

Summary
The dual function protein ACAD9 catalyzes α,β-dehydrogenation of fatty acyl-CoA thioesters in fatty acid β-oxidation and is an essential chaperone for mitochondrial respiratory complex I (CI) assembly. ACAD9, ECSIT, and NDUFAF1 interact to form the core mitochondrial CI assembly complex. Current studies examine the molecular mechanism of ACAD9/ECSIT/NDUFAF1interactions. ACAD9 binds to the carboxy-terminal half and NDUFAF1 to the amino-terminal half of ECSIT. Binary complexes are unstable and aggregate easily, while the ACAD9/ECSIT/NDUFAF1 ternary complex is soluble and highly stable. Molecular modeling and small-angle X-ray scattering studies identified intra-complex interaction sites and binding sites for other assembly factors. Binding of ECSIT at the ETF binding site in the amino-terminal domain of ACAD9 is consistent with observed loss of FAD and enzymatic activity and demonstrates that the two functions of ACAD9 are mutually exclusive. Mapping of 42 known pathogenic mutations onto the homology-modeled ACAD9 structure provides structural insights into pathomechanisms of CI deficiency.
Subject areas: Biological sciences, Molecular biology, Structural biology
Graphical abstract
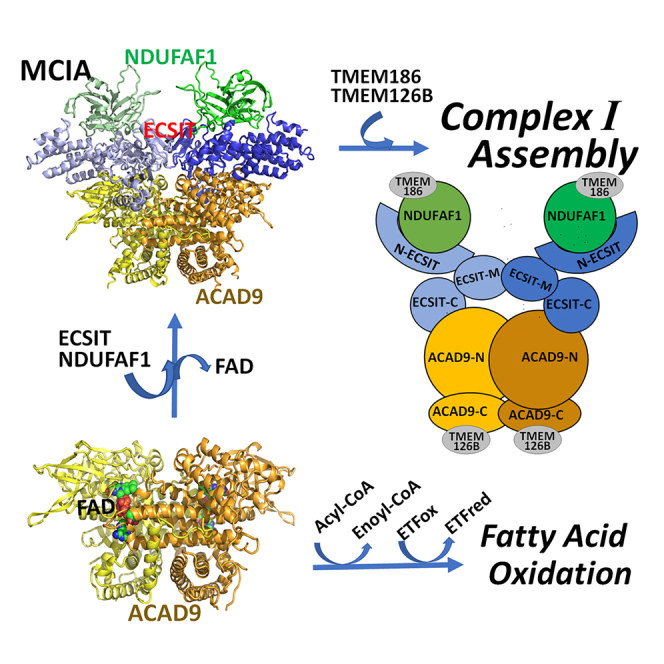
Highlights
-
•
ECSIT bridges ACAD9 and NDUFAF1 in the ACAD9/ECSIT/NDUFAF1 heterohexamer of MCIA
-
•
The NH2-domain of ECSIT binds to NDUFAF1, whereas the COOH-domain binds to ACAD9
-
•
ECSIT binds to ACAD9 near the FAD binding site and deflavinates ACAD9
-
•
Dehydrogenase and complex 1 assembly functions of ACAD9 are mutually exclusive
Biological sciences; Molecular biology; Structural biology
Introduction
ACAD9 was first identified as a member of the acyl-CoA dehydrogenase family catalyzing the α, β-dehydrogenation of fatty acyl-CoA thioesters, the first step of the fatty acid β-oxidation cycle (Zhang et al., 2002). Like its closest family member, very long chain acyl-CoA dehydrogenase (VLCAD), ACAD9 is a homodimer, contains FAD as the cofactor, and carries out the dehydrogenation of long-chain fatty acids, with optimum chain-length specificity for 16 carbons or longer (Ensenauer et al., 2005; Zhang et al., 2002). However, following reports that ACAD9-deficient patients demonstrated dysfunction of both fatty acid oxidation and oxidative phosphorylation (He et al., 2007), ACAD9 has also been shown to be essential for assembly of the mitochondrial oxidative phosphorylation complex I (CI, NADH-ubiquinone oxidoreductase), the first enzyme complex of the mitochondrial respiratory chain (Formosa et al., 2020; Formosa and Ryan, 2018; Nouws et al., 2010). The 45 subunit, 980kDa mammalian CI, requires 15 currently known assembly factors for formation of the properly functioning mature complex and its dysfunction is the most common oxidative phosphorylation disorder in humans (Elurbe and Huynen, 2016; Formosa et al., 2020; Giachin et al., 2016; Guerrero-Castillo et al., 2017; Koopman et al., 2012; Mimaki et al., 2012; Sanchez-Caballero et al., 2016). ACAD9, ECSIT, and NDUFAF1 form the core mitochondrial CI assembly (MCIA) complex that, together with TMEM126B and TMEM186, is critical for assembly of the CI ND2 module (Formosa et al., 2020).
In the current study, we have cloned and expressed in E. coli wild-type and mutant forms of human ACAD9, full-length ECSIT and its C-terminal domain, and NDUFAF1, and characterized the biochemical and biophysical properties of the individual proteins and their binary and ternary complexes. Here we describe interactions between ECSIT, ACAD9, and NDUFAF1 in binary and ACAD9/ECSIT/NDUFAF1 ternary complexes. While this manuscript was in preparation, Giachin et al. published studies on binary interactions between ACAD9, ECSIT, and NDUFAF1 employing yeast two-hybrid and bimolecular fluorescence complementation methods and determination of a 15Å-resolution cryo-EM structure for the C-terminal domain of ECSIT bound to ACAD9 (Giachin et al., 2021). Our studies provide physical evidence for binary and ternary interactions between the full-length proteins and confirm their finding of deflavination of ACAD9 upon binding of the C-terminal domain of ECSIT. In addition, we have prepared and examined properties of the ACAD9/ECSIT/NDUFAF1 ternary complex and propose an alternative model for ECSIT binding to ACAD9. Characterizing the structural and mechanistic bases of the role of ACAD9 in CI assembly provides insights into regulation of the dual activities of ACAD9 in MCIA and fatty acid β-oxidation.
Results
Expression, purification, and characterization of the core MCIA components
ACAD9 and its variants
ACAD9 proteins, including wild type ACAD9 (both ACAD9 and ACAD9-His6) and variants, were expressed in E. coli and purified to homogeneity as shown by size exclusion chromatography (SEC). ACAD9 was eluted from the SEC column as a dimer with a molecular weight of 113 kDa, at the same position as VLCAD (Figure S1A), consistent with its sequence and structural similarity.
Although purified VLCAD contains 1 mol FAD/mol (monomer) of VLCAD, the FAD content of purified wild type ACAD9 protein (non-tagged ACAD9 and ACAD9-His6) was approximately 70%, comparable with flavoproteins with low micromolar or sub-micromolar binding affinity for FAD (Ruprecht et al., 2015; Shen and Kasper, 2000; Xia et al., 2011). As purified, the wild-type ACAD9 protein has a dehydrogenation activity of 83 min−1, attributable to the FAD-containing holo-form of the protein (Table 1). Incubation of the protein with a 100-fold excess of FAD prior to the assay increased dehydrogenation activity to 128 min−1, suggesting that FAD can bind to ACAD9 molecules lacking FAD (apo-ACAD9) and restore enzymatic activity. However, this activity is only 18% of VLCAD activity (Table 1) (Nouws et al., 2010).
Table 1.
ACAD9 dehydrogenation activity and effects of FAD and ECSIT
| ACAD9 (no His6) | ACAD9-His6 | R532W- His6 | R518H- His6 | R469W- His6 | E426Q- His6 | Loop12- His6 | VLCAD (N- His6) | |
|---|---|---|---|---|---|---|---|---|
| Activity | 83 ± 5 | 80 ± 5 | 62 ± 5∗ | 95 ± 5 | 89 ± 4 | 8 ± 7 | 40 ± 4∗ | 985 ± 75 |
| Activity +100XFAD | 128 ± 5∗∗ | 132 ± 3 | 133 ± 7 | 130 ± 5 | 132 ± 4 | 8 ± 5 | 142 ± 4 | 995 ± 91 |
| Activity + 5X ECSIT | 8 ± 6 | 8 ± 3 | 11 ± 3 | 10 ± 7 | 9 ± 4 | 7 ± 2 | 10 ± 1 | 940 ± 36 |
Reactions contained 0.1 μM purified ACAD9 or VLCAD protein, 50 μM palmitoyl-CoA, and 200 μM ferricenium hexafluorophosphate (15), in a final volume of 0.5 mL. Activity is expressed as μmol ferrocene produced/min/μmol enzyme (min−1). Values are expressed as mean ± SD (n = 3).
∗, p < 0.007 compared to wildtype ACAD9 in the absence of excess FAD.
∗∗, p < 0.0001 compared to wildtype ACAD9 in the absence of excess FAD. See also Figure S2.
To further characterize the FAD affinity of apo-ACAD9, we titrated the purified ACAD9 with varying amounts of exogenous FAD. The concentration dependence for stimulation of ACAD9 activity by exogenous FAD exhibits half-maximal activity at 0.75 μM FAD (Figure S2), suggesting that the FAD binding affinity of the apo form is less than that of the purified holo-ACAD9.
