

Abstract
Aims
Endothelial cell vascular endothelial growth factor receptor 2 (VEGFR‐2) plays a pivotal role in angiogenesis, which induces physiological cardiomyocyte hypertrophy via paracrine signalling between endothelial cells and cardiomyocytes. We investigated whether a decrease in circulating soluble VEGFR‐2 (sVEGFR‐2) levels is associated with poor prognosis in patients with chronic heart failure (HF).
Methods and results
We performed a multicentre prospective cohort study of 1024 consecutive patients with HF, who were admitted to hospitals due to acute decompensated HF and were stabilized after initial management. Serum levels of sVEGFR‐2 were measured at discharge. Patients were followed up over 2 years. The outcomes were cardiovascular death, all‐cause death, major adverse cardiovascular events (MACE) defined as a composite of cardiovascular death and HF hospitalization, and HF hospitalization. The mean age of the patients was 75.5 (standard deviation, 12.6) years, and 57% were male. Patients with lower sVEGFR‐2 levels were older and more likely to be female, and had greater proportions of atrial fibrillation and anaemia, and lower proportions of diabetes, dyslipidaemia, and HF with reduced ejection fraction (<40%). During the follow‐up, 113 cardiovascular deaths, 211 all‐cause deaths, 350 MACE, and 309 HF hospitalizations occurred. After adjustment for potential clinical confounders and established biomarkers [N‐terminal B‐type natriuretic peptide (NT‐proBNP), high‐sensitivity cardiac troponin I, and high‐sensitivity C‐reactive protein], a low sVEGFR‐2 level below the 25th percentile was significantly associated with cardiovascular death [hazard ratio (HR), 1.79; 95% confidence interval (CI), 1.16–2.74] and all‐cause death (HR, 1.43; 95% CI, 1.04–1.94), but not with MACE (HR, 1.11; 95% CI, 0.86–1.43) or HF hospitalization (HR, 1.03; 95% CI, 0.78–1.35). The stratified analyses revealed that a low sVEGFR‐2 level below the 25th percentile was significantly associated with cardiovascular death (HR, 1.76; 95% CI, 1.07–2.85) and all‐cause death (HR, 1.49; 95% CI, 1.03–2.15) in the high‐NT‐proBNP group (above the median), but not in the low‐NT‐proBNP group. Notably, the patients with high‐NT‐proBNP and low‐sVEGFR‐2 (below the 25th percentile) had a 2.96‐fold higher risk (95% CI, 1.56–5.85) for cardiovascular death and a 2.40‐fold higher risk (95% CI, 1.52–3.83) for all‐cause death compared with those with low‐NT‐proBNP and high‐sVEGFR‐2.
Conclusions
A low sVEGFR‐2 value was independently associated with cardiovascular death and all‐cause death in patients with chronic HF. These associations were pronounced in those with high NT‐proBNP levels.
Keywords: Heart failure, Biomarker, Angiogenesis, Lymphangiogenesis, Mortality
Introduction
Despite current advances in therapy, heart failure (HF) remains a leading cause of hospitalization and mortality worldwide, and its prevalence is progressively increasing in aging societies. 1 HF is a clinical syndrome with several underlying aetiologies. Basic studies have demonstrated that dysfunction of angiogenesis is a major cause of the pathogenesis and progression of advanced HF, regardless of the aetiology 2 : cardiac tissue growth is angiogenesis‐dependent, and disruption of coordinated cardiac hypertrophy and angiogenesis causes the transition from adaptive cardiac hypertrophy to decompensated HF. 3 , 4
Vascular endothelial growth factor (VEGF) is a key cytokine in angiogenesis. 5 The VEGF family consists of five members [VEGF (or VEGF‐A), placental growth factor, VEGF‐B, VEGF‐C, and VEGF‐D] and three tyrosine kinase receptors (VEGFR‐1, ‐2, and ‐3). Among the pathways involving these members, the VEGF/VEGFR‐2 pathway plays a major role in angiogenesis. A recent study demonstrated that crosstalk between the endothelial cell VEGFR‐2 and cardiac myocyte ErbB signalling pathways coordinates cardiac myocyte hypertrophy with angiogenesis, and thereby contributes to physiological cardiac growth. 6 A soluble form of VEGFR‐2 (sVEGFR‐2) is generated by proteolytic hydrolysis of membrane‐bound VEGFR‐2 or by alternative splicing, 7 , 8 and sVEGFR‐2 can be measured in serum and plasma by immunoassay. 9 Circulating levels of sVEGFR‐2 were reported to serve as a surrogate biomarker of VEGF‐mediated tumour growth in patients with various cancers. 10 , 11 , 12 However, the clinical significance of sVEGFR‐2 in patients with chronic HF (CHF) is unknown. We therefore performed a multicentre prospective cohort study to investigate the prognostic value of serum sVEGFR‐2 levels in patients with CHF.
Methods
Study population
The PREHOSP‐CHF study (Development of Novel Biomarkers to Predict REHOSPitalization in Chronic Heart Failure) is a nationwide, multicentre prospective cohort study to determine the predictive value of possible novel biomarkers related to angiogenesis or lymphangiogenesis for cardiovascular (CV) events in patients with CHF (UMIN Clinical Trials Registry: UMIN000021657). We enrolled 1065 patients with HF, who were admitted to hospitals due to acute decompensated HF and were stabilized after initial management, between December 2015 and October 2017 in the 21 National Hospital Organization institutions across Japan. The present study was conducted by nationally certified cardiologists. Acute decompensated HF was defined by the modified Framingham criteria. The exclusion criteria are described in the Supporting Information (Supplemental Methods section). After excluding 41 patients who were subsequently found to be ineligible (31 patients) or who withdrew consent (10 patients), a total of 1024 patients were included in the analyses. The study was approved by the central ethics committee of the National Hospital Organization headquarters and each institution's ethical committee. All of the patients provided written informed consent.
