

Abstract
Aims
Transthyretin amyloid cardiomyopathy (ATTR‐CM) is a progressive, fatal disorder that remains underdiagnosed. The Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR‐ACT) was the first large clinical trial to include both wild‐type (ATTRwt) and hereditary (ATTRv) patients. A description of the natural history of ATTR‐CM, utilizing data from placebo‐treated patients in ATTR‐ACT, will provide a greater understanding of presentation and progression of ATTR‐CM and may aid in disease awareness, earlier diagnosis and treatment monitoring.
Methods and results
Changes in clinical endpoints (mortality, cardiovascular [CV]‐related hospitalizations, 6‐min walk test [6MWT] distance and Kansas City Cardiomyopathy Questionnaire Overall Summary [KCCQ‐OS] score) from baseline to Month 30 in the 177 patients (134 ATTRwt, 43 ATTRv) who received placebo in ATTR‐ACT were assessed. ATTRwt patients tended to have less severe disease at baseline. Over the duration of ATTR‐ACT, there were 76 (42.9%) all‐cause deaths, and 107 (60.5%) patients had a CV‐related hospitalization. There was a lower proportion of all‐cause deaths in ATTRwt (49, 36.6%) than ATTRv (27, 62.8%). There was a similar, steady decline in mean (SD) 6MWT distance from baseline to Month 30 in ATTRwt (93.9 [93.7] m) and ATTRv (89.1 [107.2] m) patients. The decline in mean (SD) KCCQ‐OS score was less severe in ATTRwt (13.8 [20.7]) than ATTRv (21.0 [26.4]) patients.
Conclusions
Patients with ATTR‐CM experience a severe, progressive disease. In ATTR‐ACT, placebo‐treated patients with ATTRv, compared with ATTRwt, had more severe disease at baseline, and their disease progressed more rapidly as shown by mortality, hospitalizations and quality of life over time.
Keywords: Transthyretin amyloid cardiomyopathy, Clinical trial, Progression, Variant, Hereditary, Wild‐type
Introduction
Transthyretin amyloid cardiomyopathy (ATTR‐CM) is caused by the accumulation of wild‐type (ATTRwt) or variant (ATTRv) transthyretin amyloid fibrils in the myocardium, leading to cardiomyopathy and symptoms of heart failure. 1 , 2 , 3 Onset of ATTRwt is typically later in life (>60 years of age) with the large majority of patients being male. 2 In contrast, symptom onset in patients with ATTRv can occur at a younger age. 1 , 2 , 3
ATTR‐CM is a progressive, fatal disease, but it remains both underdiagnosed and misdiagnosed, and its prevalence remains unknown. In studies in older patients with heart failure with preserved ejection fraction, as many as 15% of patients had evidence of previously undiagnosed ATTRwt. 4 , 5 , 6 Median survival for patients with ATTR‐CM varies with respect to genotype and disease stage at diagnosis. 3 , 7 Without treatment, survival has been reported as approximately 2.5 years for patients with ATTRv caused by the Val122Ile mutation 7 , 8 , 9 and 3.6 years for patients with ATTRwt. 8 , 10 Greater awareness of ATTR‐CM, and what to expect during the course of disease, could aid earlier diagnosis and improve patient management and treatment outcomes. 11
The Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR‐ACT) was the first large clinical trial to include both patients with ATTRwt and patients with ATTRv and represents one of the largest, and longest, collections of natural history data in patients with ATTR‐CM. 12 Patients were enrolled in ATTR‐ACT for up to 30 months, providing a detailed assessment of patients over a meaningful period of time. Following on from prior publications showing the efficacy of tafamidis in patients with ATTR‐CM, 12 in both patients with ATTRwt and patients with ATTRv, 13 this analysis aims to describe the natural history of ATTR‐CM utilizing data from placebo‐treated patients in ATTR‐ACT.
Methods
Trial design and patients
ATTR‐ACT was a Phase 3, multicentre, international, three‐arm, parallel‐design, placebo‐controlled, double‐blind, randomized study (ATTR‐ACT) for which the design has been previously published (NCT01994889). 12 , 14 Briefly, those eligible to enrol were patients aged ≥18 and ≤90 years with ATTR‐CM defined by the presence of either ATTRv or ATTRwt and a medical history of heart failure. The trial was approved by the independent review boards or ethics committee at each participating site and was conducted in accordance with the provisions of the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines. All patients provided written informed consent. This is the largest published clinical trial of placebo‐treated patients with ATTR‐CM to date. This analysis describes those patients who received placebo during the 30 months of the trial.
