

Abstract
Objectives
Lung cancer in never-smokers is a distinct disease associated with a different genomic landscape, pathogenesis, risk factors, and immune checkpoint inhibitor responses compared to those observed in smokers. This study aimed to identify novel single nucleotide polymorphisms (SNPs) of programmed death-1 (encoded by PDCD1) and its ligands, programmed death ligand 1 (CD274) and 2 (PDCD1LG2), associated with lung cancer risk in never-smoking women.
Materials and Methods
During September 2002 and July 2012, we enrolled never-smoking female patients with lung adenocarcinoma (LUAD) (n=1153) and healthy women (n=1022) from six tertiary hospitals in Taiwan. SNP data were obtained and analyzed from the genome-wide association study dataset and through an imputation method. The expression quantitative trait loci (eQTL) analysis was performed in both tumor and non-tumor tissues for the correlation between genetic expression and identified SNPs.
Results
A total of 12 PDCD1LG2 SNPs related to LUAD risk were identified in never-smoking women, including rs2381282, rs4742103, rs4237162, rs4742104, rs12237624, rs78096119, rs6476988, rs7857315, rs10975178, rs7854413, rs56001683, and rs7858319. Among them, six tagged PDCD1LG2 SNPs rs2381282, rs4742103, rs4237162, rs4742104, rs78096119, and rs56001683 were significantly associated with LUAD risk. Specifically, two PDCD1LG2 SNPs, rs12237624 and rs78096119, were associated with previous pulmonary tuberculosis infection in relation to LUAD susceptibility. Through an eQTL assay, we found that rs2381282 (p < 0.001), rs12237624 (p = 0.019), and rs78096119 (p = 0.019) were associated with the expression levels of programed death ligand 2.
Conclusions
Novel SNPs of programed death ligand 2 associated with lung adenocarcinoma risk were identified. Among them, two SNPs were associated with pulmonary tuberculosis infection in relation to lung adenocarcinoma susceptibility. These SNPs may help to stratify high-risk populations of never-smokers during lung cancer screening.
Keywords: programmed death ligand-2, single nucleotide polymorphism, lung adenocarcinoma, pulmonary tuberculosis, carcinogenesis
Introduction
Lung cancer is a growing global health concern (1), and cigarette smoking is a well-known risk factor for lung carcinogenesis (2). Nevertheless, approximately 25% of lung cancer cases are not attributable to tobacco smoking, and over 50% of female patients have been reported as never-smokers (3, 4). The prevalence of smoking among women in East Asia is lower compared to that in western countries (5). In Taiwan, more than 90% of female lung cancer patients are never-smokers (6, 7). Lung cancer in never smokers is considered a distinct disease entity with a proteogenomic landscape and oncogenic mechanisms different from those in smokers (8). Therefore, identifying genetic and environmental factors associated with lung cancer risk in never-smokers is urgently needed, especially in Asia.
Inflammation is considered one of the hallmarks of cancer, promoting tumorigenesis and neoplastic progression (9). Chronic infection and inflammation are strongly correlated with cancer risk (10). In addition to cigarette smoking, other environmental factors including chronic inflammation and particle/pollutant inhalation may also play a role in cancer developments (11, 12). However, limited data on the association between chronic infection/inflammation and lung carcinogenesis in never smokers are available.
Inflammation, including immune responses to chronic infection, may help to eliminate abnormal cells and prevent tumorigenesis (13, 14). However, tumors may overcome immune surveillance through mechanisms of immune evasion (9). The programmed death-ligand 1 (PD-L1) or programmed death-ligand 2 (PD-L2) on cancer cells would bind to programmed death-1 (PD-1) on immune cells, which could inhibit T cell activation and proliferation (15–19). PD-1 as a transmembrane protein, which is expressed on activated lymphocytes (T cells, B cells, and tumor specific T cells), natural killer cells, monocytes, and macrophages, involves in the tumorigenesis by restraining immune response (20, 21). The expression of PD-L1 is induced by oncogenes’ expression and various proinflammatory molecules and inhibited by the tumor suppressor genes expression, such as PTEN alternations (22). Therefore, activation of PD-1/PD-L1 pathway could lead to immune suppression and promote tumor growth in various cancer types (23, 24).
Further, high PD-L1 expression is not only found to accelerate skin carcinogenesis (25) but also associated with tumor differentiation, vascular invasion, and resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor treatment in non-small cell lung cancer (NSCLC) (26, 27). In NSCLC, the PD-1/PD-L1 pathway involving tumor proliferation and interacting with tumor microenvironment were greatly investigated. However, the role of PD-L2 in biological function of tumors was rarely studied.
Previous studies reported the association of PD-L1 gene polymorphisms with NSCLC risk mostly in smokers (28, 29). In never-smokers, the roles of PD-1, PD-L1, and PD-L2 gene polymorphisms in lung carcinogenesis remain unclear. Therefore, we conducted a case-control study in never-smoking women to explore the effects of PDCD1 (encoding PD-1), CD274 (encoding PD-L1), and PDCD1LG2 (encoding PD-L2) single nucleotide polymorphisms (SNPs) on lung carcinogenesis. The coding regions of these genes were of particular interest. In this study, chronic obstructive pulmonary disease (COPD), pulmonary tuberculosis (TB) infection, cooking fume exposure, and environmental tobacco smoking were defined as inflammation-related environmental exposures. We specifically investigated the interactions between these environmental factors and SNPs with regard to lung cancer susceptibility.
Materials and Methods
Subject Enrollment
This study is a part of the multi-center, case-control Genetic Epidemiological Study of Lung Adenocarcinoma (GELAC) in Taiwan (30–33), which enrolled subjects with lung cancer from six tertiary hospitals between September 2002 and July 2012. Cancer-free individuals were also enrolled as the controls from the health screening centers/clinics of these six hospitals during the recruitment period. Cases diagnosed with primary lung adenocarcinoma (LUAD) confirmed through cytologic or pathologic examination were recruited. Subjects younger than 18 years old, a prior history of other than primary lung cancer, or lack of suitable blood specimen were excluded in this study. We focused on never-smoking female subjects in the GELAC study population. We defined a never smoker as someone who had never smoked or not been smoking at least once a day for more than 6 months at any period during the lifetime. A total of 1153 female LUAD patients and 1022 healthy women were enrolled. The study was approved by the research ethics committees of these six hospitals and the National Health Research Institute in Taiwan.