Exposures, sample collection, and biomarker measurement
The primary predictor was the serum level of sVEGFR‐2 at the time of nearest discharge. The serum levels of sVEGFR‐2, VEGF, and high‐sensitivity C‐reactive protein (hs‐CRP) were measured with specific, commercially available ELISA kits according to the manufacturers' instructions: sVEGFR‐2 and VEGF were measured using a Quantikine kit (R&D Systems, Minneapolis, MN), and hs‐CRP was measured with a CycLex kit (Medical & Biological Laboratories [MBL], Nagano, Japan). 13 These assays were performed by an investigator masked to the sources of the samples. The serum levels of N‐terminal pro‐brain natriuretic peptide (NT‐proBNP) were measured using a validated, sandwich electrochemiluminescence immunoassay (Elecsys; Roche Diagnostics, Indianapolis, IN). The sensitivities of the assays for VEGF, sVEGFR‐2, and hs‐CRP were 5.0, 4.6, and 28.6 pg/mL, respectively. The inter‐assay/intra‐assay coefficients of variation of ELISA for VEGF, sVEGFR‐2, and hs‐CRP were <9%/<7%, ≤7%/<5%, and <6%/<4%, respectively. The sensitivity of the assay for NT‐proBNP was 5 pg/mL, and the assay coefficients of variation at values of the measuring range (5–35 000 pg/mL) were <10%. The high‐sensitivity cardiac troponin I (hs‐cTnI) values were measured using a cardiac troponin assay (Architect Stat High‐sensitive Troponin I; Abbott Laboratories, Abbott Park, IL). The limit of detection in this assay is 1.9 pg/mL (range, 0–50 000 pg/mL) and the 99th percentile cut‐off is 26.2 pg/mL. Additional details are described in the Supporting Information (Supplemental Methods section).
Study endpoints
The endpoints in the analyses were CV death, all‐cause death, major adverse cardiovascular events (MACE) defined as a composite of CV death and HF‐related hospitalization, and HF‐related hospitalization. Causes of death were adjudicated after consideration of all the available information and were classified according to the following pre‐specified groups: CV death and non‐CV death. CV death included death related to HF, acute coronary syndrome, stroke, and other vascular disease, and sudden death. Sudden death was defined as death related to fatal arrhythmia or unexplained death in a previously stable patient. Patients were followed up over 2 years. At the end of the follow‐up period (Day 720), survival status and detailed information about MACE were available in 1009 patients (follow‐up rate, 98.5%).
Statistical analysis
We divided the patients into quartiles according to their baseline sVEGFR‐2 levels. The categorical variables are presented as numbers and percentages and were compared using a χ 2 test. The continuous variables are expressed as mean with standard deviation or median with interquartile range. On the basis of their distributions, the continuous variables were compared using ANOVA or the Kruskal–Wallis test. The P values for trends across the groups were calculated using the Cochran–Armitage test for categorical variables and the Jonckheere–Terpstra test for continuous variables. The cumulative incidences of clinical outcomes were estimated by the Kaplan–Meier method and Cox proportional hazard regression. For competing risk analyses, we used the Fine and Gray model to estimate the subdistribution hazard ratio (HR). The relationships between the baseline biomarker levels and the outcomes were investigated with the use of Cox proportional hazard regression. In our prior exploratory study of 254 CHF patients, the incidence of MACE over a 2 year follow‐up period was 29.1% in a low sVEGFR‐2 group (below the median) and 17.3% in a high sVEGFR‐2 group. To realize 99% power for MACE, we estimated that a sample size of 785 patients was required. We increased this sample size by 20% to account for potential loss to follow‐up, arriving at a final sample size of 1050 patients with an expected MACE incidence of 242 patients. We therefore included no more than 24 variables for the statistical analysis, because that was the highest number of variables supported by the anticipated incidence of MACE. We used four sets of models to confirm the consistency of the association between sVEGFR‐2 levels and clinical events: Model 1, adjusted for age, sex, body mass index, and traditional CV risk factors (i.e. hypertension, diabetes, and dyslipidaemia), as well as established risk factors for HF [i.e. prior HF hospitalization, left ventricular dysfunction defined as an ejection fraction (EF) < 40%, and NYHA class 3/4]; Model 2, adjusted for the covariates included in Model 1 and other CV risk factors [i.e. coronary artery disease, old myocardial infarction, atrial fibrillation (AF), chronic kidney disease (CKD, defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2), anaemia (defined as haemoglobin levels of less than 13 g/dL in male participants and 12 g/dL in female participants), chronic obstructive pulmonary disease, and cerebrovascular disease]; Model 3, adjusted for the covariates included in Model 2 and prescription of renin–angiotensin system inhibitors (RAS‐I), beta blockers, loop diuretics, and mineral corticoid receptor antagonists (MRA); and Model 4, adjusted for the covariates included in Model 3 and CV biomarkers [NT‐proBNP, hs‐cTnI, and hs‐CRP (>1 mg/L)].
Subgroup analyses were performed based on the independent determinants for sVEGFR‐2 level in the multiple regression analysis, EF category (HFpEF: HF with preserved EF (≥50%); HFmrEF: HF with mid‐range EF (40–49%); and HFrEF: HF with reduced EF [<40%]), and aetiology of HF (ischaemic or non‐ischaemic).
All statistical tests were two‐sided, and P < 0.05 was considered significant. Analyses were performed using JMP version 12 (SAS Institute, Cary, NC) and R, version 3.4.4 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Baseline characteristics
The distribution of sVEGFR‐2 values is shown in Figure S1 . We divided the patients into four groups based on the quartile of the sVEGFR‐2 levels (Quartile 1: sVEGFR‐2 < 5259 pg/mL; Quartile 2: 5259 pg/mL ≤ VEGFR‐2 < 6120 pg/mL; Quartile 3: 6120 pg/mL ≤ sVEGFR‐2 < 7210 pg/mL; and Quartile 4: sVEGFR‐2 ≥ 7210 pg/mL). The baseline characteristics of the entire cohort and of the patients divided into quartiles of sVEGFR‐2 levels are shown in Tables 1 and S1 . Patients with Quartile 1 sVEGFR‐2 levels were older and had higher rates of female sex, lower body mass index, AF, and anaemia, while they had lower prevalence of diabetes and dyslipidaemia compared with those with Quartiles 2–4 sVEGFR‐2 levels. Prevalence of HFpEF was higher in those with Quartile 1 sVEGFR‐2 levels than in those with Quartiles 2–4 sVEGFR‐2 levels.
Table 1.