Clinical evaluations and statistical analyses
Clinical evaluations in ATTR‐ACT included all‐cause mortality, cardiovascular (CV)‐related mortality, CV‐related hospitalizations, functional capacity (assessed by 6‐min walk test [6MWT] distance), health status and quality of life (assessed by Kansas City Cardiomyopathy Questionnaire Overall Summary [KCCQ‐OS] score), changes in N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) levels and echocardiographic measures of cardiac structure and function (interventricular septal wall thickness, left ventricular [LV] posterior wall thickness, LV ejection fraction, LV stroke volume and LV strain). As prespecified in the study protocol, 6MWT distance and KCCQ‐OS score were assessed at baseline and every 6 months for the duration of the trial. NT‐proBNP was assessed at baseline and at Month 12 and Month 30. Echocardiographic measures were assessed at baseline and at Months 6, 18 and 30. Vital status was collected for every patient at Month 30. Heart transplant and implantation of a cardiac mechanical assist device were counted as death/mortality for this description; actual deaths and heart transplants are also shown separately.
This is a descriptive analysis of the changes in clinical endpoints from baseline to Month 30 in patients who received placebo. Clinical endpoints are also described in patients by genotype (ATTRwt compared with ATTRv) and NYHA class at baseline (Class I and II compared with Class III). Some data on the effect of treatment with tafamidis (compared with placebo) on these outcomes have been previously published. 12 , 13
Results
Patient characteristics at baseline
A total of 177 patients received placebo; the majority were male, with a mean age of 74 years (Table 1 ). Genotypes of the 43 patients with ATTRv were Val122Ile (23 patients [53.5%]); Thr60Ala (6 [14.0%]); Val30Met (6 [14.0%]); Ile68Leu (4 [9.3%]); and Asp18Glu, Glu54Leu, Glu89Gln, and Pro24Ser (one patient each [2.3%]). At baseline, patients with ATTRwt tended to be older, but with less severe disease, as shown by the smaller proportion of patients in NYHA Class III and longer mean 6MWT distance (Table 1 ). In contrast, KCCQ‐OS scores and NT‐proBNP levels were similar in patients with ATTRwt and patients with ATTRv. Patients with ATTRwt tended to be more likely to have atrial fibrillation, coronary artery disease, sleep apnoea syndrome and Type 2 diabetes mellitus at enrolment than patients with ATTRv. The majority of patients were receiving diuretics at baseline (69.5% of patients), although use of beta blockers (29.9%) and agents acting on the renin‐angiotensin system (27.1%) was less because they are poorly tolerated or considered harmful in patients with ATTR‐CM. 15
Table 1.
Demographic and clinical characteristics at baseline
| ATTRwt (n = 134) | ATTRv (n = 43) | |
|---|---|---|
| Age, years | ||
| Mean (SD) | 74.9 (6.0) | 71.4 (8.1) |
| Median | 75.0 | 73.0 |
| Min, max | 57, 89 | 51, 86 |
| Sex, n (%) | ||
| Male | 128 (95.5) | 29 (67.4) |
| Female | 6 (4.5) | 14 (32.6) |
| Race, n (%) | ||
| White | 124 (92.5) | 22 (51.2) |
| Black | 5 (3.7) | 21 (48.8) |
| Asian | 5 (3.7) | 0 |
| BMI, mean (SD) | 26.6 (3.6) | 25.4 (5.8) |
| mBMI a , mean (SD) | 1085.9 (180.2) | 1005.6 (225.1) |
| NYHA class, n (%) | ||
| I | 11 (8.2) | 2 (4.7) |
| II | 79 (59.0) | 22 (51.2) |
| III | 44 (32.8) | 19 (44.2) |
| 6MWT distance (metres), mean (SD) | 366.7 (126.2) | 311.2 (117.1) |
| KCCQ‐OS score, mean (SD) | 65.1 (21.3) | 68.4 (23.1) |
| NT‐proBNP (pg/mL), mean (SD) | 3826.3 (2840.2) | 3905.4 (3384.2) |
| Comorbidities b , n (%) | ||
| Atrial fibrillation | 71 (53.0) | 18 (41.9) |
| Hypertension | 61 (45.5) | 23 (53.5) |
| Hyperlipidaemia | 44 (32.8) | 14 (32.6) |
| Cardiac failure, congestive | 38 (28.4) | 11 (25.6) |
| Chronic kidney disease | 31 (23.1) | 10 (23.3) |
| Coronary artery disease | 35 (26.1) | 5 (11.6) |
| Osteoarthritis | 32 (23.9) | 7 (16.3) |
| Benign prostatic hyperplasia | 30 (22.4) | 7 (16.3) |
| Sleep apnoea syndrome | 32 (23.9) | 4 (9.3) |
| Gastro‐oesophageal reflux disease | 26 (19.4) | 6 (14.0) |
| Gout | 21 (15.7) | 4 (9.3) |
| Constipation | 19 (14.2) | 5 (11.6) |
| Insomnia | 17 (12.7) | 6 (14.0) |
| Hypothyroidism | 15 (11.2) | 6 (14.0) |
| Type 2 diabetes mellitus | 19 (14.2) | 0 |
| Dyspnoea | 13 (9.7) | 6 (14.0) |
| Carpal tunnel syndrome | 15 (11.2) | 3 (7.0) |
| Fatigue | 15 (11.2) | 3 (7.0) |
| Echocardiography measures, mean (SD) | ||
| IVST, mm | 16.1 (3.3) | 16.5 (4.1) |
| LV posterior wall thickness, mm | 16.6 (3.9) | 17.0 (4.8) |
| LV ejection fraction, % | 48.9 (8.7) | 47.9 (11.3) |
| LV stroke volume, mL | 46.5 (16.4) | 40.8 (17.9) |
| Circumferential mid‐global strain, % | −17.2 (10.0) | −15.9 (8.5) |
| Radial mid‐global strain, % | 18.6 (10.8) | 14.9 (9.0) |
| Global longitudinal strain, % | −9.6 (3.6) | −8.6 (3.6) |
Some demographic data have been previously published. 13
6MWT, 6‐minute walk test; ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; BMI, body mass index; IVST, interventricular septal wall thickness; KCCQ‐OS, Kansas City Cardiomyopathy Questionnaire Overall Summary; LV, left ventricular; mBMI, modified body mass index; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association; SD, standard deviation.