Table 1 shows the dehydrogenation activities of wild-type and ACAD9 variants in the absence and presence of exogenous FAD. We have previously shown that many ACAD9 variants associated with human ACAD9 deficiency exhibit defects in protein folding and/or stability (Schiff et al., 2015). However, three variants, R469W, R518H, and R532W, could be purified to homogeneity. We also investigated the importance of the linker between the two domains of ACAD9 (residues between Thr450 to Asp480) using two variants, Loop8-ACAD9 and Loop12-ACAD9, in which the linker was replaced by 8 and 12 residues of GlyGlySer repeats, respectively. Activities and stimulation of activity by exogenous FAD of the R518H and R469W variants were similar to wild type. Activities of the R532W and Loop12-ACAD9 variants in the presence of exogenous FAD were the same as FAD-supplemented wild-type activity, but only 75 and 48%, respectively, of wild-type levels in the absence of exogenous FAD. The Loop8-ACAD9 variant was unstable, producing only inclusion bodies. Complete lack of activity of the E426Q protein confirms Glu426 as the catalytic base found in most ACAD enzymes.
ECSIT and N-terminal and C-terminal domains of ECSIT
Initial attempts to express and purify full-length ECSIT yielded multiple peaks in SEC, revealing the protein is poly disperse, i.e., in multiple aggregated states, as reported by Giachin et al. (2021). To obtain non-aggregated, mono-disperse protein, His-tagged ECSIT was eluted from the Ni-affinity column with a stepwise imidazole concentration gradient. The fraction eluting at 80 mM imidazole had the largest amount of the apparent-low molecular-weight protein and was further purified by SEC to homogeneity, as shown by SDS-PAGE and MALDI-TOF mass spectra (Figure S3). ECSIT purified by SEC had an apparent molecular weight of ∼110 kDa, consistent with its homodimeric form. The purified, dimeric ECSIT protein had a tendency to aggregate at high protein concentrations and so was concentrated to ≤0.5 mg/mL.
While expression of the N-terminal domain of ECSIT (Ser49-Ser269) resulted in formation of inclusion bodies, the C-terminal domain of ECSIT (Leu249-Ser431, termed C-ECSIT) could be expressed and purified to homogeneity using the same purification procedures used for the full-length ECSIT. It is more stable than the dimeric full-length ECSIT, judging from the fact that the protein was more soluble and did not aggregate as easily as the full-length protein. In contrast to full-length ECSIT, C-ECSIT migrated as a tetramer, with an apparent MW of 89 kDa (Figure S1B). Presence or absence of the N-terminal His-tag did not affect stability.
NDUFAF1
Although wild type His6-NDUFAF1 could be expressed in E. coli and purified to homogeneity as assessed by SDS-PAGE, it was highly aggregated, and a large portion of the protein was eluted in the void volume on a size exclusion column. However, addition of an SUMO tag enabled expression and purification of His6-SMT3-NDUFAF1 in monomeric form and its molecular weight was estimated to be ∼48 kDa by SDS-PAGE, consistent with its expected value. However, size-exclusion chromatography gave a homogeneous single peak with an apparent molecular weight around 60 kDa, indicating it exists as a monomer in solution (Figure S1B) but is not a compact globular protein. Presence of the SUMO tag was necessary for maintenance of the globular NDUFAF1 conformation. Although the His6-SMT3 fusion tag could be cleaved by the ULP1 enzyme, the resulting tag-free NDUFAF1 aggregated easily. Therefore, His6-SMT3-NDUFAF1 protein was used in subsequent experiments unless otherwise noted.
Characterization of ACAD9, ECSIT, and NDUFAF1 interactions in the ternary complex by Ni-NTA affinity chromatography pull-down assays
To study the nature of interactions among ACAD9, ECSIT, and NDUFAF1, we performed pull-down assays using a Ni-NTA affinity column. Results shown in Figure 1 are consistent with a model in which ACAD9 binds to the C-terminal domain of ECSIT, and NDUFAF1 binds to the N-terminal domain of ECSIT. ACAD9 with a His6-tag at the C-terminus (ACAD9-His6), which has the same properties as non-tagged ACAD9 (exists as a homodimer and has similar catalytic activity) pulled down both ECSIT and C-ECSIT efficiently, at a near 1:1 monomer ratio (Figure 1, lanes 6 and 7). As a control, His6-ECSIT failed to pull down VLCAD (Figure 1, lane 13). Full-length His6-ECSIT can also pull down NDUFAF1 (Figure 1, lane 9), as well as the N-terminal domain of ACAD9 (Figure S6). However, ACAD9 does not interact with NDUFAF1 (Figure S6). When ACAD9-His6 was mixed with both tag-free ECSIT and NDUFAF1, the elution product contained all three components (Figure 1, lane 8), indicating that the ternary complex was formed.
Figure 1.

Interactions between purified ACAD9, ECSIT, and NDUFAF1 monitored by pull-down assay on a Ni-NTA affinity column
SDS-PAGE of pull-down products. Lane 1, MW Marker; 2, ACAD9-His6; 3, ECSIT; 4, C-ECSIT; 5, NDUFAF1; 6, pull-down product of ACAD9-His6+ECSIT; 7, pull-down product of ACAD9-His6+ C-ECSIT; 8, pull-down product of ACAD9-His6+ECSIT + NDUFAF1; 9, pull-down product of His6-ECSIT + NDUFAF1; 10, His6-ECSIT; 11, VLCAD; 12, 1:1 mixture of His6-ECSIT + VLCAD; and 13, pull-down product of His6-ECSIT + VLCAD. Lanes 6, 7, and 8 show that ACAD9-His6 can pull down either ECSIT or C-ECSIT alone, or both ECSIT and NDUFAF1. Lane 9 shows that His6-ECSIT can pull down NDUFAF1. As a control, Lane 13 shows that His6-ECSIT fails to pull-down VLCAD. See also Figure S6.
ECSIT deflavinates ACAD9 and abolishes its α,β-dehydrogenation activity
Although purified wild-type or variant ACAD9 in the absence of ECSIT has dehydrogenation activity, no activity was observed in the presence of an excess amount of ECSIT (Table 1). Dehydrogenation activities of the variant proteins were also abolished in the presence of excess ECSIT. When ECSIT and ACAD9 were mixed in equimolar ratio and dialyzed overnight, absorbance at 450 nm was abolished (Figure 2A), consistent with deflavination of the enzyme, in agreement with recent results of Giachin et al. (2021). ECSIT had no effect on VLCAD dehydrogenation activity or FAD content.
Figure 2.

ECSIT deflavination and loss of ACAD9 dehydrogenation activity and restoration of dehydrogenation activity by FAD
(A) UV-VIS spectra of VLCAD and ACAD9 before and after after incubation with ECSIT. Black and blue lines show VLCAD and ACAD9 (20 μM protein concentration) after overnight dialysis in 100 mM Tris, pH 7.4, 100 mM NaCl, 10% glycerol. Spectra of VLCAD or ACAD9 (20 μM protein concentration) mixed with 20 μM ECSIT in 100 mM Tris, pH 7.4, 100 mM NaCl, 10% glycerol and dialyzed in the same buffer overnight are shown in red (VLCAD + ECSIT) and green (ACAD9+ECSIT). The inset shows the same spectra with an expanded scale.
(B and C) Dehydrogenation activity, as % of maximum activity, after incubation of ACAD9 with increasing concentrations of (B) ECSIT, or (C) C-ECSIT. The dashed lines show biphasic decreases in the dehydrogenation activity with an inflection point at an ECSIT:ACAD9 ratio near 0.4:1. Values are expressed as mean ± SD.
(D) Restoration of dehydrogenation activity by addition of FAD to HPLC-purified ACAD9/ECSIT binary (red) or ACAD9/ECSIT/NDUFAF1 ternary (blue) complexes. The concentration of each complex was 0.1 μM. Numbers in parentheses indicate the molar ratio of FAD:complex. Activity is expressed as % of maximum activity. Values are expressed as mean ± SD.
We then titrated the enzyme activity of ACAD9 by the addition of varying amounts of ECSIT. In the absence of exogenous FAD, ACAD9 activity was approximately 65% of maximal FAD-supplemented activity. As shown in Figure 2B, addition of up to 0.4 mol ECSIT/mol ACAD9 (monomer:monomer) produced an 18% decrease in ACAD9 activity, while further addition of ECSIT produced a linear decrease in catalytic activity. Activity was essentially abolished at a 1:1 ratio of ECSIT to ACAD9. The observation that low levels of ECSIT did not produce appreciable decreases in dehydrogenation activity suggests that ECSIT may bind preferentially to the apo-form of ACAD9. However, upon saturation of apo-ACAD9 with FAD, ECSIT binding to holo-ACAD9 produces a linear decrease in catalytic activity. Similar results were observed with C- ECSIT (Figure 2C). ECSIT-induced loss of catalytic activity could be completely reversed by addition of excess FAD (Figure 2D), with half-maximal activity observed at 2.1 μM (1.2–3.5, 95% CI) and 2.8 μM (1.8–4.5, 95% CI), respectively, for the ACAD9/ECSIT binary and ACAD9/ECSIT/NDUFAF1 ternary complexes.
Molecular weight determination of binary and ternary complexes of ACAD9, ECSIT, and NDUFAF1 by SEC
ACAD9/ECSIT binary complexes
Figure 3A shows the elution profiles of ACAD9 alone, ECSIT alone, and a 1:1 mixture of ACAD9 and ECSIT on a size exclusion column. The ACAD9/ECSIT binary complex eluted at an apparent MW of about 270 kDa (black), which corresponds to one ACAD9 dimer (green) plus one ECSIT dimer (red). The elution profile of the ACAD9/ECSIT mixture monitored at 450 nm shows that FAD was released from ACAD9 in the ACAD9/ECSIT complex and eluted as free FAD (gold peak in Figure 3A). Similarly, Figure 3B shows ACAD9/C-ECSIT binary complex formation. Consistent with the tetrameric structure of C-ECSIT, the binary complex of ACAD9/C-ECSIT differed from the ACAD9/ECSIT complex, eluting at an apparent MW of ∼380 kDa (black) corresponding to one C-ECSIT tetramer and two ACAD9 dimers. FAD eluted at the free FAD position, consistent with deflavination.