Baseline characteristics according to quartiles of sVEGFR‐2
| Variable | Entire cohort | Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | P value | P for trend |
|---|---|---|---|---|---|---|---|
| N | 1024 | 256 | 256 | 256 | 256 | ||
| Age (years) | 75.5 (12.6) | 80 (10.7) | 77.3 (11.8) | 74.7 (11.9) | 70.2 (13.7) | <0.001 | <0.001 |
| Male gender | 601 (58.7) | 132 (51.6) | 133 (52.0) | 167 (65.2) | 169 (66.0) | <0.001 | <0.001 |
| Body mass index (kg/m2) | 22.2 (4.9) | 20.9 (4.1) | 22 (4.8) | 22.5 (4.4) | 23.3 (5.7) | <0.001 | <0.001 |
| SBP (mmHg) | 114.8 (17.8) | 112.8 (17) | 115.5 (17.3) | 114.8 (18.6) | 116.2 (18) | 0.2 | 0.07 |
| DBP (mmHg) | 65.6 (12.6) | 63.8 (12.9) | 66.7 (13.0) | 65 (12.5) | 66.7 (11.9) | 0.02 | 0.052 |
| Pulse rate (bpm) | 70.1 (13.6) | 69.5 (13.9) | 70.3 (13.6) | 69.8 (13.8) | 70.9 (13.2) | 0.7 | 0.3 |
| NYHA 3/4 | 139 (13.6) | 34 (13.3) | 38 (14.9) | 36 (14.1) | 31 (12.1) | 0.8 | 0.6 |
| HFpEF/mrEF/rEF | 429/186/409 (41.9/18.2/39.9) | 127/51/78 (49.6/19.9/30.5) | 103/45/108 (40.2/17.6/42.2) | 104/48/104 (40.6/18.8/40.6) | 95/42/119 (37.1/16.4/46.5) | 0.02 | <0.001 |
| Ischaemic aetiology | 252 (24.6) | 48 (18.8) | 69 (27.0) | 65 (25.4) | 70 (27.3) | 0.08 | 0.04 |
| Hypertension | 777 (75.9) | 189 (73.8) | 196 (76.6) | 202 (78.9) | 190 (74.2) | 0.5 | 0.8 |
| Diabetes | 396 (38.7) | 83 (32.4) | 99 (38.7) | 91 (35.5) | 123 (48) | 0.002 | 0.001 |
| Dyslipidaemia | 418 (40.8) | 81 (31.6) | 118 (46.1) | 99 (38.7) | 120 (46.9) | <0.001 | 0.005 |
| Prior HF hospitalization | 322 (31.4) | 80 (31.3) | 93 (36.3) | 83 (32.4) | 66 (25.8) | 0.08 | 0.1 |
| Coronary artery disease | 319 (31.2) | 74 (28.9) | 87 (34.0) | 77 (30.1) | 81 (31.6) | 0.6 | 0.7 |
| Old myocardial infarction | 181 (17.7) | 33 (12.9) | 48 (18.8) | 48 (18.8) | 52 (20.3) | 0.1 | 0.04 |
| AF | 533 (52.1) | 153 (59.8) | 144 (56.3) | 135 (52.7) | 101 (39.5) | <0.001 | <0.001 |
| CKD | 527 (51.5) | 128 (50.0) | 143 (55.9) | 134 (52.3) | 122 (47.7) | 0.3 | 0.5 |
| Anaemia a | 577 (56.4) | 174 (68.0) | 143 (55.9) | 138 (53.9) | 122 (47.7) | <0.001 | <0.001 |
| COPD | 77 (7.5) | 22 (8.6) | 23 (9.0) | 21 (8.2) | 11 (4.3) | 0.1 | 0.06 |
| Cerebrovascular disease | 176 (17.2) | 51 (19.9) | 45 (17.6) | 37 (14.5) | 43 (16.8) | 0.4 | 0.2 |
| RAS‐I | 725 (70.8) | 176 (68.8) | 183 (71.5) | 172 (67.2) | 194 (75.8) | 0.1 | 0.2 |
| Beta blockers | 752 (73.4) | 167 (65.2) | 189 (73.8) | 195 (76.2) | 201 (78.5) | 0.005 | <0.001 |
| Loop diuretics | 871 (85.1) | 213 (83.2) | 225 (87.9) | 212 (82.8) | 221 (86.3) | 0.3 | 0.7 |
| MRA | 441 (43.1) | 111 (43.4) | 117 (45.7) | 106 (41.4) | 107 (41.8) | 0.8 | 0.5 |
| NT‐proBNP (pg/mL) | 1493 [664–3395] | 1404 [674–3493] | 1591 [680–3560] | 1656 [706–3289] | 1389 [598–3201] | 0.7 | 0.5 |
| hs‐cTnI (pg/mL) | 21.9 [12.1–47.4] | 22.5 [13.4–47.8] | 21.8 [12.0–45.6] | 20.0 [10.5–44.6] | 23.9 [13.8–52.6] | 0.3 | 0.8 |
| hs‐CRP (mg/L) | 2.48 [0.83–8.87] | 2.74 [0.87–9.52] | 2.35 [0.78–9.68] | 2.05 [0.86–7.62] | 3.11 [0.83–8.76] | 0.4 | 0.8 |
| VEGF (pg/mL) | 377 [223–632] | 324 [189–521] | 401 [231–666] | 366 [226–636] | 438 [243–672] | <0.001 | 0.001 |
Continuous variables are expressed as the mean (standard deviation) or median [interquartile range] according to the distributions. Categorical variables are presented as numbers (percentages).
AF, atrial fibrillation; CKD, chronic kidney disease; COPD, chronic obstructive pulmonary disease; DBP, diastolic blood pressure; EF, ejection fraction; Hb, haemoglobin; HF, heart failure, HFmrEF, HF with mid‐range EF; HFpEF, HF with preserved EF; HFrEF, HF with reduced EF; hs‐CRP, high‐sensitivity C‐reactive protein; hs‐cTnI, high sensitivity cardiac troponin I; MRA, mineral corticoid receptor antagonists; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; RAS‐I, renin angiotensin system inhibitor; SBP, systolic blood pressure; VEGF, vascular endothelial growth factor; VEGFR‐2, soluble vascular endothelial growth factor receptor 2.
Anaemia was defined as a haemoglobin level <13 g/dL in male participants and <12 g/dL in female participants.
In simple regression analyses, the sVEGFR‐2 level was inversely correlated with EF, and positively correlated with the VEGF levels, but it was not significantly correlated with the levels of established CV biomarkers (NT‐proBNP, hs‐cTnI, and hs‐CRP) (Table S2 ). Stepwise multiple regression analysis revealed that independent determinants of the sVEGFR‐2 level were lower age, male gender, presence of diabetes, absence of AF and anaemia, and higher levels of NT‐proBNP (Table S2 ).
