

Abstract
Aims
Calcific aortic valve disease (CAVD) is frequent in the elderly. Telocytes (TCs) are implicated in intercellular communication by releasing extracellular vesicles (EVs). This study investigated the role of TC‐EVs in aortic valve calcification.
Methods and results
TCs were obtained and identified using enzymolysis method and flow cytometry. EVs were isolated from TCs using differential high‐speed centrifugation method and identified using transmission electron microscope, western blot, and qNano analysis. The mouse model of CAVD was established. The changes of aortic valve activity‐related indicators were analysed by ultrasound, and the expressions of TC markers CD34 and vimentin in mouse valve tissues were detected using RT‐qPCR and western blot. The model mice were injected with TC‐derived EVs. The expressions of Runx2, osteocalcin, and caspase‐3 were detected using RT‐qPCR and western blot. The calcification model of valvular interstitial cells (VICs) was established. TC‐EVs were co‐cultured with calcified VICs, and calcium deposition was detected using alizarin red S staining. miR‐30b expression in calcified valvular tissues and cells was detected after EV treatment. miR‐30b expression in TCs was knocked down and then EVs were extracted and co‐cultured with calcified VICs. The target of miR‐30b was predicted through bioinformatics website and verified using dual‐luciferase assay. The levels of Wnt/β‐catenin pathway‐related proteins were detected. ApoE−/− mice fed with a high‐fat diet showed decreased aortic valve orifice area, increased aortic transvalvular pressure difference and velocity, reduced left ventricular ejection fraction, decreased CD34 and vimentin, and increased caspase‐3, Runx2, and osteocalcin. The levels of apoptosis‐ and osteogenesis‐ related proteins were inhibited after EV treatment. TC‐EVs reduced calcium deposition and osteogenic proteins in calcified VICs. EVs could be absorbed by VICs. miR‐30b expression was promoted in calcified valvular tissues and cells after EV treatment. Knockdown of miR‐30b weakened the inhibitory effects of TC‐EVs on calcium deposition and osteogenic proteins. miR‐30b targeted Runx2. EV treatment inhibited the Wnt/β‐catenin pathway, and knockdown of miR‐30b in TCs attenuated the inhibitory effect of TC‐EVs on the Wnt/β‐catenin pathway.
Conclusion
TC‐EVs played a protective role in aortic valve calcification via the miR‐30b/Runx2/Wnt/β‐catenin axis.
Keywords: Aortic valve calcification, Telocytes, Extracellular vesicles, miR‐30b, Runx2, Wnt/β‐catenin, Apoptosis
Introduction
Calcific aortic valve disease (CAVD) is a common cardiovascular disease in the elderly population, which imposes heavy economic and social burdens. 1 CAVD is primarily characterized by abnormal accumulation of calcium rich nodules on the surface of the aorta and/or in the annular region of the valve tip, resulting in sclerosis, movement restriction, and stenosis. 2 There lacks of established drug therapies to prevent the progression of CAVD, and surgical valve replacement is the only available treatment option, which, however, is accompanied by insurmountable complications, economic burdens, and uncertainties in the long‐term prognosis. 3 The mechanism of promoting the occurrence and development of aortic valve calcification has not been fully identified, but a recent report has suggested that it is a complex process concerning molecular and cellular phenotypes, and bone formation. 4 It is worth noting that the prevalence of CAVD is still rising due to the trend of aging population. 5 Hence, a better understanding about the underlying events concerning the onset and progression of aortic valve calcification remains an urgent issue to be solved.
Telocytes (TCs) is a new type of interstitial cells, which are characterized by smaller cell body with long prolongations of uneven calibre. 6 Cardiac interstitium contains typical TCs, which establish complicated spatial relationships with myocardial cells and cardiac stem cells, confirming the regulatory role of TCs in the three‐dimensional organization of cardiac tissues. 7 In pathological state, TCs can improve cardiac functions by promoting myocardial cell regeneration, enhancing angiogenesis and reducing cardiac fibrosis. 8 A previous literature has pointed out that TCs are implicated in intercellular communication through direct gap junctions or extracellular vesicle (EV) release. 9 Currently, TC‐derived EVs have been accepted as biomarkers and transferring tools for drug, vaccine, and gene of cardiovascular diseases. 10 New evidence has suggested that calcification‐competent EVs from valvular interstitial cells (VICs) are regulators of calcification in diseased aortic valves and atherosclerotic plaques. 11 More importantly, the intercellular communication mediated by EVs can also be achieved through the delivery of genetic information, such as microRNAs (miRs). 10
miRs are highly conserved non protein‐coding RNAs that maintain intracellular homeostasis through negative gene regulation. 12 Deregulation of miRs is extensively reported in the progression and prognosis of CAVD. 13 , 14 But the profiling and role of miR in TC‐EVs in CAVD is less studied. Based on these findings, we investigated the role of TC‐EVs in CAVD, along with its underlying miR mechanism, which shall provide impetus for the clinical management of CAVD.
Methods
Ethics statement
The study was approved by the Ethical Committee of The First Affiliated Hospital of Nanjing Medical University. All experimental procedures were performed based on the Ethical Guidelines for the study of experimental pain in conscious animals.
Preparation of cardiac telocytes
Female C57BL/6 mice aged 8 weeks and weighed 15–20 g [SYXK (Guangdong) 2019‐0203, Ruijian Xingze Biological Medicine Co., Ltd, Dongguan, Guangdong, China] were treated with 3% pentobarbital sodium. Hearts were dissected under aseptic conditions and put into 50 mL centrifuge tubes containing cold phosphate buffer saline (PBS) added with 1% penicillin and streptomycin. Then the hearts were placed in the cell culture room immediately. Following rinsing with fresh PBS to remove the blood, the hearts were sliced into millimetre‐sized pieces in a aseptic culture dish containing Dulbecco's modified Eagle's medium (DMEM)/F12 (12400‐024, Gibco, Grand Island, NY, USA) added with 1% penicillin and streptomycin. These pieces were rinsed twice and resuspended in PBS. Next, the enzymatic digestion medium containing 1.5 mg/mL collagenase type 2 (V900892, Sigma‐Aldrich, Merck KGaA, Darmstadt, Germany), DMEM/F12, and 1% penicillin and streptomycin was supplemented and the mixture was centrifuged at 180 rpm and 37°C for 40 min. The solution was filtered through a 41‐μm nylon mesh (Millipore, Billerica, MA, USA), and the collected cell suspension was centrifuged at 300× g for 10 min. Thereafter, the cells were seeded in aseptic culture dishes containing 10 mL DMEM/F12, 10% EV‐depleted fetal bovine serum (FBS) (System Biosciences, Mountain View, CA, USA) and 1% penicillin and streptomycin, followed by incubation in humidified air at 37°C for 1 h for fibroblast attachment. Unattached cells were transplanted into a fresh dish containing the above medium, and the medium was refreshed every 2 days.