Figure 3.

Monitoring of complex formation by size-exclusion column chromatography
Chromatographic elution profiles of equimolar mixtures of the indicated proteins are shown. Absorbance at 280 nm is shown in black and absorbance at 450 nm in gold. For clarity, absorbance at 450 nm is shown on a 10-fold expanded scale. Elution profiles of ACAD9, His6-ECSIT or His6-C-ECSIT, and His6-SMT3-NDUFAF1 alone are shown in green, red, and cyan, respectively. The standard MW protein peaks (gray) from left to right include thyroglobulin (670 kDa), apoferritin (443 kDa), IgG (158 kDa), BSA (67 kDa), ovalbumin (43 kDa), and cytochrome c (12.3 kDa).
(A) ACAD9/His6-ECSIT binary complex.
(B) ACAD9/His6-C-ECSIT binary complex.
(C) His6-ECSIT/His6-SMT3-NDUFAF1 binary complex.
(D) ACAD9/His6-ECSIT/His6-SMT3-NDUFAF1 ternary complex.
(E) Increased stability of NDUFAF1 in the ACAD9/His6-ECSIT/His6-SMT3-ACAD9/NDUFAF1 ternary complex. HPLC elution profiles, recorded at 280 nm, of the 1:1:1 ACAD9/His6-ECSIT/His6-SMT3-NDUFAF1 (black) and tag-free ACAD9/His6-ECSIT/NDUFAF1 (red) ternary complex, obtained by digesting the purified ACAD9/His6-ECSIT/His6-SMT3-NDUFAF1 complex (eluting at 25–30 min, lane 1 in (F)) with ULP1 to cleave the His6-SMT3 tag (lane 2 in (F)). The yellow curve is the HPLC profile at 450nm of the tag-free ternary complex (20X expanded scale). The small peaks between 45 and 50 min in the His6-SMT3 tag-free HPLC profile (red) are nonspecific contaminants.
(F) SDS-PAGE of the ACAD9/ECSIT/NDUFAF1 ternary complex. Lane 1, the HPLC-purified ACAD9/His6ECSIT/His6-SMT3-NDUFAF1 ternary complex eluting between 25 and 30 min, corresponding to the black curve in (E). Lane 2, the tag-free ternary complex obtained by digesting the ACAD9/His6ECSIT/His6-SMT3-NDUFAF1 complex overnight with ULP1, corresponding to the red curve in (E). Lane 3, the ternary ACAD9/His6ECSIT/His6-SMT3-NDUFAF1 complex digested overnight with both ULP1 and thrombin. The double band at ∼33 kDa is NDUFAF1 (lower band) and the product of His6-SMT3-NDUFAF1 cleaved by thrombin due to a non-specific cleavage of the SMT3 tag (upper band). Protein markers are shown at the right. Molecular weights from top to bottom are 70, 60, 50, 40, 30, 25, 20, and 15kDa. Positions of ACAD9, His6-SMT3-NDUFAF1, ECSIT, NDUFAF1, and the His6-SMT3 tag are indicated at the right.
(G–J) Chromatographic elution profiles showing ternary complex formation of ACAD9-His6 variants. (G) R469W, (H) R518H, (I) Loop12-ACAD9, and (J) E426Q.
ECSIT/NDUFAF1 binary complex
To study interactions between ECSIT and NDUFAF1, we used His6-SMT3-NDUFAF1, due to the aggregation of untagged NDUFAF1. The broad elution peak of the His6-SMT3-NDUFAF1/ECSIT complex (Figure 3C, black profile), indicates that the binary complex is relatively polydisperse. In addition, the relatively sharp tall peak of excess free His6-SMT3-NDUFAF1 suggests that, at the concentrations used for complex formation, a portion of ECSIT existed as higher oligomers rather than dimers and consequently lost its ability to interact with NDUFAF1. Nevertheless, the His6-SMT3-NDUFAF1/ECSIT binary complex has an apparent MW of 250 kDa based on SEC analysis. This MW is about 30% larger than the calculated MW of 197 kDa, based on the binary complex of one ECSIT dimer and two His6-SMT3-NDUFAF1 monomers, indicating the non-globular nature of the binary complex.
ACAD9/ECSIT/NDUFAF1 ternary complex
Figures 3D and 3E shows that mixing of the three purified, individual proteins forms a ternary complex, ACAD9/ECSIT/NDUFAF1, with a 1:1:1 molar (monomer) ratio (Figure 3F). ACAD9, ECSIT, and His6-SMT3-NDUFAF1 co-eluted from the SEC column at an apparent MW of 620 kDa (black profile). Remarkably, the ternary complex was very stable and did not aggregate, even in the presence of the critical micelle concentration of β-octylglucoside (25 mM). Although the His6-SMT3 tag was necessary to prevent aggregation of NDUFAF1 alone or in the ECSIT/NDUFAF1 binary complex, it could be safely cleaved from the ternary complex without precipitation or aggregation, yielding a complex having the same elution profile as the tagged protein (Figures 3E and 3F). As with the binary ACAD9/ECSIT complex, FAD was released from ACAD9 upon ternary complex formation (gold profile). The 620 kDa apparent molecular weight of the ternary complex implies that it contains four monomers each of ACAD9, ECSIT, and His6-SMT3-NDUFAF1.
The five ACAD9 variants examined also bound ECSIT (Table 1) and formed the ternary complex, with release of FAD, indicating that these residues were not required for ECSIT binding and ternary complex formation. Representative SEC profiles are shown in Figures 3G–3J.
SEC-SAXS studies of ACAD9 and its binary and ternary complexes
Small-angle X-ray scattering (SAXS) has emerged as a very powerful technique for the study of flexible and less compact proteins, providing important information including the overall size and shape of macromolecules in solution. Figure S4 displays SEC-SAXS profiles, experimental SAXS profiles, normalized Kratky plots, and P(r) fittings (pair-distance distribution) of the ACAD9 dimer alone (red curves), ACAD9/ECSIT binary complex (green curves), and ACAD9/ECSIT/His6/SMT3/NDUFAF1 ternary complex (gray curves). Kratky plots indicate that ACAD9 and the binary and ternary complexes are all globular in nature and their maximum dimensions derived from P(r) distribution fittings are approximately 100, 140, and 200 Å, respectively. The overall parameters obtained from the scattering data are presented in Table 2. The molecular weights of ACAD9 and the binary and ternary complexes are in line with their molecular weights estimated by SEC and calculated according to their amino acid sequences.
Table 2.
Parameters of ACAD9 and its complexes calculated from the SEC-SAXS data
| Samples | Rg(nm) | Dmax(nm) | Vporod(nm3) | MW (kDa) By sequence |
MW (kDa) By HPLC |
MW (kDa)a By SAXS |
|---|---|---|---|---|---|---|
| ACAD9 | 3.41 | 10.3 | 230 | 130 | 110 | 140 |
| ACAD9/ECSIT | 5.02 | 16.0 | 494 | 223 | 270 | 320 |
| ACAD9/ECSIT/His6-SMT3-NDUFAF1 | 6.78 | 21.3 | 1340 | 637 | 620 | 710 |
The abbreviations used are as follows: Rg, radius of gyration; Dmax, maximum size of the particle; VP, excluded volume of the hydrated particle, Experimental mass, experimental molecular mass of the solute.
SAXS MW estimates were obtained from the Primus program according to Bayesian Inference (Konarev et al., 2003). See also Figure S4.
Homology modeling of ACAD9 and the ACAD9/ECSIT/NDUFAF1 ternary complex
No experimentally determined structures of ACAD9, ECSIT, or NDUFAF1 are available to date. We have generated the structures of the individual proteins using both homology modeling and molecular modeling. A model of the ACAD9/ECSIT/NDUFAF1 ternary complex was then obtained by docking these three molecules using ClusPro followed by fitting to SAXS data.
ACAD9
ACAD9 shares 47% sequence identity and 77% similarity to VLCAD, suggesting similar overall structures (Nouws et al., 2010; Schiff et al., 2013). Results of our SEC and SAXS characterization are consistent with this hypothesis. Like VLCAD, ACAD9 is a homodimer, and each monomer consists of two large domains. The N-terminal domain (∼400 residues) harboring FAD is responsible for enzyme activity and is connected to the C-terminal domain (∼180 residues), by a ∼30-residue linker region (Thr450 to Asp480). Figure 4A shows a ribbon representation of the human ACAD9 structure modeled after the VLCAD crystal structure ((McAndrew et al., 2008), PDB: 3B96), showing the sites of reported human complex I deficient variants. The model fits well with our SAXS data (Figures 4B and S4), further validating our homology-modeled structure.
Figure 4.

Structures of ACAD9, ACAD9 complexes, and VLCAD
(A) Homology-modeled structure of ACAD9 homodimer with locations of 42 human CI deficient mutations. The monomers are colored blue or green. The N-terminal domains are shaded lighter and the C-terminal domains are shown in darker shades. Dotted lines indicate the position of the linker. Mutations discussed in the text are labeled. The mutations near the surface in the N-terminal domain that might be involved in ECSIT binding (I87N, L98S, A170V, G172R, A190V, A220V, I222N, T243R, C271G, L286P) are shown as blue balls. Mutations that might decrease ACAD9 stability, including those involved in the dimer interface interactions (R127Q, R266Q, S268F, L314P, A326P, K330Q, A390T, E413K, R414C/S, R417C, D418G, R532W/Q, S602F, L606H, Y611C), are shown as red balls. The mutants E413K, R414C/S, R417C, and D418G are located on the Y413-I423 helix that destabilizes the ACAD9 dimer relative to VLCAD. Mutations in the C-terminal domain (F44I, R469W, R518H/C, V546L, R549Q, S551G, D562N, H563D, E564K, C572Y, N579K) that are unlikely to be involved in the dimer interface interactions are shown as yellow balls. For clarity, mutation sites are marked only on the green monomer.
