

Abstract
Background
Increased left ventricular (LV) mass is associated with adverse cardiovascular events including heart failure (HF). Both increased LV mass and HF disproportionately affect Black individuals. To understand underlying mechanisms, we undertook a proteomic screen in a Black cohort and compared the findings to results from a white cohort.
Methods
We measured 1305 plasma proteins using the SomaScan® platform in 1772 Black participants (mean age 56 years, 62% women) in the Jackson Heart Study (JHS) with LV mass assessed by 2D echocardiography. Incident HF was assessed in 1600 participants. We then compared protein associations in JHS to those observed in white participants from the Framingham Heart Study (FHS, mean age 54 years, 56% women).
Results
In JHS, there were 110 proteins associated with LV mass and 13 proteins associated with incident HF hospitalization with false discovery rate <5% after multivariable adjustment. Several proteins showed expected associations with both LV mass and HF, including N-terminal pro-BNP (β = 0.04, p = 2 × 10−8; HR = 1.48, p = 0.0001). The strongest association with LV mass was novel: Leukotriene A-4 hydrolase (LKHA4) (β = 0.05, p = 5 × 10−15). This association was confirmed on an alternate proteomics platform and further supported by related metabolomic data. Fractalkine/CX3CL1 showed a novel association with incident HF (HR = 1.32, p = 0.0002). While established biomarkers such as cystatin C and N-terminal pro-BNP showed consistent associations in Black and white individuals, LKHA4 and fractalkine were significantly different between the two groups.
Conclusions
We identified several novel biological pathways specific to Black adults hypothesized to contribute to the pathophysiologic cascade of LV hypertrophy and incident HF including LKHA4 and fractalkine.
Keywords: proteomics, heart failure, left ventricular hypertrophy, left ventricular mass, race
Journal Subject Terms: Hypertrophy, Race and Ethnicity
Introduction
Left ventricular hypertrophy (LVH) represents a major worldwide disease burden: it complicates 18-41% of the estimated 65 million cases of hypertension.1 Despite the fact that hypertension is a notable LVH risk factor, LVH represents an independent mortality risk beyond blood pressure alone, raising mortality risk six- to eightfold, even in the presence of a normal ejection fraction.2,3 LVH has a diverse range of risk factors in addition to hypertension including age, weight, diabetes, obesity, hyperlipidemia, and smoking. Race is also associated with risk; Black individuals in particular suffer from an increased burden of LVH. Findings from HyperGEN and the Dallas Heart Study suggest that the prevalence of LVH is at least two-fold higher in Black individuals compared to white individuals.4
Specific cardiovascular outcomes are similarly heterogeneous. Namely, LVH is an independent risk factor for myocardial ischemia, stroke, heart failure (HF), and sudden cardiac death. However, not all individuals with LVH suffer from all of these complications.5 Furthermore, the mechanisms by which LVH progresses to HF remain unclear. Treatment response is also diverse; while it has been shown that anti-hypertensive treatment can reduce LVH over time, and that this reduction improves mortality, in the LIFE trial, 23% of participants still had LVH after 5 years.6 The heterogeneity of outcomes and treatment effects likely stems from complex interplay between environmental and genetic effects, particularly given the fact that the pathophysiologic mechanisms of LVH involve multiple molecular pathways at the level of the myocytes, kidneys, and the sympathetic nervous system.7-9
Given the heterogeneity of risks, outcomes, and treatment effects, particularly when comparing Black and white individuals, it is essential that we explore the underlying pathophysiologic processes of LVH in more depth specifically in a Black population. Proteomic screening provides a way to interrogate multiple biologic pathways simultaneously. Recent advances in aptamer-based proteomic technologies enable high throughput profiling of low abundance analytes in large epidemiological cohorts.10-12 While some proteomic screens have been performed in mouse and rat models of LVH and in white human populations,13-16 there is a paucity of data describing the associations between the human proteome and LVH in Black populations, despite their increased burden of disease.
We therefore undertook a proteomic screen of the Jackson Heart Study (JHS), a large population-based cohort comprised of Black participants from the Jackson, MS community in order to better understand the pathophysiology of LVH and incident HF. We then compared our findings to those from the Framingham Heart Study (FHS) to test for potential differences. Finally, we leveraged genetic and metabolomic data from JHS to corroborate these findings.
Methods
Data availability
All proteomics data is available through dbGaP: JHS accession phs000964/phs002256.v1.p1 and FHS accession phs000007.v31.p12, requiring access request. JHS metabolomics data have been submitted to the JHS Data Coordinating Center and to dbGaP; until posted in dbGaP, all JHS data are available from the JHS Data Coordinating Center on request.
Study approval
The human study protocols were approved by the Institutional Review Boards of Beth Israel Deaconess Medical Center, Boston University Medical Center, and University of Mississippi Medical Center, and all participants provided written informed consent.
Full methods are available as a supplement.
Results
Baseline characteristics in JHS
A total of 2,145 individuals from JHS had proteomic profiling (Figure 1). These individuals were clinically similar to JHS as a whole (N = 5,306, Supplemental Table I). Of this group with profiling, 1772 had echocardiography. After indexing to height, 301 (17%) had LVH by American Society of Echocardiography criteria (Table 1). Individuals with LVH were more likely to be older, female, hypertensive, overweight, have diabetes, have a history consistent with coronary heart disease (CHD), and have lower estimated glomerular filtration rate (eGFR). Ejection fraction and low-density lipoprotein (LDL) cholesterol levels were similar.
Figure 1.