Immunofluorescence staining
The phenotype of sorted cells was identified using immunofluorescence staining. In brief, the cells were seeded on cover glasses for 2 days, fixed with 4% para‐formaldehyde for 10 min, and permeabilized with 0.5% Triton X‐100 for 10 min. The cover glasses were cultured with the primary antibodies against α‐smooth muscleactin (α‐SMA), von Willebrand factor (vWF), and vimentin, and then incubated with the fluorescent‐conjugated secondary antibodies. Images were captured under a fluorescence microscope (Carl Zeiss, Jena, Germany).
Preparation of extracellular vesicles
The supernatants of cultured TCs were collected on ice and centrifuged at 10 000× g for 30 min to remove cells and debris. Then, the supernatants were transferred to a fresh tube, filtered through a 0.22‐μm membrane, and centrifuged at 120 000× g and 4°C for 2 h. The isolated EV pellet was washed once with aseptic PBS and resuspended in 200 μL PBS. In some samples, an Amicon ultra filter (Millipore) was used to reduce the volume from 50 to 1 mL to concentrate the supernatant, with a 100 000 molecular weight cutoff. Subsequently, EVs were isolated from the supernatants using the differential high‐speed centrifugation method.
The quality of EVs was identified using qNano analysis (Izon Science, Burnside, New Zealand). The protein of EVs was quantified using the bicinchoninic acid (BCA) assay kit (Thermo Fisher Scientific, Rockford, IL, USA). EVs were observed using transmission electron microscopy (TEM) (CM120, Philips, Andover, MA, USA). The expression levels of CD63, CD81, and calnexin were detected using western blot. The supernatant supplemented with GW4869 acted as the negative control (NC) of EVs.
Extracellular vesicles used in this study were assigned into four groups: NC group (the supernatant of TCs supplemented with GW4869), TC‐EVs group, in‐miR‐30b/TC‐EVs group (TCs were transfected with miR‐30b inhibitor and then EVs were extracted), and in‐NC/TC‐EVs group (TCs were transfected with inhibitor NC and then EVs were extracted). For the cell transfection, TCs in the logarithmic growth phase were seeded into 6‐well plates (1.0 × 105 cells per well), and then transfected with miR‐30b inhibitor or inhibitor NC using Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA, USA) upon 60% confluence. The cells were harvested for subsequent experiments 48 h later. miR‐30b inhibitor and inhibitor NC were synthesized and purified by GenePharma (Shanghai, China).
Reverse transcription quantitative polymerase chain reaction
The total RNA were extracted from tissues and cells using the TRIzol reagent (Invitrogen) and reverse transcribed into cDNA using the Toyoba reverse transcription kit (Fermentas Inc., Hanover, MD, USA). RT‐qPCR was performed in line with the instructions of the ABI PRISM 7900 system (Applied Biosystems, Carlsbad, CA, USA). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and U6 acted as the internal reference for genes and miR‐30b, respectively. PCR mixture included the SYBR Green/Fluorescein QPCR Master Mix (2X) (Fermentas), cDNA, and the age‐miR‐30b LNA™ PCR primer set (UniRT) for miR‐30b. The primer sequences are illustrated in Table 1 . The relative gene expression was calculated based on the 2−ΔΔCt method. The experiments were conducted three times independently.
Table 1.
Primer sequence for RT‐qPCR
| Gene | Primer sequence |
|---|---|
| miR‐29a | F: 5′‐ACCAAGTTTCAGTTCATGTAAAC‐3′ |
| R: 5′‐TCCAAGTCAGCTGAAGTAAAC‐3′ | |
| CD34 | F: 5′‐TCTGGAGTTCCCATCACTCC‐3′ |
| R: 5′‐TATGGCAGCTAGGAGGCACT‐3′ | |
| Vimentin | F: 5′‐CAGATGCGTGAGATGGAAGA‐3′ |
| R: 5′‐TCCAGCAGCTTCCTGTAGGT‐3′ | |
| Caspase‐3 | F: 5′‐AGAGCTGGGGCTCAAGTGTA‐3′ |
| R: 5′‐AGAACCACTGCCCTATGTGG‐3′ | |
| Runx2 | F: 5′‐ATGGCATCAAACAGCCTCTTCAG‐3′ |
| R: 5′‐TCAATATGGTCGCCAAACAGAT‐3′ | |
| Osteocalcin | F: 5′‐ATGAGAGCCCTCACACTCCTCG‐3′ |
| R: 5′‐CACCCTAGACCGGGCCGTAGAAG‐3′ | |
| GAPDH | F: 5′‐GGGAGCCAAAAGGGTCAT‐3′ |
| R: 5′‐GAGTCCTTCCACGATACCAA‐3′ | |
| U6 | F: 5′‐CGCTTCGGCAGCACATATAC‐3′ |
| R: 5vAATATGGAACGCTTCACGA‐3′ |
Western blot analysis
The total protein was extracted from tissues and cells using radio‐immunoprecipitation assay buffer (Shanghai Beyotime Biotechnology Co. Ltd., Shanghai, China) containing protease inhibitors (Roche, Basel, Switzerland). The concentration of protein was evaluated using the BCA assay kit (Beyotime). Then, the protein was separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. The membranes were blocked for 1 h and cultured with primary antibodies CD14 (1/10 000, ab221678, Abcam), vimentin (1/10 000, ab92547, Abcam), caspase‐3 (0.7 μg/mL, ab184787, Abcam), Runx2 (1 μg/mL, ab23981, Abcam), osteocalcin (1/1000, ab133612, Abcam), Wnt3 (1/10 000, ab172612, Abcam), and β‐catenin (0.5 μg/mL, ab16051, Abcam) at 4°C overnight. Afterwards, the membranes were cultured with the secondary antibody immunoglobulin G (IgG) (1/2000, ab205718, Abcam). The membranes were developed and analysed using Image Lab™ software (BioRad Laboratories, Hercules, CA, USA).