(B) Cartoon model structures of ACAD9, ACAD9/ECSIT and ACAD9/ECSIT/NDUFAF1 fit into the ab initio envelope models derived from SAXS experiments. The ACAD9 dimer is colored orange/yellow, ECSIT-C green, ECSIT-M red, N-ECSIT blue, NDUFAF1 cyan, and SMT3 tag gray. See also Figure S5.
(C) Docking model between ACAD9 dimer (yellow and gold monomers) and ECSIT-C. Docking results obtained from the online protein docking program ClusPro are shown in multiple colors. FAD is shown as green sticks.
(D) Docking model of ETF bound to ACAD9 mol B, based on the structure of the MCAD/ETF complex. ACAD9 monomers are shown in yellow and gold. The ETF α-N domain is shown in black and the β-subunit and the α-subunit C-domain are in gray. ETF FAD is shown as purple sticks.
(E) Superposition of C and D. Colors are as in (C) and (D) showing overlap of ETF and ECSIT-C.
(F) 90° rotation of (E) showing details of the overlap of ECSIT-C and the ETF-α N-terminal domain. Colors are as in (C) and (D).
(G) Comparison of the surface electrostatic potential maps of VLCAD and ACAD9. ECSIT-C and ECSIT- M binding sites are indicated by black ovals. The corresponding views are shown with the ribbon diagram. The ACAD9 dimer is colored orange/yellow, with ECSIT-C shown in green. The electrostatic potential scale (±5 kT) is shown at the bottom. ACAD9 has more positive charges at the middle of the surface.
Mutation sites associated with human ACAD9 deficiency can be divided into three categories according to their locations in the 3D structure (Figure 4A): (1) mutation sites located on the surface of the N-terminal domain that are predicted to be involved in interactions with ECSIT (blue balls), (2) mutation sites in the N-terminal domain predicted to be involved in ACAD9 stability, including those directly involved in the dimer interface interactions (red balls); and (3) mutation sites in the C-terminal domain that are predicted to have no interaction with ECSIT (yellow balls).
Examination of the location of pathogenic variants reveals that, with the exception of R532W, those located at the dimer interface (blue balls) were unstable and could not be purified to homogeneity. In contrast, the variants, R469W and R518H, in the C-terminal domain (yellow balls), are relatively stable and have FAD contents and dehydrogenation activities similar to those of wildtype ACAD9. As predicted, these mutants form stable ternary complexes with ECSIT and NDUFAF1 (Table 1; Figure 3), but are CI-deficient, suggesting they are involved in interactions with other CI subunits or assembly factors such as TMEM126B (Nouws et al., 2010).
The lower affinity of ACAD9 for FAD compared with VLCAD is not obvious from casual inspection of the two structures. Almost all the residues that directly interact with FAD in ACAD9 and VLCAD are identical. The ribityl-isoalloxazine ring portion of FAD binds to one monomer of the ACAD9 homodimer, while the ADP moiety binds to the other monomer near the homodimer interface. Therefore, the lower FAD-binding affinity of ACAD9 is most likely due to the higher flexibility of the homodimer interface and FAD may act to stabilize the ACAD9 homodimer. This flexibility may contribute to the observation of two ACAD9 conformations, holo- and apo-, with differing FAD binding affinities, and is consistent with the decreased dehydrogenation activity of the R532W variant in the absence of exogenous FAD (Table 1). Upon comparing their interface interactions, Asp391 in VLCAD, which forms important salt bridges with Arg416 and Arg419 of the other monomer of VLCAD, is replaced by Ser395 in ACAD9, while Arg419 of VLCAD is replaced by Leu423 in ACAD9. These residues reside at the center of the helix containing residues Tyr412 through Leu423, situated at the ACAD9 dimer interface, decreasing dimer stability and FAD binding affinity.
NDUFAF1
Little structural information is currently available for either NDUFAF1 or ECSIT. For these two proteins, we used the web-based Robetta programs (Kim et al., 2004) for domain prediction and model construction. The C-terminal domain of NDUFAF1 (Leu117-Lys327), comprising most of the protein, was constructed based on its homology of the galactose-binding domain-like protein, exo-β-agarase Aga50D from the marine bacterium Saccharophagus degradans (PDB: 4BQ5), with a confidence score of 0.643. The five homology models calculated from Robetta aligned well except for the last twenty residues (Figure S5). The N-terminal domain of NDUFAF1 (Tyr25-Leu117) has no good homology model, and Robetta gave only a 0.208 confidence score with an uncharacterized protein (PDB: 3H36). Therefore, five models were calculated by the de novo method (Figure S5, only first model shown).
ECSIT
For the ECSIT structure, we used two web-based program packages, Robetta (Kim et al., 2004) and Phyre2 (Kelley et al., 2015). The N-terminal domain (N-ECSIT) was assigned to residues Ser49-Gly266, excluding the 48-residue mitochondrial targeting signal. On the basis of the Robetta domain prediction, the C-terminal domain of ECSIT from Leu249 to Ser431 was divided into two sub-domains, termed ECSIT-M (Ile267 to Gly340), and ECSIT-C (Tyr341 to Ser431). The structure of N-ECSIT was calculated from the structure of pentatricopeptide repeat protein (PDB: 4OE1) with a confidence score of 0.249. Although this domain contained only helices, the overall structure based on the five models is relatively flexible (Figure S5). The ECSIT-C domain (Tyr341-Ser431) was predicted to have an isomerase fold (PDB: 4R3U) with a homology confidence score of 0.282. All five calculated models contained two helices and four β-strands with slightly different conformations (Figure S5). On the other hand, the ECSIT-M domain, Ile267-Gly340, had no good homology model, giving a 0.211 confidence score with a mammalian transport protein (PDB: 3EGD). However, when this domain was modeled as a homodimer using the GalaxyWeb server (Lee et al., 2013), it shared a 27% sequence identity with Bacillus subtilis YojF homodimer protein (PDB: 1NJH). Thus, this domain is most likely responsible for ECSIT homodimer formation. These Robetta-based results agree well with those obtained for the N-ECSIT domain using the Phyre2 online server (Kelley et al., 2015). However, Phyre2 was not able to find a good homology model for the C-ECSIT domain (Ile267-Ser431), predicting it to be distantly related to the pleckstrin homology domain, and was not able to predict the dimer formation of ECSIT via the ECSIT-M domain. Figure S5 shows the overall domain prediction and structural models obtained by Robetta.
ACAD/ECSIT/NDUFAF1 ternary complex
Once the structures of all individual domains were modeled, the docked models between the ECSIT N-terminal domain and the NDUFAF1 C-terminal domain and between the ECSIT C-terminal domain and the ACAD9 dimer were obtained using the ClusPro online server (Ignatov et al., 2019; Kozakov et al., 2013; Vajda et al., 2017). These partial models were then manually placed onto the corresponding DAMMIN-refined ab initio models using the CHIMERA program (Pettersen et al., 2004). Figure 4B shows the possible structures of ACAD9 alone, the ACAD9/ECSIT binary complex, and the ACAD9/ECSIT/His6-SMT3-NDUFAF1 ternary complex, fit onto the beads models derived from the SAXS data. The ternary complex contains two heterohexamers, interacting with each other through NDUFAF1. Each heterohexamer comprises one ACAD9 dimer, one ECSIT dimer, and two NDUFAF1 monomers, in agreement with SEC results (Figure 3D). Examination of docking results of ACAD9 and ECSIT-C using ClusPro (Kozakov et al., 2017; Vajda et al., 2017), show that 16 of the 22 final models place ECSIT-C (Tyr341-Ser431) in the N-terminal domain, bound to the ACAD9 surface (Figure 4C), at the same position as the binding site for electron transfer protein (ETF) (Figures 4D–4F). This binding site is also near the FAD binding site, indicating that the C-terminal domain of ECSIT is directly responsible for the FAD depletion of ACAD9.
Comparison of the electrostatic surface maps of ACAD9 and VLCAD, shown in Figure 4G, provides a basis for ECSIT binding to ACAD9 but not VLCAD. More positively charged residues in ACAD9 compared with VLCAD include Arg85 (Ala80, mature VLCAD residue number in parenthesis), Arg195 (Ala191), Lys258 (Pro255), Arg317 (Gly314), Lys330 (Thr327), Lys334 (Glu331), Arg335 (Lys332), Arg408 (Lys404), and Arg609 (Gly605). Some of these positively charged residues may directly interact with the highly negatively charged ECSIT C-terminal domain. Based on its amino acid sequence, ECSIT is negatively charged with a calculated pI value of 5.5; however, the negative charges are more concentrated on the C-terminal domain. The calculated pI of the N-terminal domain (Ser49-Gly266) is 9.4, while that of C-terminal domain (Trp341-Ser431) is 4.1.
Discussion
Although first identified as a member of the acyl-CoA dehydrogenase family involved in fatty acid oxidation, ACAD9 has been demonstrated to be critical for stability of the MCIA complex, which is essential for biosynthesis of mitochondrial complex I (Formosa et al., 2020; Guerrero-Castillo et al., 2017). Mutations in ACAD9 result in defective complex I assembly and deficiencies in oxidative phosphorylation in addition to defects in fatty acid oxidation (Nouws et al., 2010, 2014; Schiff et al., 2015). Unlike ECSIT (Evolutionarily Conserved Signaling Intermediate in Toll pathway), which acts in the cytoplasm as a signaling protein in the Toll BMP pathways (Kopp et al., 1999) (Vogel et al., 2007), both ACAD9 functions are localized to the same organelle, the mitochondrion.