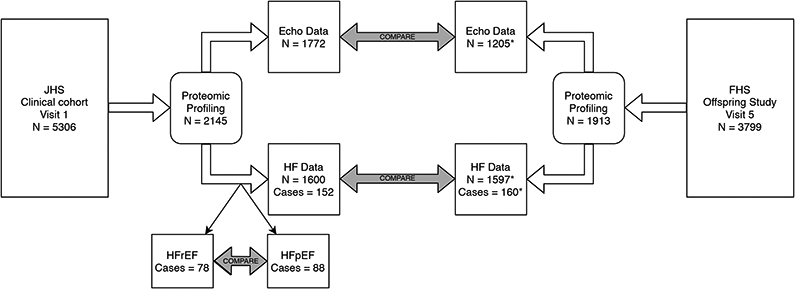
Flowchart of analytical approach to proteomic data. A subset of participants in each study had proteomic profiling. Individuals with a complete complement of clinical data and available echocardiography were included in models of left ventricular mass. Individuals with a complete complement of clinical data, free of heart failure at baseline, and able to be followed for incident heart failure were included in models of incident heart failure. *As samples from Framingham were run on two separate platforms, some proteins are available in fewer individuals: N=694 for some proteins in LV mass analyses and N=891 (72 cases) for some proteins in incident heart failure analyses. JHS = Jackson Heart Study, FHS = Framingham Heart Study, HF = heart failure, HFrEF = heart failure with reduced ejection fraction, HFpEF = heart failure with preserved ejection fraction.
Table 1.
Cohort Characteristics by Left Ventricular Hypertrophy Status and Incident Heart Failure Status
| JHS | FHS | |||||||
|---|---|---|---|---|---|---|---|---|
| Characteristic1 | No LVH N = 1471 |
LVH N = 301 |
No HF N = 1448 |
HF N = 152 |
No LVH N = 925 |
LVH N = 280 |
No HF N = 1437 |
HF N = 160 |
| Age, y | 55 (12) | 62 (11) | 55 (12) | 65 (11) | 53 (10) | 57 (10) | 54 (10) | 63 (8) |
| Female | 868 (59%) | 238 (79%) | 900 (62%) | 101 (66%) | 492 (53%) | 185 (66%) | 774 (54%) | 71 (44%) |
| Hypertension, n | 876 (60%) | 256 (85%) | 866 (60%) | 123 (81%) | 237 (26%) | 138 (49%) | 456 (32%) | 106 (66%) |
| Systolic blood pressure, mmHg | 126 (17) | 135 (21) | 126 (18) | 134 (19) | 123 (18) | 132 (20) | 125 (18) | 140 (20) |
| Hypertensive medication, n | 730 (50%) | 239 (79%) | 731 (50%) | 112 (74%) | 126 (14%) | 70 (25%) | 239 (17%) | 75 (47%) |
| Diabetes, n | 275 (19%) | 93 (31%) | 246 (17%) | 66 (43%) | 33 (3.6%) | 27 (9.6%) | 75 (5.2%) | 46 (29%) |
| Hemoglobin A1c, % | 6.0 (1.2) | 6.3 (1.5) | 5.9 (1.2) | 6.5 (1.7) | 5.3 (0.8) | 5.6 (1.0) | 5.4 (0.9) | 6.3 (1.7) |
| Body mass index, kg/m2 | 31 (7) | 35 (8) | 32 (7) | 33 (8) | 26 (4) | 29 (5) | 27 (5) | 30 (6) |
| Estimated glomerular filtration rate, ml/min/1.73m2 | 84 (19) | 78 (23) | 85 (18) | 74 (24) | 91 (19) | 87 (22) | 90 (19) | 81 (23) |
| LDL-cholesterol, mg/dL | 127 (37) | 126 (37) | 127 (37) | 129 (42) | 125 (33) | 127 (34) | 126 (33) | 128 (35) |
| History of CAD, n | 74 (5.0%) | 37 (12%) | 57 (3.9%) | 15 (9.9%) | 13 (1.4%) | 11 (3.9%) | 18 (1.3%) | 17 (11%) |
| Ejection fraction, % | 62 (7) | 63 (10) | 62 (7) | 62 (8) | 67 (7) | 67 (9) | 67 (7) | 62 (13) |
| Left ventricular mass index, g/m2.7 | 32.9 (6.0) | 54.2 (9.9) | 35.4 (9.5) | 42.0 (12.1) | 36.1 (6.2) | 51.9 (6.7) | 39.3 (8.8) | 44.8 (10.5) |
| Current smoker, n | 183 (12%) | 37 (12%) | 160 (11%) | 24 (16%) | 165 (18%) | 50 (18%) | 286 (20%) | 25 (16%) |
Statistics presented: mean (SD); n (%)
Of 1600 individuals with proteomic profiling, clinical covariates, and without prevalent HF at baseline, 152 (10%) were hospitalized for incident HF during a median of 11 years of follow up. The pattern of comorbidity burden in the HF group was similar to that of the LVH group. (Table 1)
Protein Associations with Left Ventricular Mass
We used linear models to assess the association between 1305 proteins and LV mass. In an age and sex adjusted model, 381 proteins were associated with LV mass with false discovery rate (FDR) < 5% (Supplemental Table II). After adjustment for age, sex, body mass index, eGFR, systolic blood pressure, presence of hypertension, presence of diabetes, total/high-density lipoprotein cholesterol, history of myocardial infarction (MI), current smoking status, 110 proteins were associated with LV mass with FDR < 5% (Figure 2, Supplemental Table III). As expected, N-terminal pro-BNP (β = 0.04 [0.02, 0.05], p = 2 × 10−8) was strongly associated with LV mass. This association held true among obese individuals (data not shown). Many of the protein-LVH associations were novel, including LKHA4 (change in ln[LV mass] per standard deviation change in protein level, β = 0.05 [0.03, 0.06], p = 5 × 10−15), and cAMP-dependent protein kinase catalytic subunit alpha (PRKACA, β = 0.03 [0.02, 0.04], p = 6 × 10−8). Proteins with the strongest inverse association with LV mass included carbonic anhydrase (β = −0.03 [−0.04, −0.02], p = 8 × 10−8) and 6-phosphogluconate dehydrogenase (β = −0.03 [−0.04, −0.02], p = 4 × 10−7). Modeling LV mass indexed to height2.7 did not significantly affect the results (Supplemental Table IV).