Genotyping Analyses
Genomic DNA extracted from blood samples of the study participants was genotyped using an Illumina SNP array (310K, 610K, or 660K). All subjects were included in our previous genome-wide association study (GWAS) (30–33). Furthermore, SNP array and questionnaire data were jointly analyzed for quality control, as done in our previous GWAS. We calculated the relatedness coefficient (PI-HAT) in PLINK (34) and obtained 2175 unrelated samples (PI-HAT < 0.05 for any two samples). SNPs in PDCD1, CD274, and PDCD1LG2 were analyzed.
In addition to the retrieved genome-scale genotype data, an imputation was performed by using IMPUTE v.2 and data from the 1000 Genomes Project as the reference panel, so as to obtain more PDCD1, CD274, and PDCD1LG2 genotype data (35, 36). After imputation, SNPs derived from the previous GWAS genotype data were filtered in accordance with quality control criteria, including posterior probability > 0.5 and minor allele frequency > 1%. SNPs with a p-value < 0.05 in association analysis were selected and annotated as intron, transcript, untranslated region (UTR), missense, or synonymous by using information from the website of University of California, Santa Cruz (https://genome.ucsc.edu/cgi-bin/hgGateway). Micro (mi)RNA-related SNPs were identified using the miRNASNP database (http://bioinfo.life.hust.edu.cn/miRNASNP2/index.php, release 2.0). Tagged SNPs were selected by using Haploview 4.2 (37), a software used to analyze patterns of linkage disequilibrium and haplotypes from genotyping results.
Clinical Data Collection
All subjects provided written informed consent before collection of blood samples and clinical data. The patients’ clinical characteristics and related information were previously described (30–33). Clinical data were obtained from medical records as well as through personal interviews based on questionnaires and included age, education levels, body mass index (BMI, kg/m2), smoking status (including active and passive smoking), COPD, previous pulmonary TB infection, cumulative duration of hormone replacement therapy and contraceptive medications, and cooking fume exposure.
The body weight of healthy controls was recorded to adjust for the interference of cancer-related weight loss in BMI estimation. The BMI values were categorized into five levels (< 18.5, 18.5-24, 24-27, 27-30, and ≥ 30) and treated as categorical variables, following the guideline of Taiwan’s Administration of Health Promotion. A subject who had been smoking cigarettes regularly for at least 6 months, regardless of whether she had now quit or not, was defined as an “ever-cigarette smoker”. Otherwise, the subject was defined as a never-smoker (38, 39).
Supplementary therapy with synthetic estrogen or/and progesterone for a period of more than 90 days was defined as hormone replacement use. Contraceptive use was defined as the use of relevant medication for over 90 days on a cumulative basis. Cooking fume exposure was defined as a history of continuous cooking for more than 180 days. Furthermore, the cumulative cooking fume exposure (the duration of cooking is defined in years) was calculated by multiplying the number of cooking times every day by the number of cooking years. Cooking without a fume extractor was defined as the subject being continuously exposed to cooking fumes for at least 6 months without using a fume extractor.
Educational degree was considered a variable with 6 levels of value: 1 for lower than elementary school, 2 for elementary school graduate, 3 for junior high school graduate, 4 for senior high school graduate, 5 for college graduate, and 6 for postgraduate education. Exposure to environmental tobacco smoking (ETS) was categorized as being from parents or spouse, other relatives, and workplace, which were also assessed and stratified.
Quantitative Trait Loci Expression
The identified SNPs associated with LUAD risk were assessed for their association with the mRNA levels of the respective genes by quantitative trait loci (eQTL) assay. This cis eQTL analysis was performed using the Lung Cancer Tissue Cohort of Never-smokers, which included 115 never-smoking LUAD patients from the China Medical University Hospital in Taiwan. We collected their tumor tissues, adjacent non-tumor tissues, blood, and clinical information. Microarray gene expression experiments were performed, and genome-scale genotype data based on buffy coat DNA were obtained. Details are available from our previous study (GSE46539) (33).
Statistical Analysis
Logistic regression models were applied to assess the relationship between each selected covariate and LUAD risk. To investigate the correlation between individual SNPs and LUAD risk, we introduced clinical variables with a p-value less than 0.05 into multivariate logistic regression analysis. We coded the genotypes as additive by using the counts of the minor allele for each SNP.
For categorical clinical risk factors with more than two levels (more than one p-value in a model, such as BMI), the factor was retained if one of the p-values was less than 0.05. Two-tailed tests were used to determine significance in all analyses. A p-value less than 0.05 was considered statistically significant for identifying correlations between SNPs and LUAD risk. The interactions were estimated by including additional interaction terms (each SNP × inflammation-related environmental factors) in the logistic models. Statistical tests were performed by using R, a free software of the GNU project.
Results
Clinical Characteristics
A significant association with LUAD risk was observed for low education levels (p < 0.001), any first-degree family member with a history of lung cancer (p < 0.001), previous pulmonary TB infection (p < 0.001), cooking time in years (p = 0.037), cooking fume exposure (p < 0.001), and exposure to ETS from relatives or workplace (p < 0.001) ( Table 1 ). Covariates, including age, education levels, BMI levels, any first-degree family with a history of lung cancer, previous pulmonary TB infection, cooking time, cooking with a fume extractor, and ETS exposure, were thus introduced into multivariate analyses.
Table 1.
