

Abstract
In patients with glioblastoma, resistance to the chemotherapeutic temozolomide (TMZ) limits any survival benefits conferred by the drug. Here we show that the convection-enhanced delivery of nanoparticles containing disulfide bonds (which are cleaved in the reductive environment of the tumour) and encapsulating an oxaliplatin prodrug and a cationic DNA intercalator inhibit the growth of TMZ-resistant cells from patient-derived xenografts, and hinder the progression of TMZ-resistant human glioblastoma tumours in mice without causing any detectable toxicity. Genome-wide RNA profiling and metabolomic analyses of a glioma cell line treated with the cationic intercalator or with TMZ showed substantial differences in the signalling and metabolic pathways altered by each drug. Our findings suggest that the combination of anticancer drugs with distinct mechanisms of action with selective drug release and convection-enhanced delivery may represent a translational strategy for the treatment of TMZ-resistant gliomas.
Despite rapid advances in cancer research during the past decades, glioblastoma (GBM) remains the most aggressive brain tumour in adults with a rate of 15,000 deaths every year in the United States alone, and a 5-year survival rate of less than 10%1–3. While temozolomide (TMZ) increases the survival rate of GBM patients by methylating DNA and inducing toxicity in tumour cells, its therapeutic benefits are limited by resistance, which arises via numerous mechanisms, including the acquisition of mismatch repair (MMR) defects and re-expression of O6-methylguanine-DNA-methytransferase (MGMT)4–8. Anticancer agents with alternative mechanisms of action are needed to treat TMZ-resistant GBM patients.
Platinum-based compounds—such as the third generation of platinum anticancer drug oxaliplatin and a cationic platinum DNA intercalator (5,6-dimethyl-1,10-phenanthroline) (1S,2S-diaminocyclohexane) platinum(II)] (56MESS)—have been shown to possess potent anticancer properties with negligible cross-resistance to DNA-alkylating agents9,10. Different from TMZ, oxaliplatin forms interstrand and intrastrand crosslinks with DNA that cannot be reversed by MGMT, and also is active in the setting of MMR defects11,12. A recent study demonstrates that oxaliplatin also induces ribosome biogenesis stress and leads to cell death in a p53-dependent manner13,14. 56MESS, on the other hand, intercalates DNA, disrupts intracellular iron and copper metabolism, suppresses the biosynthesis of sulfur-containing amino acids, and inhibits tumour cell proliferation10,15,16. Despite their remarkable antitumour efficacy, the therapeutic applications of oxaliplatin and 56MESS are hindered by toxicity15,17. Here we propose to address this problem by encapsulating these agents in reduction-responsive nanoparticles (NPs)15,16,18–20, an approach widely utilized for the delivery of chemotherapeutics21.
As an emerging class of nanocarriers, reduction-responsive polymers possess great potential for tumour-specific delivery of bioactive molecules22–26. Reduction-responsive polymers usually incorporate disulfide bonds that are sufficiently stable in the extracellular space, but are rapidly cleaved in the reductive environment of tumours23,27. It is reported that the glutathione (GSH) concentration in tumour tissue is 4-fold higher than that in non-neoplastic tissue27. Moreover, TMZ-resistant glioma cell lines show even higher levels of GSH than TMZ-sensitive cell lines28. Such differences make reduction-responsive NPs especially attractive for GBM chemotherapy28.
The blood–brain barrier (BBB), which is impermeable to most drugs29–31, is another obstacle to GBM therapy. Recent clinical trials show that convection-enhanced delivery (CED) safely bypasses the BBB and directly delivers drugs to target brain regions32. Using CED, the drugs can diffuse to a broader region compared to bolus injection or implants where the diffusion is solely driven by the concentration gradient33,34. Combining the advantages of these technologies, here we demonstrate that CED of reduction-responsive NPs containing highly potent platinum agents serves as a promising therapeutic strategy for TMZ-resistant GBM.
Polymer synthesis and nanoparticle formulation
To encapsulate OxaPt(IV) and 56MESS, we synthesized and characterized a reduction-responsive polymer, poly(1,2,4,5-cyclohexanetetracarboxylic dianhydride-co-hydroxyethyl disulfide)-polyethylene glycol (poly (CHTA-co-HD)-PEG), which contained disulfide bonds and pendent pairwise carboxylic acids (Fig. 1a, Supplementary Figs. 1–4). With nanoprecipitation, this polymer forms: (1) spherical NPs encapsulating OxaPt(IV) through hydrophobic interaction (Fig. 1c) and (2) NPs incorporating positively charged 56MESS through electrostatic complexation (Fig. 1d and Supplementary Fig. 5). The resulting NPs inhibit the growth of TMZ-resistant GBM cells through mechanisms illustrated in Fig. 1e.
Figure 1 |. Synthesis of the reduction-responsive polymer and formation of NPs.
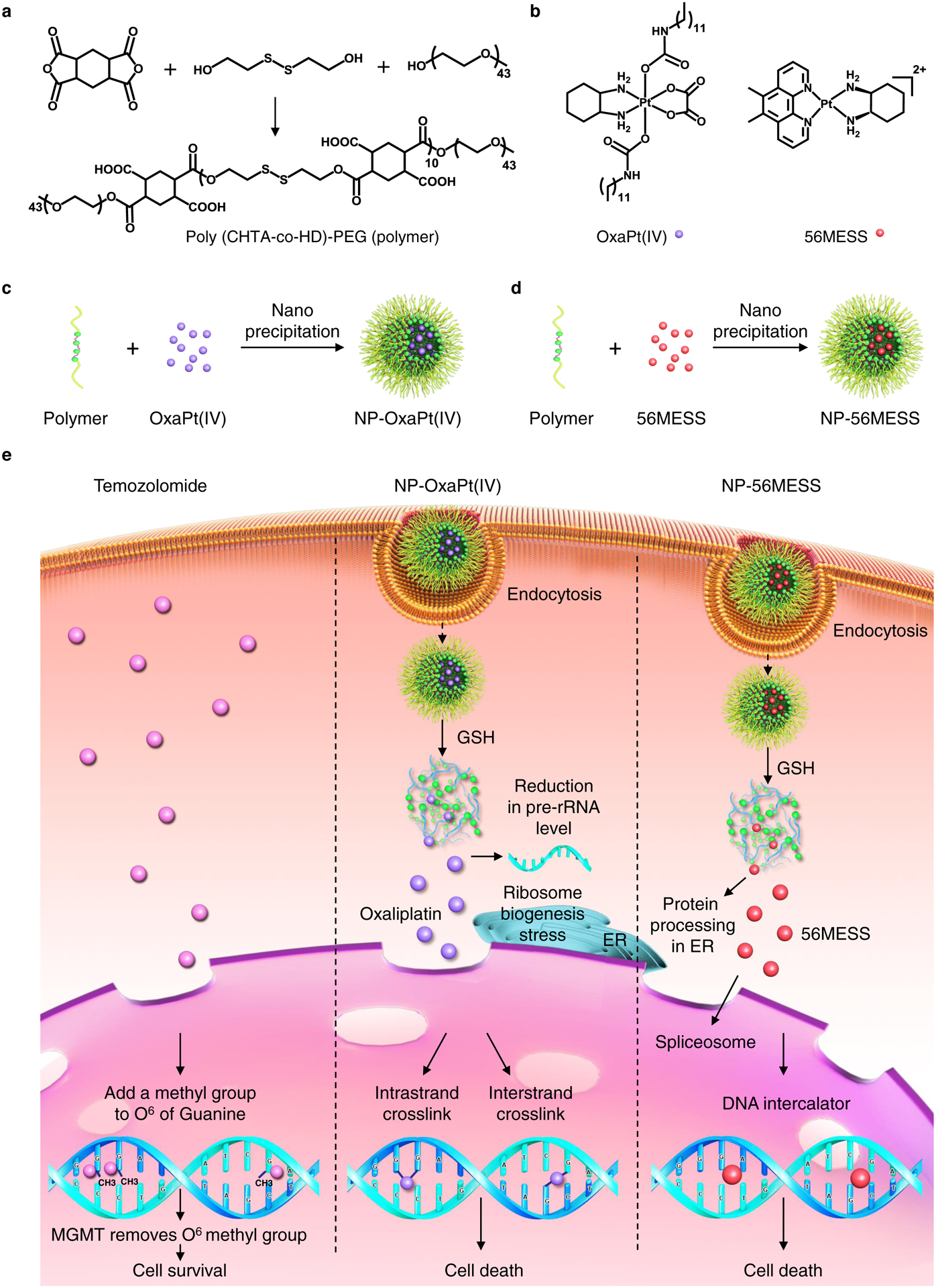
a, Synthesis of poly (CHTA-co-HD)-PEG. b, Structures of OxaPt(IV) and 56MESS. c, d, Formation of NPs by nanoprecipitation. c, Formation of NP-OxaPt(IV). d, Preparation of NP-56MESS. e, A schematic illustrating that oxaliplatin and 56MESS induce cell death after GSH-mediated drug release. ER, endoplasmic reticulum; rRNA, ribosomal RNA.
Characterization of the NPs
The critical micelle concentration of a polymer is a good predictor for NP stability35,36. The critical micelle concentration for our polymer was measured at 0.018 mg ml−1 via the Nile red assay (Fig. 2a, b), which predicts a slow dissociation rate37. The size and surface charge of NPs influence intracellular uptake38–41. The hydrodynamic diameter of OxaPt(IV)-loaded NPs (NP-OxaPt(IV)) in artificial cerebrospinal fluid (aCSF) was measured to be 105 ± 15 nm, and that of 56MESS-loaded NPs (NP-56MESS) was 105 ± 2.5 nm (Fig. 2c and Supplementary Fig. 5); the polydispersity indexes (PDI) for the NPs were 0.19 and 0.15, respectively (Fig. 2d); both NP types were negatively charged, with zeta-potentials of −26 mV and −22 mV, respectively (Fig. 2e); these parameters were within the range of optimal internalization identified previously38–40. In addition, we observed that the encapsulation efficiency of OxaPt(IV) (36.2%) was lower than that of 56MESS (63.4%, Fig. 2f).
