

Abstract
Lysine acylations are reversible and ubiquitous post-translational modifications that play critical roles in regulating multiple cellular processes. In the current study, highly abundant and dynamic acetylation, besides succinylation, was uncovered in a soil bacterium, Streptomyces coelicolor. By affinity enrichment using anti–acetyl-lysine antibody and the following LC−MS/MS analysis, a total of 1298 acetylation sites among 601 proteins were identified. Bioinformatics analyses suggested that these acetylated proteins have diverse subcellular localization and were enriched in a wide range of biological functions. Specifically, a majority of the acetylated proteins were also succinylated in the tricarboxylic acid cycle and protein translation pathways, and the bimodification occurred at the same sites in some proteins. The acetylation and succinylation sites were quantified by knocking out either the deacetylase ScCobB1 or the desuccinylase ScCobB2, demonstrating a possible competitive relationship between the two acylations. Moreover, in vitro experiments using synthetically modified peptides confirmed the regulatory crosstalk between the two sirtuins, which may be involved in the collaborative regulation of cell physiology. Collectively, these results provided global insights into the S. coelicolor acylomes and laid a foundation for characterizing the regulatory roles of the crosstalk between lysine acetylation and succinylation in the future.
Keywords: Streptomyces coelicolor, post-translational modification, lysine acetylation, lysine succinylation, crosstalk, sirtuin
Abbreviations: GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PTM, post-translational modification; SIRT, sirtuin; TCA, tricarboxylic acid; TSB, tryptic soy broth
Graphical Abstract
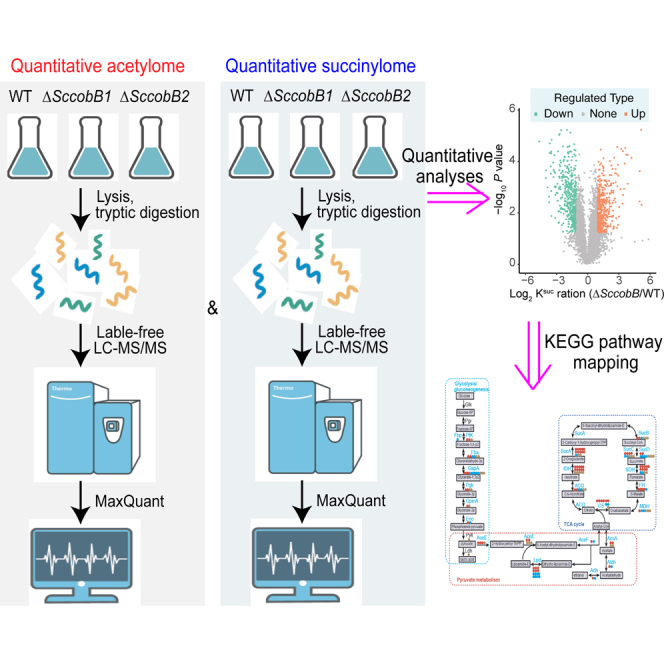
Highlights
-
•
A highly abundant and dynamic acetylation is discovered in Streptomyces coelicolor.
-
•
Quantitative acetylome and succinylome analyses in Streptomyces coelicolor.
-
•
The bimodification proteins are enriched in multiple metabolic pathways.
In Brief
The bimodification of lysine acylation has been observed in multiple organisms, while the understanding of acylation crosstalk remains at an early stage. In the current study, by implementing proteome-wide analyses, an extensive overlap of lysine acetylation and succinylation was revealed in S. coelicolor. Moreover, the modification sites were quantified by knocking out either the deacetylase ScCobB1 or the desuccinylase ScCobB2, demonstrating a possible competitive relationship between the acetylation and succinylation in vivo. Further analyses suggested that these bimodification proteins were enriched in multiple metabolic pathways including the tricarboxylic acid cycle and protein translation pathways.
The reversible protein post-translational modifications (PTMs) such as acetylation and succinylation function as efficient regulatory mechanisms that fine-tune protein activity, stability, subcellular localization, and others (1, 2). Among the 20 amino acid residues in proteins, lysine (K) is one of the most frequent targets for different PTMs. With the rapid development of high-resolution MS and immunoaffinity purification technology, diverse acylations on lysine, including acetylation (3, 4), succinylation (5), malonylation (6), glutarylation (7), propionylation (8), butyrylation (8), and lactylation (9), have been discovered in the past decade.
