

Abstract
Purpose:
The etiology of postmenopausal recurrent urinary tract infection (UTI) is not completely known, but the urinary microbiome is thought to be implicated. We compared the urinary microbiome in menopausal women with recurrent UTIs to age-matched controls, both in the absence of acute infection.
Materials and Methods:
This is a cross-sectional analysis of baseline data from 64 women enrolled in a longitudinal cohort study. All women were using topically applied vaginal estrogen. Women >55 years of age from the following groups were enrolled: 1) recurrent UTIs on daily antibiotic prophylaxis; 2) recurrent UTIs not on antibiotic prophylaxis; and 3) age-matched controls without recurrent UTIs. Catheterized urine samples were collected at least 4 weeks after last treatment for UTI and at least 6 weeks after initiation of vaginal estrogen. Samples were evaluated using expanded quantitative urine culture (EQUC) and 16S rRNA gene sequencing.
Results:
With EQUC, there were no significant differences in median numbers of microbial species isolated among groups (p=0.96), even when considering Lactobacilli (p=0.72). However, there were trends towards different Lactobacillus species between groups. With 16S rRNA sequencing, the majority of urinary samples contained Lactobacilli, with non-significant trends in relative abundance of Lactobacilli among groups. Using a Bayesian analysis, we identified significant differences in anaerobic taxa associated with phenotypic groups. Most of these differences centered on Bacteroidales and the family Prevotellaceae, though differences were also noted in Actinobacteria and certain genera of Clostridiales.
Conclusions:
Associations between anaerobes within the urinary microbiome and postmenopausal recurrent UTI warrants further investigation.
Keywords: Urobiome, female urinary microbiome, postmenopausal recurrent UTI, microbiota
INTRODUCTION
Recurrent urinary tract infections (UTIs) are common among post-menopausal women.1 UTIs are thought to arise either from repeated ascending bacteria originating outside of the urinary tract, or from reinfection by intracellular bacterial communities within the urothelium.2 Despite the source, the urinary microbiome (or “urobiome”) is a necessary intermediary. The urobiome contains microbiota considered to be commensals as well as those considered uropathogens.3–6 As such, investigators are keenly interested in the role that the urobiome plays in recurrent UTI. In general, we know that urinary and vaginal microbiota are highly associated,7, 8 that Lactobacilli are less abundant after menopause,9–11 and that vaginal estrogen will lead to increased Lactobacilli in both niches.11–12
Vaginal estrogen and daily prophylactic antibiotics are used in evidence-based treatment of postmenopausal women with recurrent UTIs.13 We hypothesized that women taking antibiotics would have fewer Lactobacilli and less microbial diversity compared to those not taking antibiotics, as well as age-matched controls without UTIs. Due to the potentially significant effects of estrogen on microbial communities, we selected a control group using vaginal estrogen for other therapeutic reasons. Our overall objective was to characterize how Lactobacilli and other constituents of the urobiome differ in postmenopausal women with and without recurrent UTIs.
MATERIALS AND METHODS
Clinical Procedures and Sample Acquisition:
This is a cross-sectional analysis of baseline data from a longitudinal cohort. After Duke University Institutional Review Board approval was granted, women from Urogynecology and Urology clinics were enrolled between 12/2017 and 3/2019 into one of three groups: 1) Recurrent UTI on antibiotic prophylaxis (rUTI+antibiotics); 2) Recurrent UTI not on antibiotic prophylaxis (rUTI); and 3) Age-matched controls without recurrent UTIs (controls). A minimum age of 55 was selected to ensure menopausal status, and because this threshold was associated with differences in the urobiome.14 See Table 1 for detailed inclusion and exclusion criteria. Participants were sampled at least 4 weeks after the most recent treatment for UTI, at least 6 weeks after initiating prophylactic antibiotics or vaginal estrogen, and after a 6-week wash-out period if taking UTI prevention supplements.
Table 1:
Detailed inclusion and exclusion criteria
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
|
Culture proven UTI requires urinary symptoms (e.g. dysuria, suprapubic pain, increased urinary frequency, hematuria) and a urine culture showing ≥ 104 colony forming units (CFU) of a dominant organism. Though in some instances, there can be multiple organisms with ≥ 104 colony forming units, mixed flora cultures are not considered positive.
Antibiotic was selected based on clinical indications; acceptable antibiotics included trimethoprim, nitrofurantoin, or cephalexin.
At the sampling visit, participants provided demographic and medical history, completed the Urinary Distress Inventory short form (UDI-6),15 and additional questions regarding diet, bowel incontinence, and sexual activity. A transurethral catheterized urine sample was obtained and processed as specified in Figure 1. If there was evidence of symptomatic UTI, the participant was treated and returned after 4 weeks. Those with symptomatic UTI at three sampling visits in a row were excluded.
Figure 1: Urine Sample Processing.