Figure 2.

Volcano Plot of proteins associated with left ventricular mass in Jackson Heart Study. Beta estimates and their p-values are shown for 1305 proteins evaluated by multivariable adjusted linear models predicting ln(left ventricular mass).
Protein Associations with Incident Heart Failure
The association between protein levels and incident HF was assessed using Cox proportional hazards models. In an age and sex adjusted model, 111 proteins were associated with incident HF with FDR < 5%. In a multivariable adjusted model (same covariates as above as well as interim MI), 13 proteins were associated with FDR < 5%. (Figure 3, Supplemental Table V & VI). These included well-described biomarkers such as cystatin C (Hazard ratio [HR] = 1.66 [1.35, 2.04], p = 1 × 10−6) and N-terminal pro-BNP (HR = 1.48 [1.22, 1.81], p = 0.0001). Cardiotrophin-1, angiopoeitin-2, troponin I, and troponin T were all associated with incident HF as well, as expected, though at levels of significance below the stated threshold in adjusted models (p=0.001, p=0.001, p=0.004, p=0.006, respectively). Growth hormone receptor (HR = 0.68 [0.56, 0.83], p = 0.0001) and galectin-3 (HR = 0.74 [0.62, 0.87], p = 0.0004) showed negative associations with incident HF. More novel findings included fractalkine (HR = 1.32 [1.14, 1.53], p = 0.0002). Only N-terminal pro-BNP, galectin-3, cystatin C, and beta-2 microglobulin had an association with both LV mass and incident HF in adjusted models at FDR < 5%.
Figure 3.

Volcano Plot of proteins associated with incident heart failure in Jackson Heart Study. Hazard ratios for incident heart failure and their p-values for 1305 proteins are shown for multivariable adjusted Cox-proportional hazard models.
Comparing protein associations between JHS and FHS
To understand differences in protein associations between Black and white cohorts, we compared the effect sizes of those proteins associated with LV mass or incident HF in JHS (at FDR < 5% in multivariable adjusted models) to FHS. In the subset of FHS patients that could be evaluated for incident HF (N=1597, Figure 1), subjects had notably less hypertension and hypertension treatment, less diabetes, less obesity, but overall more CHD (Table 1). Over a median of 21 years, 160 patients developed heart failure.
For proteins associated with LV mass in JHS, the majority showed directionally consistent associations in FHS (Supplemental Figure I). Figure 4 shows proteins associated with LV mass in JHS with a Bonferroni-adjusted difference in effect size when compared to FHS. Leukotriene A-4 hydrolase, alanine aminotransferase 1, heat shock 70kDa protein 1A (HSP70), cysteine and glycine rich protein 3, and neurexophilin-1 were associated with increased LV mass and had effect sizes that differed from FHS at a Bonferroni adjusted level of significance. Serine/threonine-protein kinase 17B, matrilysin (MMP7), matrix metalloproteinase-9 (MMP9), bactericidal permeability-increasing protein, peptidyl-prolyl cis-trans isomerase D, and high mobility group protein B1 also had significant differences from FHS, but were inversely associated with LV mass. For comparison, renin and N-terminal pro-BNP showed no significant differences: renin was inversely associated with LV mass in both cohorts, while N-terminal pro-BNP was directly associated with LV mass.
Figure 4.

Proteins which associated with left ventricular (LV) mass in multivariable adjusted models in JHS at FDR<5% are compared to FHS. Any proteins below Bonferroni adjusted p value for difference are shown above, as well as renin and N-terminal pro-BNP, which did not differ between cohorts, for comparison.
*Proteins available only on the 1.3K SomaScan® platform which was not run in batch 1 of FHS, therefore N=694 rather than N =1205.
Given the significant differences, we leveraged data from an alternate, antibody-based proteomics platform to support these findings. Of the 11 proteins which differed from FHS, 5 are measured by the Olink® platform and were available in a subset of 458 individuals from JHS. Despite reduced power (due to one-quarter the samples), LKHA4, MMP7, and HSP70 showed significant and consistent associations with LV mass while MMP9 was directionally consistent; neurexophilin-1’s association was inconsistent with the aptamer-based data (Supplemental Table VII).
Fewer differences between the cohorts were seen for proteins associated with incident HF (Figure 5). Growth arrest-specific protein 1 and fractalkine/CX3CL1 levels were associated with a higher risk of incident HF in JHS compared to FHS, while growth hormone receptor was associated with a lower risk of incident HF in JHS compared to FHS. However, only growth arrest-specific protein 1 met the Bonferroni-adjusted significance threshold. These relationships were not evaluated using Olink® given the smaller sample size and reduced HF events, limiting power.
Figure 5.

Proteins which associated with incident heart failure (HF) in multivariable adjusted models in JHS at FDR<5% are compared to FHS and shown above.