Figure 2 |. Characterization of NPs.
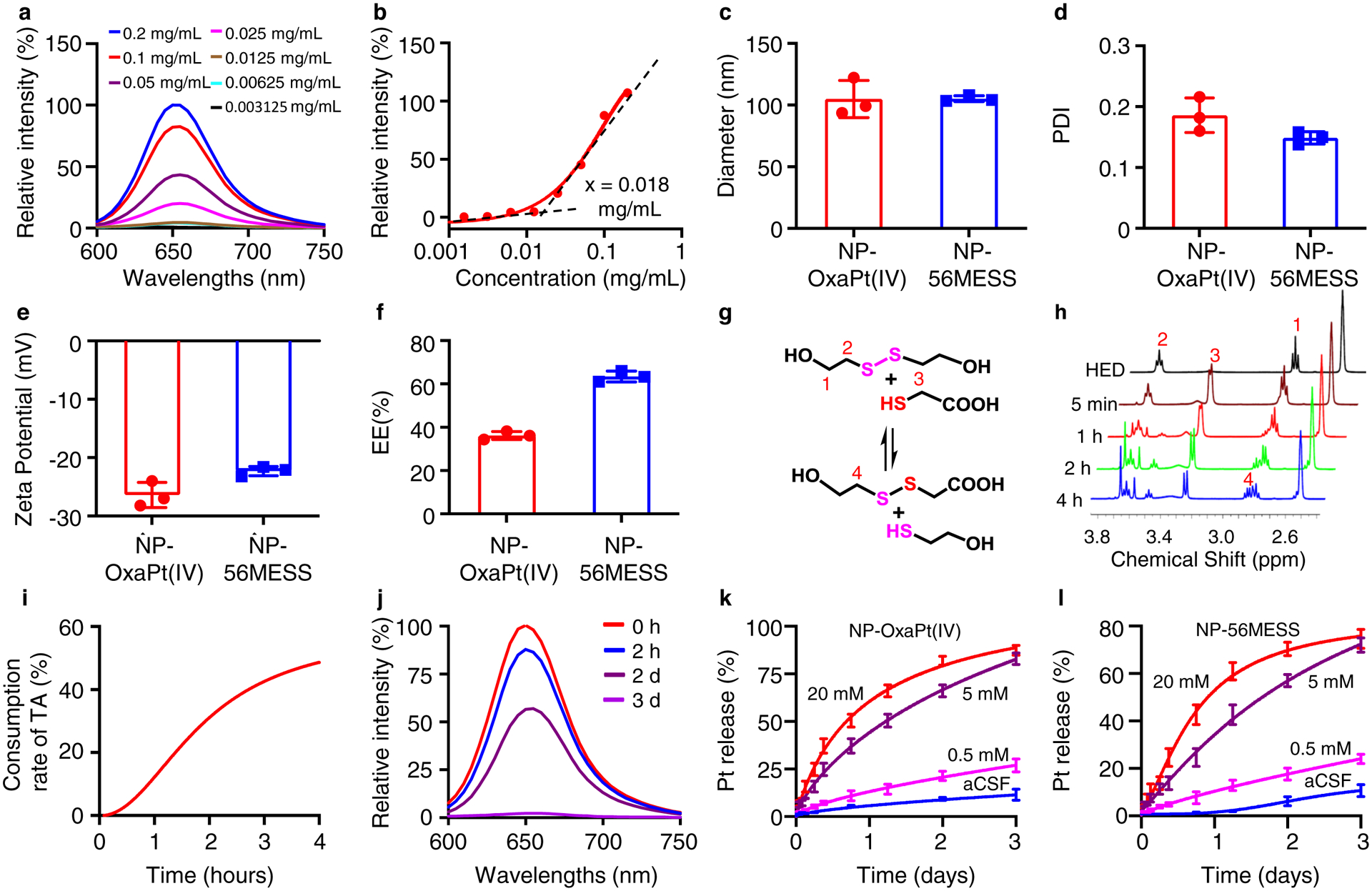
a, b, Critical micelle concentration for the polymer. Relative fluorescence intensity at different concentrations of polymer (a). The minimal concentration for the polymer to form micelles is approximately 0.018 mg ml−1(b). c-f, Characterization of NPs, including hydrodynamic diameters in aCSF (c), PDI in aCSF (d), surface charges in aCSF (e) and encapsulation efficiencies in deionized water (f). n=3, data are mean ± s. d. c, Mean diameter: 105 nm (NP-OxaPt(IV)), 105 nm (NP-56MESS). d, Mean PDI: 0.19 (NP-OxaPt(IV)), 0.15 (NP-56MESS). e, Mean zeta potential: −26 mV (NP-OxaPt(IV)), −22 mV (NP-56MESS). f, Mean encapsulation efficiency: 36% (NP-OxaPt(IV)), 63% (NP-56MESS). g, A schematic of a thiol–disulfide exchange reaction: 2-hydroxyethyl disulfide reacts with thioglycolic acid to produce 2-mercaptoethan-1-ol and 2-((2-hydroxyethyl)disulfanyl) acetic acid. h, 1 H NMR of the products from the thiol–disulfide exchange reaction. HED, 2-hydroxyethyl disulfide. i, Consumption rate of thioglycolic acid (TA). j, NP dissociation kinetics monitored by Nile red assay in aCSF. k, l, The triggered release of OxaPt(IV) (k) and 56MESS (l) from NPs in the presence three different concentrations of GSH in aCSF. n=3, data are mean ± s. d.
To test whether our polymer was reduction-sensitive, we designed a thiol–disulfide exchange reaction using thioglycolic acid and found that disulfide bonds broke quickly in the presence of a reducing agent (Fig. 2g, h). The consumption rate of thioglycolic acid is displayed in Fig. 2i. It has been reported that the GSH concentration is 2–20 μM in extracellular space42,43 and 0.1–10 mM in the cytosol42,44,45. To evaluate the reductive responsiveness of NPs in the cell, we performed a Nile red assay46, revealing complete dissolution of NPs in 5 mM GSH solution within 3 d (Fig. 2j).
Research has shown that the GSH concentration in tumour tissue is fourfold higher than in normal tissue27 and that TMZ-resistant glioma cell lines possess higher levels of GSH than TMZ-sensitive cell lines28. To investigate the triggered release of platinum drugs, we incubated the NPs in aCSF and GSH-aCSF solutions with different GSH concentrations – 0.5 mM, 5 mM and 20 mM – and observed that approximately 83% of platinum was released from the NP-OxaPt(IV) with continuous incubation in 5 mM GSH solution over 3 d, whereas only 11.3% OxaPt(IV) was released during incubation in aCSF over the same time period (Fig. 2k). Similarly, approximately 72% of platinum was released from NP-56MESS in 5 mM GSH solution, whereas only 9.1% was released in aCSF (Fig. 2l). Both types of NPs dissociated faster in 20 mM GSH solution and more slowly in 0.5 mM GSH solution. These data suggest that NPs are responsive to reductive conditions.
To evaluate intracellular uptake of NPs, we formulated NPs loaded with a fluorescent tracer, Dil (NP-Dil). NP-Dil physical characteristics were similar to NP-OxaPt(IV) and NP-56MESS (hydrodynamic diameter 90 ± 2 nm, PDI 0.13, and zeta potential −22.7mV) (Fig. 3b–d). Two human GBM cell lines, TMZ-resistant LN229 (LN229-TR) and cells from a patient-derived xenograft (PDX), were incubated with NP-DiI. The fluorescence intensity in both cell lines increased markedly within the first a few hours. This suggests that NPs could be taken up rapidly by GBM cells (Fig. 3e–g), which we also visualized using confocal microscopy (Fig. 3h and Supplementary Fig. 7). Notably, the majority of endocytosed NPs were outside of endosomes (Fig. 3i–m).
Figure 3 |. Intracellular uptake of dye-loaded NPs.