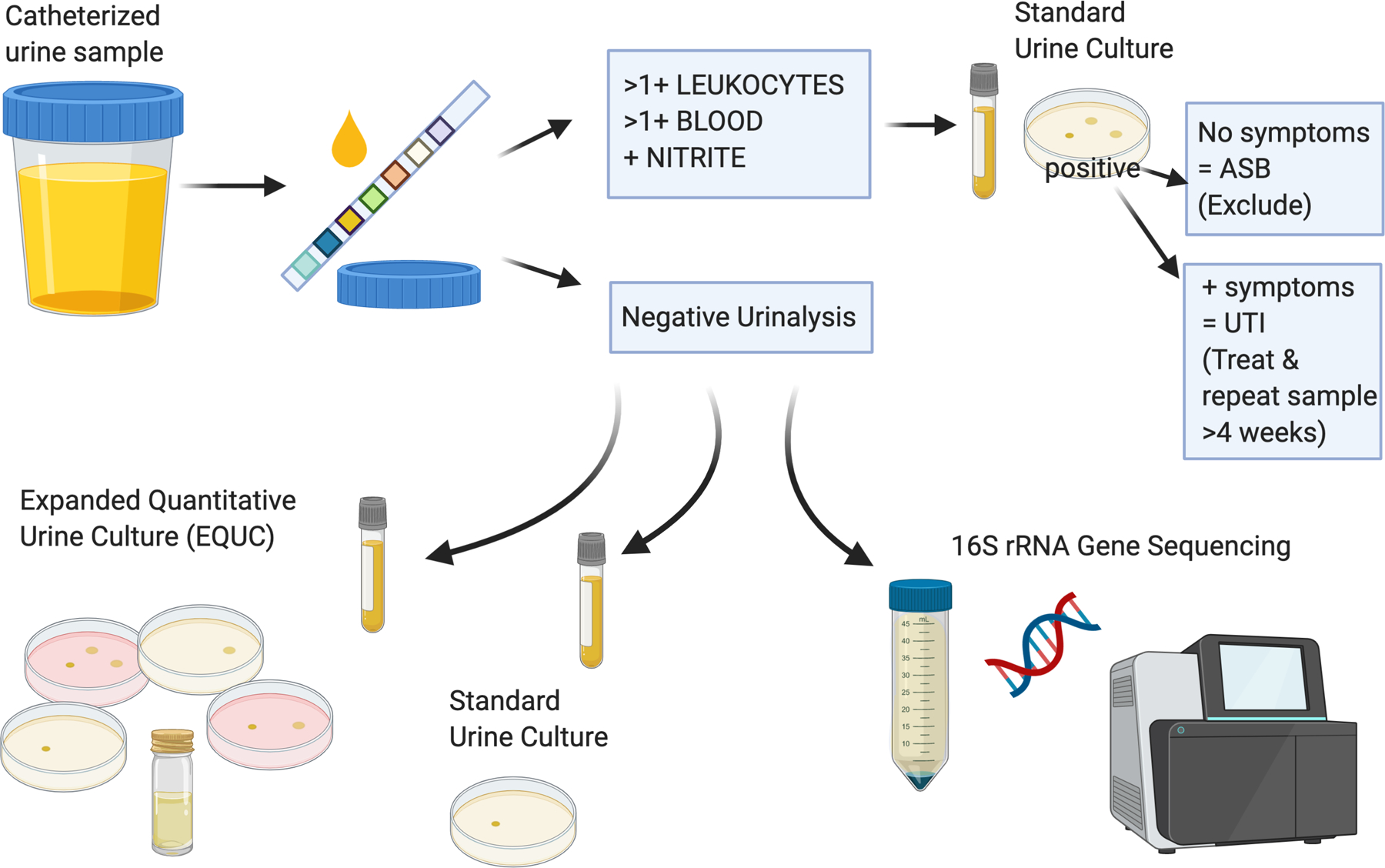
Catheterized urine was obtained using a standard research protocol. Urine was initially tested in the clinic with dipstick urinalysis (UA). If UA was suspicious for bacteruria, it was sent for standard urine culture only. If the participant had a negative (no growth) standard urine culture after suspicious UA, they were invited to provide a repeat sample. If the participant had a positive standard urine culture, the research documentation was reviewed for presence or absence of any symptoms. At this point, the participant was either: 1) excluded if there was asymptomatic bacteruria (ASB); or 2) treated for symptomatic urinary tract infection (UTI) and invited to provide a repeat sample in 4 weeks. For samples where office UA was negative, two 5mL samples were prepared and transferred to the Duke Clinical Microbiology Laboratory for standard and expanded quantitative urine culture. The remaining urine was poured into 50mL conical tubes prepared with Assay Assure, refrigerated, and transferred to the research laboratory for further processing and 16S rRNA gene sequencing. Created with BioRender.com.
Laboratory Procedures and DNA Isolation:
We used two complementary techniques to assess microbial communities. The first was expanded quantitative urine culture (EQUC),16 which allows for detection of live bacteria at the genus and species level. All urine cultures (standard and EQUC) were performed in the Duke Clinical Microbiology Laboratory with confirmation of bacterial species using mass spectrometry; EQUC was performed using the previously published protocol by Hilt et al.16
The second technique was 16S rRNA gene sequencing, a culture-independent method that allows for more thorough characterization of microbiota, but often with less resolution at the genus and species level. Supplemental Table 1 includes detailed sample processing methods. Urine samples intended for sequencing were placed into a nucleic acid protectant (Assay Assure™; Thermo Fisher Scientific, Waltham, MA), refrigerated, and processed as recommended for urinary microbiome studies.17
After all samples were collected, and multiple positive and negative controls were prepared, DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). The samples were submitted to the Duke Microbiome Shared Resource for library preparation and 16S rRNA gene sequencing via polymerase chain reaction (PCR) amplification of the V4 hypervariable region. Paired-end 250 base-pair sequencing was performed with Illumina MiSeq.
Bioinformatics & Statistical Analysis of Sequencing Data:
Raw sequences were processed into amplicon sequence variants (ASVs) with a DADA2 pipeline (v 1.14.0),18 then mapped to the SILVA reference database (v 132) for taxonomic identification using the RDP classifier. Decontam (v 1.2.1) was used to identify potential contaminant ASVs.19 Data were further processed and visualized in R using phyloseq (v. 1.26.1).20
Prior to statistical analyses, further filtering and normalization was performed (Supplemental Table 1). For the family Lactobacillaceae, we categorized samples with greater or less than 20% abundance, a threshold extrapolated from other published reports.10, 21
We used non-metric multidimensional scaling (NMDS) to cluster microbial communities. Unifrac distances were compared among groups using permutational multivariate analysis of variance (PERMANOVA) while incorporating covariates (Supplemental Table 1) including clinically relevant variables and those that were statistically significant in univariate analysis. Further pairwise comparisons were performed with PERMANOVA with the same covariates.
We further analyzed sequencing data using Bayesian graphical compositional regression (BGCR), a technique that more accurately models the distribution of microbiome data, incorporates phylogenetic relationships, and allows for the inclusion of other variables to adjust for potential confounders.22 BGCR returns the posterior joint alternative probability (PJAP) and inherently controls for multiple testing (see Supplemental Table 1). Larger PJAPs (up to 1) indicate stronger evidence for a cross-group difference. BGCR also returns the posterior marginal alternative probability (PMAP) at each split of the phylogenetic tree, that quantifies evidence for cross-group differences in the sub-composition at that split.
Other statistical analyses:
Demographic and baseline characteristics were compared using one-way analysis of variance (ANOVA), t-tests, chi-square, Fisher’s exact and Linear-by-Linear association tests, as appropriate. Median microbial species recovered from EQUC were compared using Kruskal-Wallis. Multivariable logistic regression was performed when assessing EQUC results. A separate model was created for variable selection (Supplemental Table 2). The final regression model included BMI, sexual activity status and history of diabetes mellitus as covariates, as these were the only covariates that had a p-value of <0.1 in either the full regression model or bivariate analysis. Data were analyzed using IBM SPSS Statistics version 24.0 (SPSS Inc., Armonk, NY) and R packages phyloseq (version 1.32.0) and BGCR (version 0.1.0).
