University, Atlanta, Georgia 30322, United States; Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Emory School of Medicine, Atlanta, Georgia 30322, United States.
Christopher C Porter, Department of Pediatrics, Emory School of Medicine, Atlanta, Georgia 30322, United States; Winship Cancer Institute of Emory University, Atlanta, Georgia 30322, United States; Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Emory School of Medicine, Atlanta, Georgia 30322, United States.
Erik C Dreaden, Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30322, United States; Department of Pediatrics, Emory School of Medicine, Atlanta, Georgia 30322, United States; Winship Cancer Institute of Emory University, Atlanta, Georgia 30322, United States; Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta and Emory School of Medicine, Atlanta, Georgia 30322, United States; Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, United States.
REFERENCES
- (1).Baeuerle PA; Reinhardt C Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009, 69 (12), 4941–4944. [DOI] [PubMed] [Google Scholar]
- (2).Goebeler M-E; Bargou RC T cell-engaging therapies — BiTEs and beyond. Nat. Rev. Clin. Oncol 2020, 17, 418–434. [DOI] [PubMed] [Google Scholar]
- (3).Bardhi A; Wu Y; Chen W; Li W; Zhu Z; Zheng JH; Wong H; Jeng E; Jones J; Ochsenbauer C; Kappes JC; Dimitrov DS; Ying T; Goldstein H Potent In Vivo NK Cell-Mediated Elimination of HIV-1-Infected Cells Mobilized by a gp120-Bispecific and Hexavalent Broadly Neutralizing Fusion Protein. J. Virol 2017, 91 (20), e00937–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brozy J; Schlaepfer E; Mueller CKS; Rochat M-A; Rampini SK; Myburgh R; Raum T; Kufer P; Baeuerle PA; Muenz M; Speck RF Antiviral Activity of HIV gp120-Targeting Bispecific T Cell Engager Antibody Constructs. J. Virol 2018, 92 (14), e00491–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Schooten WV; Juelke K; Iyer S; Buelow B; Buelow R; Volk D 293 A novel CD3/BCMA bispecific antibody selectively kills plasma cells in bone marrow of healthy individuals with improved safety. Lupus Sci. Med 2019, 6 (Suppl 1), A213–A213. [Google Scholar]
- (6).Vallera DA; Felices M; McElmurry R; McCullar V; Zhou X; Schmohl JU; Zhang B; Lenvik AJ; Panoskaltsis-Mortari A; Verneris MR; Tolar J; Cooley S; Weisdorf DJ; Blazar BR; Miller JS IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res 2016, 22 (14), 3440–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Scott EM; Jacobus EJ; Lyons B; Frost S; Freedman JD; Dyer A; Khalique H; Taverner WK; Carr A; Champion BR; Fisher KD; Seymour LW; Duffy MR Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunotherapy Cancer 2019, 7 (1), 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liddy N; Bossi G; Adams KJ; Lissina A; Mahon TM; Hassan NJ; Gavarret J; Bianchi FC; Pumphrey NJ; Ladell K; Gostick E; Sewell AK; Lissin NM; Harwood NE; Molloy PE; Li Y; Cameron BJ; Sami M; Baston EE; Todorov PT; Paston SJ; Dennis RE; Harper JV; Dunn SM; Ashfield R; Johnson A; McGrath Y; Plesa G; June CH; Kalos M; Price DA; Vuidepot A; Williams DD; Sutton DH; Jakobsen BK Monoclonal TCR-redirected tumor cell killing. Nat. Med 2012, 18 (6), 980–987. [DOI] [PubMed] [Google Scholar]
- (9).Schütz C; Varela JC; Perica K; Haupt C; Oelke M; Schneck JP Antigen-specific T cell Redirectors: a nanoparticle based approach for redirecting T cells. Oncotarget 2016, 7 (42), 68503–68512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yuan H; Jiang W; von Roemeling CA; Qie Y; Liu X; Chen Y; Wang Y; Wharen RE; Yun K; Bu G; Knutson KL; Kim BYS Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat. Nanotechnol 2017, 12 (8), 763–769. [DOI] [PubMed] [Google Scholar]
- (11).Vaidya T; Straubinger RM; Ait-Oudhia S Development and Evaluation of Tri-Functional Immunoliposomes for the Treatment of HER2 Positive Breast Cancer. Pharm. Res 2018, 35 (5), 95. [DOI] [PubMed] [Google Scholar]
- (12).Staerz UD; Bevan MJ Hybrid hybridoma producing a bispecific monoclonal antibody that can focus effector T-cell activity. Proc. Natl. Acad. Sci. U. S. A 1986, 83 (5), 1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Burris HA; Giaccone G; Im S-A; Bauer TM; Oh D-Y; Jones SF; Nordstrom JL; Li H; Carlin DA; Baughman JE; Lechleider RJ; Bang Y-J Updated findings of a first-in-human, phase I study of margetuximab (M), an Fc-optimized chimeric monoclonal antibody (MAb), in patients (pts) with HER2-positive advanced solid tumors. J. Clin. Oncol 2015, 33 (15_suppl), 523–523.25584007 [Google Scholar]