Establishment of a mouse model of calcific aortic valve disease
ApoE−/− mouse model of atherosclerosis induced by a high‐fat diet was used as the reference to establish the CAVD model. Briefly, ApoE−/− mice aged 7–8 weeks were fed with a high‐fat diet containing 1.25% cholesterol and 7.5% saturated fat (CAVD group). Wild type C57BL/6 mice fed with a normal diet and a high‐fat diet were set as the control (WT group). After 10 months of feeding, the mice were subjected to echocardiography examination. Additionally, after the mice were fed with 10 months of high‐fat diet for calcification induction, they were injected with 50 μg TC‐EVs via tail vein (CAVD + TC‐EVs group), and the mice injected with equal amounts of cell supernatants containing GW4869 were set as the controls (CAVD + NC group). After 7 days of normal feeding, the mice were euthanized by intraperitoneal injection of pentobarbital sodium ≥100 mg/kg. In this study, six C57BL/6 mice and 24 ApoE−/− mice were used for the establishment of CAVD model, with six mice in each group. After euthanasia, the valve tissues were collected for RT‐qPCR and western blot analysis.
Echocardiography
Echocardiography was performed using the cardiac ultrasound instrument (Vivid E95, General Electric Company, Schenectady, NY, USA), with the transducer frequency of 2.0–5.0 MHz. The mice were anaesthetised with ether. The chest and upper abdomen of mice were depilated, coated with conductive adhesive, and fixed on the test table. The left, right, and apex of mouse sternum were scanned. The valve orifice area, aortic transvalvular pressure difference and velocity, and left ventricular ejection fraction (LVEF) were recorded. Vivid paediatric heart ultrasound probe M5Sc‐D was used. Echocardiography parameters were evaluated by radiologist independently in a blind manner.
Calcification induction of valvular interstitial cells
The VICs at passage 3–7 (Beijing Qbioscience Biotechnology Co., Ltd, Beijing, China) were detached with trypsin to prepare cell suspension. Cells were seeded in 6‐well plates at 1 × 105/mL. The interstitial cells in the blank group were cultured in the standard medium (DMEM + 10% FBS), and interstitial cells in the CAVD group were incubated in the calcification medium (0.1% FBS, 50 ng/mL BMP‐2, 100 nM dexamethasone, 50 μg/mL ascorbic acid and 5 mM β‐glycerophosphate). The medium was refreshed every 2 days for totally 7 days of culture. Additionally, the CAVD + NC group and CAVD + TC‐EVs group were set, and NC was the cell supernatant supplemented with GW4869.
Valvular interstitial cells used in this study were assigned into six groups: blank group, CAVD group, CAVD + TC‐Evs group (CAVD VICs were treated with 50 μg TC‐Evs), CAVD + NC group (CAVD VICs were treated with equal amounts of NC), CAVD + in‐miR‐30b/TC‐EVs group (CAVD VICs were treated with equal amounts of in‐miR‐30b/TC‐EVs), and CAVD + in‐NC/TC‐EVs group (CAVD VICs were treated with equal amounts of in‐NC/TC‐EVs). After 24‐h incubation, the cells were harvested for subsequent experiments.
Von Kossa staining
The valve tissue sections were immersed in 2% silver nitrate for 20 min under good sunlight or ultraviolet lamp, washed with distilled water for 5 min, treated with 5% sodium thiosulfate solution for 2 min, and counterstained with 0.1% nuclear solid red solution for 1 min. Following washing, the calcified tissues were black under the light microscope.
Alizarin red S staining
After staining with alizarin red S solution (40 mM; Sigma‐Aldrich), the calcified VICs were placed for 30 min. The cells were washed and then observed under the light microscope.
Dual‐luciferase reporter gene assay
The 3′UTR sequences of Runx2 containing miR‐30b binding site were synthesized. Wild‐type (WT) plasmids (Runx2‐WT) and mutant‐type plasmid (Runx2‐MUT) were constructed. Then the constructed vectors were mixed with mimic‐NC or miR‐30b mimic and transfected into 293T cells (American Type Culture Collection, Manassas, Virginia, USA). After 48 h of transfection, luciferase activities were detected using the dual‐luciferase reporter Gene assay kit (BioVision, SanFrancisco, CA, USA) on the Glomax20/20 luminometer (Promega, WI, USA).
Statistical analysis
SPSS 21.0 software (IBM Corp., Armonk, NY, USA) was utilized for data analysis. Data were expressed as mean ± standard deviation. Comparisons of data between two groups were analysed using the t‐test. One‐way analysis of variance (ANOVA) was employed for comparisons among multi‐groups, followed by Tukey's multiple comparison test. The P value was obtained from a two‐tailed test, and a value of P < 0.05 meant a statistical difference.
Results
Culture of telocytes and extraction of extracellular vesicles
Single cell suspension was obtained using enzymatic hydrolysis, and the cells were seeded in the culture flask after adherence and gradient centrifugations. After 96 h of culture, a large amount of cell debris could be observed under the light microscope. After 7 days, the cell monolayer showed clonal phenomenon with the size of 10–30 μM. After 14 days, the cells reached 80% confluence and cell populations arranged in the form of paving stones; podoms and podomers appeared alternately with uneven length (Figure 1 A ). The cardiac cells expressing CD34 and CD117 were sorted using flow cytometry (Figure 1 B ). The level of vimentin was detected using immunofluorescence staining. Telopodes and podomers also showed vimentin‐positive, and the nucleus was blue after DAPI staining (Figure 1 C ).
Figure 1.
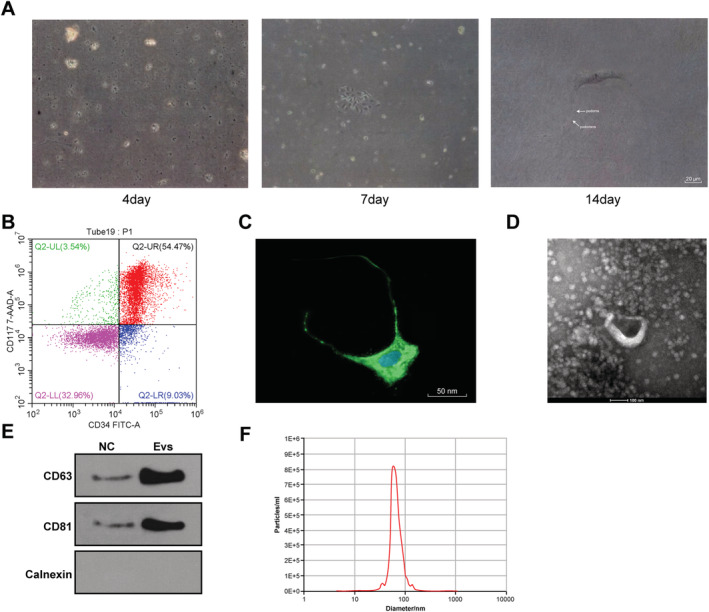
Incubation of TCs and extraction of EVs. (A) Morphology of TCs was observed under the light microscope on the 4th, 7th, and 14th day; (B) levels of CD34 and CD117 on the surface of primary cells were identified using flow cytometry; (C) level of vimentin in cardiac cells was observed using the laser confocal scanning microscope; (D) morphology of EVs was observed under the TEM; (E) makers of EVs were detected using western blot analysis; (F) diameter of EVs was detected using qNano analysis.