RESULTS
Baseline characteristics:
We assessed the first 66 women enrolled in a longitudinal cohort. Two samples did not have sufficient DNA for sequencing and were excluded, leaving a study population of 64 women with mean age of 70.5 ± 7.5 years and mean BMI of 29.8 ± 6.3 kg/m2. Of these, 17 (27%) were in the recurrent UTI on antibiotic prophylaxis group (rUTI+antibiotics), 24 (38%) were in the recurrent UTI on vaginal estrogen only group (rUTI), while 23 (36%) were in the age-matched control group (controls). There were no significant differences in multiple baseline characteristics (Table 2) though there were trends towards higher BMI and fewer sexually active participants in the rUTI+antibiotics group.
Table 2:
Baseline characteristics of study population
| rUTI+abx N=17 |
rUTI N=24 |
Controls N=23 |
p-value | |
|---|---|---|---|---|
| Age (years) | 71.1±7.1 | 71.2±8.7 | 69.3±6.6 | 0.65* |
| BMI (kg/m2) | 32.5±8.7 | 29.8±5.7 | 27.8±3.8 | 0.06* |
| Caucasian | 17 (100%) | 23 (95.8%) | 21 (91.3%) | 0.28† |
| African American | 0 (0%) | 1 (4.2%) | 2 (8.7%) | 0.21† |
| Diabetes | 3 (17.6%) | 6 (25.0%) | 1 (4.3%) | 0.12† |
| Diet - daily yogurt | 4 (23.5%) | 9 (37.5%) | 6 (26.1%) | 0.56† |
| Symptoms of FI | 2 (11.8%) | 5 (20.8%) | 5 (21.7%) | 0.51† |
| Sexually active | 5 (29.4%) | 9 (37.5%) | 11 (47.8%) | 0.07† |
| Prior UI surgery | 4 (25%) | 9 (37.5%) | 9 (39.1%) | 0.45† |
| Prior back surgery | 4 (23.5%) | 3 (12.5%) | 2 (8.7%) | 0.24† |
| History of OAB | 4 (23.5%) | 11 (45.8%) | 6 (26.1%) | 0.74† |
| Total UDI-6 score | 16.7[11.5,30.2] | 20.8[12.5,29.2] | 12.5[8.3,20.8] | 0.84# |
Data are presented as mean±standard deviation, n (%), median[interquartile range]
Diabetes and BMI were not collinear in our study population (Pearson correlation coefficient 0.19)
rUTI, recurrent urinary tract infection; abx, antibiotics; NA, not applicable; BMI, body mass index; FI, fecal incontinence; UI, urinary incontinence; OAB, overactive bladder
A priori pairwise analyses were planned for any significant results that were identified in ANOVA or linear association test. Since none were significant, no pairwise analyses were performed.
ANOVA test
Linear-by-linear association test
Kruskal Wallis test
Culture results:
Thirty-eight women (59.4%) were culture-positive via EQUC in the setting of a negative standard urine culture (Table 3). There were no significant differences among groups in culture-positive or median numbers of microbial species detected by EQUC, with 2 [IQR 1,2.5] in rUTI+antibiotics, 1 [IQR 1,2] in rUTI, and 1 [IQR 1,3] in the control group (Kruskal-Wallis test p=0.91).
Table 3:
Comparison of organisms recovered using standard urine culture and expanded quantitative urine culture.
| Standard Urine Culture | Organisms Recovered on EQUC |
|---|---|
| Actinomyces turicensis | |
| Actinotignum schaalii | |
| Anaerobic gram positive rods | |
| Aspergillus spp* | |
| Bifidobacterium spp* | |
| Candida glabrata | |
| No growth | Corynebacterium coyleae |
| C. riegelii | |
| Diphtheroids spp* | |
| Aerococcus urinae | |
| Enterococcus faecalis | |
| Escherichia coli | |
| Gardnerella vaginalis | |
| Lactobacillus species | |
| L. casei/paracasei/rhamnosus | |
| L. crispatus | |
| L. delbrueckii | |
| L. fermentum | |
| L. gasseri/acidophilus | |
| L. hominis | |
| L. iners | |
| L. jensenii | |
| Leuconostoc spp* | |
| Pseudomonas aeruginosa | |
| Staphylococcus species | |
| S. epidermidis | |
| S. hominus | |
| S. lugdunensis | |
| Streptococcus species | |
| S. anginosus | |
| S. gallolyticus gallolyticus | |
| S. gallolyticus pasteurianus | |
| S. mitis | |
| S. oralis | |
| S. parasanguinis | |
| S. sanguinis | |
| S. spp* | |
| Lactobacillus spp. | Bifidobacterium spp |
| Gardnerella vaginalis | |
| Lactobacillus species | |
| L. casei/paracasei/rhamnosus | |
| L. gasseri/acidophilus | |
| L. jensenii | |
| Staphylococcus epidermidis | |
| Streptococcus anginosus |
EQUC, expanded quantitative urine culture; spp, species
Microbe could not be identified beyond genus level
In EQUC, the proportions of urine samples with Lactobacilli were not significantly different (29.4% rUTI+antibiotics, 41.7% rUTI, 39.1% controls, χ2 p=0.76). History of diabetes was associated with increased odds of growing Lactobacilli (OR 5.56, 95% CI 1.14–27.02). In multivariable logistic regression when controlling for this variable as well as BMI and sexual activity, there were still no differences in growth of Lactobacilli between groups. Of the three most prevalent Lactobacillus species, there were trends toward fewer L. gasseri and L. crispatus in the rUTI+antibiotics group, but these did not reach statistical significance (Figure 2a).
Figure 2: Comparison of the most common organisms recovered in EQUC.
