

Abstract
The synthesis of tetracyclic indole alkaloid (±)-decursivine was accomplished using BINOL-phosphoric acid catalyzed tandem oxidative cyclization as a key step with (bis(trifluoroacetoxy)iodo)benzene (PIFA) as an oxidizing agent. This represents one of the shortest and highest yielding routes for the synthesis of (±)-decursivine from readily available starting materials.
Subject terms: Natural product synthesis, Synthetic chemistry methodology
Introduction
Decursivine 1, an indole alkaloid, was isolated in the optically active form from the leaves and stems of Rhaphidophora decursiva Schott (Araceae) by Fong’s group1 in 2002. Decursivine is structurally related to serotobenine 2 (isolated as a racemic mixture in 1985 by Sato et al.)2 with a unique tetracyclic framework containing a trans-dihydrobenzofuran, an indole, and an eight-membered lactam that bridges the indole 3- and 4-positions (Fig. 1). Decursivine 1 exhibits antimalarial activity1 against the D6 and W2 isolates of Plasmodium falciparum with IC50 values of 3.93 and 4.41 µg/mL, respectively, whereas serotobenine 2 exhibits no activity against Plasmodium falciparum.
Figure 1.
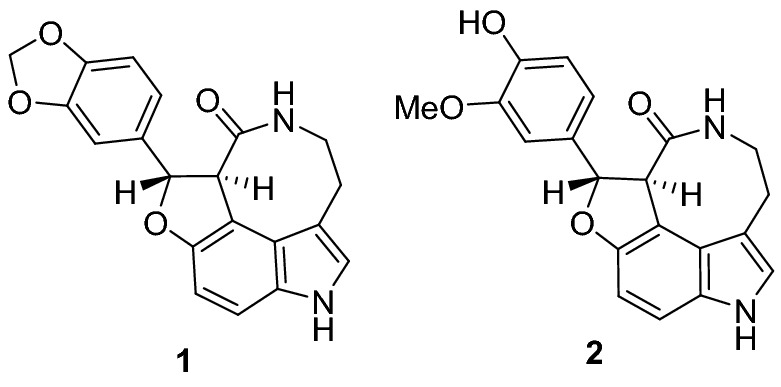
Chemical structures of naturally occurring (+)-decursivine 1 and (±)-serotobenine 2.
Owing to its novel structural features and potent antimalarial activity, decursivine has been the target of many synthetic efforts over the last decade3–11. The first total synthesis of (±)-decursivine was reported in 2007 by Kerr and co-workers4. The synthesis was completed in 19 steps with a 3.7% overall yield and featured Diels–Alder / Plieninger indolization reactions as key transformations. In 2011, Jia5 and Mascal6 independently and simultaneously developed a 4-step total synthesis of (±)-decursivine through a cascade photocyclization/elimination/O-Michael addition protocol with overall yields of 47.6% and 53.3%, respectively. In 2013, Jia’s group7 extended this cascade via a Witkop photocyclization approach wherein (+)- and (−)-decursivine were obtained with overall yields of 9.5% and 1.6% in 9 and 8 steps, respectively. The first asymmetric total synthesis of (+)-decursivine was developed in 2011 by Li and co-workers8 that involves an intramolecular [3 + 2] cycloaddition as the main step with an overall yield of 16.7% over 11 steps. In 2014, Jia’s group9 reported the synthesis of (±)-decursivine via a cascade C-H activation/oxidation approach in 4 steps with an overall yield of 19.3%. Subsequently, in 2015, Jia’s group10 broadened this cascade approach, implementing C-H activation/oxidation, to synthesize (-)-decursivine in 11 steps with a 6.5% overall yield. More recently, Xia and co-workers11 reported an 11-step total synthesis of (+)-decursivine in 2016 using an iron-catalyzed oxidative radical coupling protocol with an overall yield of 17.7%.
Results and discussion
In continuation of our work towards developing antimalarial heterocyclic compounds12,13 and indole-containing natural products14,15, we report a 5-step total synthesis of (±)-decursivine, an antimalarial indole alkaloid, from inexpensive and commercially available starting materials. Our retrosynthetic plan is illustrated in Fig. 2. We envisaged that decursivine 1 could be obtained from 3 via tandem oxidative cyclization, which in turn could be prepared by a simple coupling reaction from readily available starting materials, serotonin hydrochloride 4 and 3,4-(methylenedioxy)cinnamic acid 5.