*Proteins available only on the 1.3K SomaScan® platform, which was not run in batch 1, therefore N=891 with Cases = 72 rather than N=1597 with cases = 160
HFpEF vs HFrEF
We compared heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction (HFrEF), to explore proteins more strongly associated with specific subtypes of HF. Of the 1836 individuals without prevalent HF in whom HF status could be followed, there were 78 cases of HFrEF and 88 cases of HFpEF (Figure 1). Fourteen cases of HF had an indeterminate type and were excluded. In competing risk analysis, 324 proteins were associated with at least one subtype of HF at p < 0.05 (Supplemental Table VIII). When compared, 41 proteins showed a difference in risk estimate between HFpEF and HFrEF at FDR < 10% (Figure 6). Hexokinase-2 levels were associated with a decreased risk of HFpEF but with increased risk of HFrEF (HR for HFrEF 1.22 [1.07-1.38] per standard deviation change in protein level, p = 0.003; HR for HFpEF 0.80 [0.67-0.95], p-value = 0.01, p-value for difference = 6 × 10−4). Alternatively, killer cell immunoglobulin-like receptor 3DS1 showed the opposite pattern of association (HR for HFpEF 1.24 [1.07-1.43], p-value 0.004; HR for HFrEF 0.73 [0.56-0.96], p-value = 0.02, p-value for difference = 8 × 10−4). Proteins including cystatin C and N-terminal pro-BNP were associated with increased risk for both types of HF, and therefore were not significantly different between the two groups (Figure 6).
Figure 6.

Proteins which predicted either heart failure with preserved ejection fraction (HFpEF) or heart failure with reduced ejection fraction (HFrEF) in age, sex, and batch adjusted competing risk models (N of aptamers = 324) were then compared to one another. Displayed are proteins whose competing risk estimate significantly differed between the two subtypes at an FDR<10%, as well as cystatin C and N-terminal pro-BNP, which do not differ between subtypes, for comparison.
Associations between arachidonic acid oxidation products and cardiac phenotypes in JHS
Leukotriene A-4 hydrolase catalyzes the conversion of leukotriene A4 to leukotriene B4 in a reaction which lies downstream of the conversion of arachidonic acid to multiple signaling molecules. Using available metabolomics profiling in JHS, we identified six metabolites downstream of arachidonic acid metabolism. While leukotriene A4 and B4 could not be measured due to low abundance, multiple eicosanoids were measured. We determined each metabolite’s partial correlation with LKHA4 after adjustment for age and sex as well as their association with both LV mass and incident HF after adjustment for all of the previously noted clinical covariates. With the exception of Arachidonic acid, all metabolites were significantly correlated with LKHA4. Much like LKHA4, each metabolite was directly associated with LV mass, but none were associated with incident HF (Table 2). The strongest association was that of the mono-Hydroxyeicosatetraenoic acids, which, during profiling, group together as a single peak (Pearson correlation with LKHA4 = 0.27, p = 1 × 10−23; beta for association with ln(LV mass) = 0.03, p = 5 × 10−5; HR for incident HF = 1.17, p = 0.20).
Table 2.
Arachidonic acid oxidation products and their associations with Leukotriene A-4 Hydrolase, Left Ventricular Mass, and Incident Heart Failure
| Metabolite | Pearson Correlation with LKHA4 (95% CI) |
p-value | Beta for association with ln (LV mass) (95% CI) |
p-value | HR for incident HF (95% CI) |
p-value |
|---|---|---|---|---|---|---|
| Arachidonic acid | 0.02 (−0.03, 0.07) | 0.44 | 0.02 (0.01, 0.04) | 5.0e-4 | 0.98 (0.76, 1.26) | 0.89 |
| 5-Oxo-ETE | 0.18 (0.13, 0.23) | 6.4e-11 | 0.02 (0.00, 0.03) | 0.02 | 1.07 (0.82, 1.39) | 0.62 |
| 12-Oxo-ETE | 0.20 (0.15, 0.25) | 2.4e-13 | 0.02 (0.01, 0.03) | 0.006 | 1.11 (0.85, 1.45) | 0.44 |
| 15-Oxo-ETE | 0.21 (0.16, 0.26) | 1.3 e-14 | 0.02 (0.01, 0.03) | 0.003 | 1.14 (0.87, 1.49) | 0.35 |
| 14(15)-EET | 0.26 (0.20, 0.30) | 4.2e-21 | 0.02 (0.00, 0.03) | 0.01 | 1.15 (0.89, 1.49) | 0.29 |
| 11(12)-EET | 0.27 (0.22, 0.32) | 1.8e-23 | 0.02 (0.01, 0.03) | 0.005 | 1.15 (0.89, 1.48) | 0.28 |
| HETE | 0.27 (0.22, 0.32) | 1.3e-23 | 0.03 (0.01, 0.04) | 4.6e-5 | 1.17 (0.92, 1.49) | 0.20 |
ETE = eicosatetraenoic acid, EET = epoxyeicosatrienoic acid, HETE = hydroxyeicosatetraenoic acid, CI = Confidence Interval
Discussion
Both LV hypertrophy and incident HF are associated with a myriad of upstream risk factors and downstream complications. Increased risk is associated with race,4 though the combined impact of social/structural, environmental, and genetic factors on pathobiology remains unclear.17 While proteomic profiling of HF has been performed in white populations,15,16,18 similar work is lacking in Black populations. Comparative profiling is critical to our understanding of the disease and its underlying pathways. To this end, our study utilizes the largest proteomic profiling of a Black population both in number of subjects and number of proteins profiled – 1,305 proteins in 2,145 individuals – and compares it to an equally large and well profiled white population. These individuals had high quality echocardiographic phenotyping and rigorous adjudication of HF events, as well as detailed profiling of their comorbidities allowing for statistical adjustment in these heterogeneous disease states. Taken together, these results can form the basis for deeper exploration into the biology of LVH and incident HF.