N-ε-Lysine acylation consists of two reversible reactions—the acylation, in which the acyl groups are added to the ε-amino of lysine residues by specific acyltransferases (2) or nonenzymatic reactions (10), and the deacylation, which is the removal of the acyl groups from the acylated proteins by deacylases (11, 12). The deacylations by sirtuins play dominant regulatory roles in cell physiology. Seven sirtuins (SIRT1–SIRT7) were identified in human cells that modulate multiple cellular processes (13), whereas only one sirtuin (CobBL) was reported in Escherichia coli as a bifunctional deacetylase and desuccinylase (14).
Acetylation among different acylations is one of the most well-studied lysine modifications that has gained increasing attentions. It has been reported to be involved in the regulation of multiple cellular processes, like gene transcription, cellular metabolism, DNA damage response, and aging (15, 16, 17, 18, 19, 20). Besides acetylation, succinylation is another well-studied lysine PTM, which was reported to regulate the tricarboxylic acid (TCA) cycle, protein translation, and energy metabolism (21, 22). Both acetylation and succinylation use the abundant cellular metabolites, that is, acetyl-CoA and succinyl-CoA, respectively, as their acylation donors, and can occur nonenzymatically in some circumstances in vivo (23, 24) (Fig. 1A). Also, a small high-energy compound, acetyl phosphate, was reported as the donor for nonenzymatic acetylation in E. coli (10). Previous studies suggested that acetylation and succinylation targeted different lysine residues and showed a preference for different residues surrounding the modified lysine sites (25). However, stochastic modifications on the same lysine residue were also observed, which were probably because of the nonenzymatic reactions (26). The combined analyses of cellular acetylome and succinylome have been reported in rice, Pseudomonas aeruginosa, and Vibrio parahemolyticus (27, 28, 29). Nevertheless, the understanding of acylation coordination remains at an early stage both in vivo and in vitro.
Fig. 1.

Highly abundant and dynamic acetylation in Streptomyces coelicolor.A, lysine acetylation by acetyl-CoA and lysine succinylation by succinyl-CoA are shown in detail. B, the acetylation level in S. coelicolor was determined using Western blotting, displaying highly abundant and dynamic modification in cells. Cell lysates were separated by 12% SDS-PAGE, and the pan anti–acetyl-lysine antibody was used (left panel). The relative acetylation levels were normalized to the loading protein levels and quantified with ImageJ (right panel). The normalized value from the first line of the gel (sample of the second day) was set as 100%.
Because of the lack of identification of specific sirtuins, the understanding of acylations and their coordination has long been elusive in microorganisms. Our recent work in Streptomyces coelicolor identified an evolutionary divergent desuccinylase (ScCobB2), which was different from the known deacetylase, providing a suitable model to study the crosstalk between acetylation and succinylation in bacteria (22). The desuccinylase ScCobB2 regulates protein biosynthesis and carbon metabolism in S. coelicolor (22). The other sirtuin ScCobB1 was identified as a deacetylase previously (30) and was shown to modulate the activity of chromosome segregation protein ParB (15). Here, we presented the profile of the lysine acetylome in S. coelicolor by combining the methods of immunoaffinity enrichment and high-accuracy MS. A large portion of the acetylated proteins was also found to be succinylated in metabolic pathways such as the TCA cycle and protein translation. The interplay between lysine acetylation and succinylation was globally explored and quantified in vivo and further tested by treatments with ScCobB1 and ScCobB2 in vitro. The results enriched our understanding of acylation regulation in bacteria and provided new insights into acylation coordination in cell metabolism.
Experimental Procedures
Bacterial Strains, Media, and Materials
The wildtype S. coelicolor M145 was cultured in tryptic soy broth (TSB) liquid medium (50 mM glucose plus) or on MS solid medium at 30 °C. E. coli BL21 (DE3) was used as the host strain to express the proteins. The pan anti–acetyl-lysine and pan anti–succinyl-lysine antibodies used in this study were obtained from Jingjie PTM Biolab (Hangzhou) Co Ltd. The unmodified, acetylated, and succinylated peptides (listed in supplemental Table S1) were synthesized in Guoping Pharmaceutical (Hefei), with >98% purity.
Experimental Design and Statistical Rationale
To determine the acetylome of S. coelicolor, the wildtype S. coelicolor sample with three biological replicates and two technical replicates was prepared and analyzed using Q Exactive HF-X (Thermo Fisher Scientific). To determine the quantitative acetylome and succinylome, three samples (wildtype, ΔSccobB1, and ΔSccobB2) with three biological replicates were prepared and analyzed using label-free MS. Similar procedures were followed for the cell culture, sample preparation, peptide affinity enrichment, and LC–MS/MS analysis. The modified sites were quantified using paired t tests where p < 0.05 were considered significant. The Gene Ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway/domains that presented a corrected p < 0.05 were considered significant.