Clinical characteristics of lung adenocarcinoma patients and healthy controls.
| n (%) | Cases | Controls | p b |
|---|---|---|---|
| (n = 1153) | (n = 1022) | ||
| Age (years old), mean (SD) | 59.63 (11.34) | 58.96 (11.06) | 0.162 |
| Education levels | < 0.001 | ||
| Lower than elementary school | 255 (22.14) | 157 (15.38) | |
| Elementary school graduate | 383 (33.25) | 326 (31.93) | |
| Junior high school graduate | 136 (11.81) | 144 (14.10) | |
| Senior high school graduate | 215 (18.66) | 201 (19.69) | |
| College graduate | 154 (13.37) | 167 (16.36) | |
| Postgraduate | 9 (0.78) | 26 (2.55) | |
| BMI a | |||
| < 18.5 | 47 (4.17) | 29 (2.88) | Baseline |
| 18.5–24 | 597 (52.97) | 519 (51.59) | 0.159 |
| 24–27 | 307 (27.24) | 290 (28.83) | 0.088 |
| 27–30 | 112 (9.94) | 124 (12.33) | 0.030 |
| ≥ 30 | 64 (5.68) | 44 (4.37) | 0.724 |
| First-degree family with lung cancer | < 0.001 | ||
| Yes | 134 (12.96) | 43 (5.93) | |
| No | 900 (87.04) | 682 (94.07) | |
| COPD | 0.174 | ||
| Yes | 26 (2.28) | 15 (1.48) | |
| No | 1112 (97.72) | 1001 (98.52) | |
| History of pulmonary TB infection | < 0.001 | ||
| Yes | 50 (4.39) | 17 (1.67) | |
| No | 1088 (95.61) | 1001 (98.33) | |
| HRT use | 0.399 | ||
| Yes | 197 (18.12) | 188 (19.58) | |
| No | 890 (81.88) | 772 (80.42) | |
| Contraceptive use | 0.630 | ||
| Yes | 77 (7.15) | 75 (7.71) | |
| No | 1000 (92.85) | 898 (92.29) | |
| Cooking time in years, mean (SD) | 74.29 (53.22) | 69.59 (50.47) | 0.037 |
| Cooking without fume extractor | < 0.001 | ||
| Yes | 68 (5.9) | 23 (2.25) | |
| No | 1085 (94.1) | 999 (97.75) | |
| ETS exposure | < 0.001 | ||
| Yes | 856 (77.05) | 668 (66.73) | |
| No | 255 (22.95) | 333 (33.27) |
BMI, body mass index; COPD, chronic obstructive pulmonary disease; ETS, environmental tobacco smoking; HRT, hormone replacement therapy; SD, standard deviation; TB, tuberculosis.
Body weight (kg)/body height (m2).
p values for continuous variables (age, education levels, and cooking time in years) were determined via two-sample t-test; p-values for binary variables were determined via Chi-square test. For BMI, a categorical clinical risk factor with five levels, p-values were determined from the multivariate logistic regression model with BMI < 18.5 as the baseline.
PDCD1LG2 SNPs Are Associated With LUAD Risk
A flowchart for the identification of PDCD1, CD274, and PDCD1LG2 SNPs associated with LUAD risk is presented in Figure 1 . There were 36, 58, and 137 genotyped SNPs located within PDCD1, CD274, and PDCD1LG2, respectively, all of which met the genotype control criteria ( Supplementary Table S1 ). No PDCD1 SNPs were associated with LUAD risk. One CD274 SNP rs144841978 was related to LUAD risk with borderline significance (p = 0.051) and annotated in the 3′-UTR as a non-coding transcript variant. A total of 12 PDCD1LG2 SNPs were significantly associated with the LUAD risk, including rs2381282, rs4742103, rs4237162, rs4742104, rs12237624, rs78096119, rs6476988, rs7857315, rs10975178, rs7854413, rs56001683, and rs7858319 ( Table 2 ). Among them, rs2381282, rs4742103, rs4237162, rs4742104, rs78096119, and rs56001683 were further identified as tagged SNPs ( Figure 2A ). The linkage disequilibrium (LD) patterns of these tagged SNPs are shown in Figure 2B .
Figure 1.

A flowchart for the identification of single nucleotide polymorphisms (SNPs) associated with the risk of lung adenocarcinoma in PDCD1, CD274, and PDCD1LG2.
Table 2.
PDCD1LG2 SNPs associated with lung adenocarcinoma risk.
| SNP | Allele a | MAF | β | OR (95% CI) | p b | Annotation |
|---|---|---|---|---|---|---|
| rs2381282# | T/C | 0.357 | 0.149 | 1.160 (1.001, 1.345) | 0.049 | intron_variant |
| rs4742103# | C/T | 0.304 | -0.221 | 0.802 (0.676, 0.950) | 0.011 | intron_variant |
| rs4237162 | C/T | 0.221 | 0.235 | 1.264 (1.058, 1.511) | 0.010 | intron_variant |
| rs4742104# | C/T | 0.425 | -0.185 | 0.831 (0.715, 0.966) | 0.016 | intron_variant |
| rs12237624# | C/T | 0.043 | 0.390 | 1.476 (1.033, 2.111) | 0.033 | intron_variant |
| rs78096119# | A/G | 0.047 | 0.365 | 1.440 (1.027, 2.020) | 0.035 | intron_variant |
| rs6476988# | A/G | 0.273 | 0.166 | 1.181 (1.003, 1.390) | 0.046 | intron_variant |
| rs7857315# | T/C | 0.273 | 0.176 | 1.192 (1.013, 1.404) | 0.035 | intron_variant |
| rs10975178# | A/G | 0.294 | 0.178 | 1.195 (1.019, 1.402) | 0.028 | intron_variant |
| rs7854413 | C/T | 0.101 | -0.294 | 0.745 (0.588, 0.945) | 0.015 | missense_variant I (ATA) –> T (ACA) synonymous_variant D (GAT) –> D (GAC) |
| rs56001683# | G/T | 0.109 | -0.240 | 0.786 (0.627, 0.987) | 0.038 | intron_variant |
| rs7858319# | C/A | 0.108 | -0.241 | 0.786 (0.625, 0.988) | 0.039 | intron_variant |
CI, confidence interval; MAF, minor allele frequencies; OR, odds ratio; SNP, single nucleotide polymorphism; UTR, untranslated region.
Minor/major allele.