Then, EVs were isolated from TCs using high‐speed centrifugation and identified using TEM, western blot analysis, and qNano analysis. The isolated EVs were cup‐shaped or round vesicles (Figure 1 D ). EV markers CD63 and CD81 were enriched, but calnexin (an endoplasmic reticulum protein) and β‐actin (a protein encoded by the host gene of the cell) were absent (Figure 1 E ). The diameter of most EVs ranged from 50 to 150 nm (Figure 1 F ).
Telocyte‐extracellular vesicles prevented aortic valve calcification and apoptosis of valvular interstitial cells
ApoE−/− mice were fed with a high‐fat diet for the establishment of CAVD model. After 10 months of high‐fat diet feeding for calcification induction, echocardiographic analysis was performed, and the results showed that the aortic valve orifice area was decreased, the aortic transvalvular pressure difference and velocity were increased, and LEVF was reduced significantly (Figure 2 A ), indicating that the mouse model of CAVD was successfully established. Moreover, the expressions of TC markers CD34 and vimentin in mouse valve tissues were detected using RT‐qPCR and western blot, and the results showed after 10 months of calcification induction in ApoE−/− mice. The expressions of CD34 and vimentin in mouse valve tissues were significantly decreased (Figure 2 B,C ), indicating that TCs played a positive role in myocardial injury.
Figure 2.
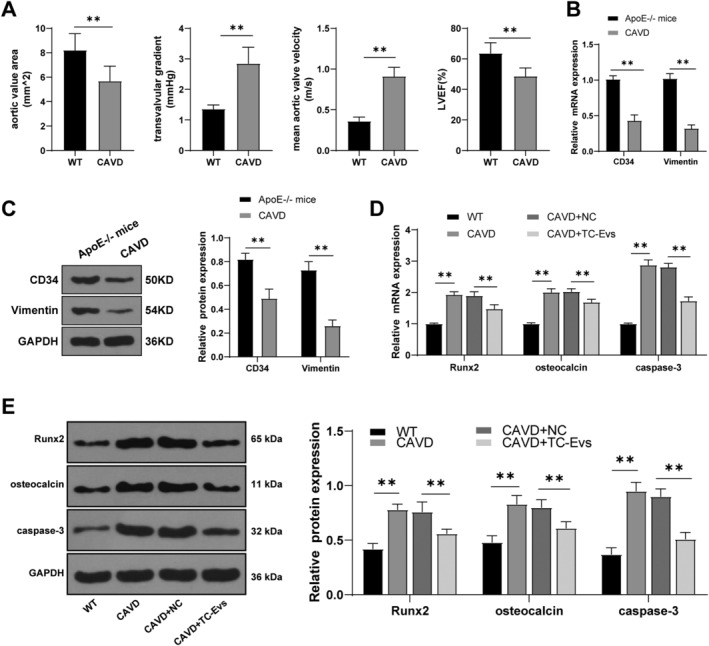
TC‐EVs reduced aortic valve calcification and apoptosis of valve interstitial cells. (A) Aortic valve orifice area, aortic transvalvular pressure difference and velocity, and left ventricular ejection fraction of mice were detected using echocardiography; (B, C) expressions of TC markers CD34 and vimentin in mouse valve were detected using RT‐qPCR and western blot analysis; (D, E) expressions of Runx2, osteocalcin, and caspase‐3 in calcified valves of mice were detected using RT‐qPCR and western blot analysis. The cell experiment was repeated three times independently. N = 6. Data are presented as mean ± standard deviation. Data in panels A/B/C were analysed using t‐test, and data in panels D/E were analysed using one‐way ANOVA, followed by Tukey's multiple comparison test, **P < 0.01.
Telocyte‐extracellular vesicles can reduce myocardial fibrosis, improve cardiac function, and increase angiogenesis. 15 Hence, we hypothesized that TC‐EVs had an effect on aortic valve calcification. To confirm this hypothesis, model mice were treated with TC‐EVs, and then the expression levels of Runx2, osteocalcin, and caspase‐3 were detected using RT‐qPCR and western blot. The results revealed that the expressions of Runx2, osteocalcin, and caspase‐3 in calcified valves of model mice were notably higher than those in EV‐treated mice (Figure 2 D,E ), indicating that TC‐EVs treatment reduced the calcification of aortic valve and alleviated the apoptosis of VICs.
Telocyte‐extracellular vesicles prevented valves from transforming into contractile and osteogenic phenotypes
To further determine the effect of TC‐EVs on valve calcification, we conducted in vitro experiments. A calcification model of VICs was established in vitro, and the appearance and phenotype changes of calcified VICs were observed. VICs were cultured and identified. After 12 h of culture, the primary VICs began to adhere to the wall and formed a continuous monolayer in about 1 week (Figure 3 A ). VICs were positive for α‐SMA and vimentin and negative for vWF, as immunofluorescence staining indicated (Figure 3 B ). The aforementioned markers were quantified using flow cytometry. Vimentin was highly expressed in VICs (99%), and α‐SMA‐positive cells accounted for about 50% of all cells (Figure 3 C ) (both P < 0.01). Subsequently, calcification induction was performed. The growth morphology of the cells induced by calcification was similar to that of control cells, with spindle‐shape, clear boundary, and radial or swirling growth arrangement (Figure 3 D ). TC‐EVs were cultured with calcified valve cells, and then the calcium deposition was detected. The results showed that EV treatment reduced the formation of calcium deposition significantly in calcified valve cells (Figure 3 E ). The levels of Runx2 and osteocalcin were increased in calcified valve cells, and EV treatment notably decreased the levels of osteogenic‐related proteins in calcified valve cells (Figure 3 F,G ) (all P < 0.01).
Figure 3.