The Determination of Cellular Acetylation and Succinylation Levels in S. coelicolor
The S. coelicolor wildtype cells were harvested with the interval of 1 day during the 6-day cultivation, and the whole cell proteins were then extracted. Approximately 15 μg of protein samples were separated using 12% SDS-PAGE and visualized via Coomassie brilliant blue staining. For the Western blotting assay, similar amounts of proteins were transferred onto the nitrocellulose membranes, and standard procedures were followed for the detection of acetylation levels. Briefly, 100 mM Tris–HCl (pH 7.5) with 0.5% (v/v) Tween-20 and 1% peptone was used for blocking, and 100 mM Tris–HCl (pH 7.5) with 0.05% (v/v) Tween-20 and 0.1% peptone was used as the primary and secondary antibody buffer. The membranes were incubated overnight with pan anti–acetyl-lysine antibody at a dilution of 1:1000 in the primary antibody buffer at 4 °C. Nitrocellulose membranes were then treated with an ECL Chemiluminescent Substrate reagent and visualized using an ImageQuant LAS 4000 mini (GE Healthcare).
Similar procedures were followed for the detection of acetylation and succinylation levels in wildtype S. coelicolor and its derivatives ΔSccobB1 and ΔSccobB2. The pan anti–succinyl-lysine antibody with 1:2000 dilution in the primary antibody buffer was used in these studies.
Protein Extraction and Trypsin Digestion for Acylome Analysis
The wildtype S. coelicolor M145 and its derivatives ΔSccobB1 and ΔSccobB2 were cultured on MS solid medium at 30 °C for 3 days. Fresh spores were collected with a cotton swab and transferred to TSB liquid medium supplemented with 50 mM glucose. After growing for 60 h, the cells were transferred (10%) to the new TSB medium containing 50 mM glucose and harvested at the log phase after 2 days. The collected cells were first frozen and grinded into powder using liquid nitrogen and then transferred to 50-ml centrifuge tubes for sonication in lysis buffer (8 M urea, 2 mM EDTA, 5 mM DTT, 1% Protease Inhibitor Cocktail III, 3 μM trichostatin A, and 50 mM nicotinamide). Cell debris was removed by centrifugation at 12,000g at 4 °C for 10 min. Finally, the protein was precipitated using cold 15% trichloroacetic acid for 2 h at −20 °C. After centrifugation at 4 °C for 10 min, the supernatant was discarded. The remaining precipitate was washed with cold acetone thrice. The protein was dissolved in a buffer (8 M urea, 100 mM TEAB, and pH 8.0), and the protein concentration was determined using a 2-D Quant kit (GE Healthcare) according to the manufacturer's instructions.
For digestion, trichloroacetic acid was added slowly to make a final concentration of 20% and then the mixture was eddy mixed and precipitated at 4 °C for 2 h. The supernatant was discarded after centrifugation at 4500g for 5 min. The precipitate was then washed with precooled acetone 2 to 3 times. After the precipitate was dried, TEAB, with a final concentration of 200 mM, was added. The precipitate was dispersed by ultrasonication, and trypsin was added at a mass ratio of 1:50 (trypsin to protein) overnight. Then, the peptide solution was reduced with 5 mM DTT for 30 min at 37 °C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in the dark.
Affinity Enrichment of Acylated Peptides
To enrich Kac or Ksuc peptides, tryptic peptides dissolved in immunoprecipitation buffer (50 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40; pH 8.0) were incubated with prewashed acetylation and succinylation antibody beads (PTM Biolabs) at 4 °C overnight with gentle shaking, respectively. The beads were washed four times with immunoprecipitation buffer and twice with double-distilled water. The bound peptides were eluted from the beads using 0.1% trifluoroacetic acid. The eluted fractions were vacuum dried, and the resulting peptides were desalted using C18 ZipTips (Millipore) according to the manufacturer's instructions, followed by LC–MS/MS analysis.