- (14).Löffler A; Kufer P; Lutterbüse R; Zettl F; Daniel PT; Schwenkenbecher JM; Riethmüller G; Dörken B; Bargou RC A recombinant bispecific single-chain antibody, CD19 × CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95 (6), 2098–2103. [PubMed] [Google Scholar]
- (15).Stadler CR; Bähr-Mahmud H; Celik L; Hebich B; Roth AS; Roth RP; Karikó K; Türeci Ö; Sahin U Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med 2017, 23 (7), 815–817. [DOI] [PubMed] [Google Scholar]
- (16).Speck T; Heidbuechel JP; Veinalde R; Jaeger D; von Kalle C; Ball CR; Ungerechts G; Engeland CE Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin. Cancer Res 2018, 24 (9), 2128–2137. [DOI] [PubMed] [Google Scholar]
- (17).Yeku OO; Purdon TJ; Koneru M; Spriggs D; Brentjens RJ Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep 2017, 7 (1), 10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sampei Z; Haraya K; Tachibana T; Fukuzawa T; Shida-Kawazoe M; Gan SW; Shimizu Y; Ruike Y; Feng S; Kuramochi T; Muraoka M; Kitazawa T; Kawabe Y; Igawa T; Hattori K; Nezu J Antibody engineering to generate SKY59, a long-acting anti-C5 recycling antibody. PLoS One 2018, 13 (12), No. e0209509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Perkel JM The computational protein designers. Nature 2019, 571, 585–587. [DOI] [PubMed] [Google Scholar]
- (20).Zuch de Zafra CL; Fajardo F; Zhong W; Bernett MJ; Muchhal US; Moore GL; Stevens J; Case R; Pearson JT; Liu S; McElroy PL; Canon J; Desjarlais JR; Coxon A; Balazs M; Nolan-Stevaux O Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell–recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res 2019, 25 (13), 3921–3933. [DOI] [PubMed] [Google Scholar]
- (21).Kantarjian H; Stein A; Gökbuget N; Fielding AK; Schuh AC; Ribera J-M; Wei A; Dombret H; Foà R; Bassan R; Arslan O; Sanz MA; Bergeron J; Demirkan F; Lech-Maranda E; Rambaldi A; Thomas X; Horst H-A; Brüggemann M; Klapper W; Wood BL; Fleishman A; Nagorsen D; Holland C; Zimmerman Z; Topp MS Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med 2017, 376 (9), 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rabe JL; Gardner L; Hunter R; Fonseca JA; Dougan J; Gearheart CM; Leibowitz MS; Lee-Miller C; Baturin D; Fosmire SP; Zelasko SE; Jones CL; Slansky JE; Rupji M; Dwivedi B; Henry CJ; Porter CC IL-12 abrogates calcineurin-dependent immune evasion during leukemia progression. Cancer Res 2019, 79 (14), 3702–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Berraondo P; Etxeberria I; Ponz-Sarvise M; Melero I Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. 2018, 24 (12), 2716–2718. [DOI] [PubMed] [Google Scholar]
- (24).Mansurov A; Ishihara J; Hosseinchi P; Potin L; Marchell TM; Ishihara A; Williford J-M; Alpar AT; Raczy MM; Gray LT; Swartz MA; Hubbell JA Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed Eng 2020, 4, 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Momin N; Mehta NK; Bennett NR; Ma L; Palmeri JR; Chinn MM; Lutz EA; Kang B; Irvine DJ; Spranger S; Wittrup KD Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci. Transl. Med 2019, 11 (498), eaaw2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Braun M; Ress ML; Yoo Y-E; Scholz CJ; Eyrich M; Schlegel PG; Wölfl M IL12-mediated sensitizing of T-cell receptor-dependent and -independent tumor cell killing. OncoImmunology 2016, 5 (7), No. e1188245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Goplen NP; Saxena V; Knudson KM; Schrum AG; Gil D; Daniels MA; Zamoyska R; Teixeiro E IL-12 Signals through the TCR To Support CD8 Innate Immune Responses. J. Immunol 2016, 197 (6), 2434–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Aigner M; Feulner J; Schaffer S; Kischel R; Kufer P; Schneider K; Henn A; Rattel B; Friedrich M; Baeuerle PA; Mackensen A; Krause SW T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia 2013, 27 (5), 1107–1115. [DOI] [PubMed] [Google Scholar]
- (29).Chames P; Baty D Bispecific antibodies for cancer therapy. mAbs 2009, 1 (6), 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dreier T; Lorenczewski G; Brandl C; Hoffmann P; Syring U; Hanakam F; Kufer P; Riethmuller G; Bargou R; Baeuerle PA Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100 (6), 690–697. [DOI] [PubMed] [Google Scholar]
- (31).Hoffmann P; Hofmeister R; Brischwein K; Brandl C; Crommer S; Bargou R; Itin C; Prang N; Baeuerle PA Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 2005, 115 (1), 98–104. [DOI] [PubMed] [Google Scholar]