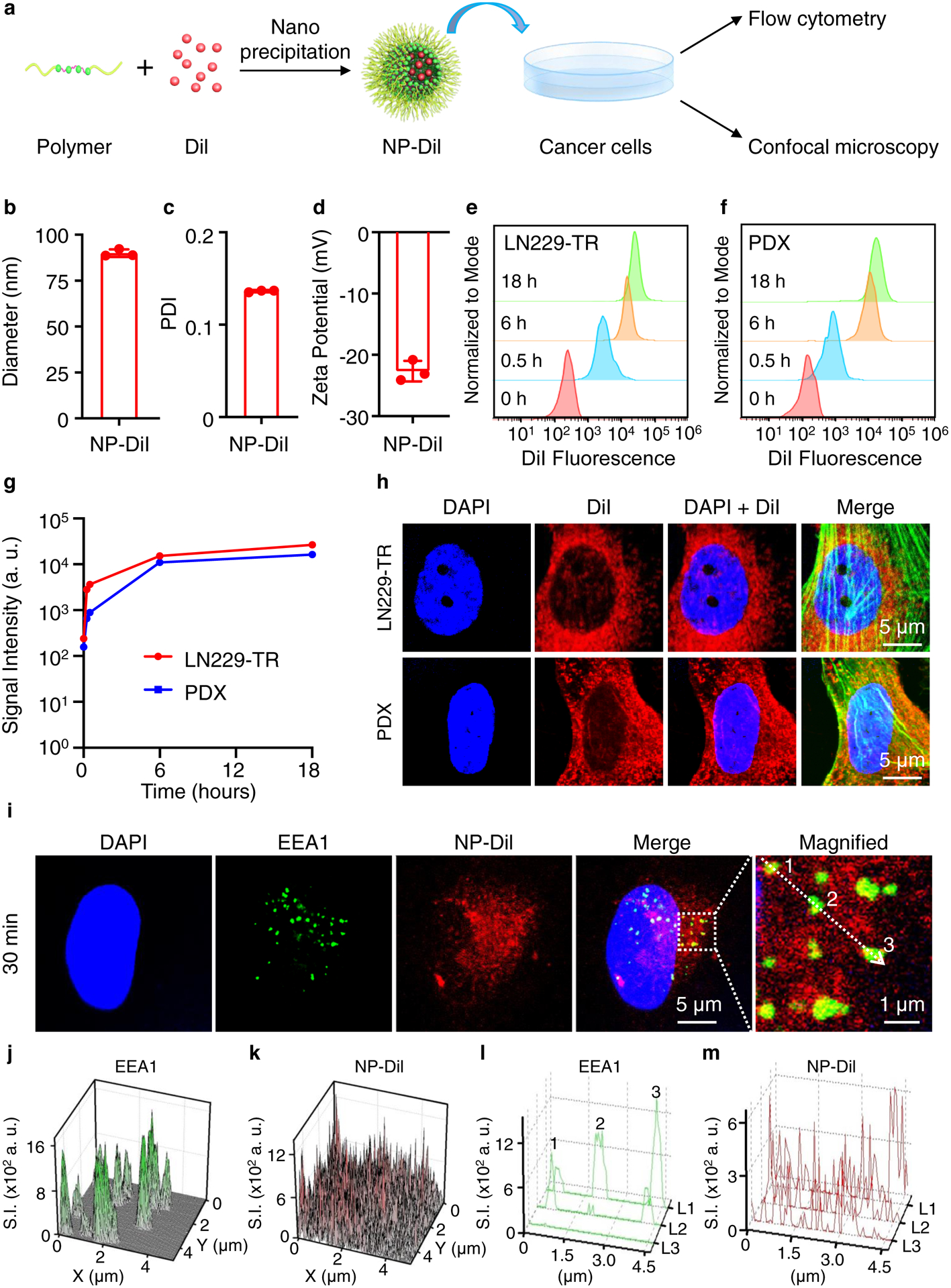
a, Schematic of experimental procedure. b-d, Characterization of NPs containing DiI (NP-DiI), including the hydrodynamic diameter (b; mean=90 nm), PDI (c; mean=0.14) and zeta potential (d; mean=−23 mV). n=3, data are mean ± s. d. e, f, The intracellular uptake of NP-DiI by LN229-TR (e) and PDX (f) measured by flow cytometry. The mean fluorescence intensity of the cells increases over time. n=3, data are mean ± s. d. g, Quantification of mean fluorescence intensity over time. h, NP-DiI NPs exhibit perinuclear localization in both LN229-TR cells and PDX cells 12 hours after incubation. F-actin is labelled with a phalloidin antibody (green). Scale bars, 5 μm. i-m, Subcellular localization of NP-Dil. LN229-TS cells are stained with early endosome antigen 1 (EEA1) antibodies. The spatial signal in the dashed square in i is quantified and presented in j (EEA1) and k (NP-DiI). The signal along the dotted arrow in i is measured and plotted in l (EEA1) and m (NP-DiI). L1, L2 and L3 refer to image layers 1, 2 and 3 from confocal imaging.
Antitumour efficacy of nanoparticles in vitro
To test the anticancer activities of OxaPt(IV) and 56MESS, we performed growth-delay assays using human GBM cell lines including TMZ-sensitive LN229 (LN229-TS), TMZ-resistant LN229 (LN229-TR), PDX and U87 (Fig. 4b–e). The half-inhibitory concentration (IC50) of TMZ in LN229-TS was 2.0 μM (Fig. 4b), whereas the IC50 of TMZ in LN229-TR was 162.6 μM (Fig. 4c), confirming an approximately 81-fold resistance to TMZ in the LN229-TR cell line. Both OxaPt(IV) and 56MESS showed higher potencies than TMZ in all cell lines tested, especially in TMZ-resistant cells (Table 1). After loading into NPs, the IC50 of both drugs decreased in most cell lines, except in the LN229-TS cell line, where the IC50 values were similar for both free drugs and their NP-loaded forms. (Fig. 4f–i and Table 1).
Figure 4 |. NP-OxaPt(IV) and NP-56MESS inhibit the growth of GBM cells.
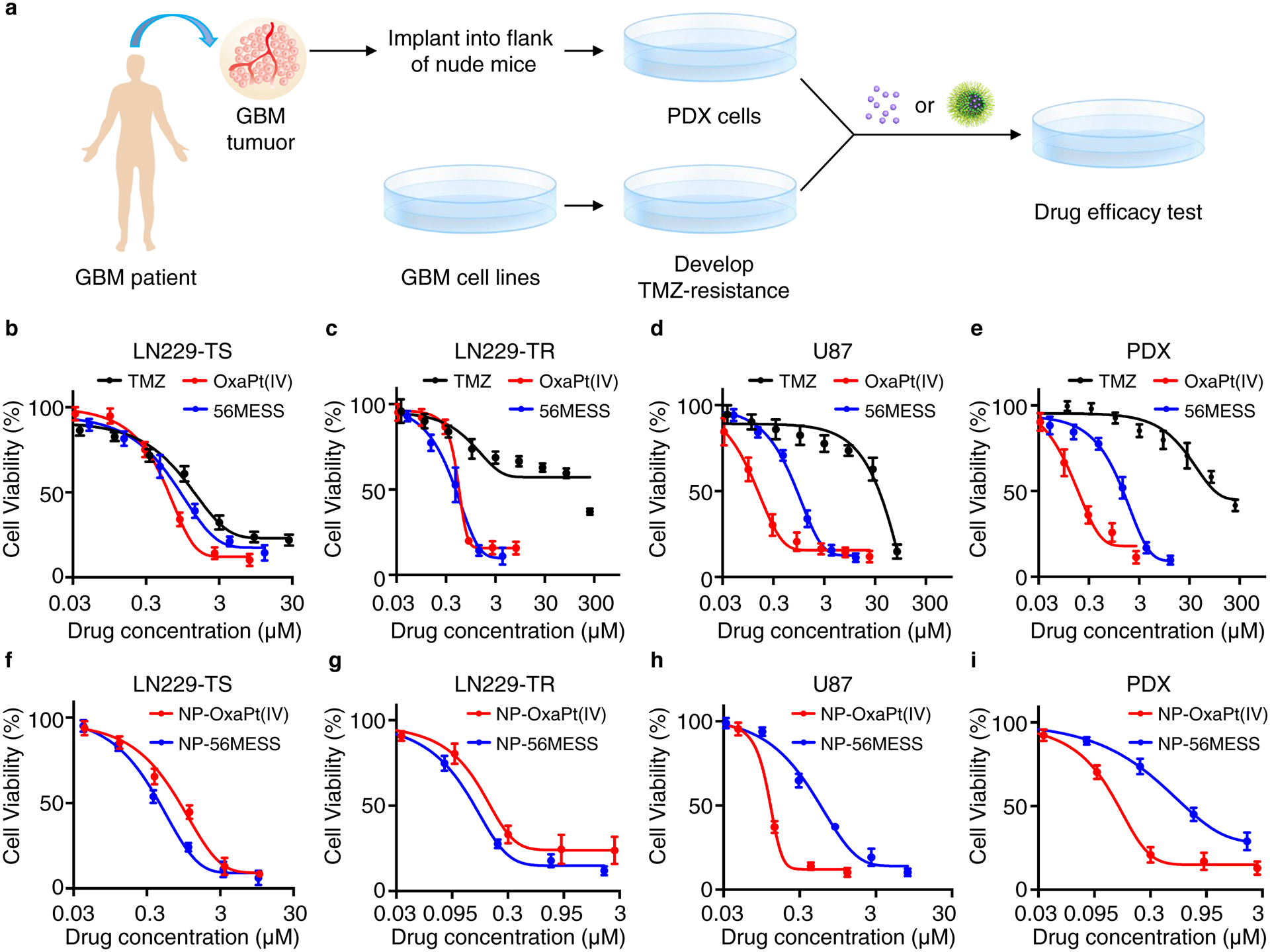
a, Schematic of experimental procedure. b-e, Viability of GBM cells LN229-TS (b), LN229-TR (c), PDX (d), U87 (e) following treatment with TMZ or OxaPt(IV) drugs. f-i, Viability of GBM cells LN229-TS (f), LN229-TR (g), PDX (h), U87 (i) following treatment with NP-OxaPt(IV) or NP-56MESS. The assay was repeated 4 times (n=4), data are mean ± s. d. IC50 values were determined using the ‘[inhibitor] vs. response – variable slope (four parameters)’ regression equation in GraphPad Prism. The P values for relevant comparisons are included in Supplementary Dataset 2.
Table 1.
IC50 (in μM) values of TMZ, OxaPt(IV) and 56MESS in different cell lines.
| Drug/Cell line | LN229-TS | LN229-TR | U87 | PDX |
|---|---|---|---|---|
| TMZ | 2.0 | 162.6 | 39.9 | 189.6 |
| OxaPt(IV) | 0.7 (2.9) | 0.6 (271) | 0.2 (199.5) | 0.3 (632) |
| 56MESS | 1.1 (1.8) | 0.5 (325.2) | 1.1 (36.3) | 1.7 (111.5) |
| NP-OxaPt(IV) | 1.0 (2.0) | 0.2 (813) | 0.1 (399) | 0.2 (948) |
| NP-56MESS | 0.6 (3.3) | 0.2 (813) | 0.7 (57) | 0.7 (270.9) |
The numbers within parentheses represent the radios of the IC50 values of TMZ to those of each drug.