LC–MS/MS Analysis
The peptides were dissolved in mobile phase A of LC and separated using the EASY-nLC 1200 UPLC system. Mobile phase A was an aqueous solution containing 0.1% formic acid and 2% acetonitrile. Mobile phase B was an aqueous solution containing 0.1% formic acid and 90% acetonitrile. Liquid phase gradient settings were 0 to 36 min, 9 to 25% B; 36 to 54 min, 25 to 35% B; 54 to 57 min, 35 to 80% B; and 57 to 60 min, 80% B. The flow rate was maintained at 500 nl/min. The resulting peptides were analyzed using a Q Exactive HF-X hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific).
The peptides were subjected to a nanospray ionization source followed by MS/MS in Q Exactive HF-X coupled online with the UPLC. Intact peptides were detected using the Orbitrap at a resolution of 120,000. Peptides were selected for MS/MS using normalized collision energy setting 28, and ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 5E4 in the MS survey scan with a 15-s dynamic exclusion. The electrospray voltage applied was 2.1 kV. Automatic gain control was used to prevent overfilling of the ion trap, and 1E5 ions were accumulated to generate MS/MS spectra. For MS scans, the m/z scan range was 350 to 1600. The fixed first mass was set as 100 m/z.
Data Processing and Database Searches
The peak list was generated using Xcalibur 4.2.47 (Thermo Fisher). The resulting data were processed using MaxQuant (version 1.6.15.0, Max Planck Institute of Biochemistry) with the integrated Andromeda search engine. Tandem mass spectra were searched against a local UniProt S. coelicolor database concatenated with a reverse decoy database (Blast_Streptomyces_coelicolor_strain_ATCC_BAA-471_100226_PR_20201113.fasta; release date, November 13, 2020; 8038 sequences). Trypsin/P was specified as the cleavage enzyme allowing up to four missing cleavages, five modifications per peptide, and five charges. The mass error tolerance was set to 20 ppm for the first search, 4.5 ppm for the main search, and 20 ppm for fragment ions. Carbamidomethylation on Cys was specified as a fixed modification, and oxidation on Met, acetylation on Lys, and acetylation on protein N termini were specified as variable modifications. The false discovery rate thresholds for protein, peptide, and modification sites were specified at 1%. The minimum peptide length was set at 7. All other parameters in MaxQuant were set to default values. The site localization probability was set as >0.75.
Bioinformatics Analysis
Functional Enrichment Analysis
The GO derived from the UniProt-GOA database (http://www.ebi.ac.uk/GOA/) was used to annotate the functions of proteins in the acetylome. The database of KEGG was used to annotate the pathways. To identify the enriched GO/KEGG pathways, the Functional Annotation Tool of DAVID Bioinformatics Resources (31) and InterPro database (32) were used against the background of S. coelicolor proteome. A two-tailed Fisher's exact test was employed to test the protein enrichments.
Motif Analysis for Lysine Acetylation Substrates
Programs Motif-X (Harvard Medicine School) (33) and iceLogo (Universiteit Gent) (34) were used to analyze the model of identified acetylation sequences of acetyl-21-mers (10 amino acids upstream and downstream of the acetylation site). The S. coelicolor proteome sequences were used as the background database parameter, and other parameters were set to default values. In addition, an in-house R-package script (35) was used to generate the position-specific heat map by plotting the log10 ratio values of frequencies.
Protein–Protein Interaction Network
The acetylated proteins were searched against the STRING database version 9.1 (36) for protein–protein interactions. Only interactions between proteins belonging to the searched dataset were selected. The interaction network from STRING was visualized using Cytoscape (National Resource for Network Biology) (37). A graph obtained from the clustering algorithm, molecular complex detection (MCODE) (38), was utilized to analyze densely connected regions. MCODE is a plug-in in the network analysis kit, and the graph was visualized using Cytoscape.
Cloning, Expression, and Purification of ScCobB1, ScCobB2, and CobBL
The plasmids pET28b-ScCobB1/ScCobB2/CobBL were constructed using a previously reported method (22). The correct plasmids were checked using PCR and then transformed into BL21. A single colony was picked and transferred into a 5-ml LB medium containing kanamycin overnight and then 1:100 diluted into a 500-ml LB liquid medium at 37 °C. When the absorbance at 600 nm reached about 0.6, the cells were transferred to 16 °C and induced with 0.5 mM IPTG overnight.