Covariates of age, education levels, body mass index, first-degree family with a history of lung cancer, history of pulmonary tuberculosis infection, cooking time in years, cooking with fume extractor, and environmental tobacco smoking exposure were used as adjusted variables.
#Imputed SNP.
Figure 2.
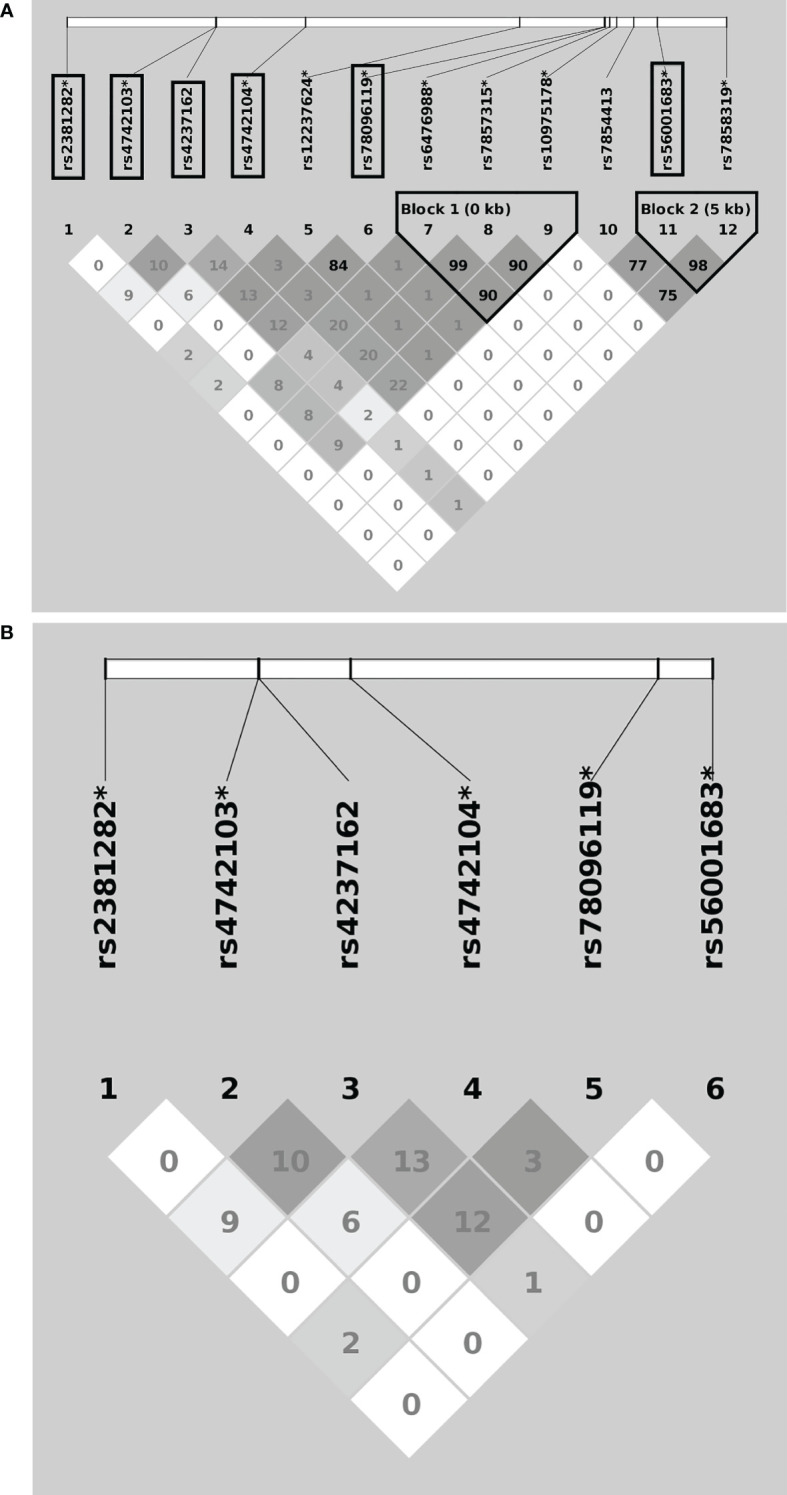
Linkage disequilibrium patterns of the overall 12 (A) and the 6 (B) tagged PDCD1LG2 single nucleotide polymorphisms (SNPs) associated with lung adenocarcinoma risk. The “*” indicates imputed SNPs, and the frames indicate the tagged SNPs.
PDCD1LG2 SNPs rs12237624 and rs78096119 Were Associated With Previous Pulmonary TB Infection in Relation to LUAD Susceptibility
Since the PD1/PD-L1/PD-L2 pathway plays a critical role in the anti-tumor immune response, we further investigated the interaction between identified SNPs and inflammation-related environmental factors, including COPD, history of pulmonary TB infection, cooking time, cooking with a fume extractor, and ETS exposure.
Associations between previous pulmonary TB infection and PDCD1LG2 SNPs rs12237624 and rs78096119 in relation to LUAD risk were observed ( Supplementary Table S2 ). The LD between these two SNPs was 0.84 (R2). No subject had two minor alleles for these SNPs and a history of pulmonary TB infection. We treated the presence of two minor alleles as a single category during association analysis (the dominant model). For rs12237624, this risk allele was significantly associated with an increased LUAD risk among patients with a history of pulmonary TB (ORTT = 3.605, 95% CI = 1.688 - 7.699) (p < 0.001) ( Table 3 ), as was the case for SNP rs78096119 (ORGG = 4.075, 95% CI =1.842 - 9.014) (p < 0.001) ( Table 3 ). Otherwise, no significant association with the other inflammation-related environmental factors was observed.
Table 3.