Figure 2.

Retrosynthetic pathway of (±)-decursivine 1.
Our initial efforts towards the synthesis of (±)-decursivine 1 is described in Fig. 3. Coupling of serotonin hydrochloride 4 with 3,4-(methylenedioxy)cinnamic acid 5 using HBTU afforded key intermediate 3. Direct conversion of 3 into 1 via tandem oxidative cyclization (oxidation through single-electron transfer followed by cycloaddition) was unsuccessful via both photochemical and electrochemical approaches despite varying oxidizing agents and reaction conditions. In many attempts, 3 underwent decomposition (Table 1).
Figure 3.

Attempted synthesis of (±)-decursivine 1.
Table 1.
Tandem oxidative cyclization of compound 3.
| Sr. No. | Reaction conditions | Time (h) | Yield of 1 (%) |
|---|---|---|---|
| 1 | Ru(bpz)3(PF6)2 (0.1 equiv), (NH4)2S2O8 (2 equiv), CH3CN, Ar, blue LED, r.t | 18 | Decomposed |
| 2 | Acridinium (0.1 equiv), TBHP (2 equiv), CH3CN, Ar, blue LED, r.t | 48 | Decomposed |
| 3 | CAN (2.5 equiv), CH3CN, Ar, 0 °C | 2 | Decomposed |
| 4 | Mn(OAc)3·2H2O (4 equiv), CH3CN, Ar, reflux | 5 | Decomposed |
| 5 | PIFA (1.2 equiv), HFIP, Ar, r.t | 4 | Decomposed |
| 6a | Glassy carbon anode, Glassy carbon cathode, 0.1M LiClO4, 1.25 V | 18 | – |
| 7a | Platinum anode, Glassy carbon cathode, 0.1MLiClO4,1.25 V | 10 | – |
aPassivation of electrode observed.
These failures motivated us to protect the indole and amide –NH groups (Fig. 4). The hydroxy group of 3 was first protected using TBSCl, then the indole and amide –NH groups were protected by heating a mixture of 6, Boc2O, and DMAP in THF at reflux. Silyl group deprotection of compound 7 by treatment with TBAF yielded hydroxy derivative 8. With compound 8 on hand, we investigated tandem oxidative cyclization reaction under various conditions (Table 2).
Figure 4.

Total synthesis of (±)-decursivine 1.
Table 2.
Tandem oxidative cyclization of compound 8.
| Sr. No. | Reaction conditions | Time (h) | Yielda of 1 (%) |
|---|---|---|---|
| 1 | I2 (0.1 equiv), TBHP (2 equiv), EtOH, Ar, reflux | 48 | No reaction |
| 2 | CAN (2.5 equiv), CH3CN, Ar, 0 °C | 2 | Decomposed |
| 3 | Mn(OAc)3·2H2O (4 equiv), CH3CN, Ar, reflux | 5 | Decomposed |
| 4 | KHMDS (1.05 equiv), THF, Ar, 0 °C to r.t | 2 | Decomposed |
| 5 | Ru(bpz)3(PF6)2 (0.1 equiv), (NH4)2S2O8 (2 equiv), CH3CN, Ar, blue LED, r.t | 24 | Decomposed |
| 6 | [Ir(dtbbpy)(ppy)2[PF6] (0.1 equiv), BrCCl3 (2 equiv), CH3CN, Ar, blue LED, r.t | 36 | Decomposed |
| 7 | Acridinium (0.1 equiv), TBHP (2 equiv), CH3CN, Ar, blue LED, r.t | 48 | Decomposed |
| 8 | PIFA (1.2 equiv), HFIP, Ar, r.t | 6 | 47 |
| 9 | PIFA (1.2 equiv), H3PO4 (0.1 equiv), HFIP, Ar, r.t | 4 | Trace amounts |
| 10 | PIFA (1.2 equiv), (±)-BINOL phosphoric acid (0.05 equiv), HFIP, Ar, r.t | 3 | 74 |
aIsolated yield after column chromatography.