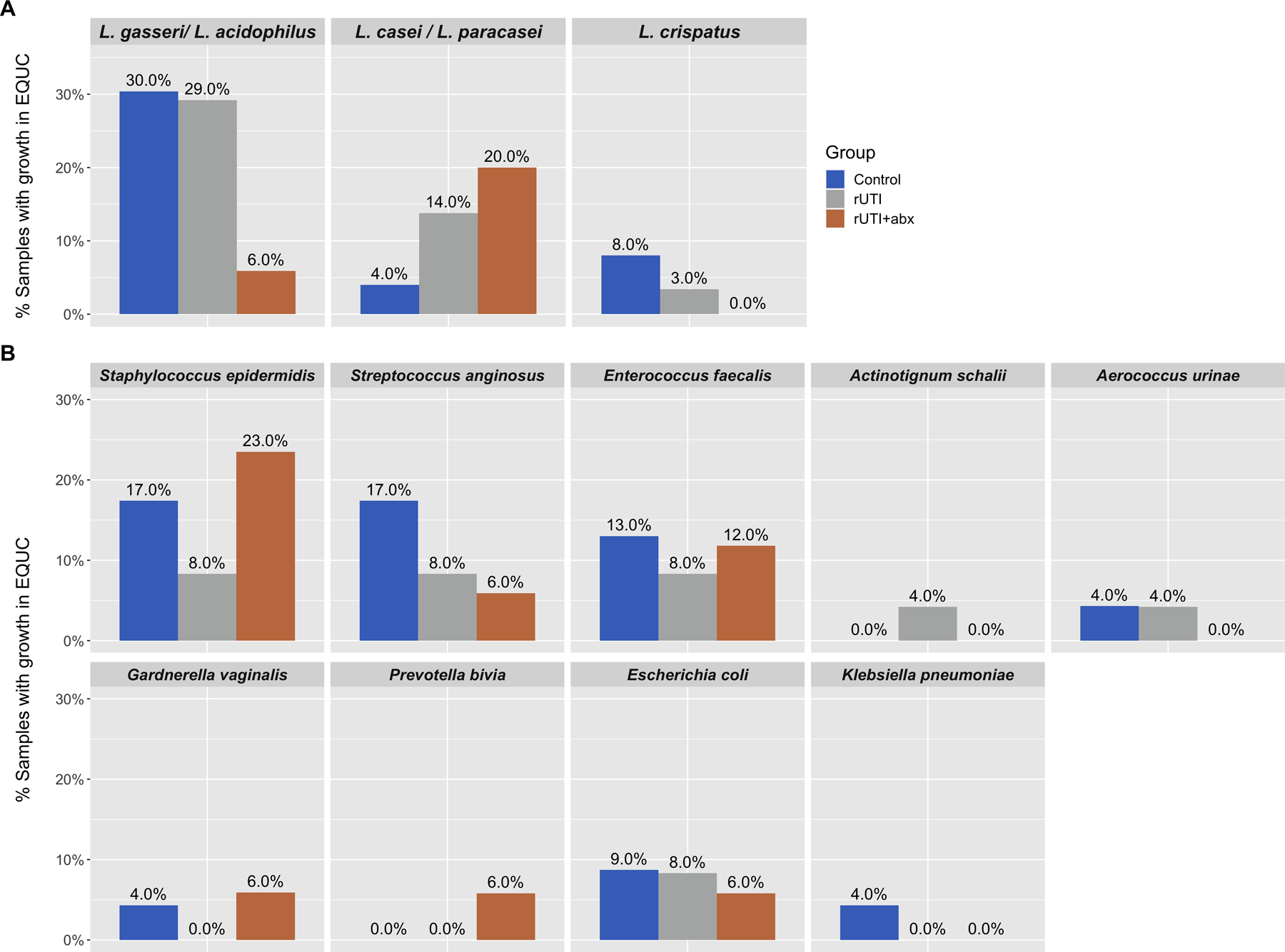
a) Lactobacilli: Of 64 total samples, 38 (59%) demonstrated growth of one or more organisms in expanded quantitative urine culture (EQUC). The majority of these organisms were Lactobacilli. The three most commonly isolated Lactobacillus species were L. gasseri/L. acidophilus (isolated in n=15 samples), L. casei/L. paracasei (n=6 samples), and L. crispatus (n=3 samples). Proportions of urine samples from each phenotypic group with the three most common Lactobacillus species in EQUC are depicted in the figure. For example, in women with recurrent UTI taking antibiotics, 6% contained L. gasseri/L. acidophilus in their EQUC samples compared to 29–30% in the other two phenotypic groups. These differences in proportions were not statistically significant (p=0.14, p=0.39, p=0.43, respectively). b) The most common non-Lactobacillus organisms from EQUC are identified. Proportions of urine samples from each phenotypic group where the microbe was are depicted; there were no statistically significant differences.
We also compared proportions of urine samples that grew known or potential uropathogens. There were no statistically significant differences among groups, though interesting trends were noted in S. epidermidis and S. anginosus (Figure 2b).
16S rRNA gene sequencing:
Taxa identified through 16S rRNA gene sequencing were highly variable (Figure 3). The family Lactobacillaceae were recovered in 97% of samples but were not taxonomically classified at higher resolutions. When Lactobacillaceae comprised <20% of the total microbiota, this was considered “low” relative abundance. Based on this threshold, there was moderate to high Lactobacillaceae in 6/17 (35%) of the rUTI+antibiotics group, 9/24 (37.5%) of the rUTI group, and 13/23 (56.5%) of the control group, with no statistically significant differences (χ2 p=0.3).
Figure 3: Stacked bar plot depicting relative abundance of all microbiota per sample.
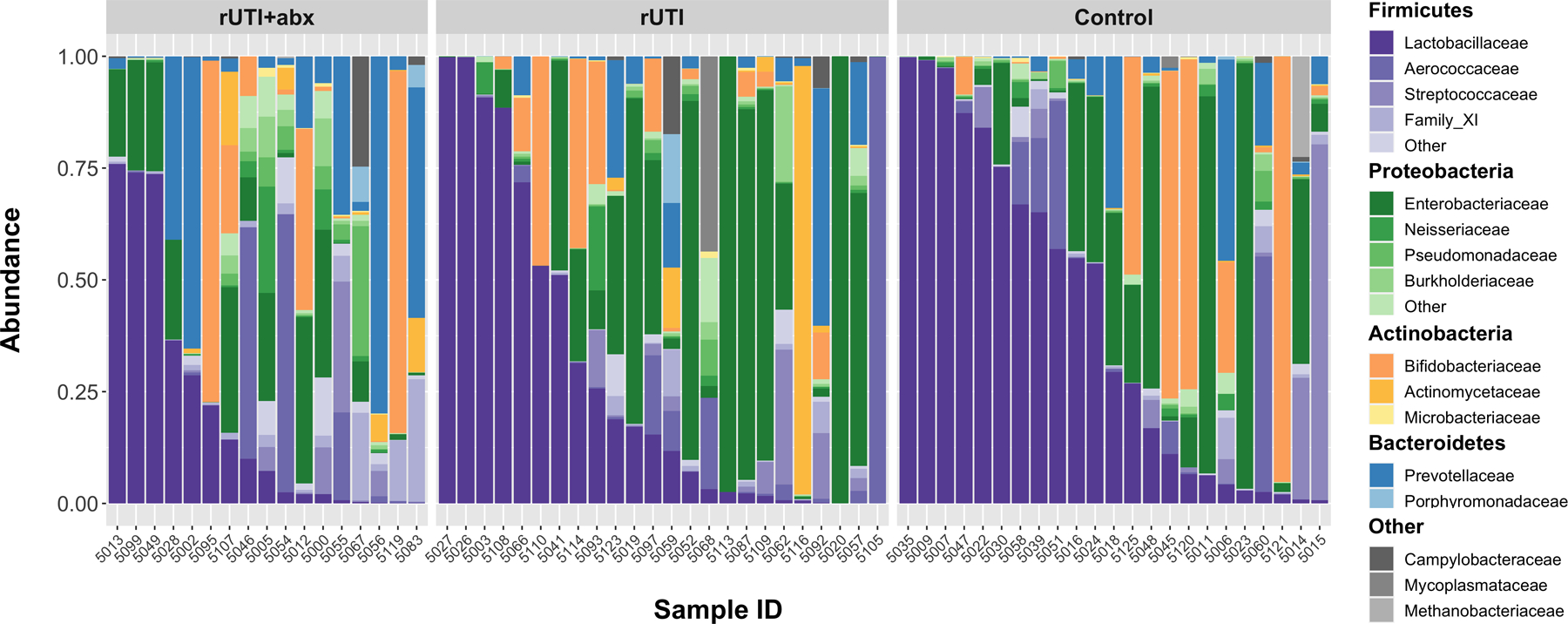
Stacked bar plot where each vertical bar depicts the relative abundance of adjusted sequence variants (ASVs) and associated taxa that were recovered per sample. For each color group, the darkest shade represents the most common family identified with a phylum, with lighter shades representing other families within the same phylum. For example, the darkest shade of purple represents Lactobacillaceae while lighter purple shades represent other Firmicutes. Results obtained from V4 amplicon sequencing of the 16S rRNA gene, processed with DADA2 and mapped to the SILVA reference database.