Correlation between PDCD1LG2 SNPs and pulmonary TB for lung adenocarcinoma susceptibility.
| SNP | Genotype/TB | Case | Control | Odds Ratio | Adjusted p-value a | p-value for correlation b |
|---|---|---|---|---|---|---|
| rs12237624 | TT + no TB | 987 (86.88%) | 930 (91.45%) | 1 | 0.040 | |
| CT/CC + no TB | 99 (8.71%) | 70 (6.88%) | 1.975 (0.392 - 9.938) | 0.409 | ||
| TT + TB | 46 (4.05%) | 13 (1.28%) | 3.605 (1.688 - 7.699) | < 0.001 | ||
| CT/CC + TB | 4 (0.35%) | 4 (0.39%) | NA | NA | ||
| rs78096119 | GG + no TB | 978 (86.09%) | 925 (90.95%) | 1 | 0.031 | |
| AG/AA + no TB | 109 (9.60%) | 75 (7.37%) | 1.957 (0.392 - 9.772) | 0.413 | ||
| GG + TB | 46 (4.05%) | 11 (1.08%) | 4.075 (1.842 - 9.014) | < 0.001 | ||
| AG/AA + TB | 4 (0.35%) | 5 (0.49%) | NA | NA |
NA, data not available; SNP, single nucleotide polymorphism; TB, tuberculosis.
Adjusted for education level, cooking time, and passive smoking.
The p-values were obtained from the additive model ( Supplementary Table S2 ). Covariates of age, education levels, body mass index, first-degree family with a history of lung cancer, history of pulmonary tuberculosis infection, cooking time in years, cooking with fume extractor, and environmental tobacco smoking exposure were used as adjusted variables.
Expression of Quantitative Trait Loci
eQTL analyses were performed for the aforementioned PDCD1LG2 SNPs ( Supplementary Table S3 ). The significance of eQTL results in tumor or non-tumor tissues was determined on the basis of p < 0.05 as the threshold. Among the PDCD1LG2 SNPs, rs2381282 (p < 0.001), rs12237624 (p = 0.019), and rs78096119 (p = 0.019) risk alleles were negatively associated with PD-L2 expression in non-tumor tissues.
Discussion
In this multi-center case-control study, a total of 12 PDCD1LG2 SNPs (rs2381282, rs4742103, rs4237162, rs4742104, rs12237624, rs78096119, rs6476988, rs7857315, rs10975178, rs7854413, rs56001683, and rs7858319) associated with LUAD risk in never-smoking women were identified. Among them, rs2381282, rs4742103, rs4237162, rs4742104, rs78096119, and rs56001683 were recognized as tagged SNPs. Furthermore, PDCD1LG2 SNPs rs12237624 and rs78096119 had significant associations with a history of pulmonary TB infection related to LUAD susceptibility. The PDCD1LG2 SNPs rs12237624, rs78096119, and rs2381282 were associated with PD-L2 expression via eQTL analysis. To our knowledge, this is the first study identifying novel PD-L2 gene polymorphisms associated with lung carcinogenesis in female never-smokers.
Among the 12 PDCD1LG2 SNPs, the clinical significance of rs7854413 was the most commonly reported in the previous literatures. A cohort study in south India reported that patients with the PDCD1LG2 SNP rs7854413 and lymphatic filariasis infection were susceptible to chronic lymphatic pathologies (40), and rs7854413 polymorphism was related to advanced fibrosis and development of hepatocellular carcinoma from patient with non-alcoholic steatohepatitis (41). Notably, rs7854413 was also associated with recurrence in patients with early-stage NSCLC (42). During the process of literature review, no studies on these PDCD1LG2 SNPs other than rs7854413 were reported. The functional role of these SNPs in lung carcinogenesis warrants further investigation.
Immune checkpoint blockade through inhibition of the PD-1/PD-L1 pathway is a state-of-the-art cancer immunotherapy (43). In contrast, the clinical significance of PD-L2 is seldom investigated (44). The role of PD-L2 in modulating the anti-tumor immune response remains controversial (19). PD-L2 inhibits the Crohn-like lymphoid reaction and adaptive immune response during colorectal carcinogenesis (18). In addition, PD-L2 was reportedly upregulated in myeloid-derived suppressor cells with the potential to inhibit anti-tumor immunity and promote tumor growth (45). Previous analyses of The Cancer Genome Atlas (TCGA) dataset revealed that the expression of PD-L2, rather than PD-L1, was positively associated with immune-related gene expression in renal cell carcinoma and lung squamous cell carcinoma (46). Furthermore, PD-L2 was expressed independently of PD-L1 expression, providing limited value for the prediction of anti-PD-1/PD-L1 therapy responses during cancer treatment (19). Although the constitutive expression and binding affinity of PD-L2 are low (24, 47, 48), our findings support that PD-L2 may play an important role in lung carcinogenesis.
Positive correlations between Mycobacterium tuberculosis infection and lung cancer risk were previously reported (49, 50). TB infection can cause chronic inflammation, which may not only lead to innate and adaptive immune responses but may also be associated with immune-related gene expression (51). In this study, pulmonary TB infection was an environmental exposure associated with LUAD risk in never-smoking women. Furthermore, PDCD1LG2 SNPs rs12237624 and rs78096119 had a significant correlation with pulmonary TB infection in relation to lung carcinogenesis. The underlying mechanisms potentially bridging the immune response to TB infection with lung carcinogenesis require further investigation, especially in the TB-endemic areas. Importantly, this finding highlights the importance of gene-environment interaction in relation to lung carcinogenesis for never-smokers.
In our study, the risk alleles of PDCD1LG2 rs2381282, rs12237624, and rs78096119 were negatively associated with PD-L2 expression in non-tumor tissue, but not in tumor tissue. The expression and prognostic value of PD-L2 expression in lung cancer have been previously reported (52–54). The interaction between PD-L2 and PD-1 inhibits strong B7-CD28 signals at low antigen concentrations. At high antigen concentrations, the interaction between PD-L2 and PD-1 reduced cytokine production but did not inhibit T cell proliferation (55). The correlation between these PDCD1LG2 SNPs and PD-L2 expression requires further investigation, which might provide further insight into the PD-1/PD-L2 axis in lung carcinogenesis.