Incidence of outcomes and Cox regression analyses
During the 720 day follow‐up, a total of 113 (11.0%) CV deaths, 211 (20.6%) all‐cause deaths, 350 (34.2%) MACE, and 309 (30.2%) HF‐related hospitalizations occurred (Table 2 ). Figure 1 shows the cumulative incidence of outcomes and unadjusted Cox proportional HRs according to quartiles of sVEGFR‐2. The patients with Quartile 1 sVEGFR‐2 levels showed the greatest risks of CV death [P = 0.01; HR, 1.54; 95% confidence interval (CI), 0.96–2.51 (vs. Quartile 2); HR, 1.75; 95% CI, 1.07–2.91 (vs. Quartile 3); HR, 2.32; 95% CI, 1.37–4.09 (vs. quartile 4)] and all‐cause death [P = 0.004; HR, 1.51; 95% CI, 1.06–2.18 (vs. Quartile 2); HR, 1.57; 95% CI, 1.09–2.26 (vs. Quartile 3); HR, 1.94; 95% CI, 1.32–2.87 (vs. quartile 4)], but did not show a significantly higher risk of MACE (P = 0.7) or HF‐related hospitalization (P = 0.97) than the other quartile groups. In contrast, quartiles of VEGF were not significantly associated with the risk of CV death, all‐cause death, MACE, or HF‐related hospitalization (Figure S2 ). Because there was an apparent threshold effect between Quartile 1 and Quartile 2 in the incidence of all‐cause death and CV death, sVEGFR‐2 was modelled as a dichotomous variable in subsequent analyses by applying a threshold of Quartile 1 vs. Quartiles 2–4. In competing risk analyses, the patients with Quartile 1 sVEGFR‐2 also showed significantly higher incidence of CV death than those with Quartiles 2–4 (HR, 1.77; 95% CI 1.21–2.56), but not that of MACE (HR, 1.13; 95% CI, 0.89–1.43) or HF‐related hospitalization (HR, 1.00; 95% CI, 0.77–1.29) (Figure S3 ).
Table 2.
Incidence of events according to quartiles of sVEGFR‐2
| Event | Entire cohort | Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 |
|---|---|---|---|---|---|
| CV death | 113 (11.0) | 41 (16.0) | 28 (10.9) | 25 (9.8) | 19 (7.4) |
| All‐cause death | 211 (20.6) | 72 (28.1) | 50 (19.5) | 49 (19.1) | 40 (15.6) |
| MACE (CV death + HF hospitalization) | 350 (34.2) | 94 (36.7) | 85 (33.2) | 85 (33.2) | 86 (33.6) |
| HF hospitalization | 309 (30.2) | 77 (30.1) | 74 (28.9) | 77 (30.1) | 81 (31.6) |
Variables are presented as numbers (percentages).
CV, cardiovascular; HF, heart failure; MACE, major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization; sVEGFR‐2, soluble vascular endothelial growth factor receptor 2.
Figure 1.
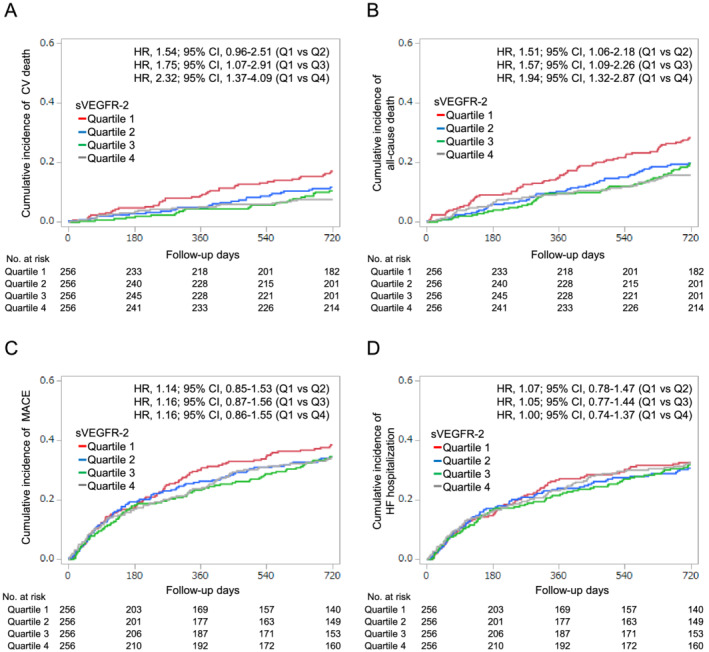
Incidence of CV death (A), all‐cause death (B), MACE (C), and HF‐related hospitalization (D) according to the quartiles of baseline sVEGFR‐2 levels during the follow‐up period. CI, confidence interval; CV, cardiovascular; HF, heart failure; HR, hazard ratio, MACE, major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization; Q, quartile; sVEGFR‐2, soluble vascular endothelial growth factor receptor 2.
Figure 2 shows the unadjusted and adjusted HRs of low sVEGFR‐2 levels below the 25th percentile for the various outcomes. After adjusting for traditional risk factors, a low sVEGFR‐2 level below the 25th percentile was significantly associated with the risk of CV death, but not with the risk of all‐cause death, MACE, or HF‐related hospitalization (Figure 2 , Model 1). After additional adjustment for other potential clinical confounders, a low sVEGFR‐2 level below the 25th percentile was significantly associated with the risk of CV death, but not with the risk of all‐cause death, MACE, or HF‐related hospitalization (Figure 2 , Model 2). Even after additional adjustment for medications for HF, these associations were consistent (Figure 2 , Model 3). Importantly, after additional adjustment for established CV biomarkers, a low sVEGFR‐2 level below the 25th percentile was significantly associated with the risk of CV death (HR, 1.79; 95% CI, 1.16–2.74) and all‐cause death (HR, 1.43; 95% CI, 1.04–1.94), but not with the risk of MACE (HR, 1.11; 95% CI, 0.86–1.43) or HF‐related hospitalization (HR, 1.03; 95% CI, 0.78–1.35) (Figure 2 , Model 4, Table S3 ).
Figure 2.
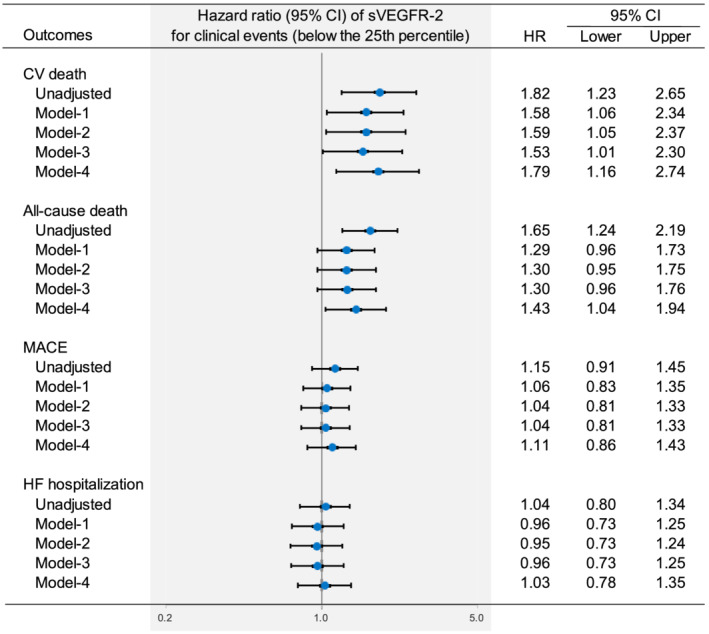
Multivariate Cox proportional hazard analysis for CV death, all‐cause death, MACE, and HF‐related hospitalization. Model 1: adjusted for age, sex, body mass index, and traditional cardiovascular risk factors (hypertension, diabetes, and dyslipidaemia), as well as established risk factors for HF [prior HF hospitalization, left ventricular dysfunction (ejection fraction < 40%), and NYHA class 3/4]. Model 2: adjusted for the covariates included in Model 1 and other CV risk factors (CAD, old myocardial infarction, AF, CKD, anaemia, chronic obstructive pulmonary disease, and cerebrovascular disease). Model 3: adjusted for the covariates included in Model 2 and prescription of RAS‐I, beta blockers, loop diuretics, and MRA. Model 4: adjusted for the covariates included in Model 3 and CV biomarkers [NT‐proBNP, hs‐cTnI, and hs‐CRP (>1 mg/L)]. AF, atrial fibrillation; BMI, body mass index; CAD, coronary artery disease; CKD, chronic kidney disease; hs‐CRP, high sensitivity C‐reactive protein; hs‐cTnI, high sensitivity cardiac troponin I; MRA, mineral corticoid receptor antagonists; NT‐proBNP, N‐terminal pro‐brain natriuretic peptide; RAS‐I, renin angiotensin inhibitor. Other abbreviations are defined in Figure 1 .