Oxidation of Boc-protected compound 8 through single-electron transfer, followed by cyclization using different oxidizing agents or photochemical approaches (entries 1–7) did not yield the desired product. Instead, compound 8 underwent decomposition. We then turned our attention towards a two-electron oxidation/cyclization approach using a hypervalent iodine reagent. In the literature, hypervalent iodine has been used for the oxidative [3 + 2] cycloaddition of various phenols and styrenes to yield 2,3-dihydrobenzofuran derivatives16–20. Based upon these findings, we treated compound 8 with a hypervalent iodine reagent, (bis(trifluoroacetoxy)iodo)benzene (PIFA), and product 1 was obtained in 47% yield (entry 8). The moderate yield of the product could be due to partial decomposition of the quinone intermediate (formed in situ) before undergoing the cycloaddition. Masson’s group21 recently reported the use of chiral phosphoric acid to catalyze the intermolecular oxidative [3 + 2] cycloaddition for the asymmetric synthesis of 3-aminodihydrobenzofurans. With the idea of stabilizing the in situ formed quinone intermediate21 via hydrogen bonding, we attempted the [3 + 2] cycloaddition with H3PO4 (entry 9) as a catalyst. Under these conditions, the reaction was sluggish, forming trace amounts of product that was only observed by LC–MS. Using (±)-BINOL phosphoric acid (entry 10), the reaction was faster and the product was obtained in higher yield. The proposed mechanism for the (±)-BINOL phosphoric acid-catalyzed [3 + 2] cycloaddition is shown in Fig. 5.
Figure 5.

Proposed mechanism of tandem oxidative cyclization.
PIFA oxidizes 8 to form quinone intermediate 9 which may be stabilized by (±)-BINOL phosphoric acid through hydrogen bonding21 to give adduct 10. Intramolecular cyclization of adduct 10 forms eight-membered lactam intermediate 11 that loses a proton to generate phenolic species 12. During this process, (±)-BINOL phosphoric acid is released for the next catalytic cycle. Finally, annulation of 12 leads to the formation of 2,3-dihydrobenzofuran containing compound 13 (N-Boc-protected decursivine)8.
Next, we investigated the use of chiral BINOL phosphoric acid catalysts, shown in Table 3, to perform an asymmetric version of the aforementioned reaction. We employed (S)-BINOL phosphoric acid (entry 1) and the bulky (R)-3,3’-bis(2,4,6-triisopropylphenyl)-BINOL phosphoric acid (entry 2) to induce chirality during oxidative cyclization. Unfortunately, negligible (˂ 2%) or no enantioselectivity was observed and the racemic product was obtained in 74% and 67% yield, respectively.
Table 3.
Attempted asymmetric synthesis of decursivine using chiral BINOL phosphoric acid.
|
| |||
|---|---|---|---|
| Sr. No. | Reaction conditions | Yielda (%) | eeb (%) |
| 1 | PIFA (1.2 equiv), (S)-BINOL phosphoric acid (0.05 equiv), HFIP, Ar, r.t., 3 h | 74 | – |
| 2 | PIFA (1.2 equiv), (R)-3,3'-bis(2,4,6-triisopropylphenyl)-BINOL phosphoric acid (0.05 equiv), HFIP, Ar, r.t., 3 h | 67 | < 2% |
aIsolated yield after column chromatography.
bDetermined by HPLC.
Conclusion
In conclusion, we have developed a concise total synthesis of (±)-decursivine, an antimalarial natural product via a cascade oxidative cyclization using PIFA as an oxidizing agent and (±)-BINOL phosphoric acid as a catalyst with a good overall yield of 43.8%.
Methods
General remarks
Reagents and solvents were purchased from commercial suppliers (Fisher Scientific {Hampton, New Hampshire, USA}, Sigma-Aldrich {St. Louis, Missouri, USA}, Strem Chemicals {Newburyport, Massachusetts, USA} and TCI America {Portland, Oregon, USA}) and used without further purification unless otherwise stated. Melting points were recorded on MEL-TEMP laboratory device. 1H and 13C NMR spectra were recorded at ambient temperature on a Varian Mercury NMR spectrometer (Palo Alto, California, USA) operating at 400 MHz (1H NMR) and 100 MHz (13C NMR) in the solvent indicated with the signal of the residual solvent (Chloroform-d δ 7.26 ppm or Methanol-d4 δ 3.31 ppm or Pyridine-d5 δ 8.74, 7.58, 7.22 ppm) as internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet), coupling constant (Hz) and integration. Thin-layer chromatography (TLC) was performed with silica gel 60 F254 pre-coated plates and visualized with exposure to UV light (254 nm) or by iodine staining. Flash chromatography was performed on Biotage (Model: Isolera, Sweden). HPLC analyses were performed using multiwavelength detector (Agilent Technologies 1200 Series). The chiral column used for the HPLC analysis was Lux 5 µm Cellulose-4 (Phenomenex, 250 × 4.6 mm). ESI–MS were recorded on Agilent Technologies 6120 Quadrupole spectrometer (Santa Clara, California, USA). HRMS were recorded on MDS Analytical Technologies AB CSIEX TOF/TOF 5800 spectrometer (Sunnyvale, California, USA). Electrochemical Experiments were performed with an EZstat Pro potentiostat galvanostat (NuVant Systems, Crown Point, IN, USA).