In the current study, we have expressed, purified, and characterized human full-length ACAD9, NDUFAF1, and ECSIT, and characterized their interactions in binary complexes and the ternary, core MCIA complex. ACAD9 variants associated with human CI deficiency have also been examined. Wild-type and variant ACAD9 proteins, with or without a His tag, purified as dimers, with a flavin content of approximately 0.7 mol/mol protein. All variants examined formed the ternary complex, consistent with binding of ECSIT to the N-terminal domain of ACAD9, and, with the exception of the E426Q catalytic residue mutant, exhibited dehydrogenation activity. Taken together, no direct relationship was observed between the catalytic activity and CI assembly function; thus, these two functions are completely distinct.
The MCIA core complex
Figure 5 shows a simplified diagram of our proposed model of the ACAD9/ECSIT/NDUFAF1 ternary complex, showing that ECSIT bridges between NDUFAF1 and ACAD9 through its N- and C-terminal domains, respectively. Pull-down assays and similar half-maximal FAD concentrations for restoration of dehydrogenation activity to ACAD9/ECSIT and ACAD9/C-ECSIT complexes show that ACAD9 interacts with both ECSIT and C-ECSIT, with similar affinities, consistent with binding of the ECSIT C-terminal domain to ACAD9. SEC results indicate the ACAD9/ECSIT binary complex is composed of a single ACAD9 dimer bound to one ECSIT dimer, and this stoichiometry is maintained in the ternary complex. In contrast, the ACAD9/C-ECSIT binary complex consists of one C-ECSIT tetramer and two ACAD9 dimers. Although full-length ESCIT interacts with NDUFAF1 directly, neither ACAD9 nor C-ESCIT interact with NDUFAF1. Additionally, the amino acid sequence of the linker between the N- and C-terminal domains of ACAD9 is not important for ECSIT binding or dehydrogenation activity, but a minimum length of ∼12 residues is required for proper alignment of the two domains to form a functional ACAD9 molecule.
Figure 5.

Cartoon model of the ternary complex ACAD9/ECSIT/NDUFAF1
The ECSIT C-terminal domain binds to the N-terminal domain of ACAD9 and the N-terminal domain of ECSIT binds to NDUFAF1. Gray circles indicate proposed binding sites of TMEM126B and TMEM186.
In vitro, the ACAD9/ECSIT/NDUFAF1 ternary complex exists as a dimer of heterohexamers, with each heterohexamer composed of one ACAD9 dimer, one ECSIT dimer, and 2 NDUFAF1monomers, in a head-to-head orientation (see Figures 3D and 4B). In each heterohexamer, interactions between the three proteins stabilize the ternary complex. Interactions between His6-SMT3-NDUFAF1 and the N-terminal domain of ECSIT stabilize the His6-SMT3-NDUFAF1/ESCIT binary complex, consistent with the previous observation that ECSIT is required for the stabilization of NDUFAF1 (Vogel et al., 2007). Protein-protein interactions in the ternary complex further stabilize NDUFAF1, permitting removal of the His6-SMT3 tag.
However, in vivo, the ternary complex most likely exists as a single heterohexamer, with NDUFAF1 interacting with another assembly factor. Presence of a heterohexamer is consistent with profiling of CI assembly intermediates (Guerrero-Castillo et al., 2017). The Pp-b intermediate (also referred to as ND2 module (Formosa et al., 2020)), containing subunits ND2, ND3, ND4L, ND6, NDUFC1, and NDUFC2, and assembly factors ACAD9, ECSIT, NDUFAF1, COA1, TMEM186, and TMEM126B was reported to migrate at ∼ 436 kDa in blue native PAGE, close to the 465 kDa predicted by the presence of the heterohexamer containing one ACAD9 dimer, two ECSIT molecules, and two NDUFAF1 molecules. Similarly, the calculated molecular weight of the Q/PP intermediate containing the ACAD9/ECSIT/NDUFAF1 heterohexamer, 815 kDa, is similar to the ∼800 kDa obtained by blue native PAGE.
These observations provide a physical basis for ACAD9 as a primary determinant of MCIA complex stability, consistent with previous knockdown and knockout studies. ACAD9 or ECSIT knockdown decreased NDUFAF1 protein levels (Dunning et al., 2007; Nouws et al., 2010; Vogel et al., 2007), while Formosa et al. have demonstrated that NDUFAF1 knockout decreases ECSIT levels and vice versa, with no effect on ACAD9 levels (Formosa et al., 2020).
ACAD9-ECSIT interaction
FAD is absent in the complex between ACAD9 and full-length ECSIT, and catalytic activity is lost upon ECSIT binding, as previously shown for C-ECSIT (Giachin et al., 2021). The concentration dependence for ECSIT-induced loss of catalytic activity suggests preferential binding of ECSIT to ACAD9 lacking FAD (apo-ACAD9); however, addition of a stoichiometric amount of ECSIT leads to complete loss of catalytic activity.
ECSIT also deflavinated the variants R469W, R518H, R532W, and Loop12-ACAD9, with loss of enzyme activity (Table 1). Since FAD is bound to the N-terminal domain of ACAD9, and these mutation sites are all located in the C-terminal domain of ACAD9, it is most likely that ECSIT interacts with the N-terminal domain of the ACAD9 homodimer. Docking results also suggest that C-ECSIT binds to the ACAD9 surface near the FAD binding site (Figures 4B and 4C). Thus, ECSIT binding to the N-terminal domain provides a basis for direct deflavination of ACAD9 by ECSIT and reversal of deflavination by excess FAD.
Upon inspection of the ACAD9-ECSIT interface in the modeled complex (Figures 4C–4F), it should be noted that the C-terminal domain of ECSIT binds to ACAD9 at the ETF binding site in the N-terminal domain, assuming that human ETF binds to ACAD9 in the same manner as it binds to medium chain acyl-CoA dehydrogenase (Toogood et al., 2005). ETF is the electron carrier from various acyl-CoA dehydrogenases (including ACAD9) to ETF-ubiquinone oxidoreductase, linking the fatty acid oxidation function of ACAD9 to the main mitochondrial respiratory chain. In the dehydrogenation reaction, ETF interacts with both ACAD9 and VLCAD transiently. On the other hand, ACAD9, but not VLCAD, binds to ECSIT very tightly, forming the MCIA complex. Mutually exclusive binding of ETF or ECSIT implies that, at a given time, ACAD9 can interact with only one of these two partners, i.e., functioning in either CI assembly or as a dehydrogenase in fatty acid metabolism.
With the exception of deflavination of ACAD9 by C-ECSIT, our conclusions regarding interaction of full-length ECSIT with ACAD9 differ from those obtained from studies using C-ECSIT (Giachin et al., 2021). We find that dehydrogenation activity can be restored to both the ACAD9/ECSIT complex and the ACAD9/ECSIT/NDUFAF1 ternary complex by addition of excess FAD. We observe a single ACAD9/ECSIT binary complex, with one ACAD9 dimer bound to one ECSIT dimer. For ACAD9/C-ECSIT complex, we observe two ACAD9 dimers bound to a C-ECSIT tetramer, while Giachin et al. reported multiple species, ascribed to 3 or 4 C-ECSIT monomers per ACAD9 dimer. Based on mutation and modeling studies, we propose that ECSIT binds to the ACAD9 N-terminal domain rather than the C-terminal domain suggested by cryo-EM results. Binding of ECSIT to the N-terminal domain of ACAD9 and competition for ETF also provides an explanation for the observation of Giachin et al. that FAD was unable to restore catalytic activity of the C-ECSIT/ACAD9 complex when ETF was the electron acceptor.
ACAD9 vs. VLCAD
ACAD9 has about 20% of VLCAD’s catalytic activity and, unlike VLCAD, is a CI assembly factor. What are the differences between these two ACADs that make each protein unique? What is the mechanism by which ACAD9 participates in CI assembly? To address these questions, we have constructed an ACAD9 homology model based on the VLCAD structure (PDB: 3B96, (McAndrew et al., 2008)).
VLCAD is a homodimer and each monomer contains an MCAD-like N-terminal domain and a C-terminal domain linked by a highly mobile arginine-rich region (McAndrew et al., 2008). We have previously reported that both VLCAD and ACAD9 have dehydrogenation activity toward long chain acyl-CoAs and their trypsin digestion patterns are also very similar (Schiff et al., 2015). In the presence of trypsin, the arginine-rich linker region between the N-terminal and C-terminal domains in ACAD9 was easily cleaved, as was observed in VLCAD. Both the N-terminal and C-terminal domains were relatively stable, and yet they were still bound to each other after limited trypsin digestion, retaining 90% of the enzyme activity (Schiff et al., 2015). However, the ACAD9 dimer is less stable and more susceptible to trypsin cleavage than VLCAD. This may account for the decreased FAD content. Unlike VLCAD, which contains the full stoichiometric amount of FAD, ACAD9 contains at best only about 70% FAD. Our homology model shows that, despite structural similarities, amino acid substitutions on the Tyr412-Ile423 helix disrupt salt bridges, likely leading to higher flexibility of the homodimer interface and decreased affinity for FAD. Mutations associated with human CI deficiency (E413K, R414C, R417C, and D418G) are also located on this helix, severely decreasing dimer stability (Figure 4A) (Schiff et al., 2015).