The current study has several limitations. First, this multi-center study was hospital-based. The number of participants was considerably smaller than those in population-based studies. Second, no independent data validation was carried out. Since the proportion of never-smokers in most population-based studies on lung carcinogenesis has been relatively small, large studies in never-smokers are necessary to validate the current findings. Third, our healthy controls were recruited from the health examination departments of six hospitals, which may result in a healthy volunteer effect. Therefore, the current results should be interpreted cautiously.
In conclusion, we identified novel PDCD1LG2 SNPs significantly correlated with LUAD risk in never-smoking women. Of note, some of the identified SNPs interacted with pulmonary TB infection in relation to lung carcinogenesis. These findings may help stratify a high-risk population in never-smokers for early detection of lung cancer.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material .
Ethics Statement
The protocol of the study was approved by Research Ethics Committee, National Taiwan University Hospital, Taipei, Taiwan; Institutional Review Board, Taipei Veterans General Hospital, Taipei, Taiwan; Chang Gung Medical Foundation Institutional Review Board, Taipei, Taiwan; Institutional Review Board, Taichung Veterans General Hospital, Taichung, Taiwan; Institutional Review Board, National Cheng Kung University Hospital, Tainan, Taiwan; Institutional Review Board, Kaohsiung Medical University Chung-Ho Memorial Hospital, Kaohsiung, Taiwan; Research ethics committee, National Health Research Institutes, Zhunan, Taiwan; Institutional Review Board, China Medical University Hospital, Taichung, Taiwan. The obtained consents from all participants were written in the study. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CH, I-SC, W-CS, K-YC, Y-HT, C-JC, and P-CY conceived the study and designed the experiments. CH, I-SC, K-YC, L-HC, S-KL, H-HH, and P-CY contributed to study design and statistical analyses. H-CL, W-TF, and S-SJ contributed to the bioinformatics analysis. CH, W-CS, G-CC, K-YC, Y-MC, M-SH, Y-HT, and P-CY contributed reagents and materials. S-KL, L-HC, and K-YC wrote the first draft of the manuscript. C-YC contributed to the collection of tumor specimens for eQTL analysis. W-CS, G-CC, K-YC, Y-MC, M-SH, Y-HT, and P-CY contributed to the recruitment of subjects. All authors contributed to the article and approved the submitted version.
Funding
The GELAC study was granted by the National Health Research Institutes (NHRI) and by the Ministry of Sciences and Technology of Taiwan (grant no. MOST 106-2319-B-400-001).
Conflict of Interest
S-KL received honoraria for speech from Roche, AstraZeneca, Pfizer, Merck Sharp & Dohme, Novartis, and Boehringer Ingelheim. K-YC received honoraria for speech from Pfizer, Novartis, Merck Sharp & Dohme, AstraZeneca, Roche, Boehringer Ingelheim, Eli Lilly, Chugai Pharmaceutical, and Bristol-Myers Squibb, as well as travel/accommodation/meeting expenses from Merck Sharp & Dohme, Chugai Pharmaceutical, and Boehringer Ingelheim. Y-MC served on advisory boards of Merck Sharp & Dohme, Ono Pharmaceutical, Astra-Zeneca, Roche, Boehringer Ingelheim, and Bristol-Myers Squibb.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank members of seven medical centers in Taiwan, including National Taiwan University Hospital, Taipei Veterans General Hospital, Linkou Chang Gung Memorial Hospital, Taichung Veterans General Hospital, National Cheng Kung University Hospital, Kaohsiung Medical University Chung-Ho Memorial Hospital, and China Medical University Hospital, for participants recruitment and specimens collection. We would also like to thank the National Health Research Institutes (NHRI) for data storage, statistical and facility support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.753788/full#supplementary-material
Abbreviations
COPD, chronic obstructive pulmonary disease; EGFR, epidermal growth factor receptor; ETS, environmental tobacco smoking; eQTL, expression quantitative trait loci; GELAC, Genetic Epidemiological Study of Lung Adenocarcinoma; GWAS, genome-wide association study; LD, linkage disequilibrium; LUAD, lung adenocarcinoma; NSCLC, non-small cell lung cancer; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PD-L2, programmed death-ligand 2; SNP, single nucleotide polymorphism; TB, tuberculosis; TCGA, The Cancer Genome Atlas.
References
- 1. Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol (2019) 5(12):1749–68. doi: 10.1001/jamaoncol.2019.2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Khuder SA. Effect of Cigarette Smoking on Major Histological Types of Lung Cancer: A Meta-Analysis. Lung Cancer (2001) 31(2-3):139–48. doi: 10.1016/S0169-5002(00)00181-1 [DOI] [PubMed] [Google Scholar]
- 3. Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung Cancer in Never Smokers–A Review. Eur J Cancer (2012) 48(9):1299–311. doi: 10.1016/j.ejca.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 4. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 5. Zhou F, Zhou C. Lung Cancer in Never Smokers-The East Asian Experience. Transl Lung Cancer Res (2018) 7(4):450–63. doi: 10.21037/tlcr.2018.05.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen KY, Chang CH, Yu CJ, Kuo SH, Yang PC. Distribution According to Histologic Type and Outcome by Gender and Age Group in Taiwanese Patients With Lung Carcinoma. Cancer (2005) 103(12):2566–74. doi: 10.1002/cncr.21087 [DOI] [PubMed] [Google Scholar]