Synthesis of compound 3
Serotonin hydrochloride 4 (306 mg, 1.44 mmol) and 3,4-(methylenedioxy)cinnamic acid 5 (276 mg, 1.44 mmol) was dissolved in DMF (10 mL). To this solution was added DIPEA (375 µL, 2.15 mmol), HOBt∙3H2O (326 mg, 1.72 mmol), and HBTU (655 mg, 1.72 mmol) and the mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with EtOAc (50 mL) and washed with saturated aqueous NH4Cl, H2O and brine. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. Crude product was purified using flash chromatography on a Biotage Snap Cartridge (KP-Sil 25 g) using a gradient solvent system (40% to 90% ethyl acetate in hexanes) to give product 3 (434 mg, 86% yield). White solid. Mp 96–98 °C. Rf = 0.33 (70% ethyl acetate in hexanes). 1H NMR (400 MHz, Methanol-d4) δ 9.97 (s, 1H), 8.09 (s, 1H), 7.42 (d, J = 15.7 Hz, 1H), 7.15 (d, J = 8.6 Hz, 1H), 7.08–6.83 (m, 4H), 6.79 (d, J = 7.9 Hz, 1H), 6.66 (d, J = 9.0 Hz, 1H), 6.38 (d, J = 15.6 Hz, 1H), 5.95 (s, 2H), 3.54 (d, J = 7.5 Hz, 2H), 2.91 (t, J = 7.3 Hz, 2H). 13C NMR (100 MHz, Methanol-d4) δ 167.7, 149.9, 149.4, 148.6, 140.2, 129.5, 128.3, 123.9, 123.1, 118.7, 111.5, 111.2, 108.2, 105.9, 102.3, 101.7, 48.1, 47.8, 47.6, 40.3, 25.3. ESI–MS: m/z 351 [M + H]+. HRMS (MALDI-TOF) calcd for C20H18N2O4Na [M + Na]+ 373.1159, found 373.1129.
Synthesis of compound 6
To a solution of compound 3 (296 mg, 0.84 mmol) in DMF (10 mL) was added imidazole (172 mg, 2.53 mmol) and TBSCl (152 mg, 1.01 mmol) and the mixture was stirred at room temperature overnight. Reaction mixture was diluted with EtOAc (50 mL) and washed with saturated aqueous NH4Cl, H2O and brine. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. Crude product was purified using flash chromatography on a Biotage Snap Cartridge (KP-Sil 25 g) using a gradient solvent system (30% to 80% ethyl acetate in hexanes) to give product 6 (345 mg, 88% yield). White solid. Mp 68–72 °C. Rf = 0.47 (70% ethyl acetate in hexanes). 1H NMR (400 MHz, Chloroform-d) δ 7.91 (s, 1H), 7.51 (d, J = 15.5 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H), 7.02 (dd, J = 9.4, 2.3 Hz, 2H), 6.98–6.91 (m, 2H), 6.78 (dd, J = 8.6, 2.3 Hz, 2H), 6.11 (d, J = 15.5 Hz, 1H), 5.98 (s, 2H), 5.61 (s, 1H), 3.71 (q, J = 6.4 Hz, 2H), 2.98 (t, J = 6.5 Hz, 2H), 0.99 (s, 9H), 0.18 (s, 6H). 13C NMR (100 MHz, Chloroform-d) δ 166.4, 149.3, 149.2, 148.4, 140.8, 132.3, 129.4, 128.2, 124.1, 123.3, 119.1, 116.5, 111.9, 108.7, 108.4, 106.5, 101.6, 60.7, 40.1, 26.0, 25.5, 18.4, 14.4, -4.2. ESI–MS: m/z 465 [M + H]+. HRMS (MALDI-TOF) calcd for C26H32N2O4SiNa [M + Na]+ 487.2024, found 487.2028.