We observed 110 proteins to be associated with LV mass, even after adjustment for multiple co-morbidities and multiple comparisons, and 13 proteins associated with incident HF. Validating the platform, N-terminal pro-BNP was strongly associated with increased LV mass as well as incident HF. Two other well described renal markers showed a similar pattern, cystatin C and beta-2 microglobulin. These markers are known to be associated with LV mass in patients with chronic kidney disease,19-21 though the association has not previously been shown in community dwelling adults. By contrast, cystatin C is well known to be associated with incident HF and HF prognosis.22 Similar data for a relationship between beta-2 microglobulin and incident HF is weaker, though it does appear to be elevated once HF has developed.23,24 Interestingly, galectin-3, a known biomarker in HF and renal disease, showed an inverse association with LV mass and incident HF in both JHS and FHS, contrary to existing literature which include data from FHS.25,26 This suggests the measurement of galectin-3 by the aptamer may differ from that of the ELISA (enzyme-linked immunosorbent assay). The aptamer’s specificity for galectin-3 is supported by genetic and mass spectroscopy data (Supplemental Table IX), however, there are four known splice variants of galectin-3, so the specific variant detected by the aptamer may differ from ELISA. If signaling in HF is related to an increase in one variant relative to another, this may explain the result. Further investigation is certainly warranted.
Several proteins showed a strong association with LV mass, but not with incident HF. The strongest association between any protein and LV mass was LKHA4. Specifically, this association was unique to the Black population. LKHA4 hydrolyzes leukotriene A4 to leukotriene B4, which exerts pro-inflammatory effects via the G protein coupled receptor BLT1 on leukocytes. This pathway has been implicated in vascular inflammation, and LKHA4 activity has been targeted in drug trials for atherosclerotic disease.27,28 These drugs proved ineffective, possibly due to concomitant inhibition of the enzyme’s anti-inflammatory capabilities.28 Despite a strong association with LV mass, it is not clear why LKHA4 was not associated with incident HF, nor a specific subtype of HF in our analyses. However, previous work shows an association with prevalent HF in a small cohort that included nearly 25% Black participants.29 To explore the relationship more deeply, we utilized available profiling of metabolites downstream of arachidonic acid. Leukotriene A4 and leukotriene B4 are such metabolites, but are low abundance and not well measured in the blood by liquid chromatography and mass spectrometry. However, arachidonic acid (a precursor to leukotriene A4) and other oxidation products of arachidonic acid are available, including mono-hydroxyeicosatetraenoic acids (HETEs), epoxyeicosatrienoic acids (EETs), and oxoeicosatetraenoic acids (oxo-ETEs). Interestingly, these oxidation products were both correlated with LKHA4 levels and associated with LV mass, but not associated with incident HF, a similar pattern to LKHA4 itself. Furthermore, phospholipase A2, which produces arachidonic acid was not associated with either LV mass or incident HF after adjustment for clinical covariates. These data highlight pathways downstream of arachidonic acid as areas for further investigation rather than arachidonic acid metabolism as a whole. It may be that LKHA4 and related pathways are responsive to these disease states, rather than causative. Detailed profiling of the eicosanoid lipidome is of critical importance in future studies, and, given the observed data, should ideally be performed in diverse populations.
Conversely, fractalkine/CX3CL-1 was found to be associated with incident HF, but not LV mass, an association that was notably stronger in JHS compared to FHS. Fractalkine is observed in both a membrane-bound and soluble form, and is involved in chemo-attraction and binding of peripheral inflammatory cells.30 It has been implicated in atherosclerosis, ischemia-reperfusion injury, and cardiomyopathy.30-32 Our data suggest that it has an independent association with incident HF hospitalization in an ambulatory Black population. The fact that many proteins associated with either LV mass or incident HF but not both fits with data suggesting the pathobiology of each may differ: drugs that improve LVH have not been shown to have benefit in HFpEF.33,34
Comparing subtypes of HF (HFpEF vs HFrEF), while exploratory given samples sizes, highlighted a substantial difference between the proteins associated with the HF syndrome as a whole. Those proteins associated most strongly with incident HF overall did not show a predilection for either type, highlighting the syndromic similarities between each subtype. Conversely, the proteins with significant difference between the two subtypes highlight the varying pathobiology between the two phenotypes. Hexokinase 2 and growth differentiation factor (GDF) 11 both showed a stronger association with HFrEF compared to HFpEF. Hexokinase 2 is known to attenuate cardiac hypertrophy when over-expressed in mice by modulating flux through the pentose phosphate pathway, which in turn modulates the reactive oxygen species which contribute to hypertrophy.35 Similarly, GDF11, which has significant homology to GDF8/Myostatin, is thought to modulate cardiac muscle mass. Recent data from experimental studies in mice suggest that GDF8/11 levels decline with age and injection of GDF11 was able to reduce cardiac mass.36 Our data correlate with these findings, as levels of these proteins appeared to be protective of HFpEF. However, we also show the levels of these proteins associated with a risk of HFrEF, a consideration for any therapeutic targeting of these pathways. The fact that these proteins did not associate with LV mass in our data suggest that their role in humans may have less of an effect on actual cardiac mass and a greater effect on myocardial function.