mBMI is calculated by multiplying the BMI [weight (kg)/height (m2)] by serum albumin concentration (g/L).
Comorbidities present at enrolment in >10% of all patients.
Mortality and hospitalizations
In total, there were 76 (42.9%) all‐cause deaths, of which 63 (35.6% of all patients) were CV related (Table 2 and Figure 1 ). There was a lower proportion of all‐cause deaths in patients with ATTRwt than in patients with ATTRv (Table 2 and Figure 2 A ). There was also a lower proportion of all‐cause deaths in patients with baseline NYHA Class I and II than NYHA Class III (Table 2 and Figure 2 B ). CV‐related deaths were also less common in patients with ATTRwt and patients who were NYHA Class I and II (Table 2 and Figure 3 ). Overall, all‐cause mortality was greatest in patients with ATTRv who were NYHA Class III at baseline (78.9%), followed by patients with ATTRwt who were NYHA Class III (54.5%), patients with ATTRv who were NYHA Class I and II (50.0%) and patients with ATTRwt who were NYHA Class I and II (27.8%) (Table 3 ).
Table 2.
All‐cause, and CV‐related, mortality in all patients and by genotype and NYHA class at baseline
| All‐cause mortality | CV‐related mortality | |
|---|---|---|
| All patients | ||
| n | 177 | 177 |
| Mortality, n (%) | 76 (42.9) | 63 (35.6) |
| Deaths | 72 (40.7) | 59 (33.3) |
| Heart transplants | 4 (2.3) | 4 (2.3) |
| Median time to event, months | NE | NE |
| ATTRwt | ||
| n | 134 | 134 |
| Mortality, n (%) | 49 (36.6) | 41 (30.6) |
| Deaths | 46 (34.3) | 38 (28.4) |
| Heart transplants | 3 (2.2) | 3 (2.2) |
| Median time to event, months | NE | NE |
| ATTRv | ||
| n | 43 | 43 |
| Mortality, n (%) | 27 (62.8) | 22 (51.2) |
| Deaths | 26 (60.5) | 21 (48.8) |
| Heart transplants | 1 (2.3) | 1 (2.3) |
| Median time to event, months | 23.5 | 28.4 |
| NYHA Class I and I | ||
| n | 114 | 114 |
| All‐cause mortality, n (%) | 37 (32.5) | 32 (28.1) |
| Deaths | 33 (28.9) | 28 (24.6) |
| Heart transplants | 4 (3.5) | 4 (3.5) |
| Median time to event, months | NE | NE |
| NYHA Class III | ||
| n | 63 | 63 |
| All‐cause mortality, n (%) | 39 (61.9) | 31 (49.2) |
| Deaths | 39 (61.9) | 31 (49.2) |
| Heart transplants | 0 | 0 |
| Median time to event, months | 24.1 | 28.4 |
Figure 1.
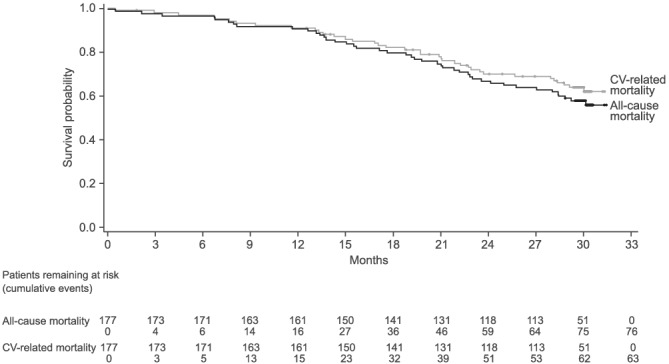
All‐cause mortality and CV‐related mortality in all patients. (Some data on all‐cause mortality have been previously published 12 ).
Figure 2.