Antitumour efficacy of nanoparticles in an animal model
In previous studies, platinum-based drugs have typically been administered intraperitoneally in doses ranging from 5mg kg−1 to 60mg kg−1 to achieve a therapeutic effect47–49. To compare the safety profiles of CED versus IP injection of platinum-based drugs, we performed whole blood cell counts (Fig. 5a–c) and examined tissue histology after drug administration (Supplementary Figs. 8 and 9). We found that at therapeutic doses, CED was safer than intraperitoneal injection: intraperitoneal injection of drug-loaded NPs reduced the number of white blood cells, platelets and red blood cells; By contrast, the white blood cell, platelets, and red blood cell counts of the CED-treated groups were within the normal ranges (Fig. 5a–c). Furthermore, no toxicity was detected in the organs from mice treated with CED, whereas intraperitoneal injection of the NPs caused splenic abnormalities (Supplementary Figs. 8 and 9) such as cells with brown-black pigment in the spleen. This pigment could be the result of macrophage engulfment of effete or damaged red blood cells during drug-induced hemolytic anemia.50–54
Figure 5 |. Antitumour efficacies of NP-OxaPt(IV) and NP-56MESS in mice bearing LN229-TR-LUC tumours.
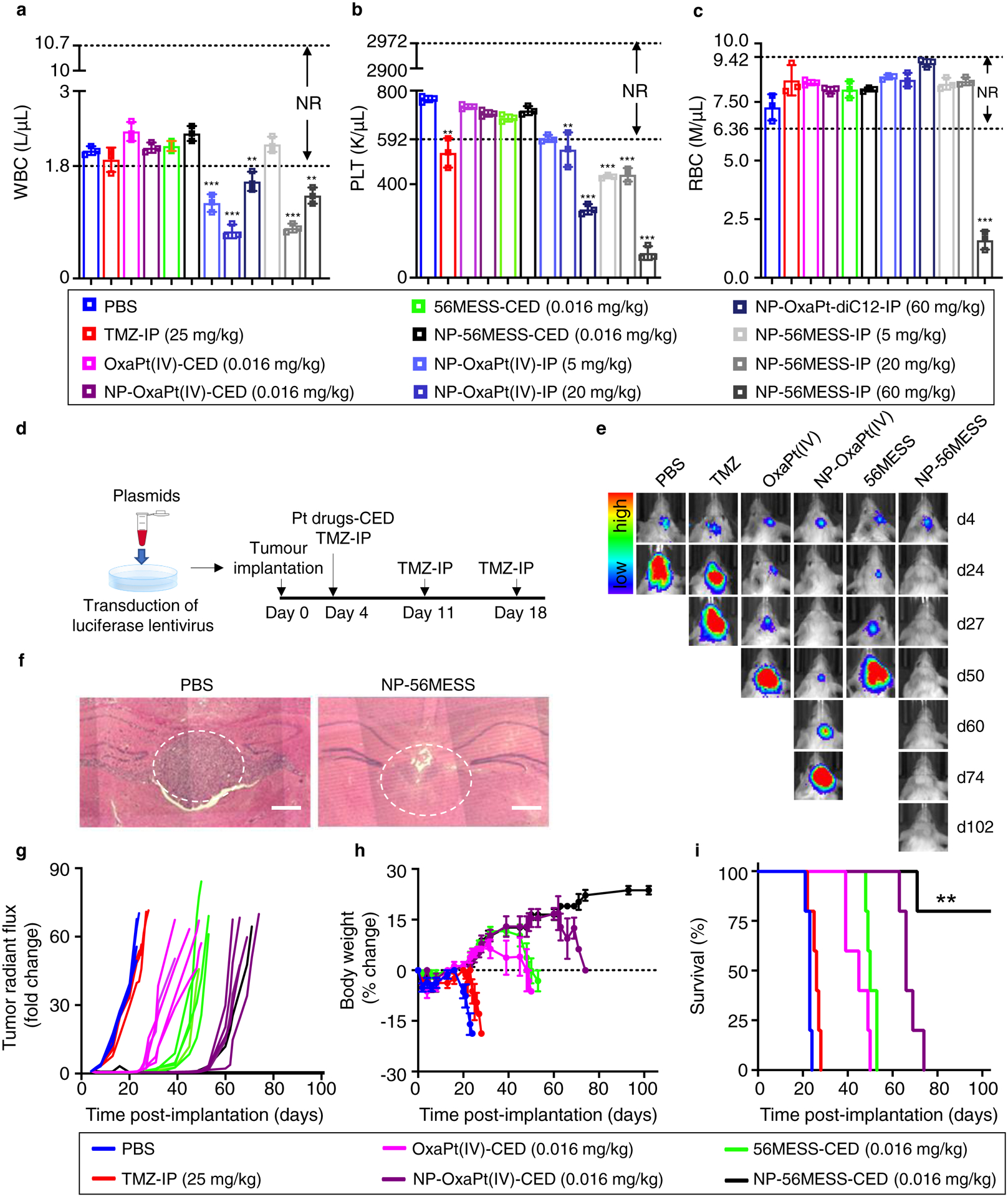
a-c, Intraperitoneal injection (IP) of NPs led to reduction in white blood cells (a), platelets (b), and red blood cells (c), whereas the numbers of these cells were in the normal range (NR) in CED groups. K μl−1, thousand per microliter. M μl−1, million per microliter. n=3 biological replicates, data are mean ± s. d. Differences among the PBS group and the treatment groups with out-of-range values are assessed by unpaired t-tests. **P < 0.01, ***P < 0.001. d, Schematic of treatment schedule. e, Bioluminescent IVIS images of representative mice. Five mice are used for each treatment group (n=5 biological replicates). f, Haematoxylin and eosin images of brain tissue from the PBS group (left) and from a long-term survivor of the NP-56MESS group (right). The white dotted circles show tumour sites. Scale bars, 250 μm. g, Changes in bioluminescence signal from the baseline (day 4). The concentrations of NP groups represent the concentrations of OxaPt(IV) or 56MESS; that is, encapsulation efficiencies are taken into account. h, Changes in the body weight compared with baseline (body weight at day 0). Data are mean ± s. d. i, Survival of mice bearing LN229-TR-LUC cells. The NP-56MESS-CED group displays a statistically significant improvement in survival compared with other groups (**P < 0.01, log-rank test).
To test the antitumour efficacy of the NPs in vivo, we first established an animal model by transducing LN229-TR cells with lentivirus to express luciferase (LN229-TR-LUC, Supplementary Fig. 10a) and implanting these cells into mice. Two weeks after the implantation, we investigated luciferin kinetics using an in vivo imaging system (IVIS) and found that the bioluminescent signal from the tumours peaked approximately 17 min after injection of luciferin (Supplementary Fig. 10b and c). We next tested the antitumour efficacy of our NP formulations in mice bearing LN229-TR-LUC tumours. Here, we observed that the TMZ-treated mice died around day 27, which was not significantly different to the survival of mice in the PBS group (around 23 d). Conversely, NP-OxaPt(IV) substantially inhibited tumour growth and tripled the survival time of mice bearing LN229-TR-LUC tumours. Most notably, 80% of NP-56MESS-treated mice were long-term survivors (surviving for more than 102 days, Fig. 5d–i). In addition, we characterized the in vivo biodistribution of NP-56MESS-FITC and observed that NP-56MESS-FITC covered the area labelled by U87 cells expressing red fluorescent protein (Supplementary Figs. 11a–d), suggesting that NPs penetrated the tumours.
Mechanisms of action examined by RNA-seq analysis
To understand the antitumour mechanisms of the drugs, we performed RNA-seq analysis and observed that the transcription of a number of genes and the corresponding signaling pathways were considerably influenced. The heat maps of TMZ and 56MESS displayed distinct patterns (Fig. 6a and Supplementary Dataset 1), indicating that their mechanisms of action were different. This is also evidenced by the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis (Fig. 6b). We found some signalling pathways to be altered in the 56MESS group (Supplementary Fig.13) but not in the TMZ group (Supplementary Fig.12), including the Faconi anemia pathway, metabolic pathways (805 out of 1,923 genes) and mTOR signalling pathways. Faconi anemia proteins have critical roles in the repair of DNA interstrand crosslinks, such as those induced by platinum-based compounds, and thus this finding is consistent with the differential DNA lesions induced by TMZ versus 56MESS55. To investigate the metabolic alterations induced by 56MESS, we conducted a metabolomic analysis and noted that histidine metabolism was markedly influenced by 56MESS treatment. No significant difference was detected comparing the 56MESS group and the NP56MESS group (Fig. 7).
Figure 6 |. Transcriptional analysis of LN229 cells treated with TMZ, 56MESS and NP-56MESS.