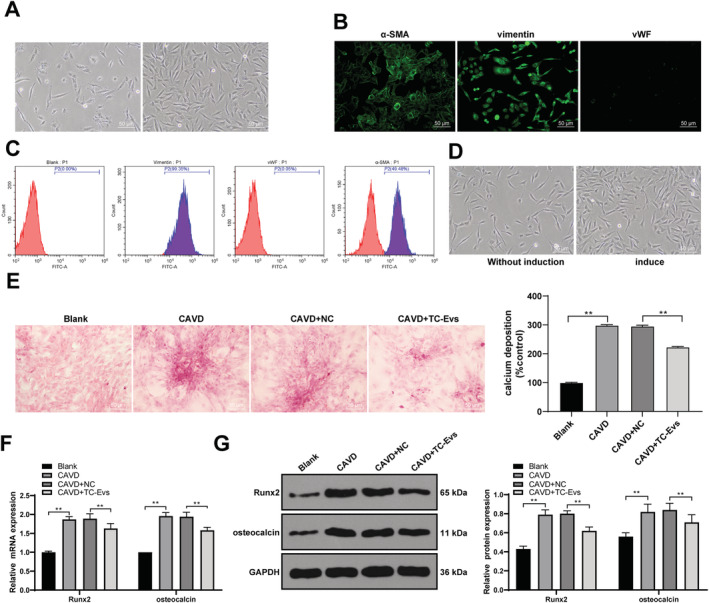
TC‐EVs prevented aortic valves from transforming into contractile and osteogenic phenotype. (A) Appearance of calcified valve interstitial cells was observed under the light microscope; (B) levels of α‐SMA, vimentin, and vWF in interstitial cells were observed; (C) quantitative analysis of vimentin and α‐SMA‐positive cells in valve interstitial cells was conducted using flow cytometry; (D) cell growth morphology before and after calcification induction was observed; (E) formation of calcium deposition was detected using Alizarin red S staining; (F, G) levels of Runx2 and osteocalcin were detected using RT‐qPCR and western blot analysis. The cell experiment was repeated three times independently. Data are presented as mean ± standard deviation. Data in panel E/F/G were analysed using one‐way ANOVA, followed by Tukey's multiple comparison test, **P < 0.01.
Telocyte‐extracellular vesicles were the communication media of miR‐30b during aortic valve calcification
It has been reported that miR‐30b has a certain effect on aortic valve calcification. 16 We speculated that TC‐EVs protected aortic valve calcification by releasing miR‐30b. PKH26‐labelled EVs were cultured with VICs for 48 h, and the cytoplasm of cells incubated with PKH26‐labelled EVs showed red fluorescence (Figure 4 A ), suggesting that TC‐EVs could be absorbed by VICs. Then, miR‐30b expression in the supernatants treated with TC‐EVs or GW4869 was detected using RT‐qPCR, along with miR‐30b expression in calcified valvular tissues and VICs after EV treatment. The results showed that miR‐30b mainly existed in EVs, and miR‐30b expression in calcified valvular tissues and VICs was notably increased after EV treatment (Figure 4 B ) (all P < 0.01). It was indicated that TC‐EVs were the communication media of miR‐30b during aortic valve calcification.
Figure 4.
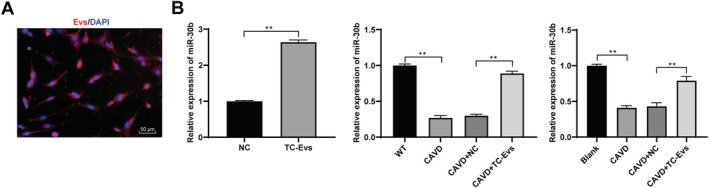
TC‐EVs were the communication media of miR‐30b in aortic valve calcification. (A) Absorption of EVs by valve interstitial cells was observed using immunofluorescence staining; (B) expression of miR‐30b in calcified valve tissues and valve interstitial cells was detected using RT‐qPCR. The cell experiment was repeated three times. Data are expressed as mean ± standard deviation. Data in panel (B) were analysed using t‐test or one‐way ANOVA, followed by Tukey's multiple comparison test, **P < 0.01.
Inhibition of miR‐30b attenuated the protective effect of telocyte‐extracellular vesicles on aortic valve calcification
We knocked down the miR‐30b expression in TCs and then extracted the EVs to confirm the protective effects of EVs on aortic valve calcification by releasing miR‐30b. miR‐30b expression in EVs extracted form TCs with knockdown of miR‐30b was significantly reduced (Figure 5 A ). EVs with low‐expressing miR‐30b were cultured with VICs, and the results showed that inhibition of miR‐30b could weaken the inhibitory effects of TC‐EVs on the formation of calcium deposition, and the levels of Runx2 and osteocalcin (Figure 5 B–D ).
Figure 5.
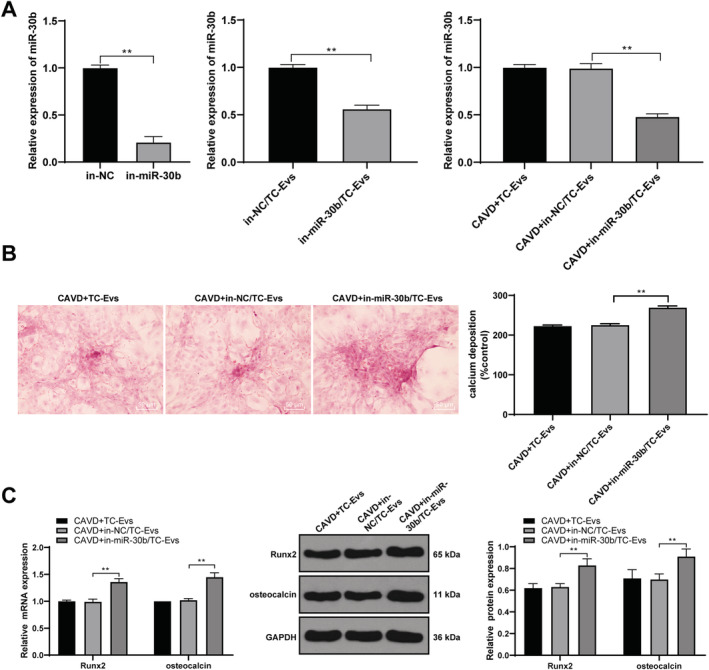
Inhibition of miR‐30b attenuated the protective effect of EVs on aortic valve calcification. (A) Expression of miR‐30b was detected using RT‐qPCR; (B) formation of calcium deposition was detected using Alizarin red S staining; (C) levels of Runx2 and osteocalcin were detected using RT‐qPCR and western blot analysis. The cell experiment was repeated three times. Data are expressed as mean ± standard deviation. Data in panels (A) and (B) were analysed using t‐test, and data in panel (C) were analysed using one‐way ANOVA, followed by Tukey's multiple comparison test, **P < 0.01.