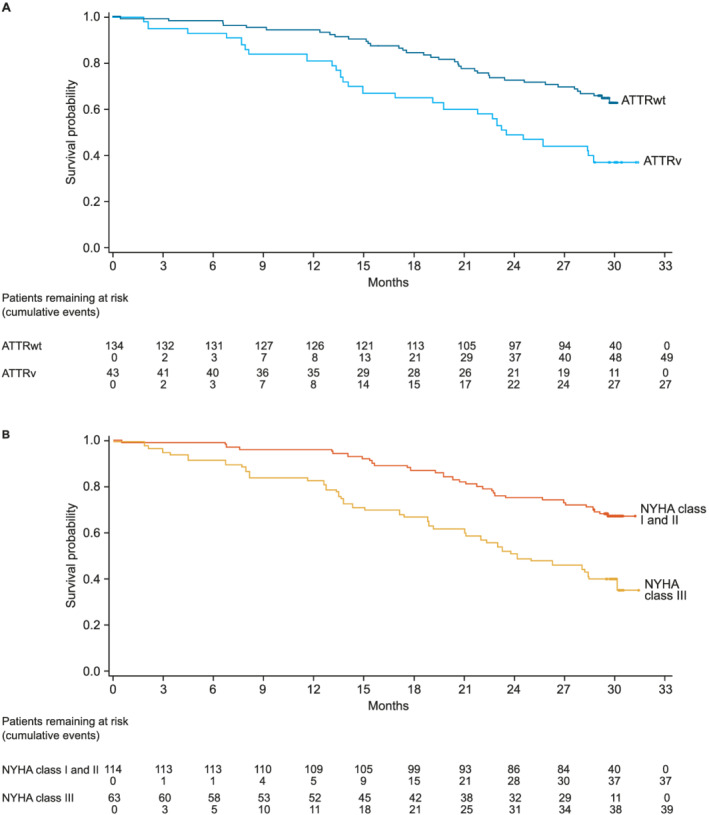
All‐cause mortality by (A) genotype and (B) NYHA class at baseline. (Some data in patients with ATTRwt and patients with ATTRv have been previously published 13 ).
Figure 3.
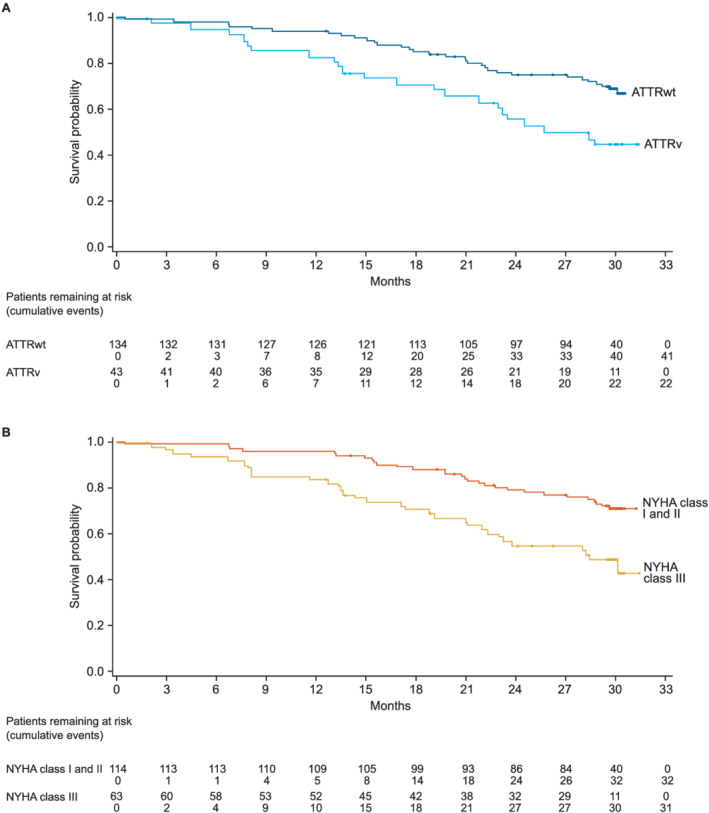
CV‐related mortality by (A) genotype and (B) NYHA class at baseline.
Table 3.
All‐cause mortality in patients with ATTRwt and patients with ATTRv by NYHA class at baseline
| ATTRwt (n = 134) | ATTRv (n = 43) | |
|---|---|---|
| NYHA Class I and II | ||
| n | 90 | 24 |
| All‐cause mortality, n (%) | 25 (27.8) | 12 (50.0) |
| Deaths | 22 (24.4) | 11 (45.8) |
| Heart transplants | 3 (3.3) | 1 (4.2) |
| Median time to event, months | NE | NE |
| NYHA Class III | ||
| n | 44 | 19 |
| All‐cause mortality, n (%) | 24 (54.5) | 15 (78.9) |
| Deaths | 24 (54.5) | 15 (78.9) |
| Heart transplants | 0 | 0 |
| Median time to event, months | 28.1 | 13.8 |
Some data in these patients have been previously published. 13
ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; NE, not estimable; NYHA, New York Heart Association.