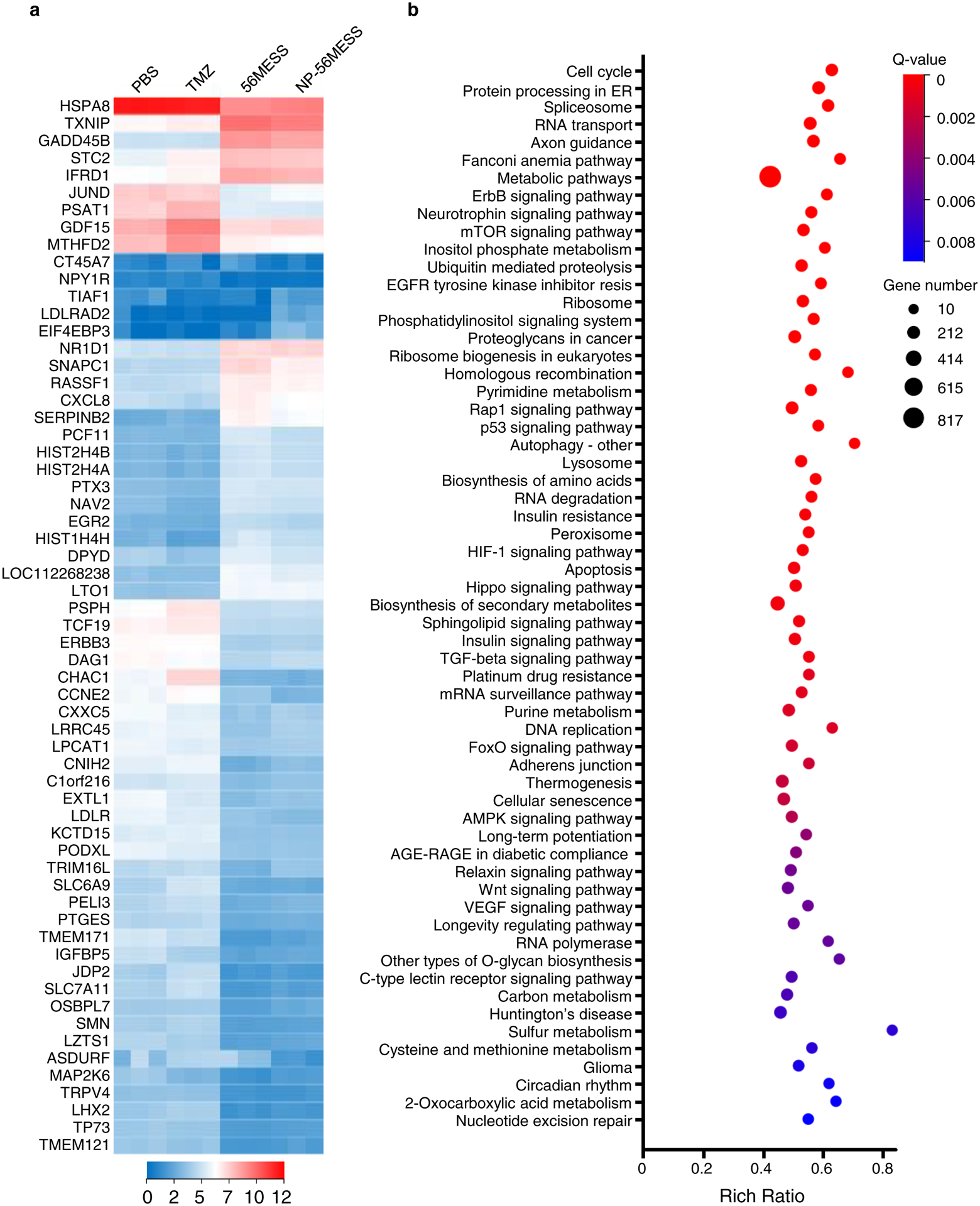
a, Heat map depicting transcriptional alterations. b, KEGG pathway enrichment analysis of differentially expressed genes between the TMZ group and the 56MESS group. Sixty pathways are arranged from top to bottom according to q values. ‘Cell cycle’ has the lowest q value. ‘Nucleotide excision repair’ has the highest q value in this chart. n=3 biological replicates. ‘Resis’ stands for resistance.
Figure 7 |. Analysis of the metabolic pathways in LN229-TS cells treated with TMZ, 56MESS and NP-56MESS.
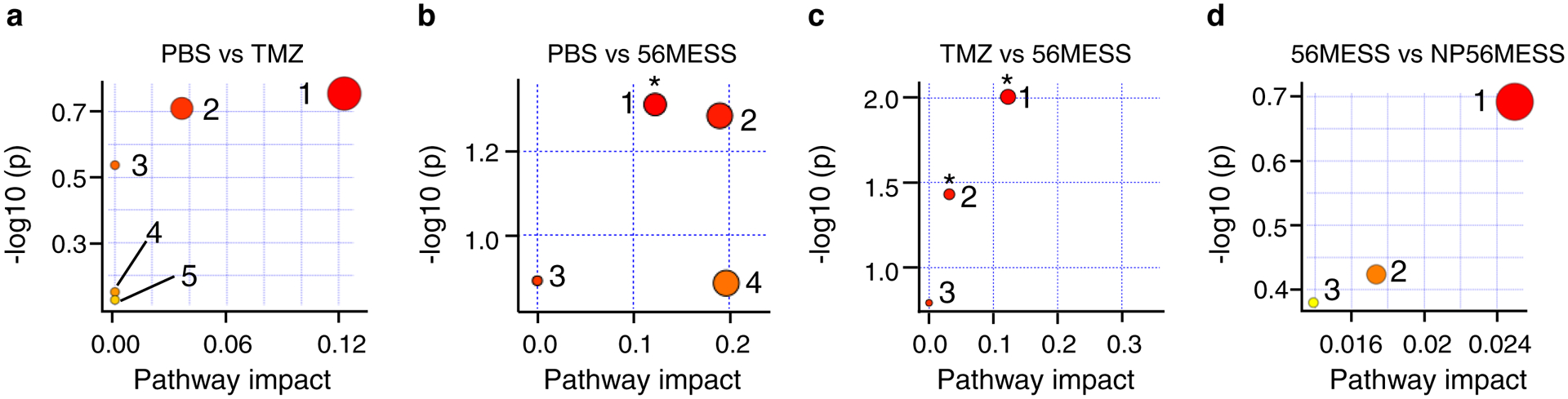
a, Comparison between the PBS group and the TMZ group (PBS versus TMZ). The top five enriched pathways are (1) histidine metabolism (P = 0.18), (2) purine metabolism, (3) thiamine metabolism, (4) lysine degradation and (5) glutathione metabolism. b, PBS versus 56MESS. Top pathways: (1) histidine metabolism (*P = 0.049), (2) arginine and proline metabolism (P = 0.05), (3) nitrogen metabolism and (4) alanine, aspartate, glutamate metabolism. c, TMZ versus 56MESS. Top pathways: (1) histidine metabolism (*P = 0.01), (2) purine metabolism (*P = 0.04) and (3) nitrogen metabolism (P = 0.16). d, 56MESS versus NP-56MESS. Top pathways: (1) purine metabolism (P = 0.2), (2) glycerophospholipid and (3) tryptophan metabolism. The colour and size of each circle is based on P values from enrichment analysis and pathway impact values from topology analysis, respectively. n=3, biological replicates.
Bioreducible polymers have been used to deliver agents for effective treatment of head and neck carcinomas56, breast cancer57, liver cancer58, ovarian cancer59 and GBM60. For example, carboxymethyl dextran derivatives linked with lithocholic acid through disulfide bonds have been synthesized for in vivo delivery of doxorubicin56. The hydrophobicity of lithocholic acid enabled the conjugates to form NPs encapsulated with doxorubicin. In addition, hydrolytically cleavable ester bonds were incorporated into polymeric nanoparticles to enhance the release of cancer stem cell-regulating microRNAs.
In this study we addressed multiple challenges in the treatment of GBM. First, we synthesized an oxaliplatin prodrug OxaPt(IV) and a cationic platinum drug 56MESS that effectively inhibited the growth of TMZ-resistant GBM cells. Moreover, poly (CHTA-co-HD)-PEG incorporating disulfide bonds and pendent pairwise carboxyl groups was synthesized through a single-step reaction for effective encapsulation of 56MESS in non-neoplastic conditions, rapid cellular uptake and selective release 56MESS in reductive environment of cancer cells. In addition, CED, a delivery approach being widely tested clinical trials, was implemented to carry drugs into the region of interest, bypassing the blood-brain barrier and enhancing drug distribution. Genome-wide RNA profiling and metabolome analysis uncovered the transcriptional and metabolic changes resulted from 56MESS treatment, confirming that its mechanism of action was distinct from that of TMZ. Together, CED of disulfide NPs with a cationic DNA intercalator substantially prolonged the survival of mice bearing TMZ-resistant GBM tumours without detectable systemic toxicity. Future research will include validation of the therapeutic efficacies of NP-56MESS with PDX models61–65, identification of its molecular target66, potential improvement of the polymer with a targeting component for GBM67 and assessment of neurotoxicity by behavioral assays68. We envision that the integrated approach presented in this proof-of-concept study could lead to promising avenues for the treatment of refractory GBM.
Methods
Materials
Dimethylformamide, dimethyl sulfoxide, oxaliplatin, hydrogen peroxide, dodecyl isocyanate, potassium tetrachloroplatinate(II), 1S,2S- diaminocyclohexane and 2-hydroxyethyl disulfide were purchased from Sigma-Aldrich. GSH (catalogue (cat.) no. 78259) and DiI (cat. no. D282) were purchased from Thermo Fisher Scientific. aCSF (cat. no. 59–7316) was procured from Harvard Apparatus. Isoflurane (SKU 029405), ketamine (SKU 056344), xylazine (SKU 061035), meloxicam (SKU 049755) and buprenorphine (SKU 055175) were purchased from Covetrus.
Bone wax (W31G) was obtained from Ethicon. Reflex 9 mm wound clips were from CellPoint Scientific. Triple antibiotic ointment (cat. no. 9004788) was obtained from Henry Schein. Polyimide microbore tubing (TPI-34×12) was bought from Professional Plastics. Epoxy 907 adhesive system was acquired from Miller-Stephenson. Betadine solution swabsticks were obtained from Betadine. Puralube vet ointment (17033-211-38) was procured from Dechra Veterinary Products. Luciferin (122799) was purchased from PerkinElmer.
Instruments
Proton nuclear magnetic resonance (1H NMR) spectra was completed using a 300 MHz NMR. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) was conducted with an Autoflex III (Bruker). The hydrodynamic diameter of each NP formulation was measured by dynamic light scattering (Malvern Panalytical). Flow cytometry experiments were performed using the Attune NxT. An Olympus confocal microscope was used for fluorescence imaging. An IVIS system (Perkin Elmer) was used to monitor tumour growth in vivo. A stereotaxic frame with UMP3 system (TAXIC-600), a mouse adaptor (cat. no. 502063) and a micro drill (503598) were obtained from World Precision Instruments. A reflex skin closure system (72–6060 to 72–6064) were purchased from Harvard Apparatus. A vacuum centrifuge concentrator (SPD120) was from procured from Thermo Fisher Scientific. The MALDI-TOF-MS instrument (Autoflex III) was acquired from Bruker.