Interestingly, when ClusPro is used to dock the ECSIT C-terminal domain with VLCAD, none of the 30 final models are bound in the ETF binding site. In fact, they are scattered over a wide range of the VLCAD molecule. Although VLCAD and ACAD9 are both able to interact transiently with ETF for fast interprotein electron transfer, VLCAD lacks positively charged residues that interact with the highly negatively charged C-ECSIT domain to stabilize the binary complex (Figure 4G).
Recycling of the MCIA complex
Release of assembly factors occurs at the final step of CI assembly, concomitant with or immediately following incorporation of the N module (Formosa et al., 2020; Guerrero-Castillo et al., 2017), and we hypothesize that the core MCIA complex is recycled for subsequent rounds of CI assembly. As discussed above, restoration of ACAD9 dehydrogenation activity requires both dissociation of the ternary complex to restore ETF accessibility and re-flavination of apo-ACAD9. The stability of the ternary complex as well as relative resistance to proteasomal degradation (Fuhrmann et al., 2018; Szczepanowska et al., 2020) argues against dissociation. Although FAD synthase has been shown to transfer the FAD cofactor to client proteins (Giancaspero et al., 2015), no mechanism for re-flavination has been identified. Thus, it is likely that apo-ACAD9 remains associated with the core MCIA complex through multiple CI assembly cycles.
ACAD9 mutation sites
Although ACAD9 mutations account for a large number of patients with CI deficiency, no clear correlations have been established between ACAD9 dehydrogenase activity or CI assembly function and disease severity. Nouws et al. (2014) and Schiff et al. (2015) have shown that ACAD9 plays a physiological role in long-chain fatty acid oxidation (Schiff et al., 2015). Thus, it has been proposed that ACAD9 plays a dual role as a long chain acyl-CoA dehydrogenase (Martínez-Limón et al., 2016) and a CI assembly factor, especially in certain organs such as brain where ACAD9 is more abundant (Ensenauer et al., 2005).
Effects of CI-deficient human variants suggest interactions of NDUFAF1 and ACAD9 with different complex I subunits and/or other assembly factor(s) during the complex I assembly process. The human ACAD9-deficient variants R469W, R518H, and R532W possess dehydrogenation activity (Table 1) and form the ternary complex with ECSIT and NDUFAF1 (Figure 3), yet are CI-deficient, suggesting that the C-terminal domain of ACAD9 interacts with other assembly factors, such as TMEM126B, required for biosynthesis of the ND2 module (Formosa et al., 2020; Heide et al., 2012). TMEM186 does not bind ACAD9 directly but may interact with NDUFAF1 (Formosa et al., 2020). The ability of the R518H protein to form the 500–850kDa intermediates (Nouws et al., 2010) suggests the C-terminal domain of ACAD9 is necessary for association with subunits in the PD module.
Since FAD is not present in the ACAD9/ECSIT/NDUFAF1 complex, it is not well understood why riboflavin supplementation is helpful for some CI-deficient patients, such as those with S602F, R532W, R518H, R418G, and R414C mutations but not others (Repp et al., 2018). With the exception of R518H, these mutations are predicted to be involved in ACAD9 dimer interface interactions (Figure 4A). Since FAD binding enhances ACAD9 dimer stability, it is possible that the beneficial effect of flavin therapy is based on improved ACAD9 protein stability (Martínez-Limón et al., 2016). More studies are needed to determine the effectiveness and mechanism of flavin therapy on patients with ACAD9 mutations.
In conclusion, we have demonstrated direct interactions between the C-terminal domain of ECSIT and the N-terminal domain of ACAD9 and interaction of the N-terminal domain of ECSIT with NDUFAF1, resulting in formation of a stable ternary complex (core MCIA complex). Localization of C-ECSIT binding to the ETF binding site, near the FAD, is consistent with FAD loss and the mutually exclusive dehydrogenation and CI assembly functions of ACAD9. Homology modeling of ACAD9 and the ACAD9/ECSIT/NDUFAF1 ternary complex provides a framework for classification of ACAD9 human mutations and rationale for riboflavin supplementation. In the ternary complex, the C-terminal domain of ACAD9 and NDUFAF1 interact with other assembly factors (including TMEM126B and TMEM186) in assembly of the ND2 module. A remaining question is the role of deflavination in regulation of the two independent functions of ACAD9. In the absence of an active re-flavination process, deflavination and interference with ETF binding irreversibly commits the ACAD9 to its CI assembly role. Facile deflavination by ECSIT coupled with the presence of excess ACAD9 over ECSIT and NDUFAF1 in tissues such as brain provides a mechanism for regulation of the balance between the dehydrogenation activity and complex I assembly, linking fatty acid metabolism and oxidative phosphorylation.
Limitations of the study
The model of the ACAD9/ECSIT/NDUFAF1 is based upon pulldown and SEC-SAXS results and homology modeling and require higher resolution structural studies for confirmation. Proposed interactions with additional assembly factors such as TMEM126B and TMEM186 require further experimental verification.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| BL21(DE3) | Thermo Fisher Scientific (Rockford, IL) | EC0114 |
| Chemicals, peptides, and recombinant proteins | ||
| Ubiquitin like protease (ULP) | Sigma-Aldrich (St. Louis, MO) | SAE0067 |
| Thrombin | Sigma-Aldrich (St. Louis, MO) | |
| Palmitoyl-coenzyme A | Avanti Polar Lipids, Inc. (Alabaster, AL) | |
| Restriction enzymes, DNA polymerases, ligase, DNA, and protein markers | Thermo Fisher Scientific (Rockford, IL) | |
| Ni-NTA resin | Clontech Laboratories (Mountain View, CA) | |
| Deposited data | ||
| SMT3 structure | PDB: 3PGE | |
| VLCAD structure | PDB: 3B96 | |
| MCAD-ETF structure | PDB: 2A1T | |
| Oligonucleotides | ||
| 5′-TAATACGACTCACTATAGGG-3′ | This work | T7 promoter |
| 5′-CCCGGC GTCGAC TCA GTG ATG ATG-GTG GTG ATG GCT CCC GCA GGT GCG GTC CAG AGG-3′ | This work | 3′ antisense |
| Recombinant DNA | ||
| ECSIT (UniProtKB-Q9BQ95) cDNA | GenScript (Piscataway, USA) | |
| NDUFAF1 (UniProtKB-Q9Y375) cDNA | GenScript (Piscataway, USA) | |
| pET21b vector | Sigma Aldrich | 69741 |
| pET28a vector | Sigma Aldrich | 69864 |
| His6-SMT3-pET28a vector | ||
| QuickChange Site-Directed Mutagenesis Kit | Agilent | 200519 |
| Software and algorithms | ||
| Protein Calculator v3.4 | http://protcalc.sourceforge.net/ | |
| ATSAS | (Franke et al., 2017) | https://www.embl-hamburg.de/biosaxs/software.html |
| PRIMUS | (Konarev et al., 2003) | https://www.embl-hamburg.de/biosaxs/software.html |
| GNOM | (Svergun, 1992) | https://www.embl-hamburg.de/biosaxs/software.html |
| DAMMIF | (Franke and Svergun, 2009) | https://www.embl-hamburg.de/biosaxs/software.html |
| DAMSTART | (Petoukhov et al., 2012) | https://www.embl-hamburg.de/biosaxs/software.html |
| DAMMIN | (Svergun, 1999) | https://www.embl-hamburg.de/biosaxs/software.html |
| SWISS-MODEL server | (Waterhouse et al., 2018) | https://swissmodel.expasy.org/ |
| Robetta web server | (Kim et al., 2004) | https://robetta.bakerlab.org/ |
| GalaxyGemini server | (Lee et al., 2013) | http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=GEMINI |
| SUPCOMB | (Kozin and Svergun, 2001) | https://www.embl-hamburg.de/biosaxs/software.html |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| FoXS server | (Schneidman-Duhovny et al., 2013; Svergun, 1999) | https://modbase.compbio.ucsf.edu/foxs/ |
| Phyre2 | (Kelley et al., 2015) | http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index |
| Graphpad PRISM | https://www.graphpad.com/company/ | |
Resource availability
SAXS data codes (www.sasdbd.org) are: SASDJ34 for ACAD9; SASDJ44 for ACAD9-ECSIT (binary complex); and SASDJ54 for ACAD9-ECSIT-NDUFAF1 (ternary complex).
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jung-Ja P. Kim, 1Department of Biochemistry, Medical College of Wisconsin, Milwaukee WI 53226, USA, jjkim@mcw.edu.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Proteins were expressed in E. coli BL21(DE3). Briefly, E. coli cells transformed with ECSIT or NDUFAF1 plasmids were grown at 30°C until the OD600 reached 0.6, then induced with 0.5mM IPTG at 16°C and allowed to grow overnight.
Method details
Materials
Palmitoyl-coenzyme A was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Restriction enzymes, DNA polymerases, ligase, DNA, and protein markers were purchased from Thermo Fisher Scientific (Rockford, IL). Ni-NTA resin was purchased from Clontech Laboratories (Mountain View, CA). All other chemicals were purchased from Sigma Aldrich (St. Louis, MO) or RPI Corp. (Mount Prospect, IL).