Synthesis of compound 7
To a solution of compound 6 (331 mg, 0.71 mmol) in THF (20 mL) was added DMAP (87 mg, 0.71 mmol), Et3N (396 µL, 2.82 mmol) and Boc2O (1.52 g, 6.97 mmol). The reaction mixture was refluxed for 3 h. Solvent was removed under reduced pressure and the residue was diluted with CH2Cl2 (30 mL) and washed with saturated aqueous NH4Cl, H2O and brine. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. Crude product was purified using flash chromatography on a Biotage Snap Cartridge (KP-Sil 25 g) using a gradient solvent system (2% to 40% ethyl acetate in hexanes) to give product 7 (398 mg, 84% yield). White solid. Mp 64–66 °C. Rf = 0.62 (40% ethyl acetate in hexanes). 1H NMR (400 MHz, Chloroform-d) δ 7.95 (s, 1H), 7.63 (d, J = 15.5 Hz, 1H), 7.41–7.29 (m, 2H), 7.09 (d, J = 2.1 Hz, 2H), 7.05 (dd, J = 8.0, 1.7 Hz, 1H), 6.87–6.77 (m, 2H), 6.00 (s, 2H), 4.07–3.95 (m, 2H), 2.92 (t, J = 7.7 Hz, 2H), 1.64 (s, 9H), 1.48 (s, 9H), 1.00 (s, 9H), 0.22 (s, 6H). 13C NMR (100 MHz, Chloroform-d) δ 168.9, 153.5, 151.6, 149.5, 148.5, 143.3, 129.9, 124.7, 124.1, 119.8, 117.8, 117.8, 115.9, 109.6, 108.7, 106.9, 101.7, 83.3, 45.1, 28.4, 28.3, 26.0, 24.7, 18.5, -4.2. ESI–MS: m/z 565 [M + H-Boc]+. HRMS (MALDI-TOF) calcd for C36H48N2O8SiNa [M + Na]+ 687.3072, found 687.3093.
Synthesis of compound 8
To a solution of compound 7 (301 mg, 0.45 mmol) in THF (10 mL) was added TBAF·3H2O (213 mg, 0.68 mmol) and the mixture was stirred at room temperature for 5 h. Solvent was removed under reduced pressure and the residue was diluted with EtOAc (30 mL) and washed with H2O and brine. The organic phase was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. Crude product was purified using Flash chromatography on a Biotage Snap Cartridge (KP-Sil 25 g) using a gradient solvent system (20% to 70% ethyl acetate in hexanes) to give product 8 (232 mg, 93% yield). White solid. Mp 74–78 °C. Rf = 0.48 (40% ethyl acetate in hexanes). 1H NMR (400 MHz, Chloroform-d) δ 7.94 (s, 1H), 7.63 (d, J = 15.5 Hz, 1H), 7.41–7.28 (m, 2H), 7.18 (d, J = 2.5 Hz, 1H), 7.07 (d, J = 1.7 Hz, 1H), 7.05–6.98 (m, 1H), 6.87 (dd, J = 8.8, 2.5 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 6.00 (s, 2H), 4.02–3.94 (m, 2H), 2.90 (t, J = 7.8 Hz, 2H), 1.63 (s, 9H), 1.49 (s, 9H). 13C NMR (100 MHz, Chloroform-d) δ 169.3, 153.4, 152.0, 148.5, 143.5, 129.8, 124.8, 124.2, 119.7, 117.5, 116.3, 113.4, 108.8, 106.9, 104.7, 101.7, 83.6, 45.2, 28.4, 28.3, 24.8. ESI–MS: m/z 452 [M + H-Boc]+. HRMS (MALDI-TOF) calcd for C30H34N2O8Na [M + Na]+ 573.2207, found 573.2204.