- 7. Tseng CH, Tsuang BJ, Chiang CJ, Ku KC, Tseng JS, Yang TY, et al. The Relationship Between Air Pollution and Lung Cancer in Nonsmokers in Taiwan. J Thorac Oncol (2019) 14(5):784–92. doi: 10.1016/j.jtho.2018.12.033 [DOI] [PubMed] [Google Scholar]
- 8. Chen YJ, Roumeliotis TI, Chang YH, Chen CT, Han CL, Lin MH, et al. Proteogenomics of non-Smoking Lung Cancer in East Asia Delineates Molecular Signatures of Pathogenesis and Progression. Cell (2020) 182(1):226–44.e17. doi: 10.1016/j.cell.2020.06.012 [DOI] [PubMed] [Google Scholar]
- 9. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 10. Coussens LM, Werb Z. Inflammation and Cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parkin DM. The Global Health Burden of Infection-Associated Cancers in the Year 2002. Int J Cancer (2006) 118(12):3030–44. doi: 10.1002/ijc.21731 [DOI] [PubMed] [Google Scholar]
- 12. Belpomme D, Irigaray P, Hardell L, Clapp R, Montagnier L, Epstein S, et al. The Multitude and Diversity of Environmental Carcinogens. Environ Res (2007) 105(3):414–29. doi: 10.1016/j.envres.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 13. Bremnes RM, Al-Shibli K, Donnem T, Sirera R, Al-Saad S, Andersen S, et al. The Role of Tumor-Infiltrating Immune Cells and Chronic Inflammation at the Tumor Site on Cancer Development, Progression, and Prognosis: Emphasis on non-Small Cell Lung Cancer. J Thorac Oncol (2011) 6(4):824–33. doi: 10.1097/JTO.0b013e3182037b76 [DOI] [PubMed] [Google Scholar]
- 14. Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, et al. Classical and Alternative Activation of Mononuclear Phagocytes: Picking the Best of Both Worlds for Tumor Promotion. Immunobiology (2006) 211(6-8):487–501. doi: 10.1016/j.imbio.2006.06.002 [DOI] [PubMed] [Google Scholar]
- 15. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its Ligands in Tolerance and Immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, et al. Programmed Death Ligand-1 Expression in Non-Small Cell Lung Cancer. Lab Invest (2014) 94(1):107–16. doi: 10.1038/labinvest.2013.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boland JM, Kwon ED, Harrington SM, Wampfler JA, Tang H, Yang P, et al. Tumor B7-H1 and B7-H3 Expression in Squamous Cell Carcinoma of the Lung. Clin Lung Cancer (2013) 14(2):157–63. doi: 10.1016/j.cllc.2012.05.006 [DOI] [PubMed] [Google Scholar]
- 18. Masugi Y, Nishihara R, Hamada T, Song M, da Silva A, Kosumi K, et al. Tumor PDCD1LG2 (PD-L2) Expression and the Lymphocytic Reaction to Colorectal Cancer. Cancer Immunol Res (2017) 5(11):1046–55. doi: 10.1158/2326-6066.CIR-17-0122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J, et al. PD-L2 Expression in Human Tumors: Relevance to Znti-PD-1 Therapy in Cancer. Clin Cancer Res: Off J Am Assoc Cancer Res (2017) 23(12):3158–67. doi: 10.1158/1078-0432.CCR-16-1761 [DOI] [PubMed] [Google Scholar]
- 20. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and are Functionally Impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 Regulates the Development, Maintenance, and Function of Induced Regulatory T Cells. J Exp Med (2009) 206(13):3015–29. doi: 10.1084/jem.20090847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cretella D, Digiacomo G, Giovannetti E, Cavazzoni A. PTEN Alterations as a Potential Mechanism for Tumor Cell Escape From PD-1/PD-L1 Inhibition. Cancers (2019) 11(9):1318. doi: 10.3390/cancers11091318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rozali EN, Hato SV, Robinson BW, Lake RA, Lesterhuis WJ. Programmed Death Ligand 2 in Cancer-Induced Immune Suppression. Clin Dev Immunol (2012) 2012:656340. doi: 10.1155/2012/656340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao Y, Zhang L, Kamimura Y, Ritprajak P, Hashiguchi M, Hirose S, et al. B7-H1 Overexpression Regulates Epithelial-Mesenchymal Transition and Accelerates Carcinogenesis in Skin. Cancer Res (2011) 71(4):1235–43. doi: 10.1158/0008-5472.CAN-10-2217 [DOI] [PubMed] [Google Scholar]
- 26. Yang CY, Lin MW, Chang YL, Wu CT, Yang PC. Programmed Cell Death-Ligand 1 Expression in Surgically Resected Stage I Pulmonary Adenocarcinoma and Its Correlation With Driver Mutations and Clinical Outcomes. Eur J Cancer (2014) 50(7):1361–9. doi: 10.1016/j.ejca.2014.01.018 [DOI] [PubMed] [Google Scholar]
- 27. Hsu KH, Huang YH, Tseng JS, Chen KC, Ku WH, Su KY, et al. High PD-L1 Expression Correlates With Primary Resistance to EGFR-TKIs in Treatment Naive Advanced EGFR-Mutant Lung Adenocarcinoma Patients. Lung Cancer (2019) 127:37–43. doi: 10.1016/j.lungcan.2018.11.021 [DOI] [PubMed] [Google Scholar]
- 28. Chen YB, Mu CY, Chen C, Huang JA. Association Between Single Nucleotide Polymorphism of PD-L1 Gene and Non-Small Cell Lung Cancer Susceptibility in a Chinese Population. Asia Pac J Clin Oncol (2014) 10(2):e1–6. doi: 10.1111/ajco.12037 [DOI] [PubMed] [Google Scholar]
- 29. Ma Y, Liu X, Zhu J, Li W, Guo L, Han X, et al. Polymorphisms of Co-Inhibitory Molecules (CTLA-4/PD-1/PD-L1) and the Risk of Non-Small Cell Lung Cancer in a Chinese Population. Int J Clin Exp Med (2015) 8(9):16585–91. [PMC free article] [PubMed] [Google Scholar]
- 30. Hsiung CA, Lan Q, Hong YC, Chen CJ, Hosgood HD, Chang IS, et al. The 5p15.33 Locus Is Associated With Risk of Lung Adenocarcinoma in Never-Smoking Females in Asia. PloS Genet (2010) 6(8):e1001051. doi: 10.1371/journal.pgen.1001051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lan Q, Hsiung CA, Matsuo K, Hong YC, Seow A, Wang Z, et al. Genome-Wide Association Analysis Identifies New Lung Cancer Susceptibility Loci in Never-Smoking Women in Asia. Nat Genet (2012) 44(12):1330–5. doi: 10.1038/ng.2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lo YL, Hsiao CF, Chang GC, Tsai YH, Huang MS, Su WC, et al. Risk Factors for Primary Lung Cancer Among Never Smokers by Gender in a Matched Case-Control Study. Cancer Causes Control (2013) 24(3):567–76. doi: 10.1007/s10552-012-9994-x [DOI] [PubMed] [Google Scholar]