We further compared microbial communities using multiple techniques including NMDS (results in Supplemental Figure 1), PERMANOVA, and BGCR modeling. While controlling for several covariates, PERMANOVA showed significant differences in Unifrac distances, or in other words, differences in microbial communities, among the three groups (p=0.045; Supplemental Table 3). Of the covariates, sexual activity was significantly associated with microbial communities (p=0.04), while age and BMI approached statistical significance (p=0.05 and p=0.06, respectively, Supplemental Table 3). In PERMANOVA pairwise comparisons there were significant differences between women with rUTI + antibiotics compared to controls (p=0.038) where again, sexual activity was the only covariate also significantly associated with microbial communities (p=0.005). Other pairwise comparisons showed non-significant differences (rUTI+antibiotics vs. rUTI, p=0.086; rUTI vs. controls p=0.12).
PERMANOVA analyses can identify differences in overall microbial compositions but have limited ability to detect the actual taxa that are responsible for those differences, and also have limited power to separate signal from noise. Thus, we performed BGCR modeling, which is a technique that is limited to only pairwise comparisons, but allows us to drill down to specific taxa that differ between phenotypic groups while adjusting for covariates.
The BGCR analysis is generally consistent with the results of PERMANOVA. When comparing women with rUTI+antibiotics versus controls, similar to PERMANOVA, a high overall probability of difference in microbial composition was identified with BGCR (PJAP = 99.9%). There are five branches of the phylogenetic tree where the probability of a difference is quite high (Fig 4). Three of these branches are downstream branches of each other and identify high probabilities of differences in the genus Prevotella, family Prevotellaceae, and order Bacteroidales (PMAP = 99.9%, 82.5%, and 80.5% respectively) between groups. Additional distinguishing features include high probabilities of differences in the order Clostridiales, family Ruminococcaceae (PMAP = 84.8%), and in different Actinobacteria (PMAP = 98.3%), summarized in Supplemental Figure 2.
Figure 4: Recurrent UTI with daily antibiotics compared to controls.
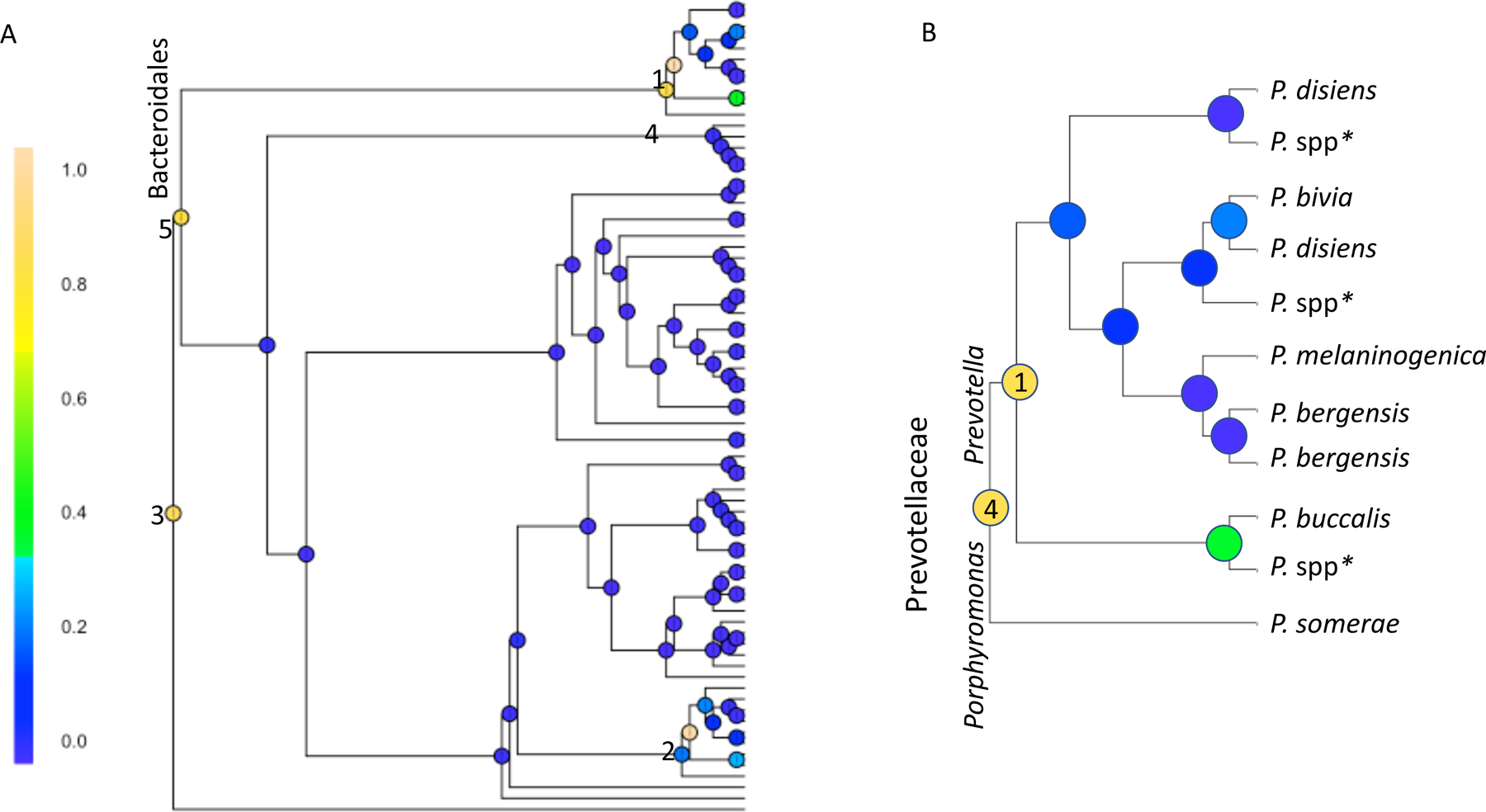