Cloning, expression, and purification of human ACAD9, ECSIT, and NDUFAF1 and variants
Cloning of human ACAD9 in the pET21a vector (ACAD9_pET21a) has been previously described (Ensenauer et al., 2005). To facilitate purification, a C-terminal His-tag (-GSHHHHHH) was introduced to the non-tagged clone (referred to as ACAD9-His6_pET21b) by PCR with the following primers: T7 promoter 5′-TAATACGACTCACTATAGGG-3′ as 5′ sense primer and 5′-CCCGGC GTCGAC TCA GTG ATG ATG-GTG GTG ATG GCT CCC GCA GGT GCG GTC CAG AGG-3′ as 3′ antisense primer. All ACAD9 mutants used in this study, including Arg469Trp, Arg518His, Arg532Trp, and Glu426Gln, were cloned in ACAD9-His6_pET21b vector using the QuickChange Site-Directed Mutagenesis Kit as previously reported (Schiff et al., 2015). Two mutants with a shortened linker were cloned similarly: Loop8-ACAD9 and Loop12-ACAD9, in which residues between Thr450 and Asp480 were replaced by 8 and 12 residues of GGS repeats, respectively. cDNAs that encode mature proteins of human mitochondrial ECSIT (UniProtKB-Q9BQ95) and NDUFAF1 (UniProtKB-Q9Y375) were purchased from as codon-optimized for E. coli expression and subcloned into pET28a vector for expression. The N-terminal domain of ESCIT (Ser49-Ser269, which excludes the 48- GenScript (Piscataway, USA) residue mitochondrial targeting sequence and is hereafter referred to as N-ECSIT), and the C-terminal domain of ESCIT (Leu249-Ser431, referred to as C-ECSIT), were also cloned into pET28a vector. To increase the solubility of expressed NDUFAF1 protein, the NDUFAF1 cDNA was subcloned into a His6-SMT3-containing pET28a vector and expressed as a SUMO fusion protein (referred to as His6-SMT3-NDUFAF1). The His6-SMT3 tag was cleaved from NDUFAF1 by protease ULP1.
Both ACAD9 and ACAD9-His6 were expressed and purified as described previously (Ensenauer et al., 2005; Schiff et al., 2015). ECSIT and NDUFAF1 proteins were expressed in BL21(DE3) cells as described above and purified in a similar manner as ACAD9-His6 (Schiff et al., 2015). All the proteins were purified by Ni-NTA affinity column to homogeneity, as judged by SDS-PAGE, and dialyzed in HPLC buffer (25 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 5% glycerol) before further purification by size exclusion chromatography. For each protein, the peak portion corresponding to ACAD9 dimer, ECSIT dimer, C-ECSIT tetramer, or His6-SMT3-NDUFAF1 monomer was collected and concentrated, and 20% glycerol was added. Purified proteins were frozen in liquid nitrogen and stored at −80°C.
Determination of protein concentration
Concentrations of all purified proteins were estimated according to their UV absorbance at 280nm. The apo-protein absorbance extinction coefficient at 280 nm was calculated according to the online Protein Calculator v3.4 (C. Putnam, The Scripps Research Institute, La Jolla, CA). Calculated molar extinction coefficients at 280 nm of ECSIT, C-ECSIT, and His6-SMT3-NDUFAF1 are 43.8 mM−1⋅cm−1, 30.4 mM−1⋅cm−1 and 49.5 mM−1⋅cm−1, respectively. The calculated molar extinction coefficient for apo-ACAD9 (not including FAD) is 31.9 mM−1⋅cm−1. To estimate the ACAD9 protein concentration and its FAD content, FAD molar extinction coefficients of 12 mM−1⋅cm−1 at 450 nm and 22 mM−1⋅cm−1 at 280 nm were used.
Measurement of α,β-dehydrogenation of fatty acyl-CoA
α,β-Dehydrogenation activity of ACAD9 or VLCAD for fatty acyl-CoA was assayed using 0.1 μM purified ACAD9 protein, 50 μM palmitoyl-CoA as the substrate and 200 μM ferricenium hexafluorophosphate as the final electron acceptor, in a final volume of 0.5 mL, as described by Lehman et al. (1990). One unit of the enzyme activity is defined as 1 μmole of ferrocene produced in one minute. To test the effect of exogenous FAD on dehydrogenation activity, 10 μM protein was preincubated with varying concentrations of FAD and then diluted directly into the assay mix. To examine the ECSIT effect on the dehydrogenation activity of ACAD9, varying amounts of ECSIT were added to the ACAD9 protein solution and incubated on ice for 30 minutes prior to the activity assay. The effect of exogenous FAD on dehydrogenation activity of ACAD9 in the ACAD9-ECSIT binary complex or ACAD9-ECSIT-NDUFAF1 ternary complex was determined by incubating the HPLC purified complexes (10 μM) with varying concentrations of FAD prior to dilution into the assay mix.
Ni-NTA pull-down assay
Proteins were mixed (each at 5 μM final concentration) in 0.5 mL buffer B (25 mM Tris-HCl pH 8.0, 300 mM NaCl, and 5% glycerol) and incubated on ice for 30 minutes. Ni-NTA resin (50 μL) was then added to the protein mixture and further incubated at 4°C for 1 hour while mixing in a cold room using a rotator. The protein-bound Ni-NTA resin was then washed three times with 500 μL of 20 mM imidazole before eluting with 100 μL of 200 mM imidazole in buffer B. The eluted fractions were directly analyzed by SDS PAGE.
Size exclusion chromatography
An HPLC size-exclusion column (Bio-Rad Enrich SEC 650, 10/30) was run at a flow rate of 0.4 ml/min using a Shimadzu Prominence HPLC system. For preparative chromatography, 1-5 mg protein in 0.5 ml buffer was injected, while 50-200 μg of protein in 200 μl buffer (25 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 5% glycerol) was injected for analytical chromatography. The column was calibrated with a set of standard molecular weight markers, cytochrome c (12.3kDa), ovalbumin (44kDa), bovine serum albumin (66kDa), IgG (158kDa), apoferritin (443kDa), and thyroglobulin (670kDa).
Small-angle X-ray scattering
All three proteins, ACAD9 (7.1 mg/ml, dimeric protein), ECSIT (0.41 mg/ml, dimeric), and His6-SMT3-NDUFAF1 (0.52 mg/ml, monomeric), were purified by HPLC and prepared in 25 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1 mM DTT, and 5% glycerol. For the binary complex ACAD9/ECSIT and the ternary complex ACAD9/ECSIT/His6-SMT3-NDUFAF1, the corresponding proteins were mixed in an equimolar ratio and concentrated to ≥1 mg/ml, immediately prior to the SEC-SAXS experiments. SEC-SAXS experiments were conducted at the 18-ID Biophysics Collaborative Access Team beam-line (BioCAT), Advanced Photon Source, Argonne National Laboratory (APS-ANL), Chicago, IL (Fischetti et al., 2004) during the “BioCAT Advanced SAXS Training Course,” held in October 2017. All samples were applied to a 24 ml GE Superose 6 Increase column, which was directly coupled to the SAXS cell. SAXS measurements were taken both immediately before and after the protein peaks to establish the baseline scattering. BioCAT beam-line–specific pipelines were used for data collection. The ATSAS suite (Franke et al., 2017) was used for data reduction; PRIMUS (Konarev et al., 2003) for Guinier Analysis; and GNOM (Svergun, 1992) for calculation of the radius of gyration, Rg, and the pair-distance distribution function (Svergun, 1992), P(r). The low-resolution ab initio models were first calculated in DAMMIF (Franke and Svergun, 2009) with the P2 symmetry. The 20 initial models of ACAD9, 40 initial models of the ACAD9/ECSIT binary complex, and 40 initial models of the ACAD9/ECSIT/His6-SMT3-NDUFAF1 ternary complex were clustered and averaged using DAMCLUST (Petoukhov et al., 2012). The clustered models were run on DAMSTART (Volkov and Svergun, 2003) and refined by DAMMIN (Svergun, 1999) using the DAMSTART beads model pdb file as the initial model. The homology model of ACAD9 was obtained from the SWISS-MODEL server (Waterhouse et al., 2018). Structures of NDUFAF1 and ECSIT domains were modeled using the Robetta web server (Kim et al., 2004) and the GalaxyGemini server (Lee et al., 2013). SMT3 structure was from its X-ray crystal structure (PDB: 3PGE). For ACAD9, SUPCOMB (Kozin and Svergun, 2001) was used to superimpose the homology-modeled structures onto the final SAXS ab initio beads model. For the ACAD9/ECSIT binary complex and the ACAD9/ECSIT/His6-SMT3-NDUFAF1 ternary complex, structures of ACAD9, ECSIT, NDUFAF1, and SMT3 domains were manually placed onto their SAXS ab initio beads models using the UCSF Chimera program (Pettersen et al., 2004). The SAXS curves calculated from these homology models were overlaid onto the experimental data using FoXS server (Schneidman-Duhovny et al., 2013; Svergun, 1999).
Quantification and statistical analysis
GraphPad Prism was used for determination of kinetic parameters for FAD dependence of dehydrogenation activity and statistical analysis. Statistical parameters are reported in the text or table legend.
Acknowledgments
We thank Drs. V. Jackson and L. Olson for helpful discussions and suggestions on the manuscript. We thank Dr. Srinivas Chakravarthy, BioCAT, Argonne National Laboratory, sector 18, for his assistance with SAXS data collection and processing during their fall 2017 SAXS data collection workshop, which was supported by grant to Thomas Irving 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This work was supported by National Institutes of Health grants, GM29076 (J.-J.P.K.) and DK78755 (J.V.).
Author contributions
Conceptualization, J.-J.P.K. and C.X.; methodology, J.-J.P.K., C.X., and A.L.S., formal analysis, C.X., A.L. S. and J.-J.P.K.; investigation, C.X., B.L., and Z.F.; resources, J.V. and A.-W.M.; writing-original draft, C.W., A.L.S., and J.-J.P.K.; writing-review & editing, J.-J.P.K., A.L.S., C.X., A.-W.M., and J.V., visualization, C.X., A.L.S., and J.-J.P.K., supervision and project administration, J.-J.P.K.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103153.