- 33. Chang IS, Jiang SS, Yang JC, Su WC, Chien LH, Hsiao CF, et al. Genetic Modifiers of Progression-Free Survival in Never-Smoking Lung Adenocarcinoma Patients Treated With First-Line Tyrosine Kinase Inhibitors. Am J Respir Crit Care Med (2017) 195(5):663–73. doi: 10.1164/rccm.201602-0300OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am J Hum Genet (2007) 81(3):559–75. doi: 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. International HapMap Consor. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A Second Generation Human Haplotype Map of Over 3.1 Million SNPs. Nature (2007) 449(7164):851–61. doi: 10.1038/nature06258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PloS Genet (2009) 5(6):e1000529. doi: 10.1371/journal.pgen.1000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics (2005) 21(2):263–5. doi: 10.1093/bioinformatics/bth457 [DOI] [PubMed] [Google Scholar]
- 38. Liaw KM, Chen CJ. Mortality Attributable to Cigarette Smoking in Taiwan: A 12-Year Follow-Up Study. Tob Control (1998) 7(2):141–8. doi: 10.1136/tc.7.2.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kreuzer M, Gerken M, Heinrich J, Kreienbrock L, Wichmann HE. Hormonal Factors and Risk of Lung Cancer Among Women? Int J Epidemiol (2003) 32(2):263–71. doi: 10.1093/ije/dyg064 [DOI] [PubMed] [Google Scholar]
- 40. Venugopal G, O’Regan NL, Babu S, Schumann RR, Srikantam A, Merle R, et al. Association of a PD-L2 Gene Polymorphism With Chronic Lymphatic Filariasis in a South Indian Cohort. Am J Trop Med Hyg (2019) 100(2):344–50. doi: 10.4269/ajtmh.18-0731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. El-Derany MO. Polymorphisms in Interleukin 13 Signaling and Interacting Genes Predict Advanced Fibrosis and Hepatocellular Carcinoma Development in Non-Alcoholic Steatohepatitis. Biology (2020) 9(4):75. doi: 10.3390/biology9040075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Q, Gu J, Wang L, Chang DW, Ye Y, Huang M, et al. Genetic Associations of T Cell Cancer Immune Response-Related Genes With T Cell Phenotypes and Clinical Outcomes of Early-Stage Lung Cancer. J Immunother Cancer (2020) 8(2):e000336. doi: 10.1136/jitc-2019-000336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity (2018) 48(3):434–52. doi: 10.1016/j.immuni.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 Ligands, and Other Features of the Tumor Immune Microenvironment With Response to Anti-PD-1 Therapy. Clin Cancer Res (2014) 20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Solito S, Pinton L, Mandruzzato S. In Brief: Myeloid-Derived Suppressor Cells in Cancer. J Pathol (2017) 242(1):7–9. doi: 10.1002/path.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tanegashima T, Togashi Y, Azuma K, Kawahara A, Ideguchi K, Sugiyama D, et al. Immune Suppression by PD-L2 Against Spontaneous and Treatment-Related Antitumor Immunity. Clin Cancer Res (2019) 25(15):4808–19. doi: 10.1158/1078-0432.CCR-18-3991 [DOI] [PubMed] [Google Scholar]
- 47. Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal Structure of the Complex Between Programmed Death-1 (PD-1) and its Ligand PD-L2. Proc Natl Acad Sci USA (2008) 105(30):10483–8. doi: 10.1073/pnas.0804453105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghiotto M, Gauthier L, Serriari N, Pastor S, Truneh A, Nunes JA, et al. PD-L1 and PD-L2 Differ in Their Molecular Mechanisms of Interaction With PD-1. Int Immunol (2010) 22(8):651–60. doi: 10.1093/intimm/dxq049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang HY, Li XL, Yu XS, Guan P, Yin ZH, He QC, et al. Facts and Fiction of the Relationship Between Preexisting Tuberculosis and Lung Cancer Risk: A Systematic Review. Int J Cancer (2009) 125(12):2936–44. doi: 10.1002/ijc.24636 [DOI] [PubMed] [Google Scholar]
- 50. Wu CY, Hu HY, Pu CY, Huang N, Shen HC, Li CP, et al. Pulmonary Tuberculosis Increases the Risk of Lung Cancer: A Population-Based Cohort Study. Cancer (2011) 117(3):618–24. doi: 10.1002/cncr.25616 [DOI] [PubMed] [Google Scholar]
- 51. Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, et al. Human Gene Expression Profiles of Susceptibility and Resistance in Tuberculosis. Genes Immun (2011) 12(1):15–22. doi: 10.1038/gene.2010.51 [DOI] [PubMed] [Google Scholar]
- 52. Zhang Y, Wang L, Li Y, Pan Y, Wang R, Hu H, et al. Protein Expression of Programmed Death 1 Ligand 1 and Ligand 2 Independently Predict Poor Prognosis in Surgically Resected Lung Adenocarcinoma. Onco Targets Ther (2014) 7:567–73. doi: 10.2147/OTT.S59959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shinchi Y, Komohara Y, Yonemitsu K, Sato K, Ohnishi K, Saito Y, et al. Accurate Expression of PD-L1/L2 in Lung Adenocarcinoma Cells: A Retrospective Study by Double Immunohistochemistry. Cancer Sci (2019) 110(9):2711–21. doi: 10.1111/cas.14128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liebler DC, Holzer TR, Haragan A, Morrison RD, O’Neill Reising L, Ackermann BL, et al. Analysis of Immune Checkpoint Drug Targets and Tumor Proteotypes in Non-Small Cell Lung Cancer. Sci Rep (2020) 10(1):9805. doi: 10.1038/s41598-020-66902-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 Is a Second Ligand for PD-1 and Inhibits T Cell Activation. Nat Immunol (2001) 2(3):261–8. doi: 10.1038/85330 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material .