Telocyte‐extracellular vesicles transferred miR‐30b to inhibit Runx2 and inactivate the Wnt/β‐catenin pathway
From the aforementioned results, we knew that TC‐EVs played a protective role in valve calcification by transferring miR‐30b, but the downstream mechanism of miR‐30b remained unclear. The bioinformatics website (http://starbase.sysu.edu.cn/agoClipRNA.php?source) showed that there were several target genes of miR‐30b, including Runx2. We found in the aforementioned results that miR‐30b negatively regulated Runx2, so we hypothesized that there was a target relationship between miR‐30b and Runx2. The target relationship between miR‐30b and Runx2 was verified using dual‐luciferase reporter gene assay (Figure 6 A ). Inactivation of the Wnt/β‐Catenin pathway can inhibit valve calcification, 17 and Runx‐2 acts coordinately with components of the Wnt/β‐catenin signalling pathway to enhance gene expression of both osteogenic bone markers and mineralization. 18 Hence, the levels of Wnt/β‐catenin pathway‐related proteins in tissues and cells were detected. The Wnt/β‐catenin pathway was activated in CAVD group compared with the blank group, and EV treatment inhibited the Wnt/β‐catenin pathway; knockdown of miR‐30b attenuated the inhibitory effect of TC‐EVs on the Wnt/β‐catenin pathway (Figure 6 B ).
Figure 6.
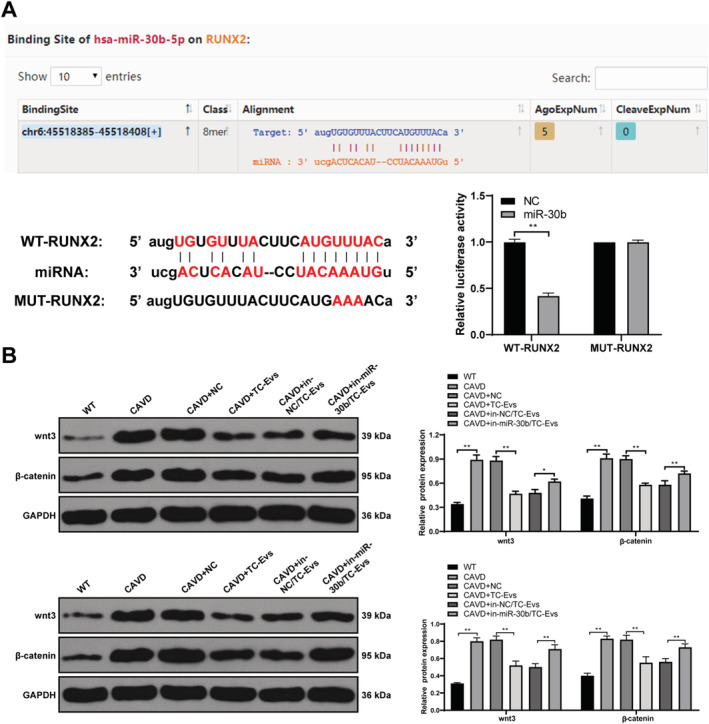
TC‐EVs transferred miR‐30b to inhibit Runx2 and inactivate the Wnt/β‐catenin pathway. (A) Binding relationship between miR‐30b and Runx2 was verified using dual‐luciferase reporter gene assay; (B) levels of the Wnt/β‐catenin pathway‐related proteins in calcified valve tissues and valve interstitial cells were detected using western blot analysis. The cell experiment was repeated three times. Data are expressed as mean ± standard deviation. Data in panel (A) were analysed using t‐test, and data in panel (B) were analysed using one‐way ANOVA, followed by Tukey's multiple comparison test, **P < 0.01.
Discussion
Telocyte‐extracellular vesicles participate in the progression of myocardial infarction, systemic sclerosis, and heart failure by delivering miRs, which can become a breakthrough in the management of CAVD. 19 , 20 Intriguingly, miRs are critical in the initiation and prognosis of CAVD. 13 Our results elucidated that TC‐EVs ameliorated aortic valve calcification via the miR‐30b/Runx2/Wnt/β‐catenin axis.
In the human heart, TCs widely exist in epicardium, myocardial interstitium, and aortic valves, where they contribute to the modulation of cardiac homeostasis and regeneration. 20 Through the release of EVs or atypical junctions, TCs form a cardiac network and manipulate the long‐distance heterologous communication in normal or diseased hearts. 21 , 22 A previous study has reported that TC‐EVs transfer macromolecular signals to adjacent cells to stimulate neovascularization in infarcted myocardium. 23 In the present study, we extracted and identified TC‐EVs, and then established the mouse model of CAVD. Our results found that 10 months of calcification induction reduced the expressions of TC markers CD34 and vimentin in mouse valve tissues, indicating that TCs played a protective role against myocardial injury. The decrease of TCs in adult hearts may be one of the reasons for the limited cardiac regeneration ability in the elderly. 20 TC‐shuttled exosomes attenuate cardiac fibrosis and reduce collagen deposition. 15 Accordingly, we hypothesized that TC‐derived EVs also exerted some effects on aortic valve calcification. Hence, model mice were treated with TC‐EVs, and the results found that TC‐EVs decreased the expressions of osteoblast specific transcription factor Runx2, the most abundant non‐collagen in bone matrix osteocalcin, and the key gene of apoptosis in calcified valves of mice caspase‐3. Briefly, the results demonstrated that TC‐EVs could reduce the degree of aortic valve calcification and the apoptosis of VICs.