In total, 107 (60.5%) patients had a CV‐related hospitalization, with similar proportions in each genotype and NYHA group (Table 4 ). The frequency of CV‐related hospitalizations per year was greater in patients with ATTRv and in NYHA Class III.
Table 4.
Hospitalizations by genotype and by NYHA class at baseline
| ATTRwt (n = 134) | ATTRv (n = 43) | |
|---|---|---|
| Patients with CV‐related hospitalizations, n (%) | 81 (60.4) | 26 (60.5) |
| Frequency of CV‐related hospitalizations per year | ||
| Mean (SD) | 0.86 (1.20) | 0.95 (1.21) |
| Median | 0.40 | 0.56 |
| Min, max | 0, 7.23 | 0, 5.40 |
| NYHA Class I and II (n = 114) | NYHA Class III (n = 63) | |
|---|---|---|
| Patients with CV‐related hospitalizations, n (%) | 70 (61.4) | 37 (58.7) |
| Frequency of CV‐related hospitalizations per year | ||
| Mean (SD) | 0.82 (1.06) | 0.99 (1.42) |
| Median | 0.40 | 0.45 |
| Min, max | 0, 5.39 | 0, 7.23 |
ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; CV, cardiovascular; NYHA, New York Heart Association; SD, standard deviation.
Functional capacity, health status and quality of life and cardiac measures
Functional capacity, as measured by 6MWT distance, declined markedly over time, with a mean (standard deviation [SD]) decline in all patients from baseline to Month 30 of 89.7 (105.2) m. The decline was similar in ATTRwt and ATTRv and in NYHA Class I and II and NYHA Class III (Figure 4 ). Health status and quality of life, as assessed by KCCQ‐OS score, also declined markedly over time, with a mean (SD) decline in all patients from baseline to Month 30 of 14.6 (21.4). The decline was slightly less severe in ATTRwt than ATTRv and in NYHA Class I and II than NYHA Class III (Figure 5 ). NT‐proBNP levels increased over the trial in all patients (Figure S1 ). Echocardiography measures worsened over time in all patients, with the worsening of radial and circumferential strain in particular more rapid, and more severe, in patients with ATTRv (Figure S2 ).
Figure 4.
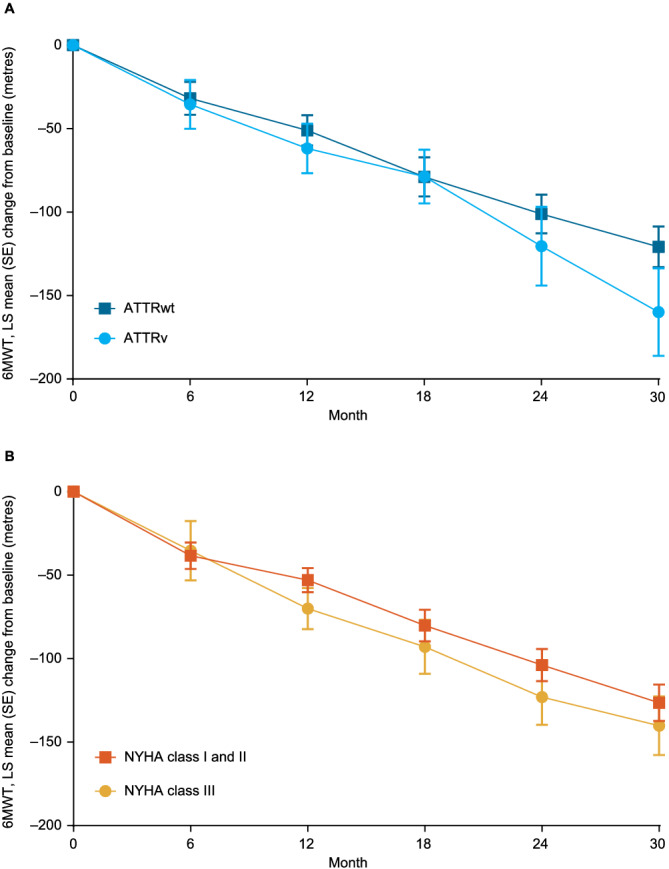
Change in 6MWT distance over time by (A) genotype and (B) NYHA class at baseline. (Some data in patients with ATTRwt and patients with ATTRv have been previously published 13 ).
Figure 5.