Subgroup analyses
Figure 3 shows the results of subgroup analyses on the association of low sVEGFR‐2 below the 25th percentile with CV death and all‐cause death. A low sVEGFR‐2 level below the 25th percentile was significantly associated with all‐cause death in men, but not in women. The risk for CV death tended to be higher in those with a low sVEGFR‐2 level below the 25th percentile compared with those with a high sVEGFR‐2 level in men (HR, 1.67; 95% CI, 0.95–2.86), but not in women (HR, 1.25; 95% CI, 0.63–2.38).
Figure 3.
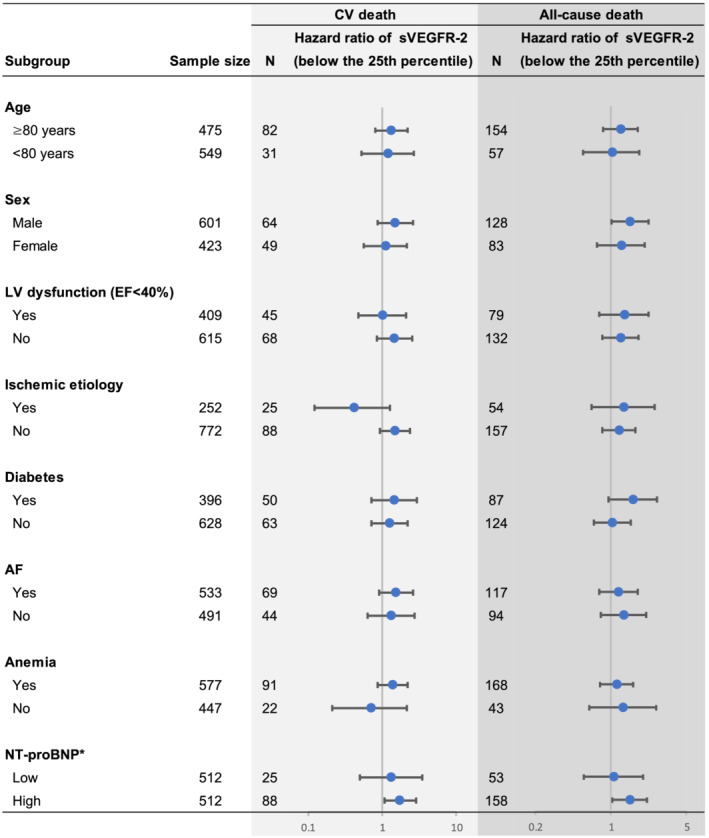
Multivariate‐adjusted stratified analyses of the associations of low sVEGFR‐2 below the 25th percentile and the risks of CV death and all‐cause death. Data were adjusted for the covariates included in Model 3 in Figure 2 . *We divided the patients according to the median of NT‐proBNP. EF, ejection fraction; LV, left ventricular. Other abbreviations are defined in Figures 1 and 2 .
Interestingly, a low sVEGFR‐2 level below the 25th percentile was significantly associated with CV death (HR, 1.76; 95% CI, 1.07–2.85) and all‐cause death (HR, 1.49; 95% CI, 1.03–2.15) in the high‐NT‐proBNP (>50th percentile) group, but not with either CV death (HR, 1.33; 95% CI, 0.50–3.52) or all‐cause death (HR, 1.07; 95% CI, 0.56–1.99) in the low‐NT‐proBNP group after adjustment for potential clinical confounders (Figure 3 ).
Figure S4 shows the results of Kaplan–Meier analysis based on the subgroups of EF category. A low sVEGFR‐2 level below the 25th percentile was associated with CV death (HR, 1.99; 95% CI, 1.20–3.22) and all‐cause death (HR, 1.78; 95% CI, 1.24–2.53) in patients with HFpEF/HFmrEF, but not in those with HFrEF (HR for CV death, 1.29; 95% CI, 0.65–2.41; HR for all‐cause death, 1.38; 95% CI, 0.84–2.21).
Figure S5 shows Kaplan–Meier analysis based on the aetiology of HF (ischaemic or non‐ischaemic). A low sVEGFR‐2 level below the 25th percentile was associated with CV death (HR, 1.99; 95% CI, 1.28–3.03) and all‐cause death (HR, 1.66; 95% CI, 1.18–2.29) in patients with non‐ischaemic aetiology, but not in those with ischaemic aetiology (HR for CV death, 0.43; 95% CI, 0.12–1.27; HR for all‐cause death, 1.32; 95% CI, 0.66–2.54).
Risk stratification by NT‐proBNP and sVEGFR‐2
Figure 4 shows the cumulative incidence (A and B) and unadjusted and adjusted HRs (C) for CV death and all‐cause death in patients divided into 4 groups based on the median of NT‐proBNP and the 25th percentile of sVEGFR‐2 levels. Notably, the high‐NT‐proBNP/low‐sVEGFR‐2 group exhibited the highest risks of CV death and all‐cause death among the four groups, even after adjustment for potential clinical confounders and the established CV biomarkers (Figure 4 C ). These findings suggest the incremental prognostic value of sVEGFR‐2 in combination with NT‐proBNP in patients with CHF.
Figure 4.