Valvular interstitial cells are the most common cells in aortic valves, which regulate normal valve functions and repair diseased valves. 24 In addition, the in vitro experiments were conducted to further determine the effect of TC‐EVs on valve calcification. TC‐EVs were co‐cultured with calcified VICs, and then the phenotypic change of VICs was observed. Valve calcification generally has a pathophysiological process involving the osteogenic differentiation of VICs, lipoprotein deposition, and inflammation. 25 The osteogenic differentiation of VICs is ultimately accounted for calcium deposition and osteogenic nodule formation. 26 Runx2, a transcription factor, is indispensable for the osteoblast differentiation and chondrocyte maturation. 27 Osteocalcin is a type of osteoblast specific secreted protein, which serves as a hormone by triggering insulin generation and promoting energy consumption in target organs. 28 The levels of osteogenic proteins, such as Runx2 and osteocalcin, are highly expressed in calcific aortic valves of human or mice. 29 EV treatment notably decreased the levels of Runx2 and osteocalcin in calcified valve cells. Consistently, Liu et al. have demonstrated that multiple myeloma‐derived EVs can lead to a reduction of Runx2 and osteocalcin in bone marrow stromal cells. 30
Extracellular vesicles derived from TCs can transfer macromolecular signals such as miRs to neighbouring cells and change their transcriptional activity. 21 It has been reported that miRs have great potentials as a biomarker for the diagnosis and treatment of CAVD. 31 miR‐30b acts as an osteogenic inhibitor and exerts effects on aortic valvular calcification and apoptosis in CAVD by modulating Runx2 and caspase‐3. 32 Vishal Nigam et al. have indicated that overexpression of miR‐30b can reduce the levels of calcification‐related genes in aortic VICs. 16 However, whether TC‐EVs play a protective role in CAVD via transferring miR‐30b remains unclear. The results of our experiments indicated that TC‐EVs could be absorbed by VICs, and miR‐30b expression in calcified valvular tissues was notably increased after EV treatment. Furthermore, inhibition of miR‐30b could attenuate the protective effect of TC‐EVs on aortic valve calcification, indicating that TC‐EVs were the communication media of miR‐30b in aortic valve calcification. Subsequently, we focused on the downstream mechanism of TC‐EVs in CAVD via transferring miR‐30b. Bioinformatics website predicted that there were multiple target genes of miR‐30b, including Runx2. It has been reported that simvastatin can reduce the calcium deposition in aortic VICs in vitro by decreasing LPS‐induced Runx2 expression, suggesting the involvement of Runx2 in aortic valve calcification. 33 The target relationship between miR‐30b and Runx2 was verified using dual‐luciferase reporter gene assay in this study. Consistently, a previous literature has also exhibited that miR‐30 family members can suppress osteoblast differentiation via inhibiting Runx2 in MC3T3‐E1 cells. 34 Wnt/β‐catenin pathway is implicated in the differentiation of VICs into osteoblast cells, eventually contributing to aortic valve calcification. 35 Cai et al. have demonstrated that Wnt/β‐catenin pathway can enhance the osteogenic differentiation and calcification of vascular smooth muscle cells by regulating Runx2. 36 Our results showed that EV treatment suppressed the Wnt/β‐catenin pathway, and EVs extracted form miR‐30b‐silencing TCs attenuated the inhibiting effects on the Wnt/β‐catenin pathway. Overall, TC‐EVs could transfer miR‐30b to inhibit Runx2 level and the Wnt/β‐catenin pathway in calcified valve tissues.
To conclude, we are the first to find that TC‐EVs alleviate valve calcification via the delivery of miR‐30b and the Runx2/Wnt/β‐catenin axis. This fundamental information may suggest potential therapy for CAVD in the clinical trial. In the future, we shall carry out more prospective trials on the feasibility and safety of TC‐EVs in the treatment of CAVD, so as to refine our clinical guidance.
Conflict of interest
The authors declared that they have no competing interests.
Author contributions
R.Y. is the guarantor of integrity of the entire study; R.Y. contributed to the study concepts, study design, definition of intellectual content, literature research, manuscript preparation, and manuscript editing and review; Y.H.T. contributed to the clinical studies; R.Y. and X.W.C. contributed to the experimental studies and data acquisition; Y.Y. contributed to the data analysis and statistical analysis. All authors read and approved the final manuscript.
Yang, R. , Tang, Y. , Chen, X. , and Yang, Y. (2021) Telocytes‐derived extracellular vesicles alleviate aortic valve calcification by carrying miR‐30b. ESC Heart Failure, 8: 3935–3946. 10.1002/ehf2.13460.
Data availability statement
All the data generated or analysed during this study are included in this published article.
References
- 1. Small A, Kiss D, Giri J, Anwaruddin S, Siddiqi H, Guerraty M, Chirinos JA, Ferrari G, Rader DJ. Biomarkers of calcific aortic valve disease. Arterioscler Thromb Vasc Biol 2017; 37: 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Otto CM, Prendergast B. Aortic‐valve stenosis—from patients at risk to severe valve obstruction. N Engl J Med 2014; 371: 744–756. [DOI] [PubMed] [Google Scholar]
- 3. Dutta P, Lincoln J. Calcific Aortic valve disease: a developmental biology perspective. Curr Cardiol Rep 2018; 20: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gu J, Lu Y, Deng M, Qiu M, Tian Y, Ji Y, Zong P, Shao Y, Zheng R, Zhou B. Inhibition of acetylation of histones 3 and 4 attenuates aortic valve calcification. Exp Mol Med 2019; 51: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hutcheson JD, Aikawa E, Merryman WD. Potential drug targets for calcific aortic valve disease. Nat Rev Cardiol 2014; 11: 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang H. Vascular telocytes. Adv Exp Med Biol 2016; 913: 377–395. [DOI] [PubMed] [Google Scholar]
- 7. Bani D. Telocytes in cardiac tissue architecture and development. Adv Exp Med Biol 2016; 913: 127–137. [DOI] [PubMed] [Google Scholar]
- 8. Tao L, Wang H, Wang X, Kong X, Li X. Cardiac telocytes. Curr Stem Cell Res Ther 2016; 11: 404–409. [DOI] [PubMed] [Google Scholar]