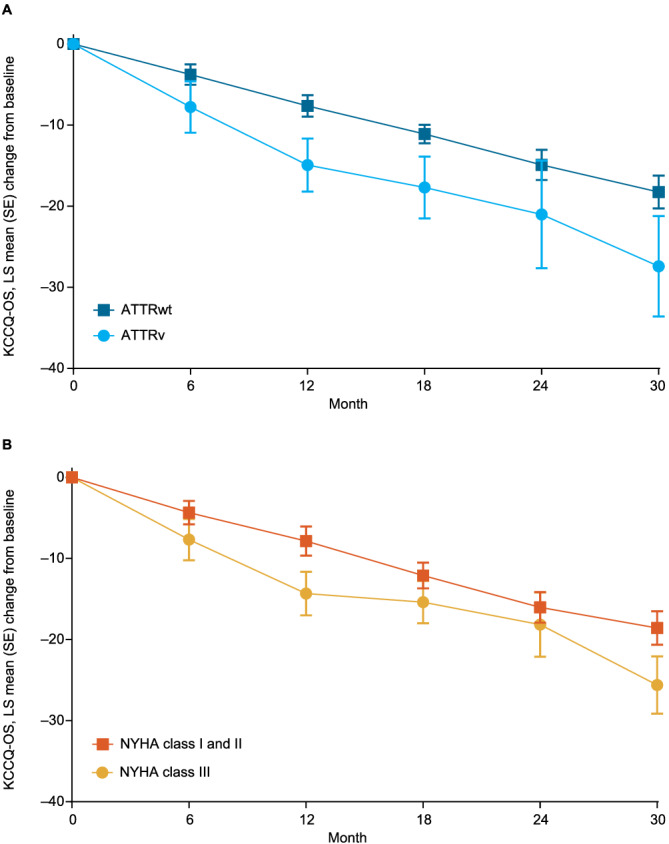
Change in KCCQ‐OS score over time by (A) genotype and (B) NYHA class at baseline. (Some data in patients with ATTRwt and patients with ATTRv have been previously published 13 ).
Discussion
Patients with ATTR‐CM experience a severe, progressive disease with high mortality and frequent hospitalizations. In ATTR‐ACT, placebo‐treated patients with ATTRv, compared with patients with ATTRwt, had more severe disease at baseline, as measured by NYHA class and functional capacity (as assessed by 6MWT distance). In addition, in patients with ATTRv, disease progressed more rapidly as shown by mortality, decline in health status and quality of life (as assessed by KCCQ‐OS score) and increase in NT‐proBNP levels over time.
These data are consistent with prior observational studies in patients with ATTR‐CM, which have demonstrated the poor prognosis of the disease, both for patients with ATTRwt (Table S1 ) and for patients with ATTRv (Table S2 ). 7 , 10 , 16 , 17 , 18 , 19 , 20 As in ATTR‐ACT, observational studies have shown that patients with ATTRv tend to have more severe disease, and poorer survival, than patients with ATTRwt. Collectively, these studies reveal a consistent population of patients with ATTRwt, the majority (>80%) male, aged approximately 75 years, with elevated NT‐proBNP/BNP levels and preserved or mildly reduced ejection fraction. The approximately one‐third mortality over 30 months in patients with ATTRwt in ATTR‐ACT was also consistent with the approximately one‐quarter to one‐third mortality over 24–48 months in observational studies. 7 , 10 , 16 , 17 , 18 , 19 , 20
There have been fewer studies, with fewer participants, in patients with ATTRv. 7 , 17 , 19 The majority of patients in these studies (67%–76%) were male, with females potentially less likely to be diagnosed with cardiac disease. 21 These studies have also demonstrated the poorer survival in patients with ATTRv, reporting median survival times consistent with the 23.5 months observed in ATTR‐ACT. Together, these data suggest that genotype, along with baseline disease severity, contributes to the prognosis of patients with ATTR‐CM.
The decline in KCCQ‐OS score over time in ATTR‐ACT was more severe in patients with ATTRv and NYHA Class III, although the difference was not large and, as with 6MWT distance, the rate of decline was largely consistent across all groups. In patients with ATTRwt, there was a decline of approximately 3.5 points every 6 months (i.e. ~0.6 per month). In patients with ATTRv, there was a decline of approximately 5.5 points each 6 months (i.e. ~0.9 per month). Patients with ATTR‐CM have been shown to have poor overall quality of life and to experience a significant burden of disease with associated financial costs. 11 , 22 , 23 The steady decline in KCCQ‐OS score was consistent with an observational study in patients with ATTR‐CM. 24
This analysis is limited by the fact that ATTR‐ACT, as a clinical trial rather than an observational study, included only patients who met the trial's inclusion criteria, 14 which, as a result, may not have been representative of the general population of patients with ATTR‐CM. Nevertheless, the population in ATTR‐ACT was broadly similar to those in observational studies (Tables S1 and S2 ) with similar demographics, but with a larger proportion of patients in NYHA Class ≥II than most observational studies.
Given the clear differences in both mortality and hospitalizations between patients with ATTRwt and patients with ATTRv and between NYHA Class I and II and NYHA Class III, it was notable that there was no difference in the decline in 6MWT distance, despite differences in 6MWT distance at baseline. The rate of decline also appeared consistent over time, with each group experiencing a decline of 25–30 m every 6 months (i.e. ~5 m per month). Decline in 6MWT distance has been suggested as a measure of disease progression. However, it should be noted that there are a number of limitations with extrapolating the decline in one group of patients (the ATTR‐ACT population) to all patients with ATTR‐CM. Furthermore, the rate of decline observed in this analysis represents a group level change in least‐squares mean and cannot necessarily be directly correlated to a specific change in any individual patient, either within the ATTR‐ACT population or in patients with ATTR‐CM more generally. The use of multiple measures will likely provide the clearest assessment of disease progression in patients with ATTR‐CM.