Bayesian GCR analysis comparing microbiota in women with recurrent UTIs taking daily antibiotics and age-matched controls (both using vaginal estrogen) while incorporating multiple clinical and technical covariates. The posterior marginal alternative probability (PMAP) was calculated at each node and shaded based on result. The highest probability of a difference between groups is at node #1 (PMAP = 99.96%), which denotes differences in Prevotella species recovered among those with rUTI+abx compared to controls. This node is a downstream branch in the taxonomic tree from two other nodes that also appear to have a high probability of differences between groups. These are identified as #4 (PMAP = 82.5%), which denotes differences in Prevotellaceae and #5 (PMAP = 80.5%), denoting differences in the order Bacteroidales. Node #2 (PMAP = 98.27%) denotes differences in Actinobacteria; this section of the tree is further exploded in Supplemental Figure 2. Node #3 (PMAP = 84.81%) denotes differences in Clostridiales, family Ruminococcaceae. Fig 4A shows the entire taxonomic tree and BCGR results; Figure 4B is an exploded section of this tree showing genus and species information.
Slightly different than in the PERMANOVA analyses, when comparing women with rUTI+antibiotics to those with rUTI not using antibiotics, the overall probability of differences in microbial composition is relatively high with BGCR (PJAP = 82.9%). Of the various splits in the phylogenetic tree, the highest probability of differences occurred within the order Clostridiales, family XI (PMAP = 66.7%, Fig 5).
Figure 5: Recurrent UTIs with compared to without daily antibiotics.
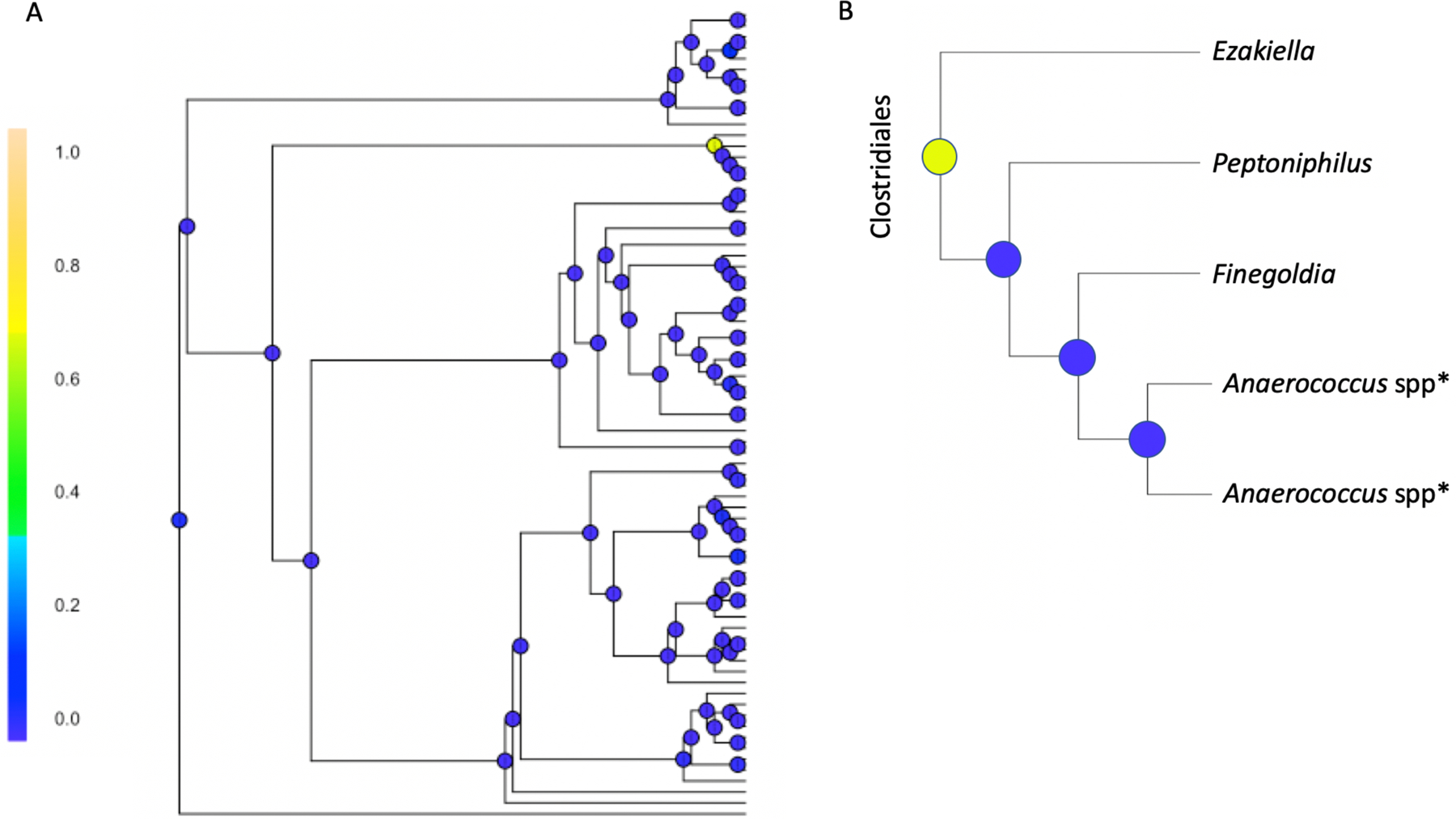
Bayesian GCR analysis comparing microbiota in women with recurrent UTIs taking daily antibiotics and women with recurrent UTIs not on antibiotics (both using vaginal estrogen)
(a) Posterior marginal alternative probability (PMAP) was calculated at each node with one area resulting in PMAP > 60%. This corresponds to the node shaded in yellow-green where PMAP = 66.73%, indicating the probability of a true difference in taxa between groups at the location specified. (b) Exploded view of the taxa where the difference was identified, specifically showing that there are differences between the genus Ezakiella, and the other genera identified in the figure.