Limitations
There are several important limitations of our work. While the proteomic profiling discussed here is the most extensive in any Black cohort to date, it does not contain the entire proteome, and important protein associations may be missed as a result. LV mass was estimated by echocardiographic measurements, which are less accurate than newer methods such as cardiac magnetic resonance imaging. However, one would expect this to bias results to the null. In JHS, HF hospitalizations were not adjudicated until 2005, so there is a chance incident HF hospitalizations were not captured (in the interval between baseline and 2005). While 152 cases of incident HF is of acceptable size, we lacked power for anything more than an exploratory analysis of HFpEF and HFrEF. The mean follow up time required to achieve comparable numbers of HF events between JHS and FHS is significant (11 years versus 21 years), which could influence the difference in protein associations, though age adjustment should account for much of that influence. These event rates illustrate the greater risk in Black individuals. Our results cannot yet be externally validated owing to the unique nature of such extensive profiling in a cohort of Black individuals. However, the numerous positive controls, evidence of aptamer specificity, and multi-omic associations we observed lend significant support to the validity of the platform and analytic approach.
Conclusions
Our results highlight several biological pathways hypothesized to contribute to the pathophysiologic cascade of LV hypertrophy and incident HF. Elements of endovascular dysfunction and inflammation – manifested in these data by associations between LKHA4 and LV mass as well as fractalkine and incident HF – may precede overt disease. Further studies are needed to validate these results and elucidate the detailed underlying mechanisms.
Supplementary Material
Acknowledgments:
The authors wish to thank the staffs and participants of the JHS. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services
Sources of Funding:
The JHS (Jackson Heart Study) is supported and conducted in collaboration with the Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I/HHSN26800001), and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I, and HHSN268201800012I) and contracts from the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute for Minority Health and Health Disparities. Dr Katz is supported by NHLBI T32 postdoctoral training grant (T32HL007374-40). Dr Tahir is supported by the Ruth L. Kirchstein postdoctoral individual National Research Award (F32HL150992). Dr Nayor is supported by NHLBI K23HL138260. Dr Cruz is supported by the KL2/Catalyst Medical Research Investigator Training award from Harvard Catalyst (National Institutes of Health [NIH]/National Center for Advancing Translational Sciences [NCATS] award TR002542). Dr Robbins and Dr Tahir are supported by the John S. LaDue Memorial Fellowship in Cardiology at Harvard Medical School. Dr Benson is supported by NHLBI K08HL145095 award. Drs Gerszten, Wang, and Wilson are supported by NIH R01 DK081572 and NIH R01 HL133870. Drs Gerszten, Wang, and Vasan are supported by NIH R01 HL132320.
Nonstandard Abbreviations and Acronyms
- BNP
B-type natriuretic peptide
- CHD
coronary heart disease
- CI
confidence interval
- EET
epoxyeicosatrienoic acid
- eGFR
estimated glomerular filtration rate
- ELISA
enzyme-linked immunosorbent assay
- ETE
eicosatetraenoic acid
- FDR
false discovery rate
- FHS
Framingham Heart Study
- GDF
growth differentiation factor
- HETE
hydroxyeicosatetraenoic acid
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- HR
hazard ratio
- HSP70
heat shock 70kDa protein 1A
- JHS
Jackson Heart Study
- LDL
low-density lipoprotein
- LKHA4
leukotriene A-4 hydrolase
- LV
left ventricle
- LVH
left ventricular mass
- MI
myocardial infarction
- MMP7
matrilysin
- MMP9
metalloproteinase-9
Footnotes
Disclosures None.
References:
- 1.Cuspidi C, Rescaldani M, Sala C, Grassi G. Left-ventricular hypertrophy and obesity. J Hypertens. 2014;32:16–25. [DOI] [PubMed] [Google Scholar]
- 2.Messerli FH, Ketelhut R. Left ventricular hypertrophy: an independent risk factor. J Cardiovasc Pharmacol. 1991;17 Suppl 4:S59–66; discussion S66-7. [PubMed] [Google Scholar]
- 3.Milani RV, Lavie CJ, Mehra MR, Ventura HO, Kurtz JD, Messerli FH. Left Ventricular Geometry and Survival in Patients With Normal Left Ventricular Ejection Fraction. Am J Cardiol. 2006;97:959–963. [DOI] [PubMed] [Google Scholar]
- 4.Kamath S, Markham D, Drazner MH. Increased prevalence of concentric left ventricular hypertrophy in African-Americans: Will an epidemic of heart failure follow? Heart Fail Rev. 2006;11:271–277. [DOI] [PubMed] [Google Scholar]
- 5.Artham SM, Lavie CJ, Milani R V, Patel DA, Verma A, Ventura HO. Clinical Impact of Left Ventricular Hypertrophy and Implications for Regression. Prog Cardiovasc Dis. 2009;52:153–167. [DOI] [PubMed] [Google Scholar]
- 6.Devereux RB, Wachtell K, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris K, Aurup P, Dahlöf B. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292:2350–6. [DOI] [PubMed] [Google Scholar]
- 7.Greenwood JP, Scott EM, Stoker JB, Mary DASG. Hypertensive left ventricular hypertrophy: Relation to peripheral sympathetic drive. J Am Coll Cardiol. 2001;38:1711–1717. [DOI] [PubMed] [Google Scholar]