- (32).Joshi NS; Cui W; Chandele A; Lee HK; Urso DR; Hagman J; Gapin L; Kaech SM Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 2007, 27 (2), 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Barrett JA; Cai H; Miao J; Khare PD; Gonzalez P; Dalsing-Hernandez J; Sharma G; Chan T; Cooper LJN; Lebel F Regulated intratumoral expression of IL-12 using a RheoSwitch Therapeutic System® (RTS®) gene switch as gene therapy for the treatment of glioma. Cancer Gene Ther. 2018, 25 (5), 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Algazi A; Bhatia S; Agarwala S; Molina M; Lewis K; Faries M; Fong L; Levine LP; Franco M; Oglesby A; Ballesteros-Merino C; Bifulco CB; Fox BA; Bannavong D; Talia R; Browning E; Le MH; Pierce RH; Gargosky S; Tsai KK; Twitty C; Daud AI Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann. Oncol 2020, 31 (4), 532–540. [DOI] [PubMed] [Google Scholar]
- (35).Luheshi N; Hewitt S; Garcon F; Burke S; Watkins A; Arnold K; Zielinski J; Martin P; Sulikowski M; Bagnall C; Lapointe J-M; Moody G; Si H; Morehouse C; Wilkinson RW; Herbst R; Frederick J Abstract 5017: MEDI1191, a novel IL-12 mRNA therapy for intratumoral injection to promote TH1 transformation of the patient tumor microenvironment. Cancer Res. 2019, 79 (13 Supplement), 5017–5017. [Google Scholar]
- (36).Lu M; Cohen MH; Rieves D; Pazdur R FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am. J. Hematol 2010, 85 (5), 315–319. [DOI] [PubMed] [Google Scholar]
- (37).McCarthy JR; Weissleder R Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Delivery Rev 2008, 60 (11), 1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ramos CA; Savoldo B; Dotti G CD19-CAR Trials. Cancer J. 2014, 20 (2), 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Frankel AE; Woo JH; Ahn C; Foss FM; Duvic M; Neville PH; Neville DM Resimmune, an anti-CD3ε recombinant immunotoxin, induces durable remissions in patients with cutaneous T-cell lymphoma. Haematologica 2015, 100 (6), 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hawe A; Hulse WL; Jiskoot W; Forbes RT Taylor Dispersion Analysis Compared to Dynamic Light Scattering for the Size Analysis of Therapeutic Peptides and Proteins and Their Aggregates. Pharm. Res 2011, 28 (9), 2302–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Klinger M; Brandl C; Zugmaier G; Hijazi Y; Bargou RC; Topp MS; Gökbuget N; Neumann S; Goebeler M; Viardot A; Stelljes M; Brüggemann M; Hoelzer D; Degenhard E; Nagorsen D; Baeuerle PA; Wolf A; Kufer P Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell–engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119 (26), 6226–6233. [DOI] [PubMed] [Google Scholar]
- (42).Schlegel P; Lang P; Zugmaier G; Ebinger M; Kreyenberg H; Witte K-E; Feucht J; Pfeiffer M; Teltschik H-M; Kyzirakos C; Feuchtinger T; Handgretinger R Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014, 99 (7), 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zimmerman Z; Maniar T; Nagorsen D Unleashing the clinical power of T cells: CD19/CD3 bi-specific T cell engager (BiTE®) antibody construct blinatumomab as a potential therapy. Int. Immunol 2015, 27 (1), 31–37. [DOI] [PubMed] [Google Scholar]
- (44).Jiang X; Chen X; Carpenter TJ; Wang J; Zhou R; Davis HM; Heald DL; Wang W Development of a Target cell-Biologics-Effector cell (TBE) complex-based cell killing model to characterize target cell depletion by T cell redirecting bispecific agents. mAbs 2018, 10 (6), 876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Minguet S; Swamy M; Alarcón B; Luescher IF; Schamel WWA Full Activation of the T Cell Receptor Requires Both Clustering and Conformational Changes at CD3. Immunity 2007, 26 (1), 43–54. [DOI] [PubMed] [Google Scholar]
- (46).Wang Q; Lin T; Tang L; Johnson JE; Finn MG Icosahedral Virus Particles as Addressable Nanoscale Building Blocks. Angew. Chem., Int. Ed 2002, 41 (3), 459–462. [DOI] [PubMed] [Google Scholar]
- (47).Gupta SS; Kuzelka J; Singh P; Lewis WG; Manchester M; Finn MG Accelerated Bioorthogonal Conjugation: A Practical Method for the Ligation of Diverse Functional Molecules to a Polyvalent Virus Scaffold. Bioconjugate Chem. 2005, 16 (6), 1572–1579. [DOI] [PubMed] [Google Scholar]
- (48).Bale JB; Gonen S; Liu Y; Sheffler W; Ellis D; Thomas C; Cascio D; Yeates TO; Gonen T; King NP; Baker D Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 2016, 353 (6297), 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.