When comparing women with rUTI who are not using antibiotics to matched controls, there was only a modest probability of an overall difference (PJAP = 63.8%) with no individual branch splits showing probabilities of differences (all PMAP < 55%). These results echo those found with PERMANOVA where there was a lack of significant differences between groups.
DISCUSSION
In this cross-sectional analysis in menopausal women using vaginal estrogen, we did not identify significant differences in abundance of Lactobacilli among those with recurrent UTIs and matched controls. In women with recurrent UTIs taking daily antibiotics, multiple analytic techniques (e.g., PERMANOVA and BGCR) identified differences in microbial compositions when compared to controls without recurrent UTIs. A BGCR analysis shows that these differences are mainly attributed to anaerobic bacteria including differences in Prevotellaceae, Clostridiales, as well as the class Actinobacteria. BGCR, which is more precise than traditional analyses such as PERMANOVA, also uniquely identified a high probability of differences in Clostridial genera between women with recurrent UTIs taking daily antibiotics compared to those not taking daily antibiotics.
There are a number of strengths inherent to our study. Due to purposeful sampling, we were able to standardize the timing of sample acquisition and collect information on potentially confounding variables. We standardized the use of vaginal estrogen, and also excluded medications that could potentially influence the urobiome. All women underwent catheterized sampling, which allows confidence that we are characterizing bladder microbiota, as described in prior studies.6 The complementary techniques of EQUC and 16S rRNA gene sequencing allow for information about viable urinary microbial species (EQUC) along with broad identification of microbiota and conclusions regarding bacterial phylogenetic relationships (16S rRNA). We also focused on clinically relevant populations, where data characterizing the urobiome are sparse.
The primary limitations of this study are inherent to the small sample size and cross-sectional design. Despite having a large clinical population of women with recurrent UTIs, we had strict entry criteria including verification of culture proven UTIs and exclusion of women taking UTI prevention supplements, which restricted the number of potential candidates. There are multiple findings, for example those pertaining to Lactobacilli abundance and differences in species, where we can see trends towards differences in groups that do not reach statistical significance, possibly due to small sample size. However, we still observed some significant differences among phenotypic groups and thus we believe this analysis still provides useful information. Many of our conclusions relate to the presence of anaerobic bacteria in the urine of women with recurrent UTIs taking daily prophylactic antibiotics. Due to the cross-sectional sampling, we are unable to determine if these differences are due to selective pressure from prolonged antibiotics, or if these anaerobes are implicated in the development of more severe recurrent UTIs that require prophylactic antibiotics. Notably, we did not standardize the antimicrobial agent used for prophylaxis. Though the majority of patients were taking trimethoprim, some were also taking nitrofurantoin or cephalexin. Finally, we chose to sample the urobiome in the absence of acute infection, and this may bias our results toward non-uropathogenic microbiota.
One microbe of interest in this study was Lactobacillus.23 In general, Lactobacilli are relatively resistant to the effects of antibiotics, but tend to decline in the absence of estrogen.24 As such, it is not surprising that microbes from the family Lactobacillaceae were found in 97% of our samples, since all women were using vaginal estrogen therapy12. Though not statistically significant, the species-level results pertaining to Lactobacilli could be biologically relevant as our data suggest that prophylactic antibiotics are associated with shifts in the Lactobacilli species that are typically found within the urobiome.25 Interestingly, we found that patients with a history of diabetes were at increased odds of having urinary Lactobacilli. This is consistent with associations seen previously in voided samples where urinary Lactobacilli were associated with increased fasting blood glucose and urinary glucose.26
Our data contribute to the growing body of literature regarding the urobiome. Using rigorous statistics we also found associations between microbial composition and several covariates including sexual activity, age, BMI, and history of diabetes. These covariates could serve as significant confounders of microbial results if not incorporated into analyses. Other groups have reported evidence for synergistic interactions amongst microbes that enhance bacterial colonization and/or persistence.2, 27 Our findings overall support the idea that it may not be one particular microbe within the urobiome that is associated with recurrent UTIs, but perhaps a shift in the balance of Lactobacilli, or particular species of Lactobacilli compared to anaerobic bacteria.
CONCLUSIONS
Within the urinary microbiome, abundance of Lactobacilli does not distinguish women with recurrent UTIs from matched controls. Associations between anaerobes within the urinary microbiome and postmenopausal recurrent UTI warrants further investigation.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the following research coordinators from the Duke Division of Urogynecology and Reconstructive Pelvic Surgery for their valuable contributions: Akira Hayes, Acacia Harris, Shantae McLean, Robin Gilliam, Claire McLaughlin, Yasmeen Bruton, and Nortorious Coleman-Taylor. The authors would also like to thank Carole Grenier, research analyst from the Murphy Laboratory at Duke University, for her expert contributions to sample processing and DNA extraction. Library preparation and DNA sequencing were performed in collaboration with the Duke Microbiome Center Core Facility. Dr. Kevin C. Hazen from the Duke Clinical Microbiology laboratory provided operational contributions and staff assistance with expanded quantitative urine culture. Finally, Erin Dahl from Oregon Health & Science University assisted with creating figures for this manuscript.
Funding sources:
This study was funded by the Duke Department of Obstetrics and Gynecology Charles B. Hammond Research Fund (MHV), the Duke Claude D. Pepper Center Older Americans Independence Center (P30 AG028716; KES & NYS), and the National Institutes of Health awards R03AG060082 (NIA) to NYS; K01DK116706 (NIDDK) to LAK; and R01GM135440 (NIGMS) to LM.
Financial Disclosure:
Dr. Siddiqui holds a research grant from Medtronic, Inc® and receives royalties from UpToDate®. Dr Amundsen receives grant support from BlueWind Medical, Inc. None of these entities provided financial support for the study. The remainder of the authors do not report any potential conflicts of interest.
Footnotes
PUBLIC DATA SHARING:
All sequences are available for download in the Sequence Read Archive under Accession Number PRJNA685466 (http://www.ncbi.nlm.nih.gov/bioproject/685466)
Contributor Information
Monique H. Vaughan, Department of Obstetrics & Gynecology, Division of Pelvic Medicine and Reconstructive Surgery, University of Virginia, Charlottesville, VA
Jialiang Mao, Department of Statistical Science, Duke University, Durham, NC.