Synthesis of OxaPt(IV)
To prepare OxaPt(IV)-OH, 0.5 g of oxaliplatin was suspended in 20 ml of H2O2 (30% w/v). The resulting solution was stirred at 50 °C until it was clear. After the solution was cooled to room temperature, needle-like crystals precipitated. The crystals were washed with acetone and dried in a desiccator. Afterwards, oxaliplatin(IV)-OH was isolated. To prepare OxaPt(IV), 400 mg of OxaPt(IV)-OH was suspended in 5ml of anhydrous dimethylformamide (DMF), followed by adding 0.744 ml of dodecyl isocyanate to the mixture. The solution was stirred at 110 °C until it was clear. The solution was then added into ice water to precipitate the reaction product. The product was washed with acetone, diethyl ether and dried under the vacuum to obtain OxaPt(IV) (45%). 1H NMR (300 MHz, DMSO) δ 9.70 (s, 2H), 8.43 (s, 2H), 6.74 (s, 2H), 2.89 (tt, J = 13.2, 6.7 Hz, 4H), 2.56 (d, J = 1.4 Hz, 2H), 2.17 (d, J = 11.8 Hz, 2H), 1.52 (d, J = 4.8 Hz, 2H), 1.27 (d, J = 20.2 Hz, 44H), 0.85 (t, J = 6.2 Hz, 6H). HRMS for C34H66N4NaO8Pt, Calculated:876.4423, observed: 876.4421.
Synthesis of 56MESS
The synthesis and characterization of 56MESS were performed as described69.
Synthesis of poly (CHTA-co-HD)-PEG
Five millilitres of DMF was used to dissolve 89 mg of 2-hydroxyethyl disulfide and 96 mg of 1,2,4,5-cyclohexanetetracarboxylic dianhydride. The reaction proceeded for 24 hours under nitrogen protection. Afterwards, 200 mg of PEG2K-OH (0.02 mmol) was added to the system. The reaction proceeded at 50 °C overnight. Five to ten millilitres of diH2O was added to the system, after which the product was dialysed for 48 hours and lyophilized.
Preparation and characterization of nanoparticles
Poly (CHTA-co-HD)-PEG (100 mg) was dissolved in 1ml of DMF. One millilitres of DMSO was used to dissolve DiI (10 mg ml−1), 56MESS (1.5 mg ml−1) and OxaPt(IV) (15 mg ml−1). Dil (100 μl), polymer (400 μl), DMF (400 μl) and DMSO(100 μl) were mixed together in a glass vial. OxaPt(IV) (66.7 μl), polymer (400 μl), DMF (400 μl) and DMSO(133.3 μl) were added to a glass vial. 56MESS (600 μl) and polymer (400 μl) were mixed. The mixture was added dropwise to 3 ml of deionized water in a glass vial with stirring at 1000 r.p.m. at room temperature, followed by stirring for 2 h in a fume hood to remove organic solvent. Afterwards, the mixture and 3 ml of deionized water were transferred to a filter (Amicon, cat. no. UFC910024) and centrifuged at 2,500g for 30 min. The NPs were resuspended in 5 ml of deionized water and centrifuged at 2,500g for 30 min. This step was repeated twice to remove organic solvent. Finally, the NPs were resuspended in 1 ml of deionized water or aCSF depending on the experiment. The hydrodynamic diameter, polydispersity index and surface charge of the nanoparticles were measured by dynamic light scattering (Malvern Panalytical). NPs were incubated at 37 °C. Ten microlitres of solution was collected at various time points to assess stability of NPs.
Release of 56MESS and OxaPt(IV) from NPs
A filter (Thermo Fisher Scientific, Slide-A-Lyzer mini dialysis device, 0.5 ml, 10 K MWCO) was placed in a well of a 24-well plate. One and a half millilitres of 0.5 mM GSH, 5 mM GSH, 20 mM GSH solution or aCSF was added into each well, followed by addition of 200 μl of NP-56MESS or NP-OxaPt(IV) to each filter. The 24-well plate was incubated at 37 °C. The release of 56MESS and OxaPt(IV) was evaluated using the same protocol. Using 56MESS as an example, 10 μl of aCSF solution was collected from the wells at various time points (0h, 1h, 2h, 3h, 6h, 9h, 18h, 30h, 48h, 72h) to measure 56MESS concentration (Ct) by inductively coupled plasma mass spectrometry (ICP-MS, Perkin Elmer ICP-MS Elan DRC-e). The volume (Vt) of the aCSF in each well was also measured at each time point. The amount of 56MESS (Wt) equals (Ct) × (Vt). The percentage of drug release at time point t equals Wt/W0 × 100%. W0 represents the weight of 56MESS in the original 200 μl of NP solution.
Cell culture
LN229 and PDX (G22) cells were acquired from R. Bindra at Yale University. LN229-TR (MGMT+) cells, engineered by transfecting LN229 cells with MGMT in the pSV2MGMT vector and selecting with 1.5 mg ml−1 G418, were obtained from B. Kaina3, which was. U87 and F98 cells were purchased from ATCC. U87-RFP cells were from H. Xiao (Chinese Academy of Sciences). Cells were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin-streptomycin at 37 °C with 5% (v/v) CO2 in a humidified atmosphere.
Generation of LN229-TR-LUC cells
The luciferase vectors were generous gifts from J. Ding (Harvard Medical School). HEK293 cells were seeded in a 10 cm tissue culture dish. Transduction was performed when the cells reached 70% confluence. Solution A was made of 500 μl of Opti-MEM (Thermo Fisher Scientific, cat. no. 31985070), 4 μg pMSCV-Lenti-Luc, 2 μg MPMG, 2 μg plasmid encoding regulator of expression of virion protein (Rev), 2 μg plasmid expressing transactivator of transcription (TAT) and 2 μg plasmid expressing the spike glycoprotein of the vesicular stomatitis virus (VSV-G). Solution B comprised 500 μl of Opti-MEM and 36 μl of Lipofectamine 2000 (Thermo Fisher Scientific, cat. no. 11668027). Solution A and B were mixed, incubated at room temperature for 20 min and added to the tissue culture dish. The culture medium was removed and replaced with fresh culture medium 12 h later. Three days later, the culture medium was collected and centrifuged at 2,000g for 5 min. LN229-TR cells were seeded into a 6-well plate. When the cells reached 30% confluence, 1 ml of culture medium containing viruses was added to each well in combination with 1 ml of fresh culture medium without penicillin-streptomycin and 1% hexadimethrine bromide. The 6-well plate was centrifuged at 2,000g for 15 min. Three days later, the culture medium was replaced with medium containing 5 μg ml−1 puromycin. Three days later, 1 μg ml−1 puromycin was used to maintain the cell line. Luciferase activity was evaluated by an in vitro assay described below.
In vitro evaluation of LN229-TR-LUC cells
A Luciferase Assay System (Promega, E1500) was used for this assay. The cells in a 10 cm tissue culture dish were rinsed with 5 ml of PBS after removal of growth medium. The cells were collected after trypsin digestion and counted. Different amounts of cells (38,500, 44,000, 49,500, and 55,000) were transferred to 1.5 ml Eppendorf tubes and centrifuged at 3,000g for 5 min. The supernatant was removed followed by addition of 20 μl of cell lysis buffer. The solution was pipetted 10 times allowing the cells to be lysed and incubated on ice for 10 min. The tubes were centrifuged at 3,000g for 5 min. Afterwards, the supernatant was transferred to a 96-well plate followed by addition of 100 μl luciferase assay reagent and measurement of bioluminescence signal with a SpectraMax microplate reader.
Growth delay assay
One-thousand cells were seeded in each well of a 96-well plate followed by addition of drugs 24 h later. The cells were cultured at 37 °C for 6 d, then fixed with 4% paraformaldehyde and stained with 0.2 μg ml−1 DAPI on day 7. The number of cells was counted with a Cytation5 image reader. The data was analysed using CellProfiler and plotted with Prism 8.
Cellular uptake of nanoparticle (NP-Dil) examined by flow cytometry.
One-hundred-thousand cells were seeded in each well of a 6-well plate. Twelve hours later, 5 μl NP-Dil was added to one well for an 18 h incubation before evaluation by flow cytometry at the end point. Eighteen hours, 6 h and 0.5 h before the end point, 5 μl NP-Dil was added to other wells respectively. NP-Dil was added in this manner to make sure all the samples were collected and assessed around the same time. The cells were washed with 1ml of PBS three times and digested with 1 ml of 0.25% Trypsin-EDTA at 37 °C for 5 min, followed by addition of 1ml of culture medium, centrifugation at 1,500g for 5 min, resuspension with 500 μl of PBS, and analysis with the channel BL3 (excitation, 549 nm; emission, 565 nm) of a flow cytometer. Gating was performed using forward scatter channel and side scatter channel to identify cells of interest and singlets.
Confocal microscopy
Cover slips (Matsunami 15 mm diameter) were placed in a 24-well plate. Ten-thousand cells were seeded in each well of the plate. Cells were incubated with NPs for 12 h followed by fixation in 4% paraformaldehyde (PFA) and staining with DAPI and Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific, cat. no. A12379). Fluorescent images were taken using an Olympus confocal microscope.
Characterization of the subcellular localization of NP-DiI
Ten-thousand LN229-TS cells were seeded into one well of a 24-well plate, followed by addition of NP-DiI (2 μl) 12 h later. Cells were rinsed with PBS twice (500 μl each) followed by fixation with 4% PFA at 30 min and 6h after addition of NP-DiI. Afterwards, the cells were rinsed with PBS three times, incubated in blocking buffer (1XPBS, 5% BSA, 0.3% Triton X-100) at room temperature for 1 h followed by three washes with PBS and staining with an EEA1 antibody (CST, cat. no. 3288S, 1:200 in Antibody Dilution Buffer (1X PBS / 1% BSA / 0.3% Triton X-100) at 4 °C for 24 h. Afterwards, the cells were washed with PBS three times (5 min each), incubated with secondary antibody at room temperature for 1.5 h and washed with PBS three times (5 min each). Prolong Gold Antifade Reagent with DAPI (CST, cat. no. 8961, 5 μl) was utilized to mount the cells. Images of cells were collected using a confocal microscope (Olympus, x100 oil objective).