Synthesis of compound (±)-decursivine 1
-
(i)
Without catalyst: To a solution of compound 8 (30 mg, 0.05 mmol) in HFIP (5 mL) was added PIFA (28 mg, 0.06 mmol) in one portion under an argon atmosphere and the mixture was stirred at room temperature for 6 h. TFA (20 µL, 0.26 mmol) was then added and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc (3 × 20 mL). The combined organic phases were dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified using flash chromatography on a Biotage Snap Cartridge (KP-Sil 10 g) using a gradient solvent system (30% to 90% ethyl acetate in hexanes) to give product 1 (9 mg, 47% yield) as beige solid. Mp ˃250 °C, literature4 Mp 260 °C (dec.). Rf = 0.15 (50% ethyl acetate in hexanes). 1H NMR (400 MHz, Pyridine-d5) 1,5,6,8 δ 12.08 (s, 1H), 8.87 (dd, J = 10.3, 4.8 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.39 (s, 1H), 7.30 (d, J = 8.5 Hz, 2H), 7.08 (t, J = 8.9 Hz, 2H), 6.94 (d, J = 7.9 Hz, 1H), 5.98 (s, 2H), 4.24–4.10 (m, 1H), 3.59 (dd, J = 15.8, 3.9 Hz, 1H), 3.22–3.16 (m, 2H). ESI–MS: m/z 349 [M + H]+. HRMS (MALDI-TOF) calcd for C20H16N2O4Na [M + Na]+ 371.1002, found 371.1006.
-
(ii)
With catalyst: To a solution of compound 8 (30 mg, 0.05 mmol) in HFIP (5 mL) was added (±)-BINOL phosphoric acid (1 mg, 0.0025 mmol) and PIFA (28 mg, 0.06 mmol) in one portion under an argon atmosphere and the reaction mixture was stirred at room temperature for 3 h. TFA (20 µL, 0.26 mmol) was then added and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with saturated aqueous NaHCO3 and extracted with EtOAc (3 × 20 mL). The combined organic phases was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. Crude product was purified using flash chromatography on a Biotage Snap Cartridge (KP-Sil 10 g) using a gradient solvent system (30% to 90% ethyl acetate in hexanes) to give product 1 (14 mg, 74% yield). 1H NMR and mass data are same as mentioned above and matches with the literature data1,5,6,8.
Supplementary Information
Acknowledgements
We thank the National Institutes of Health (GM097118 and AI090662) for financial support and Yingzhao Zhao, Xiaofan Liu (Northeastern University) for recording HRMS.
Author contributions
P.T.P. and R.M. designed the experiments. P.T.P. performed all the experiments and analyzed the data. P.T.P. and R.M. wrote the manuscript. E.S.S. helped in performing electrochemistry experiments. All authors have given approval to the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-99064-8.
References
- 1.Zhang HJ, et al. Antimalarial agents from plants—II. Decursivine, a new antimalarial indole alkaloid from Rhaphidophora decursiva. Pharm. Biol. 2002;40:221–224. doi: 10.1076/phbi.40.3.221.5832. [DOI] [Google Scholar]
- 2.Sato H, et al. Serotobenine, a novel phenolic amide from safflower seeds (Carthamus tinctorius L.) Agric. Biol. Chem. Tokyo. 1985;49:2969–2974. [Google Scholar]
- 3.Chen Z, Pitchakuntla M, Jia YX. Synthetic approaches to natural products containing 2,3-dihydrobenzofuran skeleton. Nat. Prod. Rep. 2019;36:666–690. doi: 10.1039/C8NP00072G. [DOI] [PubMed] [Google Scholar]
- 4.Leduc AB, Kerr MA. Total synthesis of (+/-)-decursivine. Eur. J. Org. Chem. 2007;2007:237–240. doi: 10.1002/ejoc.200600922. [DOI] [Google Scholar]
- 5.Qin H, Xu ZR, Cui YX, Jia YX. Total synthesis of (+/-)-decursivine and (+/-)-serotobenine: A Witkop photocyclization/elimination/o-michael addition cascade approach. Angew. Chem. Int. Edit. 2011;50:4447–4449. doi: 10.1002/anie.201100495. [DOI] [PubMed] [Google Scholar]
- 6.Mascal M, Modes KV, Durmus A. Concise photochemical synthesis of the antimalarial indole alkaloid decursivine. Angew. Chem. Int. Edit. 2011;50:4445–4446. doi: 10.1002/anie.201006423. [DOI] [PubMed] [Google Scholar]