Patients with ATTR‐CM experience a severe, progressive disease. A greater understanding of the presentation and progression of ATTR‐CM, together with the differences between patients with ATTRwt and patients with ATTRv, can help guide physicians in what to expect when treating these patients. Poorer outcomes in patients with more severe disease emphasize the need for earlier diagnosis and treatment of patients with ATTR‐CM.
Conflict of interest
JN‐N has received research grants from Akcea, Eidos Therapeutics and Pfizer and consulting fees from Akcea, Alnylam, Eidos Therapeutics and Pfizer. DPJ has received grants and funding for the trial, travel expenses and consultancy fees from Pfizer; consultancy fees from ADRx, Cytokinetics and Tenaya Therapeutics; and clinical trial funding from Array BioPharma and Eidos Therapeutics. JEH reports membership of a speakers bureau for Celgene. BG, DK and MBS are full‐time employees of Pfizer and hold stock and/or stock options. MG reports grants, advisory board and consultancy fees paid to her institution from Alnylam, Eidos Therapeutics, Prothena and Pfizer. Medical writing support was provided by Joshua Fink, PhD, of Engage Scientific Solutions and was funded by Pfizer.
Funding
This study was sponsored by Pfizer.
Data sharing
Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual anonymized participant data. See https://www.pfizer.com/science/clinical‐trials/trial‐data‐and‐results for more information.
Supporting information
Table S1. Clinical characteristics and survival in untreated patients with ATTRwt in ATTR‐ACT compared with observational studies in patients with ATTRwt.
Table S2. Clinical characteristics and survival in untreated patients with ATTRv in ATTR‐ACT compared with observational studies in patients with ATTRv.
Figure S1. LS mean change in NT‐proBNP from baseline at Months 12 and 30. ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; LS, least‐squares; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; SE, standard error.
Figure S2. LS mean change in echocardiography measures over time by genotype. ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; LS, least‐squares; LV, left ventricular; SE, standard error.
Nativi‐Nicolau, J. , Judge, D. P. , Hoffman, J. E. , Gundapaneni, B. , Keohane, D. , Sultan, M. B. , and Grogan, M. (2021) Natural history and progression of transthyretin amyloid cardiomyopathy: insights from ATTR‐ACT. ESC Heart Failure, 8: 3875–3884. 10.1002/ehf2.13541.
References
- 1. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012; 126: 1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A. Transthyretin‐related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol 2010; 7: 398–408. [DOI] [PubMed] [Google Scholar]
- 3. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state‐of‐the‐art review. J Am Coll Cardiol 2019; 73: 2872–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gonzalez‐Lopez E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐Del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, Garcia‐Pavia P. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36: 2585–2594. [DOI] [PubMed] [Google Scholar]
- 5. Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, Roger VL, Gertz MA, Dispenzieri A, Zeldenrust SR, Redfield MM. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail 2014; 2: 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hahn VS, Yanek LR, Vaishnav J, Ying W, Vaidya D, Lee YZJ, Riley SJ, Subramanya V, Brown EE, Hopkins CD, Ononogbu S, Perzel Mandell K, Halushka MK, Steenbergen C Jr, Rosenberg AZ, Tedford RJ, Judge DP, Shah SJ, Russell SD, Kass DA, Sharma K. Endomyocardial biopsy characterization of heart failure with preserved ejection fraction and prevalence of cardiac amyloidosis. JACC Heart Fail 2020; 8: 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez‐Naharro A, Quarta CC, Rezk T, Whelan CJ, Gonzalez‐Lopez E, Lane T, Gilbertson JA, Rowczenio D, Petrie A, Hawkins PN. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018; 39: 2799–2806. [DOI] [PubMed] [Google Scholar]
- 8. Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, Falk RH, Cheung KN, Patel AR, Pano A, Packman J, Grogan DR. Prospective evaluation of the morbidity and mortality of wild‐type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am Heart J 2012; 164: 222–228, e221. [DOI] [PubMed] [Google Scholar]
- 9. Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild‐type and V122I transthyretin in older adults referred to an academic medical center. Aging Health 2013; 9: 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Grogan M, Scott CG, Kyle RA, Zeldenrust SR, Gertz MA, Lin G, Klarich KW, Miller WL, Maleszewski JJ, Dispenzieri A. Natural history of wild‐type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 11. Witteles RM, Bokhari S, Damy T, Elliott PM, Falk RH, Fine NM, Gospodinova M, Obici L, Rapezzi C, Garcia‐Pavia P. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 2019; 7: 709–716. [DOI] [PubMed] [Google Scholar]