Animal survival experiment
All procedures were approved by the Yale University Institutional Animal Care and Use Committee and performed in accordance with the guidelines and policies of the Yale Animal Resource Center. Female mice from Charles River (Fox chase SCID beige, strain code 250, 4 weeks old) were used for the survival experiments. The procedures for CED were detailed in previous publications70, 71. In brief, mice were anesthetized with an intraperitoneal injection of a ketamine/xylazine mixture (100 mg kg−1 ketamine, 10 mg kg−1 xylazine), followed by a pre-emptive dose (15 min) of buprenorphine (0.06 mg kg−1) and meloxicam (0.3 mg kg−1). Mice were then restrained using a stereotaxic frame for an aseptic rodent survival surgery and craniotomy. A hole was drilled at 1 mm lateral from the bregma, 1 mm anterior and 2 mm deep from the outer border of the cranium. 1.25 × 105 LN229-TR-LUC cells were suspended in 3 μl of PBS and injected intracranially over 3 min on day 0. Three more doses of buprenorphine (0.06 mg kg−1, every 12 h) and one more dose of meloxicam (0.3 mg kg−1) were intraperitoneally administrated for post-operative care. Tumours grew for 4 d before being subjected to different therapeutic treatments. Specifically, for the mice in the TMZ group, TMZ was administered by intraperitoneal injection weekly. For the mice in other groups, a single dose of PBS or drugs (4 μl) was given through CED at 0.5 μl min−1. Tumour growth was monitored by IVIS imaging. IVIS images were collected 17 minutes after injection of luciferin (PerkinElmer, cat. no. 122799, 30 mg ml−1, 100 μl per mouse). The bioluminescent signal was recorded as photon s−1.
In vivo biodistribution of NPs
CED of NP-56MESS-FITC was performed four days after intracranial implantation of U87-RFP (125,000 cells per mouse). Mouse brains were collected, flash-frozen, and cryo-sectioned (50 μm per slide) 4 h after CED. The same slide was imaged sequentially for assessment of RFP and GFP by fluorescence microscopy and Pt by ICP-MS where brain tissue was ablated and gasified by the laser beam. Though samples could be slightly distorted due to dehydration, downstream analysis was not vastly affected. The volumes of NP-56MESS-FTIC and U87-RFP were quantified by MATLAB.
RNA-seq analysis
LN229-TS cells were treated with PBS, TMZ (2 μM), 56MESS (1.1 μM) or NP-56MESS (0.6 μM) for 36 h. Three distinct samples, 1 million cells per sample, from each treatment group were collected to purify RNA. The RNA quality was confirmed using a NanoDrop 2000/c Spectrophotometer. The sequencing data was submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Bioproject ID PRJNA668337), which will be released upon publication. BGISEQ-500 was employed for sequencing. RSEM was used to quantify the transcription levels of genes. To compare two treatment groups, the differentially expressed genes (DEGs) were identified when the fold change was greater than or equal to 2 and q value less than or equal to 0.001. Only representative genes were presented in the heat map due to limited space. The heat map was plotted by pheatmap. R phyper was used for KEGG enrichment analysis. Significant enrichments were identified when the q value was less than or equal to 0.05. Only top sixty pathways were displayed due to limited space.
Metabolic pathway analysis
LN229-TS cells were treated with PBS, TMZ (2 μM), 56MESS (1.1 μM) or NP56MESS (0.6 μM) for 36 h. Three samples, 5 million cells per sample, from each treatment group were collected. One-hundred microlitres of H2O was added to each cell pellet to resuspend the cells followed by addition of 180 μl of methanol and 120 μl of chloroform. Samples were vortexed for 1 min and incubated at room temperature for 5 min. Afterwards, 150 μl of H2O was added, followed by vortexing for 1 min and incubation at room temperature for 5 min and centrifugation at 10,000g for 10 min. Three-hundred-and-fifty microlitres of supernatant was collected and spun in a vacuum centrifuge concentrator at 225 g at room temperature for 5 h. Forty microlitres of 20 mM of ammonium acetate in H2O was added for resuspension. MALDI-TOF MS and MetabolAnalyst were used for metabolic pathway analysis72. Metabolites were identified when the fold change was greater than or equal to 2 and q value less than or equal to 0.05.
Statistics
All statistical analyses were completed using GraphPad Prism 8. Statistical tests and P values were detailed in figure legends. Error bars represent s. d.
Supplementary Material
Acknowledgements
This work was funded by grants from the US National Institutes of Health (NIH; CA149128 to W.M.S.), the National Natural Science Foundation of China (51873218 to H.X.), the National Science and Technology Major Project (2018ZX10734401 to H.X.), the Beijing Natural Science Foundation (2202071 to H.X.) and the Key Research and Development Program of Hunan Province (2019SK2251 to H.X.). A.S.P. was supported by fellowships from NIH National Research Service Awards (NRSAs; T32 GM86287 and F32 HL142144) and the Cystic Fibrosis Foundation (PIOTRO20F0). We thank J. Ding for providing luciferase vectors.
Footnotes
Competing interests
The authors declare no competing interests.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41551-021-00728-7.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary Information. The raw data generated for the RNA-seq analysis are available from the NCBI SRA database under the accession code PRJNA668337. The metabolomic dataset generated during the study is too large (2.3 GB) to be publicly shared, but the data are available for research purposes from the corresponding authors on reasonable request.
References
- 1.Stupp R et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10, 459–466, doi: 10.1016/S1470-2045(09)70025-7 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Dolecek TA, Propp JM, Stroup NE & Kruchko C CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro Oncol 14 Suppl 5, v1–49, doi: 10.1093/neuonc/nos218 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaina B, Fritz G, Mitra S & Coquerelle T Transfection and expression of human O6-methylguanine-DNA methyltransferase (MGMT) cDNA in Chinese hamster cells: the role of MGMT in protection against the genotoxic effects of alkylating agents. Carcinogenesis 12, 1857–1867, doi: 10.1093/carcin/12.10.1857 (1991). [DOI] [PubMed] [Google Scholar]
- 4.Kitange GJ et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol 11, 281–291, doi: 10.1215/15228517-2008-090 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor JW & Schiff D Treatment considerations for MGMT-unmethylated glioblastoma. Curr Neurol Neurosci Rep 15, 507, doi: 10.1007/s11910-014-0507-z (2015). [DOI] [PubMed] [Google Scholar]
- 6.Li QJ, Cai JQ & Liu CY Evolving Molecular Genetics of Glioblastoma. Chin Med J (Engl) 129, 464–471, doi: 10.4103/0366-6999.176065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat Commun 9, 2949, doi: 10.1038/s41467-018-05373-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lapointe S, Perry A & Butowski NA Primary brain tumours in adults. Lancet 392, 432–446, doi: 10.1016/S0140-6736(18)30990-5 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Xiao HH et al. Recent progress in polymer-based platinum drug delivery systems. Prog Polym Sci 87, 70–106, doi: 10.1016/j.progpolymsci.2018.07.004 (2018). [DOI] [Google Scholar]
- 10.Wang S, Higgins VJ, Aldrich-Wright JR & Wu MJ Identification of the molecular mechanisms underlying the cytotoxic action of a potent platinum metallointercalator. J Chem Biol 5, 51–61, doi: 10.1007/s12154-011-0070-x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham J, Mushin M & Kirkpatrick P Oxaliplatin. Nat Rev Drug Discov 3, 11–12, doi: 10.1038/nrd1287 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Pasetto LM, D’Andrea MR, Rossi E & Monfardini S Oxaliplatin-related neurotoxicity: how and why? Crit Rev Oncol Hematol 59, 159–168, doi: 10.1016/j.critrevonc.2006.01.001 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Golomb L, Volarevic S & Oren M p53 and ribosome biogenesis stress: the essentials. FEBS Lett 588, 2571–2579, doi: 10.1016/j.febslet.2014.04.014 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Bruno PM et al. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med 23, 461–471, doi: 10.1038/nm.4291 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pisani MJ, Wheate NJ, Keene FR, Aldrich-Wright JR & Collins JG Anionic PAMAM dendrimers as drug delivery vehicles for transition metal-based anticancer drugs. J Inorg Biochem 103, 373–380, doi: 10.1016/j.jinorgbio.2008.11.014 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Wheate NJ et al. Novel platinum(II)-based anticancer complexes and molecular hosts as their drug delivery vehicles. Dalton Trans, 5055–5064, doi: 10.1039/b704973k (2007). [DOI] [PubMed] [Google Scholar]
- 17.Di Francia R et al. Current strategies to minimize toxicity of oxaliplatin: selection of pharmacogenomic panel tests. Anticancer Drugs 24, 1069–1078, doi: 10.1097/CAD.0000000000000002 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Jiang Y et al. SOD1 nanozyme with reduced toxicity and MPS accumulation. J Control Release 231, 38–49, doi: 10.1016/j.jconrel.2016.02.038 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Jiang Y, Brynskikh AM, S. M. D & Kabanov AV, SOD1 nanozyme salvages ischemic brain by locally protecting cerebral vasculature. J Control Release 213, 36–44, doi: 10.1016/j.jconrel.2015.06.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Natarajan G et al. Nanoformulated copper/zinc superoxide dismutase exerts differential effects on glucose vs lipid homeostasis depending on the diet composition possibly via altered AMPK signaling. Transl Res 188, 10–26, doi: 10.1016/j.trsl.2017.08.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caster JM, Patel AN, Zhang T & Wang A Investigational nanomedicines in 2016: a review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip Rev Nanomed Nanobiotechnol 9, doi: 10.1002/wnan.1416 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Liechty WB, Kryscio DR, Slaughter BV & Peppas NA Polymers for Drug Delivery Systems. Annu Rev Chem Biomol 1, 149–173, doi: 10.1146/annurev-chembioeng-073009-100847 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng FH, Hennink WE & Zhong Z Reduction-sensitive polymers and bioconjugates for biomedical applications. Biomaterials 30, 2180–2198, doi: 10.1016/j.biomaterials.2009.01.026 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Guo X et al. Advances in redox-responsive drug delivery systems of tumor microenvironment. J Nanobiotechnology 16, 74, doi: 10.1186/s12951-018-0398-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y et al. A “top-down” approach to actuate poly(amine-co-ester) terpolymers for potent and safe mRNA delivery. Biomaterials 176, 122–130, doi: 10.1016/j.biomaterials.2018.05.043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kauffman AC et al. Tunability of Biodegradable Poly(amine- co-ester) Polymers for Customized Nucleic Acid Delivery and Other Biomedical Applications. Biomacromolecules 19, 3861–3873, doi: 10.1021/acs.biomac.8b00997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuppusamy P et al. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res 62, 307–312 (2002). [PubMed] [Google Scholar]