- 7.Hu WM, Qin H, Cui YX, Jia YX. Total synthesis of (+)- and (-)-decursivine and (+/-)-serotobenine through a cascade Witkop photocyclization/elimination/addition sequence: Scope and mechanistic insights. Chem.-Eur. J. 2013;19:3139–3147. doi: 10.1002/chem.201204137. [DOI] [PubMed] [Google Scholar]
- 8.Sun DQ, Zhao QW, Li CZ. Total synthesis of (+)-decursivine. Org. Lett. 2011;13:5302–5305. doi: 10.1021/ol2021669. [DOI] [PubMed] [Google Scholar]
- 9.Guo L, Zhang FY, Hu WM, Li L, Jia YX. Palladium-catalyzed synthesis of benzofurans via C–H activation/oxidation tandem reaction and its application to the synthesis of decursivine and serotobenine. Chem. Commun. 2014;50:3299–3302. doi: 10.1039/c3cc49717h. [DOI] [PubMed] [Google Scholar]
- 10.Zhang FY, Guo L, Hu WM, Jia YX. Total synthesis of (-)-decursivine and analogues via cascade sequence. Tetrahedron. 2015;71:3756–3762. doi: 10.1016/j.tet.2014.06.095. [DOI] [Google Scholar]
- 11.Liang KJ, et al. Biomimetic synthesis of moschamine-related indole alkaloids via iron-catalyzed selectively oxidative radical coupling. Org. Lett. 2016;18:1474–1477. doi: 10.1021/acs.orglett.6b00417. [DOI] [PubMed] [Google Scholar]
- 12.Neelarapu R, et al. Design and synthesis of orally bioavailable piperazine substituted 4(1H)-quinolones with potent antimalarial activity: Structure-activity and structure-property relationship studies. J. Med. Chem. 2018;61:1450–1473. doi: 10.1021/acs.jmedchem.7b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maignan JR, et al. ICI 56,780 optimization: Structure-activity relationship studies of 7-(2-phenoxyethoxy)-4(1H)-quinolones with antimalarial activity. J. Med. Chem. 2016;59:6943–6960. doi: 10.1021/acs.jmedchem.6b00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parvatkar PT, Parameswaran PS, Tilve SG. An Expeditious I-2-catalyzed entry into 6H-Indolo[2,3-b]quinoline system of cryptotackieine. J. Org. Chem. 2009;74:8369–8372. doi: 10.1021/jo901361x. [DOI] [PubMed] [Google Scholar]
- 15.Parvatkar PT, Ajay AK, Bhat MK, Parameswaran PS, Tilve SG. Iodine catalyzed one-pot synthesis of chloro-substituted linear and angular indoloquinolines and in vitro antiproliferative activity study of different indoloquinolines. Med. Chem. Res. 2013;22:88–93. doi: 10.1007/s00044-012-0015-0. [DOI] [Google Scholar]
- 16.Wang SP, Gates BD, Swenton JS. A convergent route to dihydrobenzofuran neolignans via a formal 1,3-cycloaddition to oxidized phenols. J. Org. Chem. 1991;56:1979–1981. doi: 10.1021/jo00006a003. [DOI] [Google Scholar]
- 17.Berard D, Jean A, Canesi S. Novel formal [2+3] cycloaddition between substituted phenols and furan. Tetrahedron Lett. 2007;48:8238–8241. doi: 10.1016/j.tetlet.2007.09.062. [DOI] [Google Scholar]
- 18.Berard D, Racicot L, Sabot C, Canesi S. Formal [2+3] cycloaddition between substituted phenols and allylsilane. Synlett. 2008;2008:1076–1080. doi: 10.1055/s-2008-1042924. [DOI] [Google Scholar]
- 19.Berard D, Giroux MA, Racicot L, Sabot C, Canesi S. Intriguing formal [2+3] cycloaddition promoted by a hypervalent iodine reagent. Tetrahedron. 2008;64:7537–7544. doi: 10.1016/j.tet.2008.05.114. [DOI] [Google Scholar]
- 20.Mohr AL, Lombardo VM, Arisco TM, Morrow GW. Synthesis of pterocarpan-type heterocycles via oxidative cycloadditions of phenols and electron-rich arenes. Synth. Commun. 2009;39:3845–3855. doi: 10.1080/00397910902838961. [DOI] [Google Scholar]
- 21.Gelis C, Bekkaye M, Lebee C, Blanchard F, Masson G. Chiral phosphoric acid catalyzed [3+2] cycloaddition and tandem oxidative [3+2] cycloaddition: Asymmetric synthesis of substituted 3-aminodihydrobenzofurans. Org. Lett. 2016;18:3422–3425. doi: 10.1021/acs.orglett.6b01593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.