Lisa A. Karstens, Departments of Medical Informatics and Clinical Epidemiology and Obstetrics & Gynecology Oregon Health and Science University, Portland, OR
Li Ma, Department of Statistical Science, Duke University, Durham, NC.
Cindy L. Amundsen, Department of Obstetrics and Gynecology; Division of Urogynecology & Reconstructive Pelvic Surgery, Duke University Medical Center, Durham, NC
Kenneth E. Schmader, Department of Medicine, Division of Geriatrics, Duke University Medical Center, Durham, NC; and GRECC, Durham VA Medical Center, Durham, NC
Nazema Y. Siddiqui, Department of Obstetrics and Gynecology; Division of Urogynecology & Reconstructive Pelvic Surgery; Division of Reproductive Sciences, Duke University Medical Center, Durham, NC
REFERENCES
- 1.Foxman B Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am. March 2014;28(1):1–13. doi: 10.1016/j.idc.2013.09.003 [DOI] [PubMed] [Google Scholar]
- 2.Neugent ML, Hulyalkar NV, Nguyen VH, Zimmern PE, De Nisco NJ. Advances in Understanding the Human Urinary Microbiome and Its Potential Role in Urinary Tract Infection. mBio. April 28 2020;11(2)doi: 10.1128/mBio.00218-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brubaker L, Wolfe AJ. The new world of the urinary microbiota in women. American journal of obstetrics and gynecology. November 2015;213(5):644–9. doi: 10.1016/j.ajog.2015.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. November 02 2011;11:244. doi: 10.1186/1471-2180-11-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fouts DE, Pieper R, Szpakowski S, et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J Transl Med. August 28 2012;10:174. doi: 10.1186/1479-5876-10-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfe AJ, Toh E, Shibata N, et al. Evidence of uncultivated bacteria in the adult female bladder. J Clin Microbiol. April 2012;50(4):1376–83. doi: 10.1128/JCM.05852-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Komesu YM, Dinwiddie DL, Richter HE, et al. Defining the relationship between vaginal and urinary microbiomes. American journal of obstetrics and gynecology. February 2020;222(2):154 e1–154 e10. doi: 10.1016/j.ajog.2019.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomas-White K, Forster SC, Kumar N, et al. Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat Commun. April 19 2018;9(1):1557. doi: 10.1038/s41467-018-03968-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brotman RM, Shardell MD, Gajer P, et al. Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause. November 2018;25(11):1321–1330. doi: 10.1097/Gme.0000000000001236 [DOI] [PubMed] [Google Scholar]
- 10.Komesu YM, Richter HE, Carper B, et al. The urinary microbiome in women with mixed urinary incontinence compared to similarly aged controls. International urogynecology journal. June 16 2018;doi: 10.1007/s00192-018-3683-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gliniewicz K, Schneider GM, Ridenhour BJ, et al. Comparison of the Vaginal Microbiomes of Premenopausal and Postmenopausal Women. Front Microbiol. 2019;10:193. doi: 10.3389/fmicb.2019.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas-White K, Taege S, Limeira R, et al. Vaginal estrogen therapy is associated with increased Lactobacillus in the urine of postmenopausal women with overactive bladder symptoms. American journal of obstetrics and gynecology. November 2020;223(5):727 e1–727 e11. doi: 10.1016/j.ajog.2020.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anger J, Lee U, Ackerman AL, et al. Recurrent Uncomplicated Urinary Tract Infections in Women: AUA/CUA/SUFU Guideline. The Journal of urology. August 2019;202(2):282–289. doi: 10.1097/JU.0000000000000296 [DOI] [PubMed] [Google Scholar]
- 14.Chen YB, Hochstedler B, Pham TT, Acevedo-Alvarez M, Mueller ER, Wolfe AJ. The Urethral Microbiota: A Missing Link in the Female Urinary Microbiota. The Journal of urology. August 2020;204(2):303–309. doi: 10.1097/JU.0000000000000910 [DOI] [PubMed] [Google Scholar]
- 15.Barber MD, Walters MD, Bump RC. Short forms of two condition-specific quality-of-life questionnaires for women with pelvic floor disorders (PFDI-20 and PFIQ-7). Am J Obstet Gynecol. July 2005;193(1):103–13. doi: 10.1016/j.ajog.2004.12.025 [DOI] [PubMed] [Google Scholar]
- 16.Hilt EE, McKinley K, Pearce MM, et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. J Clin Microbiol. March 2014;52(3):871–6. doi: 10.1128/JCM.02876-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung CE, Chopyk J, Shin JH, et al. Benchmarking urine storage and collection conditions for evaluating the female urinary microbiome. Sci Rep. September 16 2019;9(1):13409. doi: 10.1038/s41598-019-49823-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. July 2016;13(7):581–3. doi: 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. December 17 2018;6(1):226. doi: 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearce MM, Zilliox MJ, Rosenfeld AB, et al. The female urinary microbiome in urgency urinary incontinence. American journal of obstetrics and gynecology. September 2015;213(3):347 e1–11. doi: 10.1016/j.ajog.2015.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mao JL, Chen YH, Ma L. Bayesian Graphical Compositional Regression for Microbiome Data. J Am Stat Assoc. August 26 2019;doi: 10.1080/01621459.2019.1647212 [DOI] [Google Scholar]
- 23.Salvetti E, Torriani S, Felis GE. The Genus Lactobacillus: A Taxonomic Update. Probiotics Antimicrob Proteins. December 2012;4(4):217–26. doi: 10.1007/s12602-012-9117-8 [DOI] [PubMed] [Google Scholar]
- 24.Campedelli I, Mathur H, Salvetti E, et al. Genus-Wide Assessment of Antibiotic Resistance in Lactobacillus spp. Appl Environ Microbiol. January 1 2019;85(1)doi: 10.1128/AEM.01738-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid G, Bruce AW, Cook RL, Llano M. Effect on urogenital flora of antibiotic therapy for urinary tract infection. Scand J Infect Dis. 1990;22(1):43–7. doi: 10.3109/00365549009023118 [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Ling Z, Xiao Y, et al. Dysbiosis of urinary microbiota is positively correlated with type 2 diabetes mellitus. Oncotarget. January 17 2017;8(3):3798–3810. doi: 10.18632/oncotarget.14028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keogh D, Tay WH, Ho YY, et al. Enterococcal Metabolite Cues Facilitate Interspecies Niche Modulation and Polymicrobial Infection. Cell Host Microbe. October 12 2016;20(4):493–503. doi: 10.1016/j.chom.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.