- 28.Zhu Z et al. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J Neurochem 144, 93–104, doi: 10.1111/jnc.14250 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Efremenko EN et al. A simple and highly effective catalytic nanozyme scavenger for organophosphorus neurotoxins. J Control Release 247, 175–181, doi: 10.1016/j.jconrel.2016.12.037 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Harris NM et al. Nano-particle delivery of brain derived neurotrophic factor after focal cerebral ischemia reduces tissue injury and enhances behavioral recovery. Pharmacol Biochem Behav 150–151, 48–56, doi: 10.1016/j.pbb.2016.09.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y et al. Nanoformulation of Brain-Derived Neurotrophic Factor with Target Receptor-Triggered-Release in the Central Nervous System. Adv Funct Mater 28, doi: 10.1002/adfm.201703982 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jahangiri A et al. Convection-enhanced delivery in glioblastoma: a review of preclinical and clinical studies. J Neurosurg 126, 191–200, doi: 10.3171/2016.1.JNS151591 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bobo RH et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A 91, 2076–2080, doi: 10.1073/pnas.91.6.2076 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fung LK et al. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res 58, 672–684 (1998). [PubMed] [Google Scholar]
- 35.Kataoka K, Harada A & Nagasaki Y Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev 47, 113–131, doi: 10.1016/s0169-409x(00)00124-1 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Goodwin AP, Mynar JL, Ma Y, Fleming GR & Frechet JM Synthetic micelle sensitive to IR light via a two-photon process. J Am Chem Soc 127, 9952–9953, doi: 10.1021/ja0523035 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Su H et al. The role of critical micellization concentration in efficacy and toxicity of supramolecular polymers. Proc Natl Acad Sci U S A 117, 4518–4526, doi: 10.1073/pnas.1913655117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang L, Nienhaus K & Nienhaus GU Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology 12, 5, doi: 10.1186/1477-3155-12-5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prabha S, Arya G, Chandra R, Ahmed B & Nimesh S Effect of size on biological properties of nanoparticles employed in gene delivery. Artif Cells Nanomed Biotechnol 44, 83–91, doi: 10.3109/21691401.2014.913054 (2016). [DOI] [PubMed] [Google Scholar]
- 40.He C, Hu Y, Yin L, Tang C & Yin C Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31, 3657–3666, doi: 10.1016/j.biomaterials.2010.01.065 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Mandl HK et al. Optimizing biodegradable nanoparticle size for tissue-specific delivery. J Control Release 314, 92–101, doi: 10.1016/j.jconrel.2019.09.020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu G, Fang YZ, Yang S, Lupton JR & Turner ND Glutathione metabolism and its implications for health. J Nutr 134, 489–492, doi: 10.1093/jn/134.3.489 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Griffith OW Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med 27, 922–935, doi: 10.1016/s0891-5849(99)00176-8 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Meister A Glutathione metabolism and its selective modification. J Biol Chem 263, 17205–17208 (1988). [PubMed] [Google Scholar]
- 45.Jones DP et al. Glutathione measurement in human plasma. Evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC. Clin Chim Acta 275, 175–184, doi: 10.1016/s0009-8981(98)00089-8 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Snipstad S et al. Contact-mediated intracellular delivery of hydrophobic drugs from polymeric nanoparticles. Cancer Nanotechnol 5, 8, doi: 10.1186/s12645-014-0008-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korst AE, Boven E, van der Sterre ML, Fichtinger-Schepman AM & van der Vijgh WJ Influence of single and multiple doses of amifostine on the efficacy and the pharmacokinetics of carboplatin in mice. Br J Cancer 75, 1439–1446, doi: 10.1038/bjc.1997.247 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paraskar A, Soni S, Roy B, Papa AL & Sengupta S Rationally designed oxaliplatin-nanoparticle for enhanced antitumor efficacy. Nanotechnology 23, 075103, doi: 10.1088/0957-4484/23/7/075103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haragsim L & Zima T Protective effects of verapamil on cis-platinum and carboplatinum nephrotoxicity in dehydrated and normohydrated rats. Biochem Int 28, 273–276 (1992). [PubMed] [Google Scholar]
- 50.Suttie AW Histopathology of the spleen. Toxicol Pathol 34, 466–503, doi: 10.1080/01926230600867750 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Gupta N, Lal P, Vindal A, Hadke NS & Khurana N Spontaneous rupture of malarial spleen presenting as hemoperitoneum: a case report. J Vector Borne Dis 47, 119–120 (2010). [PubMed] [Google Scholar]
- 52.National Toxicology, P. Toxicology and carcinogenesis studies of 3,3’,4,4’-tetrachloroazobenzene (TCAB) (CAS No. 14047-09-7) in Harlan Sprague-Dawley rats and B6C3F1 mice (gavage studies). Natl Toxicol Program Tech Rep Ser, 1–206 (2010). [PubMed] [Google Scholar]
- 53.National Toxicology, P. Toxicology and carcinogenesis studies of alpha,beta-thujone (CAS No. 76231-76-0) in F344/N rats and B6C3F1 mice (gavage studies). Natl Toxicol Program Tech Rep Ser, 1–260 (2011). [PubMed] [Google Scholar]
- 54.Ward JM, Rehg JE & Morse HC 3rd. Differentiation of rodent immune and hematopoietic system reactive lesions from neoplasias. Toxicol Pathol 40, 425–434, doi: 10.1177/0192623311431467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreassen PR & Ren K Fanconi anemia proteins, DNA interstrand crosslink repair pathways, and cancer therapy. Curr Cancer Drug Targets 9, 101–117, doi: 10.2174/156800909787314011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thambi T et al. Bioreducible carboxymethyl dextran nanoparticles for tumor-targeted drug delivery. Adv Healthc Mater 3, 1829–1838, doi: 10.1002/adhm.201300691 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Son S et al. Anti-Trop2 antibody-conjugated bioreducible nanoparticles for targeted triple negative breast cancer therapy. Int J Biol Macromol 110, 406–415, doi: 10.1016/j.ijbiomac.2017.10.113 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Xia W et al. Bioreducible polyethylenimine-delivered siRNA targeting human telomerase reverse transcriptase inhibits HepG2 cell growth in vitro and in vivo. J Control Release 157, 427–436, doi: 10.1016/j.jconrel.2011.10.011 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Florinas S, Kim J, Nam K, Janat-Amsbury MM & Kim SW Ultrasound-assisted siRNA delivery via arginine-grafted bioreducible polymer and microbubbles targeting VEGF for ovarian cancer treatment. J Control Release 183, 1–8, doi: 10.1016/j.jconrel.2014.03.025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Bertoni H et al. Bioreducible Polymeric Nanoparticles Containing Multiplexed Cancer Stem Cell Regulating miRNAs Inhibit Glioblastoma Growth and Prolong Survival. Nano Lett 18, 4086–4094, doi: 10.1021/acs.nanolett.8b00390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carlson BL, Pokorny JL, Schroeder MA & Sarkaria JN Establishment, maintenance and in vitro and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr Protoc Pharmacol Chapter 14, Unit 14 16, doi: 10.1002/0471141755.ph1416s52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaubel RA et al. Genomic and Phenotypic Characterization of a Broad Panel of Patient-Derived Xenografts Reflects the Diversity of Glioblastoma. Clin Cancer Res 26, 1094–1104, doi: 10.1158/1078-0432.CCR-19-0909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tew BY et al. Patient-derived xenografts of central nervous system metastasis reveal expansion of aggressive minor clones. Neuro Oncol 22, 70–83, doi: 10.1093/neuonc/noz137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Randall EC et al. Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res 80, 1258–1267, doi: 10.1158/0008-5472.CAN-19-0638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Randall EC et al. Integrated mapping of pharmacokinetics and pharmacodynamics in a patient-derived xenograft model of glioblastoma. Nat Commun 9, 4904, doi: 10.1038/s41467-018-07334-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schenone M, Dancik V, Wagner BK & Clemons PA Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol 9, 232–240, doi: 10.1038/nchembio.1199 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reichel D et al. Near Infrared Fluorescent Nanoplatform for Targeted Intraoperative Resection and Chemotherapeutic Treatment of Glioblastoma. ACS Nano 14, 8392–8408, doi: 10.1021/acsnano.0c02509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moser VC Functional assays for neurotoxicity testing. Toxicol Pathol 39, 36–45, doi: 10.1177/0192623310385255 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Wu T et al. A Nanobody-Conjugated DNA Nanoplatform for Targeted Platinum-Drug Delivery. Angew Chem Int Ed Engl 58, 14224–14228, doi: 10.1002/anie.201909345 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Song E et al. Surface chemistry governs cellular tropism of nanoparticles in the brain. Nat Commun 8, 15322, doi: 10.1038/ncomms15322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Serwer L, Hashizume R, Ozawa T & James CD Systemic and local drug delivery for treating diseases of the central nervous system in rodent models. J Vis Exp, doi: 10.3791/1992 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia J & Wishart DS Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat Protoc 6, 743–760, doi: 10.1038/nprot.2011.319 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.