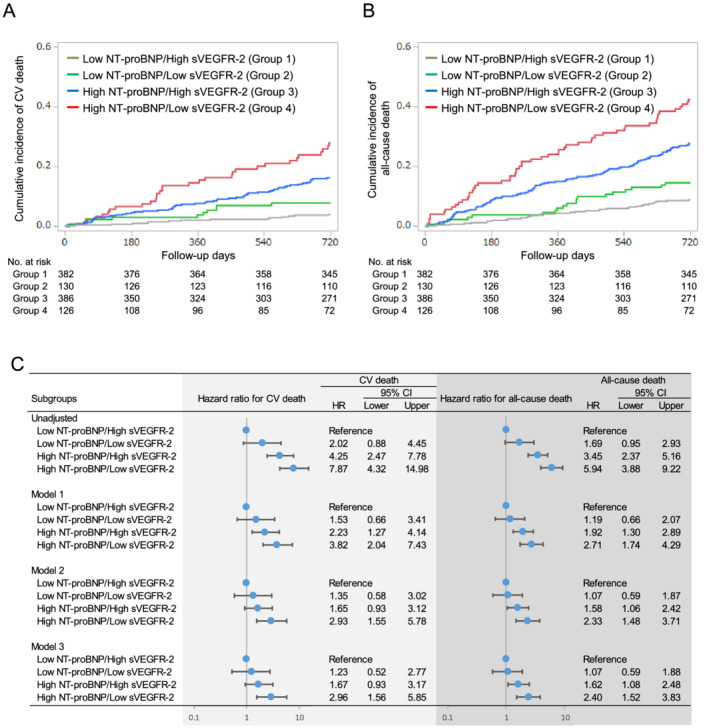
Incidence of CV death (A) and all‐cause death (B) according to the combination of baseline sVEGFR‐2 (lowest quartile) and NT‐proBNP (above the median) levels during the follow‐up period, and unadjusted and adjusted hazard ratios of the combination of sVEGFR‐2 and NT‐proBNP for CV death and all‐cause death (C). The covariates included in each model are shown in Figure 2 . Abbreviations are as defined in Figures 1 and 2 .
Discussion
This is the first dedicated and large‐scale prospective cohort study to demonstrate that a low sVEGFR‐2 level below the 25th percentile is significantly associated with the risk of CV and all‐cause mortality in patients with CHF. These associations remain significant after adjustment for potential clinical confounders and the established CV biomarkers of NT‐proBNP, hs‐cTnI, and hs‐CRP. Furthermore, the addition of a low sVEGFR‐2 level below the 25th percentile to the model with potential clinical confounders significantly improved the prediction of CV and all‐cause mortality in patients with high NT‐proBNP levels. These findings suggest that the measurement of sVEGFR‐2 provides prognostic information about CV and all‐cause mortality beyond potential clinical confounders and the established CV biomarkers in clinical settings. The strengths of our investigation include the large sample size, multicentre prospective design, and high follow‐up rate.
The underlying mechanism of the relationship between a low sVEGFR‐2 value and poor prognosis in patients with CHF should be considered. VEGFR‐2 is mainly expressed in endothelial cells, 14 and a naturally occurring sVEGFR‐2 was produced by proteolytic hydrolysis of membrane‐bound VEGFR‐2 or alternative splicing of VEGFR‐2. 7 , 15 sVEGFR‐2 protein and messenger RNA were found to be present in various tissues, including the skin, heart, spleen, kidney, ovary, and plasma in wild‐type mice. 16 The VEGF/VEGFR‐2 signalling pathway plays an important role in endothelial dysfunction, 17 which is one of the characteristic pathophysiological features of CHF. 18 Endothelium–cardiomyocyte interactions play essential roles in CV homeostasis; deranged endothelium‐related signalling pathways have been implicated in the pathophysiology of HF. 19 A recent study demonstrated that cross talk between the endothelial VEGFR‐2 and cardiomyocyte ErbB signalling pathways is required for adaptive cardiac hypertrophy. 6 On the other hand, systemic vasoconstriction associated with endothelin dysfunction has been suggested to play a central role in HF pathogenesis: dysfunction of the endothelium leads to increased vascular stiffness and impaired arterial distensibility, augmenting myocardial damage. 20 , 21 Taken together, these findings indicate that a decrease in circulating sVEGFR‐2 may reflect down‐regulation of membrane‐bound VEGFR‐2 in cardiac and systemic endothelial cells, leading to a vulnerability to decompensated HF. Further investigation is necessary to clarify the mechanistic role of sVEGFR‐2 in HF.
In the present study, the associations of low sVEGFR‐2 with CV death and all‐cause death were pronounced in patients with HFpEF. Not only cardiac microvascular rarefaction but also peripheral vascular dysfunction has been associated with impairment of cardiac reserve function in HFpEF. 22 , 23 , 24 , 25 These previous and our present findings may also support the idea that a low sVEGFR‐2 reflects both cardiac and vascular endothelial dysfunction, leading to a deterioration of the prognosis in HFpEF.
The association between a low sVEGFR‐2 value and all‐cause death was significant in men, but not in women. The risk for CV death also tended to be higher in patients with low sVEGFR‐2 compared with those with high sVEGFR‐2 in men, but not in women. These results suggest the possibility of a sex difference in the pathophysiology of sVEGFR‐2. Previous studies suggested a sex‐based difference in the endogenous expression of VEGF. 26 , 27 Thus, systemic or CV expression of VEGF may affect the circulating sVEGFR‐2 level and/or its association with prognosis in CHF. Future studies will be needed to define the mechanisms underlying the sex difference in the association between the sVEGFR‐2 level and prognosis in CHF.
The associations of a low VEGFR‐2 value with CV and all‐cause deaths became prominent when adjusting for or stratifying based on NT‐proBNP, a marker of increased left ventricular wall stress. 28 These findings may suggest that endothelial dysfunction itself does not lead to HF, but in combination with an increased left ventricular wall stress, it can lead to decompensated HF.
The VEGF/VEGFR2 signalling pathway plays a crucial role in tumour angiogenesis 29 as well as CV endothelial integrity. Since HF was subsequently noted in 2–4% of patients with cancers who were taking VEGF signalling pathway inhibitors, 30 it would be of interest to determine whether the sVEGFR‐2 level can predict incident HF in combination with NT‐proBNP among patients with cancer who are receiving VEGF signalling pathway inhibitors.
Study limitations
The present study has several limitations. First, the results were derived from a prospective observational study; therefore, they only reflect association and not causality. Second, the sources and the detailed physiological and pathological roles of sVEGFR‐2 have not been elucidated. 7 Further investigations will be needed to answer these questions. Third, we did not repeatedly measure the sVEGFR‐2 levels during the follow‐up period. The predictive significance of a change in sVEGFR‐2 levels in patients with HF is unclear. Fourth, we did not measure the levels of VEGF‐C, a central regulator of lymphangiogenesis, and the other ligand for sVEGFR‐2. Recently, we demonstrated that low VEGF‐C is inversely and independently associated with the risk of all‐cause mortality in patients with suspected or known coronary artery disease. 31 Future studies will define the prognostic value of VEGF‐C in patients with CHF. Finally, because the PREHOSP‐CHF study cohort consists exclusively of Asian individuals with HF, our results may not be generalizable to general Asian populations, or to other ethnic groups. Despite these limitations, the present study provides not only better risk stratification in patients with CHF but also deeper insight into the role of angiogenesis in the mechanisms of HF.