Supplemental information
Data and code availability
-
•
Data: Protein structure data have been deposited at Protein Data Bank and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Code: This paper does not report original code.
References
- Dunning C.J., McKenzie M., Sugiana C., Lazarou M., Silke J., Connelly A., Fletcher J.M., Kirby D.M., Thorburn D.R., Ryan M.T. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. Embo J. 2007;26:3227–3237. doi: 10.1038/sj.emboj.7601748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elurbe D.M., Huynen M.A. The origin of the supernumerary subunits and assembly factors of complex I: a treasure trove of pathway evolution. Biochim. Biophys. Acta. 2016;1857:971–979. doi: 10.1016/j.bbabio.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Ensenauer R., He M., Willard J.M., Goetzman E.S., Corydon T.J., Vandahl B.B., Mohsen A.W., Isaya G., Vockley J. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J. Biol. Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- Fischetti R., Stepanov S., Rosenbaum G., Barrea R., Black E., Gore D., Heurich R., Kondrashkina E., Kropf A.J., Wang S. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J. Synchrotron Radiat. 2004;11:399–405. doi: 10.1107/S0909049504016760. [DOI] [PubMed] [Google Scholar]
- Formosa L.E., Muellner-Wong L., Reljic B., Sharpe A.J., Jackson T.D., Beilharz T.H., Stojanovski D., Lazarou M., Stroud D.A., Ryan M.T. Dissecting the roles of mitochondrial complex I intermediate assembly complex factors in the biogenesis of complex I structure of mammalian respiratory complex I. Cell Rep. 2020;31:107541. doi: 10.1016/j.celrep.2020.107541. [DOI] [PubMed] [Google Scholar]
- Formosa L.E., Ryan M.T. Mitochondrial OXPHOS complex assembly lines. Nat. Cell Biol. 2018;20:511–513. doi: 10.1038/s41556-018-0098-z. [DOI] [PubMed] [Google Scholar]
- Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M. Atsas 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017;50:1212–1225. doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann D.C., Wittig I., Drose S., Schmid T., Dehne N., Brune B. Degradation of the mitochondrial complex I assembly factor TMEM126B under chronic hypoxia. Cell Mol. Life Sci. 2018;75:3051–3067. doi: 10.1007/s00018-018-2779-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachin G., Bouverot R., Acajjaoui S., Pantalone S., Soler-Lopez M. Dynamics of human mitochondrial complex I assembly: implications for neurodegenerative diseases. Front. Mol. Biosci. 2016;3:43. doi: 10.3389/fmolb.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachin G., Jessop M., Bouverot R., Acajjaoui S., Saïdi M., Chretien A., Bacia-Verloop M., Signor L., Mas P.J., Favier A. Assembly of the mitochondrial complex I assembly complex suggests a regulatory role for deflavination. Angew. Chem. Int. Ed. Engl. 2021;60:4689–4697. doi: 10.1002/anie.202011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancaspero T.A., Colella M., Brizio C., Difonzo G., Fiorino G.M., Leone P., Brandsch R., Bonomi F., Iametti S., Barile M. Remaining challenges in cellular flavin cofactor homeostasis and flavoprotein biogenesis. Front. Chem. 2015;3:30. doi: 10.3389/fchem.2015.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Castillo S., Baertling F., Kownatzki D., Wessels H.J., Arnold S., Brandt U., Nijtmans L. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017;25:128–139. doi: 10.1016/j.cmet.2016.09.002. [DOI] [PubMed] [Google Scholar]
- He M., Rutledge S.L., Kelly D.R., Palmer C.A., Murdoch G., Majumder N., Nicholls R.D., Pei Z., Watkins P.A., Vockley J. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am. J. Hum. Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide H., Bleier L., Steger M., Ackermann J., Drose S., Schwamb B., Zornig M., Reichert A.S., Koch I., Wittig I. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Ignatov M., Liu C., Alekseenko A., Sun Z., Padhorny D., Kotelnikov S., Kazennov A., Grebenkin I., Kholodov Y., Kolosvari I. Monte Carlo on the manifold and MD refinement for binding pose prediction of protein-ligand complexes: 2017 D3R grand challenge. J. Comput. Aided Mol. Des. 2019;33:119–127. doi: 10.1007/s10822-018-0176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.E., Chivian D., Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- Kopp E., Medzhitov R., Carothers J., Xiao C., Douglas I., Janeway C.A., Ghosh S. ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev. 1999;13:2059–2071. doi: 10.1101/gad.13.16.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D., Beglov D., Bohnuud T., Mottarella S.E., Xia B., Hall D.R., Vajda S. How good is automated protein docking? Proteins. 2013;81:2159–2166. doi: 10.1002/prot.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D., Hall D.R., Xia B., Porter K.A., Padhorny D., Yueh C., Beglov D., Vajda S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017;12:255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin M.B., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001;34:33–41. [Google Scholar]
- Lee H., Park H., Ko J., Seok C. GalaxyGemini: a web server for protein homo-oligomer structure prediction based on similarity. Bioinformatics. 2013;29:1078–1080. doi: 10.1093/bioinformatics/btt079. [DOI] [PubMed] [Google Scholar]
- Lehman T.C., Hale D.E., Bhala A., Thorpe C. An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal. Biochem. 1990;186:280–284. doi: 10.1016/0003-2697(90)90080-s. [DOI] [PubMed] [Google Scholar]
- Martínez-Limón A., Alriquet M., Lang W.H., Calloni G., Wittig I., Vabulas R.M. Recognition of enzymes lacking bound cofactor by protein quality control. Proc. Natl. Acad. Sci. U. S. A. 2016;113:12156–12161. doi: 10.1073/pnas.1611994113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew R.P., Wang Y., Mohsen A.W., He M., Vockley J., Kim J.J. Structural basis for substrate fatty acyl chain specificity: crystal structure of human very-long-chain acyl-CoA dehydrogenase. J. Biol. Chem. 2008;283:9435–9443. doi: 10.1074/jbc.M709135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimaki M., Wang X., McKenzie M., Thorburn D.R., Ryan M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Nouws J., Nijtmans L., Houten S.M., van den Brand M., Huynen M., Venselaar H., Hoefs S., Gloerich J., Kronick J., Hutchin T. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nouws J., Te Brinke H., Nijtmans L.G., Houten S.M. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum. Mol. Genet. 2014;23:1311–1319. doi: 10.1093/hmg/ddt521. [DOI] [PubMed] [Google Scholar]
- Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajda M., Gorba C., Mertens H.D., Konarev P.V., Svergun D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Repp B.M., Mastantuono E., Alston C.L., Schiff M., Haack T.B., Rotig A., Ardissone A., Lombes A., Catarino C.B., Diodato D. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018;13:120. doi: 10.1186/s13023-018-0784-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht A., Maddox J., Stirling A.J., Visaggio N., Seah S.Y. Characterization of novel acyl coenzyme A dehydrogenases involved in bacterial steroid degradation. J. Bacteriol. 2015;197:1360–1367. doi: 10.1128/JB.02420-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Caballero L., Guerrero-Castillo S., Nijtmans L. Unraveling the complexity of mitochondrial complex I assembly: a dynamic process. Biochim. Biophys. Acta. 2016;1857:980–990. doi: 10.1016/j.bbabio.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Schiff M., Haberberger B., Xia C., Mohsen A.W., Goetzman E.S., Wang Y., Uppala R., Zhang Y., Karunanidhi A., Prabhu D. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum. Mol. Genet. 2015;24:3238–3247. doi: 10.1093/hmg/ddv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff M., Mohsen A.W., Karunanidhi A., McCracken E., Yeasted R., Vockley J. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2013;109:21–27. doi: 10.1016/j.ymgme.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A.L., Kasper C.B. Differential contributions of NADPH-cytochrome P450 oxidoreductase FAD binding site residues to flavin binding and catalysis. J. Biol. Chem. 2000;275:41087–41091. doi: 10.1074/jbc.M008380200. [DOI] [PubMed] [Google Scholar]
- Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanowska K., Senft K., Heidler J., Herholz M., Kukat A., Höhne M.N., Hofsetz E., Becker C., Kaspar S., Giese H. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020;11:1643. doi: 10.1038/s41467-020-15467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood H.S., van Thiel A., Scrutton N.S., Leys D. Stabilization of non-productive conformations underpins rapid electron transfer to electron-transferring flavoprotein. J. Biol. Chem. 2005;280:30361–30366. doi: 10.1074/jbc.M505562200. [DOI] [PubMed] [Google Scholar]
- Vajda S., Yueh C., Beglov D., Bohnuud T., Mottarella S.E., Xia B., Hall D.R., Kozakov D. New additions to the ClusPro server motivated by CAPRI. Proteins. 2017;85:435–444. doi: 10.1002/prot.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R.O., Janssen R.J., van den Brand M.A., Dieteren C.E., Verkaart S., Koopman W.J., Willems P.H., Pluk W., van den Heuvel L.P., Smeitink J.A. Cytosolic signaling protein Ecsit also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev. 2007;21:615–624. doi: 10.1101/gad.408407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C., Panda S.P., Marohnic C.C., Martásek P., Masters B.S., Kim J.J. Structural basis for human NADPH-cytochrome P450 oxidoreductase deficiency. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13486–13491. doi: 10.1073/pnas.1106632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zhang W., Zou D., Chen G., Wan T., Zhang M., Cao X. Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem. Biophys. Res. Commun. 2002;297:1033–1042. doi: 10.1016/s0006-291x(02)02336-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: Protein structure data have been deposited at Protein Data Bank and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
Code: This paper does not report original code.