- 9. Kondo A, Kaestner KH. Emerging diverse roles of telocytes. Development 2019; 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cretoiu D, Xu J, Xiao J, Cretoiu SM. Telocytes and their extracellular vesicles‐evidence and hypotheses. Int J Mol Sci 2016; 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Krohn JB, Hutcheson JD, Martinez‐Martinez E, Aikawa E. Extracellular vesicles in cardiovascular calcification: expanding current paradigms. J Physiol 2016; 594: 2895–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mishra S, Yadav T, Rani V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit Rev Oncol Hematol 2016; 98: 12–23. [DOI] [PubMed] [Google Scholar]
- 13. Ni WJ, Wu YZ, Ma DH, Leng XM. Noncoding RNAs in calcific aortic valve disease: a review of recent studies. J Cardiovasc Pharmacol 2018; 71: 317–323. [DOI] [PubMed] [Google Scholar]
- 14. Tafuri E, Santovito D, de Nardis V, Marcantonio P, Paganelli C, Affaitati G, Bucci M, Mezzetti A, Giamberardino MA, Cipollone F. MicroRNA profiling in migraine without aura: pilot study. Ann Med 2015; 47: 468–473. [DOI] [PubMed] [Google Scholar]
- 15. Yang J, Li Y, Xue F, Liu W, Zhang S. Exosomes derived from cardiac telocytes exert positive effects on endothelial cells. Am J Transl Res 2017; 9: 5375–5387. [PMC free article] [PubMed] [Google Scholar]
- 16. Nigam V, Sievers HH, Jensen BC, Sier HA, Simpson PC, Srivastava D, Mohamed SA. Altered microRNAs in bicuspid aortic valve: a comparison between stenotic and insufficient valves. J Heart Valve Dis 2010; 19: 459–465. [PMC free article] [PubMed] [Google Scholar]
- 17. Fang M, Wang CG, Zheng C, Luo J, Hou S, Liu K, Li X. Mir‐29b promotes human aortic valve interstitial cell calcification via inhibiting TGF‐β3 through activation of Wnt3/β‐catenin/Smad3 signaling. J Cell Biochem 2018; 119: 5175–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yodthong T, Kedjarune‐Leggat U, Smythe C, Sukprasirt P, Aroonkesorn A, Wititsuwannakul R, Pitakpornpreecha T. Enhancing activity of Pleurotus sajor‐caju (Fr.) sing β‐1,3‐glucanoligosaccharide (Ps‐GOS) on proliferation, differentiation, and mineralization of MC3T3‐E1 Cells through the involvement of BMP‐2/Runx2/MAPK/Wnt/β‐catenin signaling pathway. Biomolecules 2020; 10: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kucybala I, Janas P, Ciuk S, Cholopiak W, Klimek‐Piotrowska W, Holda MK. A comprehensive guide to telocytes and their great potential in cardiovascular system. Bratisl Lek Listy 2017; 118: 302–309. [DOI] [PubMed] [Google Scholar]
- 20. Marini M, Ibba‐Manneschi L, Manetti M. Cardiac telocyte‐derived exosomes and their possible implications in cardiovascular pathophysiology. Adv Exp Med Biol 2017; 998: 237–254. [DOI] [PubMed] [Google Scholar]
- 21. Fertig ET, Gherghiceanu M, Popescu LM. Extracellular vesicles release by cardiac telocytes: electron microscopy and electron tomography. J Cell Mol Med 2014; 18: 1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gherghiceanu M, Popescu LM. Cardiac telocytes—their junctions and functional implications. Cell Tissue Res 2012; 348: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manole CG, Cismasiu V, Gherghiceanu M, Popescu LM. Experimental acute myocardial infarction: telocytes involvement in neo‐angiogenesis. J Cell Mol Med 2011; 15: 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu S, Gotlieb AI. Wnt3a/β‐catenin increases proliferation in heart valve interstitial cells. Cardiovasc Pathol 2013; 22: 156–166. [DOI] [PubMed] [Google Scholar]
- 25. Cai Z, Li F, Gong W, Liu W, Duan Q, Chen C, Ni L, Xia Y, Cianflone K, Dong N, Wang DW. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler Thromb Vasc Biol 2013; 33: 2345–2354. [DOI] [PubMed] [Google Scholar]
- 26. van der Ven CF, Wu PJ, Tibbitt MW, van Mil A, Sluijter JP, Langer R, Aikawa E. In vitro 3D model and miRNA drug delivery to target calcific aortic valve disease. Clin Sci (Lond) 2017; 131: 181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol 2018; 149: 313–323. [DOI] [PubMed] [Google Scholar]
- 28. Mizokami A, Kawakubo‐Yasukochi T, Hirata M. Osteocalcin and its endocrine functions. Biochem Pharmacol 2017; 132: 1–8. [DOI] [PubMed] [Google Scholar]
- 29. Gomez Stallons MV, Wirrig‐Schwendeman EE, Fang M, Cheek JD, Alfieri CM, Hinton RB, Yutzey KE. Molecular mechanisms of heart valve development and disease. In Nakanishi T., Markwald R. R., Baldwin H. S., Keller B. B., Srivastava D., Yamagishi H., eds. Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology. Tokyo; 2016. p 145–151. [Google Scholar]
- 30. Liu Z, Liu H, Li Y, Shao Q, Chen J, Song J, Fu R. Multiple myeloma‐derived exosomes inhibit osteoblastic differentiation and improve IL‐6 secretion of BMSCs from multiple myeloma. J Invest Med 2020; 68: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nader J, Metzinger L, Maitrias P, Caus T, Metzinger‐Le Meuth V. Aortic valve calcification in the era of non‐coding RNAs: the revolution to come in aortic stenosis management? Noncoding RNA Res 2020; 5: 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang M, Liu X, Zhang X, Song Z, Han L, He Y, Xu Z. MicroRNA‐30b is a multifunctional regulator of aortic valve interstitial cells. J Thorac Cardiovasc Surg 2014; 147: 1073–1080 e1072. [DOI] [PubMed] [Google Scholar]
- 33. Jarrett MJ, Yao Q, Venardos N, Weyant MJ, Reece TB, Meng X, Fullerton DA. Simvastatin down‐regulates osteogenic response in cultured human aortic valve interstitial cells. J Thorac Cardiovasc Surg 2019; 161: e261–e271. [DOI] [PubMed] [Google Scholar]
- 34. Zhang L, Li G, Wang K, Wang Y, Dong J, Wang H, Xu L, Shi F, Cao X, Hu Z, Zhang S. MiR‐30 family members inhibit osteoblast differentiation by suppressing Runx2 under unloading conditions in MC3T3‐E1 cells. Biochem Biophys Res Commun 2020; 522: 164–170. [DOI] [PubMed] [Google Scholar]
- 35. Gu GJ, Chen T, Zhou HM, Sun KX, Li J. Role of Wnt/β‐catenin signaling pathway in the mechanism of calcification of aortic valve. J Huazhong Univ Sci Technolog Med Sci 2014; 34: 33–36. [DOI] [PubMed] [Google Scholar]
- 36. Cai T, Sun D, Duan Y, Wen P, Dai C, Yang J, He W. WNT/beta‐catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp Cell Res 2016; 345: 206–217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data generated or analysed during this study are included in this published article.