- 8.Harrap SB, Dominiczak a F, Fraser R, Lever a F, Morton JJ, Foy CJ, Watt GC. Plasma angiotensin II, predisposition to hypertension, and left ventricular size in healthy young adults. Circulation. 1996;93:1148–54. [DOI] [PubMed] [Google Scholar]
- 9.Takefuji M, Wirth A, Lukasova M, Takefuji S, Boettger T, Braun T, Althoff T, Offermanns S, Wettschureck N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation. 2012;126:1972–82. [DOI] [PubMed] [Google Scholar]
- 10.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O’Sullivan JF, Keshishian H, Farrell LA, et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emilsson V, Ilkov M, Lamb JR, Finkel N, Gudmundsson EF, Pitts R, Hoover H, Gudmundsdottir V, Horman SR, Aspelund T, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science. 2018;361:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan D, Zhao Y, Zhang Y, Tang D, Wu Q. Proteomics Analysis Revealed an Altered Left Ventricle Protein Profile in a Mouse Model of Transverse Aortic Constriction. Protein Pept Lett. 2016;23:125–31. [DOI] [PubMed] [Google Scholar]
- 14.Prévilon M, Le Gall M, Chafey P, Federeci C, Pezet M, Clary G, Broussard C, François G, Mercadier J-J, Rouet-Benzineb P. Comparative differential proteomic profiles of nonfailing and failing hearts after in vivo thoracic aortic constriction in mice overexpressing FKBP12.6. Physiol Rep. 2013;1:e00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egerstedt A, Berntsson J, Smith ML, Gidlöf O, Nilsson R, Benson M, Wells QS, Celik S, Lejonberg C, Farrell L, et al. Profiling of the plasma proteome across different stages of human heart failure. Nat Commun. 2019;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nayor M, Short MI, Rasheed H, Lin H, Jonasson C, Yang Q, Hveem K, Felix JF, Morrison AC, Wild PS, et al. Aptamer-Based Proteomic Platform Identifies Novel Protein Predictors of Incident Heart Failure and Echocardiographic Traits. Circ Heart Fail. 2020;13:e006749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deere B, Griswold M, Lirette S, Fox E, Sims M. Life Course Socioeconomic Position and Subclinical Disease: The Jackson Heart Study. Ethn Dis. 2016;26:355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferreira JP, Verdonschot J, Collier T, Wang P, Pizard A, Bär C, Björkman J, Boccanelli A, Butler J, Clark A, et al. Proteomic Bioprofiles and Mechanistic Pathways of Progression to Heart Failure. Circ Heart Fail. 2019;12:e005897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakuragi S, Ichikawa K, Yamada K, Tanimoto M, Miki T, Otsuka H, Yamamoto K, Kawamoto K, Katayama Y, Tanakaya M, et al. Serum cystatin C level is associated with left atrial enlargement, left ventricular hypertrophy and impaired left ventricular relaxation in patients with stage 2 or 3 chronic kidney disease. Int J Cardiol. 2015;190:287–292. [DOI] [PubMed] [Google Scholar]
- 20.Masuda M, Ishimura E, Ochi A, Tsujimoto Y, Tahahra H, Okuno S, Tabata T, Nishizawa Y, Inaba M. Serum β2-microglobulin correlates positively with left ventricular hypertrophy in long-term hemodialysis patients. Nephron Clin Pract. 2014;128:101–106. [DOI] [PubMed] [Google Scholar]
- 21.Yılmaz A, Yılmaz B, Küçükseymen S. ß-2 microglobulin level is negatively associated with global left ventricular longitudinal peak systolic strain and left atrial volume index in patients with chronic kidney disease not on dialysis. Anatol J Cardiol. 2016;16:844–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chow SL, Maisel AS, Anand I, Bozkurt B, de Boer RA, Felker GM, Fonarow GC, Greenberg B, Januzzi JL, Kiernan MS, et al. Role of Biomarkers for the Prevention, Assessment, and Management of Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2017;135:e1054–e1091. [DOI] [PubMed] [Google Scholar]
- 23.Matsushita K, Ballew SH, Coresh J. Cardiovascular risk prediction in people with chronic kidney disease. Curr Opin Nephrol Hypertens. 2016;25:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vianello A, Caponi L, Galetta F, Franzoni F, Taddei M, Rossi M, Pietrini P, Santoro G. β2-Microglobulin and TIMP1 Are Linked Together in Cardiorenal Remodeling and Failure. Cardiorenal Med. 2015;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho JE, Liu C, Lyass A, Courchesne P, Pencina MJ, Vasan RS, Larson MG, Levy D. Galectin-3, a marker of cardiac fibrosis, predicts incident heart failure in the community. J Am Coll Cardiol. 2012;60:1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao Y, Shen D, Chen R, Ying C, Wang C, Guo J, Zhang G. Galectin-3 Predicts Left Ventricular Remodeling of Hypertension. J Clin Hypertens Greenwich Conn. 2016;18:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kristo F, Hardy GJ, Anderson TJT, Sinha S, Ahluwalia N, Lin AY, Passeri J, Scherrer-Crosbie M, Gerszten RE. Pharmacological inhibition of BLT1 diminishes early abdominal aneurysm formation. Atherosclerosis. 2010;210:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low CM, Akthar S, Patel DF, Löser S, Wong C-T, Jackson PL, Blalock JE, Hare SA, Lloyd CM, Snelgrove RJ. The development of novel LTA4H modulators to selectively target LTB4 generation. Sci Rep. 2017;7:44449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wells QS, Gupta DK, Smith JG, Collins SP, Storrow AB, Ferguson J, Smith ML, Pulley JM, Collier S, Wang X, et al. Accelerating Biomarker Discovery Through Electronic Health Records, Automated Biobanking, and Proteomics. J Am Coll Cardiol. 2019;73:2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Apostolakis S, Spandidos D. Chemokines and atherosclerosis: focus on the CX3CL1/CX3CR1 pathway. Acta Pharmacol Sin. 2013;34:1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boag SE, Das R, Shmeleva EV, Bagnall A, Egred M, Howard N, Bennaceur K, Zaman A, Keavney B, Spyridopoulos I. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest. 2015;125:3063–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escher F, Vetter R, Kühl U, Westermann D, Schultheiss H-P, Tschöpe C. Fractalkine in human inflammatory cardiomyopathy. Heart. 2011;97:733–739. [DOI] [PubMed] [Google Scholar]