- 12. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 13. Rapezzi C, Elliott P, Damy T, Nativi‐Nicolau J, Berk J, Velazquez E, Boman K, Gundapaneni B, Patterson T, Schwartz J, Sultan M, Maurer M. Efficacy of tafamidis in patients with hereditary and wild‐type transthyretin amyloid cardiomyopathy: further analyses from ATTR‐ACT. JACC Heart Fail 2021; 9: 115–123. [DOI] [PubMed] [Google Scholar]
- 14. Maurer MS, Elliott P, Merlini G, Shah SJ, Cruz MW, Flynn A, Gundapaneni B, Hahn C, Riley S, Schwartz J, Sultan MB, Rapezzi C. Design and rationale of the phase 3 ATTR‐ACT clinical trial (tafamidis in transthyretin cardiomyopathy clinical trial). Circ Heart Fail 2017; 10: e003815. [DOI] [PubMed] [Google Scholar]
- 15. Kittleson MM, Maurer MS, Ambardekar AV, Bullock‐Palmer RP, Chang PP, Eisen HJ, Nair AP, Nativi‐Nicolau J, Ruberg FL. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation 2020; 142: e7–e22. [DOI] [PubMed] [Google Scholar]
- 16. Connors LH, Sam F, Skinner M, Salinaro F, Sun F, Ruberg FL, Berk JL, Seldin DC. Heart failure resulting from age‐related cardiac amyloid disease associated with wild‐type transthyretin: a prospective, observational cohort study. Circulation 2016; 133: 282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen AV, Damy T, Plante‐Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol 2016; 68: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonzalez‐Lopez E, Gagliardi C, Dominguez F, Quarta CC, de Haro‐Del Moral FJ, Milandri A, Salas C, Cinelli M, Cobo‐Marcos M, Lorenzini M, Lara‐Pezzi E, Foffi S, Alonso‐Pulpon L, Rapezzi C, Garcia‐Pavia P. Clinical characteristics of wild‐type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J 2017; 38: 1895–1904. [DOI] [PubMed] [Google Scholar]
- 19. Damy T, Kristen AV, Suhr OB, Maurer MS, Planté‐Bordeneuve V, Yu CR, Ong ML, Coelho T, Rapezzi C. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the transthyretin amyloidosis outcomes survey (THAOS). Eur Heart J 2019. Published online ahead of print 1 April 2019. 10.1093/eurheartj/ehz173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamada T, Takashio S, Arima Y, Nishi M, Morioka M, Hirakawa K, Hanatani S, Fujisue K, Yamanaga K, Kanazawa H, Sueta D, Araki S, Usuku H, Nakamura T, Suzuki S, Yamamoto E, Ueda M, Kaikita K, Tsujita K. Clinical characteristics and natural history of wild‐type transthyretin amyloid cardiomyopathy in Japan. ESC Heart Fail 2020; 7: 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rapezzi C, Riva L, Quarta CC, Perugini E, Salvi F, Longhi S, Ciliberti P, Pastorelli F, Biagini E, Leone O, Cooke RM, Bacchi‐Reggiani L, Ferlini A, Cavo M, Merlini G, Perlini S, Pasquali S, Branzi A. Gender‐related risk of myocardial involvement in systemic amyloidosis. Amyloid 2008; 15: 40–48. [DOI] [PubMed] [Google Scholar]
- 22. Stewart M, Shaffer S, Murphy B, Loftus J, Alvir J, Cicchetti M, Lenderking WR. Characterizing the high disease burden of transthyretin amyloidosis for patients and caregivers. Neurol Ther 2018; 7: 349–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amyloidosis Research Consortium . The voice of the patient report. https://www.arci.org/voice‐of‐the‐patient/ (accessed 22 August 2019)
- 24. Lane T, Fontana M, Martinez‐Naharro A, Quarta CC, Whelan CJ, Petrie A, Rowczenio DM, Gilbertson JA, Hutt DF, Rezk T, Strehina SG, Caringal‐Galima J, Manwani R, Sharpley FA, Wechalekar AD, Lachmann HJ, Mahmood S, Sachchithanantham S, Drage EPS, Jenner HD, McDonald R, Bertolli O, Calleja A, Hawkins PN, Gillmore JD. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 2019; 140: 16–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical characteristics and survival in untreated patients with ATTRwt in ATTR‐ACT compared with observational studies in patients with ATTRwt.
Table S2. Clinical characteristics and survival in untreated patients with ATTRv in ATTR‐ACT compared with observational studies in patients with ATTRv.
Figure S1. LS mean change in NT‐proBNP from baseline at Months 12 and 30. ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; LS, least‐squares; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; SE, standard error.
Figure S2. LS mean change in echocardiography measures over time by genotype. ATTRv, variant transthyretin amyloidosis; ATTRwt, wild‐type transthyretin amyloidosis; LS, least‐squares; LV, left ventricular; SE, standard error.