Conclusions
In a multicentre prospective cohort, a low sVEGFR‐2 value was independently associated with CV death and all‐cause death among patients with CHF, especially in those with high NT‐proBNP levels.
Conflict of interest
None of the authors has any relationship relevant to the content of this paper to disclose.
Funding
The PREHOSP‐CHF study is supported by a Grant‐in‐Aid for Clinical Research from the National Hospital Organization.
Supporting information
Figure S1. Distribution of the sVEGFR‐2 levels. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2.
Figure S2. Incidence of CV death (A), all‐cause death (B), MACE (C), and HF‐related hospitalization (D) according to the quartiles of baseline VEGF levels during the follow‐up period. VEGF = vascular endothelial growth factor, CV = cardiovascular, MACE = major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization, HF = heart failure, HR = hazard ratio, CI = confidence interval.
Figure S3. Competing risk of death adjusted cumulative hazard curves for incident CV death (A), MACE (B), and HF‐related hospitalization (C) in patients with and without low sVEGFR‐2 levels below the 25th percentile during the follow‐up period. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, MACE = major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization, HF = heart failure, HR = hazard ratio, CI = confidence interval.
Figure S4. Incidence of CV death (A) and all‐cause death (B) between patients with and those without low sVEGFR‐2 levels below the 25th percentile in the HFpEF/mrEF and HFrEF groups. Adjusted HR was calculated using the covariates included in model 3. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, HR = hazard ratio, CI = confidence interval, HF = heart failure, HFpEF = HF with preserved ejection fraction (EF), HFmrEF = HF with mid‐range EF, HFrEF = HF with reduced EF.
Figure S5. Incidence of CV death (A) and all‐cause death (B) between patients with and those without low sVEGFR‐2 levels below the 25th percentile in the groups with HF with ischemic etiology and HF with non‐ischemic etiology. Adjusted HR was calculated using the covariates included in model 3. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, HR = hazard ratio, CI = confidence interval, HF = heart failure.
Table S1. Baseline characteristics according to quartiles of sVEGFR‐2.
Table S2. Simple and stepwise multiple regression analyses for the sVEGFR‐2 level.
Table S3. Multiple Cox proportional hazard analysis for clinical outcomes.
Acknowledgements
We thank Mr Shuichi Ura, who substantially contributed to this work but passed away before this manuscript was drafted. We also thank the other members, co‐operators, and participants of the PREHOSP‐CHF study for their valuable contributions. The other members of the PREHOSP‐CHF study group are listed in the Appendix of the Supporting Information.
The hs‐cTnI values were measured at Abbott Japan LLC (Minato‐ku, Tokyo, Japan).
Iguchi, M. , Wada, H. , Shinozaki, T. , Suzuki, M. , Ajiro, Y. , Matsuda, M. , Koike, A. , Koizumi, T. , Shimizu, M. , Ono, Y. , Takenaka, T. , Sakagami, S. , Morita, Y. , Fujimoto, K. , Yonezawa, K. , Yoshida, K. , Ninomiya, A. , Nakamura, T. , Funada, J. , Kajikawa, Y. , Oishi, Y. , Kato, T. , Kotani, K. , Abe, M. , Akao, M. , Hasegawa, K. , and for the PREHOSP‐CHF Study Investigators (2021) Soluble vascular endothelial growth factor receptor 2 and prognosis in patients with chronic heart failure. ESC Heart Failure, 8: 4187–4198. 10.1002/ehf2.13555.
References
- 1. Roger VL. Epidemiology of heart failure. Circ Res. 2013; 113: 646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy. Circ Res. 2014; 114: 565–571. [DOI] [PubMed] [Google Scholar]
- 3. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005; 115: 2108–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006; 47: 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taimeh Z, Loughran J, Birks EJ, Bolli R. Vascular endothelial growth factor in heart failure. Nat Rev Cardiol. 2013; 10: 519–530. [DOI] [PubMed] [Google Scholar]
- 6. Kivelä R, Hemanthakumar KA, Vaparanta K, Robciuc M, Izumiya Y, Kidoya H, Takakura N, Peng X, Sawyer DB, Elenius K, Walsh K, Alitalo K. Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2‐mediated paracrine signaling. Circulation. 2019; 139: 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stevens M, Oltean S. Modulation of receptor tyrosine kinase activity through alternative splicing of ligands and receptors in the VEGF‐A/VEGFR Axis. Cells. 2019; 8: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abou‐Fayçal C, Hatat AS, Gazzeri S, Eymin B. Splice variants of the RTK family: their role in tumour progression and response to targeted therapy. Int J Mol Sci 2017; 18: 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ebos JM, Bocci G, Man S, Thorpe PE, Hicklin DJ, Zhou D, Jia X, Kerbel RS. A naturally occurring soluble form of vascular endothelial growth factor receptor 2 detected in mouse and human plasma. Mol Cancer Res. 2004; 2: 315–326. [PubMed] [Google Scholar]
- 10. Sallinen H, Heikura T, Koponen J, Kosma VM, Heinonen S, Ylä‐Herttuala S, Anttila M. Serum angiopoietin‐2 and soluble VEGFR‐2 levels predict malignancy of ovarian neoplasm and poor prognosis in epithelial ovarian cancer. BMC Cancer. 2014; 14: 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jantus‐Lewintre E, Sanmartín E, Sirera R, Blasco A, Sanchez JJ, Tarón M, Rosell R, Camps C. Combined VEGF‐A and VEGFR‐2 concentrations in plasma: diagnostic and prognostic implications in patients with advanced NSCLC. Lung Cancer. 2011; 74: 326–331. [DOI] [PubMed] [Google Scholar]
- 12. Ebos JM, Lee CR, Bogdanovic E, Alami J, Van Slyke P, Francia G, Xu P, Mutsaers AJ, Dumont DJ, Kerbel RS. Vascular endothelial growth factor‐mediated decrease in plasma soluble vascular endothelial growth factor receptor‐2 levels as a surrogate biomarker for tumor growth. Cancer Res. 2008; 68: 521–529. [DOI] [PubMed] [Google Scholar]