- 33.Wachtell K, Bella JN, Rokkedal J, Palmieri V, Papademetriou V, Dahlöf B, Aalto T, Gerdts E, Devereux RB. Change in diastolic left ventricular filling after one year of antihypertensive treatment: The Losartan Intervention For Endpoint Reduction in Hypertension (LIFE) Study. Circulation. 2002;105:1071–1076. [DOI] [PubMed] [Google Scholar]
- 34.Yusuf S, Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJV, Michelson EL, Olofsson B, Ostergren J, CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet Lond Engl. 2003;362:777–781. [DOI] [PubMed] [Google Scholar]
- 35.McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. 2013;2:e000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poggioli T, Vujic A, Yang P, Macias-Trevino C, Uygur A, Loffredo FS, Pancoast JR, Cho M, Goldstein J, Tandias RM, et al. Circulating Growth Differentiation Factor 11/8 Levels Decline With Age. Circ Res. 2016;118:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor HA, Wilson JG, Jones DW, Sarpong DF, Srinivasan A, Garrison RJ, Nelson C, Wyatt SB. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005;15:S6-4–17. [PubMed] [Google Scholar]
- 38.Carpenter MA, Crow R, Steffes M, Rock W, Heilbraun J, Evans G, Skelton T, Jensen R, Sarpong D. Laboratory, reading center, and coordinating center data management methods in the Jackson Heart Study. Am J Med Sci. 2004;328:131–144. [DOI] [PubMed] [Google Scholar]
- 39.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An Investigation of Coronary Heart Disease in Families: The Framingham Offspring Study. Am J Epidemiol. 1979;110:281–290. [DOI] [PubMed] [Google Scholar]
- 41.Candia J, Cheung F, Kotliarov Y, Fantoni G, Sellers B, Griesman T, Huang J, Stuccio S, Zingone A, Ryan BM, et al. Assessment of Variability in the SOMAscan Assay. Sci Rep. 2017;7:14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Assarsson E, Lundberg M, Holmquist G, Björkesten J, Thorsen SB, Ekman D, Eriksson A, Rennel Dickens E, Ohlsson S, Edfeldt G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PloS One. 2014;9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith JG, Gerszten RE. Emerging affinity-based proteomic technologies for large scale plasma profiling in cardiovascular disease. Circulation. 2017;135:1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raffield LM, Zakai NA, Duan Q, Laurie C, Smith JD, Irvin MR, Doyle MF, Naik RP, Song C, Manichaikul AW, et al. D-Dimer in African Americans: Whole Genome Sequence Analysis and Relationship to Cardiovascular Disease Risk in the Jackson Heart Study. Arterioscler Thromb Vasc Biol. 2017;37:2220–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang L, Zheng Z, Qi T, Kemper KE, Wray NR, Visscher PM, Yang J. A resource-efficient tool for mixed model association analysis of large-scale data. Nat Genet. 2019;51:1749–1755. [DOI] [PubMed] [Google Scholar]
- 46.Berglund G, Elmstähl S, Janzon L, Larsson SA. The Malmo Diet and Cancer Study. Design and feasibility. J Intern Med. 1993;233:45–51. [DOI] [PubMed] [Google Scholar]
- 47.Benson MD, Yang Q, Ngo D, Zhu Y, Shen D, Farrell LA, Sinha S, Keyes MJ, Vasan RS, Larson MG, et al. The Genetic Architecture of the Cardiovascular Risk Proteome. Circulation. 2018;137:1158–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilk JB, Chen T-H, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, Borecki IB, Silverman EK, Weiss ST, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen M-H, Yang Q. GWAF: an R package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pandey A, Keshvani N, Ayers C, Correa A, Drazner MH, Lewis A, Rodriguez CJ, Hall ME, Fox ER, Mentz RJ, et al. Association of Cardiac Injury and Malignant Left Ventricular Hypertrophy With Risk of Heart Failure in African Americans: The Jackson Heart Study. JAMA Cardiol. 2019;4:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marwick TH, Gillebert TC, Aurigemma G, Chirinos J, Derumeaux G, Galderisi M, Gottdiener J, Haluska B, Ofili E, Segers P, et al. Recommendations on the Use of Echocardiography in Adult Hypertension: A Report from the European Association of Cardiovascular Imaging (EACVI) and the American Society of Echocardiography (ASE)†. J Am Soc Echocardiogr. 2015;28:727–754. [DOI] [PubMed] [Google Scholar]
- 54.Keku E, Rosamond W, Taylor HA, Garrison R, Wyatt SB, Richard M, Jenkins B, Reeves L, Sarpong D. Cardiovascular disease event classification in the Jackson Heart Study: methods and procedures. Ethn Dis. 2005;15:S6-62–70. [PubMed] [Google Scholar]
- 55.Fine JP, Gray RJ. A Proportional Hazards Model for the Subdistribution of a Competing Risk. J Am Stat Assoc. 1999;94:496–509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All proteomics data is available through dbGaP: JHS accession phs000964/phs002256.v1.p1 and FHS accession phs000007.v31.p12, requiring access request. JHS metabolomics data have been submitted to the JHS Data Coordinating Center and to dbGaP; until posted in dbGaP, all JHS data are available from the JHS Data Coordinating Center on request.