- 13. Wada H, Ura S, Kitaoka S, Satoh‐Asahara N, Horie T, Ono K, Takaya T, Takanabe‐Mori R, Akao M, Abe M, Morimoto T, Murayama T, Yokode M, Fujita M, Shimatsu A, Hasegawa K. Distinct characteristics of circulating vascular endothelial growth factor‐a and C levels in human subjects. PLoS One. 2011; 6: e29351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF‐A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal 2016; 10: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Albuquerque RJC, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, Baffi JZ, Yamada K, Kaneko H, Green MG, Chappell J, Wilting J, Weich HA, Yamagami S, Amano S, Mizuki N, Alexander JS, Peterson ML, Brekken RA, Hirashima M, Capoor S, Usui T, Ambati BK, Ambati J. Alternatively spliced vascular endothelial growth factor receptor‐2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009; 15: 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pavlakovic H, Becker J, Albuquerque R, Wilting J, Ambati J. Soluble VEGFR‐2: an antilymphangiogenic variant of VEGF receptors. Ann N Y Acad Sci. 2010; 1207: E7–E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kliche S, Waltenberger J. VEGF receptor signaling and endothelial function. IUBMB life. 2001; 52: 61–66. [DOI] [PubMed] [Google Scholar]
- 18. Alem MM. Endothelial dysfunction in chronic heart failure: assessment, findings, significance, and potential therapeutic targets. Int J Mol Sci 2019; 20: 3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lim SL, Lam CS, Segers VF, Brutsaert DL, De Keulenaer GW. Cardiac endothelium‐myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur Heart J. 2015; 36: 2050–2060. [DOI] [PubMed] [Google Scholar]
- 20. Marti CN, Gheorghiade M, Kalogeropoulos AP, Georgiopoulou VV, Quyyumi AA, Butler J. Endothelial dysfunction, arterial stiffness, and heart failure. J Am Coll Cardiol. 2012; 60: 1455–1469. [DOI] [PubMed] [Google Scholar]
- 21. Redfield MM, Jacobsen SJ, Borlaug BA, Rodeheffer RJ, Kass DA. Age‐ and gender‐related ventricular‐vascular stiffening: a community‐based study. Circulation. 2005; 112: 2254–2262. [DOI] [PubMed] [Google Scholar]
- 22. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015; 131: 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kishimoto S, Kajikawa M, Maruhashi T, Iwamoto Y, Matsumoto T, Iwamoto A, Oda N, Matsui S, Hidaka T, Kihara Y, Chayama K, Goto C, Aibara Y, Nakashima A, Noma K, Higashi Y. Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. Int J Cardiol. 2017; 231: 181–187. [DOI] [PubMed] [Google Scholar]
- 24. Lyle MA, Brozovich FV. HFpEF, a disease of the vasculature: a closer look at the other half. Mayo Clin Proc. 2018; 93: 1305–1314. [DOI] [PubMed] [Google Scholar]
- 25. Cuijpers I, Simmonds SJ, van Bilsen M, Czarnowska E, González Miqueo A, Heymans S, Kuhn AR, Mulder P, Ratajska A, Jones EAV, Brakenhielm E. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic Res Cardiol. 2020; 115: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crisostomo PR, Wang M, Herring CM, Markel TA, Meldrum KK, Lillemoe KD, Meldrum DR. Gender differences in injury induced mesenchymal stem cell apoptosis and VEGF, TNF, IL‐6 expression: role of the 55 kDa TNF receptor (TNFR1). J Mol Cell Cardiol. 2007; 42: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malamitsi‐Puchner A, Tziotis J, Tsonou A, Protonotariou E, Sarandakou A, Creatsas G. Changes in serum levels of vascular endothelial growth factor in males and females throughout life. J Soc Gynecol Investig. 2000; 7: 309–312. [PubMed] [Google Scholar]
- 28. Richards M, Troughton RW. NT‐proBNP in heart failure: therapy decisions and monitoring. Eur J Heart Fail. 2004; 6: 351–354. [DOI] [PubMed] [Google Scholar]
- 29. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005; 69: 4–10. [DOI] [PubMed] [Google Scholar]
- 30. Dobbin SJH, Cameron AC, Petrie MC, Jones RJ, Touyz RM, Lang NN. Toxicity of cancer therapy: what the cardiologist needs to know about angiogenesis inhibitors. Heart. 2018; 104: 1995–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wada H, Suzuki M, Matsuda M, Ajiro Y, Shinozaki T, Sakagami S, Yonezawa K, Shimizu M, Funada J, Takenaka T, Morita Y, Nakamura T, Fujimoto K, Matsubara H, Kato T, Unoki T, Takagi D, Ura S, Wada K, Iguchi M, Masunaga N, Ishii M, Yamakage H, Shimatsu A, Kotani K, Satoh‐Asahara N, Abe M, Akao M, Hasegawa K. VEGF‐C and mortality in patients with suspected or known coronary artery disease. J Am Heart Assoc 2018; 7: e010355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Distribution of the sVEGFR‐2 levels. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2.
Figure S2. Incidence of CV death (A), all‐cause death (B), MACE (C), and HF‐related hospitalization (D) according to the quartiles of baseline VEGF levels during the follow‐up period. VEGF = vascular endothelial growth factor, CV = cardiovascular, MACE = major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization, HF = heart failure, HR = hazard ratio, CI = confidence interval.
Figure S3. Competing risk of death adjusted cumulative hazard curves for incident CV death (A), MACE (B), and HF‐related hospitalization (C) in patients with and without low sVEGFR‐2 levels below the 25th percentile during the follow‐up period. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, MACE = major adverse cardiovascular events defined as a composite of CV death and HF‐related hospitalization, HF = heart failure, HR = hazard ratio, CI = confidence interval.
Figure S4. Incidence of CV death (A) and all‐cause death (B) between patients with and those without low sVEGFR‐2 levels below the 25th percentile in the HFpEF/mrEF and HFrEF groups. Adjusted HR was calculated using the covariates included in model 3. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, HR = hazard ratio, CI = confidence interval, HF = heart failure, HFpEF = HF with preserved ejection fraction (EF), HFmrEF = HF with mid‐range EF, HFrEF = HF with reduced EF.
Figure S5. Incidence of CV death (A) and all‐cause death (B) between patients with and those without low sVEGFR‐2 levels below the 25th percentile in the groups with HF with ischemic etiology and HF with non‐ischemic etiology. Adjusted HR was calculated using the covariates included in model 3. sVEGFR‐2 = soluble vascular endothelial growth factor receptor 2, CV = cardiovascular, HR = hazard ratio, CI = confidence interval, HF = heart failure.
Table S1. Baseline characteristics according to quartiles of sVEGFR‐2.
Table S2. Simple and stepwise multiple regression analyses for the sVEGFR‐2 level.
Table S3. Multiple Cox proportional hazard analysis for clinical outcomes.