The cells were harvested by centrifugation at 6000 rpm for 10 min at 4 °C and then suspended in a buffer containing 25 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM DTT, 10% (v/v) glycerol, and 25 mM imidazole. After lysis with the cell disruptor, cellular debris was removed by centrifugation at 13,000g for 30 min at 4 °C. The collected supernatants were loaded onto a 5-ml nickel resin column (GE Healthcare). After washing with 10 volumes of wash buffer (25 mM Tris–HCl, 150 mM NaCl, and 20 mM imidazole; pH 8.0), the proteins were eluted (elution buffer, 25 mM Tris–HCl, 150 mM NaCl, and 500 mM imidazole; pH 8.0). The proteins were subjected to the AKTA purification system (GE Healthcare), quantified, aliquoted, and stored at −80 °C.
Quantification of Peptide Deacetylation and Desuccinylation Using HPLC
The in vitro peptide deacylation system contained 50 mM Tris buffer (pH 8.0), 50 mM NaCl, 1.0 mM DTT, 6.0 mM MgCl2, 10.8 μM ScCobB1 or 3.6 μM ScCobB2/CobBL, 1.0 mM NAD+, and 540 μM acylated peptide. The reactions were carried out at 30 °C overnight in a 50-μl volume, and quenched with 1 volume of 10% (v/v) trifluoroacetic acid for 10 min at room temperature. The mixture was centrifuged at 13,000g for 10 min to discard the sediment, and the supernatant was subjected to HPLC (Agilent Technologies) using the Aeris peptide XB-C18 column (150 × 4.6 mm, 3.6 μm; Phenomenex). The mobile phase consisted of solvent A (0.1% formic acid in HPLC-grade water) and solvent B (HPLC-grade acetonitrile). Peptides were eluted in a linear gradient of 5 to 100% solvent B at a rate of 1 ml min−1 over 30 min. The column temperature was set at 25 °C, and the peptides were monitored by UV light at a wavelength of 215 nm. The deacylation activities of ScCobB1, ScCobB2, and CobBL were determined by calculating the peak area of the peptides as follows:
Results
Detection of Abundant and Dynamic Acetylation in S. coelicolor
The representative Streptomyces are a group of soil-dwelling microorganisms notable for their complex lifecycles and antibiotic production capacity (39). Our recent work in S. coelicolor discovered highly abundant and dynamic acetylation (Fig. 1B). The intensities of cellular acetylation signals varied from the culture time. Moreover, a full range of signal decrease along the cell growth was observed, suggesting possible global regulation of acetylation in S. coelicolor.
Acetylome Analyses in S. coelicolor
To acquire detailed information on acetylation regulation in S. coelicolor, acetylome analyses were performed by culturing the cells in TSB medium containing 50 mM glucose. The cells were harvested at the midlog phase, and the proteins were extracted for trypsin digestion. The acetylated peptides were then enriched by affinity purification using pan acetyl-lysine antibody and subjected to LC–MS/MS analysis with a high accuracy mass spectrometer. An overview of the experimental procedures is shown in Figure 2A. A total of 1298 lysine acetylation sites in 601 proteins were identified using the UniProt database. Detailed information for all the acetylated peptides and their matched proteins is provided in supplemental Table S2. We checked the quality of our MS data and inspected the distribution of mass errors for all the identified peptides. As presented in supplemental Figure S1, all the results suggested that the MS data met the requirements for the acetylome analysis (40).
Fig. 2.

The acetylome analysis highlights the acetylation regulation in protein translation and metabolism in Streptomyces coelicolor.A, schematic representation of the workflow used in profiling of acetyl proteomics in S. coelicolor. Gene Ontology (GO) enrichment analysis for acetylome based on biological processes (B) and molecular function analyses (C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis revealed that acetylation proteins were enriched in protein translation and metabolism pathways (D).
To better understand the lysine acetylated proteins in S. coelicolor, we performed functional annotation analysis using GO. The results showed that the acetylated proteins are widely located in S. coelicolor cells and participated in a variety of biological processes (supplemental Fig. S2 and supplemental Table S3). The classification based on biological processes showed that most acetylated proteins were involved in cell metabolism (36.5%) and cellular processes (27.2%). The classification based on molecular function showed that most acetylated proteins possessed the catalytic (47%) or DNA-binding (39.6%) activities. A high amount of acetylated proteins were located in the cell cytoplasm in comparison to other cellular components, consistent with previous reports (4, 41).
The GO enrichment analysis for the acetylated proteins showed that acetylation substrates were mostly enriched in protein biosynthesis, and the top two acetylated biological processes were protein translation and metabolism (Fig. 2B and supplemental Table S4). The function of lysine acetylated substrates included structural constituents of ribosome and RNA binding (Fig. 2C). The functional enrichment analyses were performed using the KEGG database as well. The acetylated proteins were found to be enriched in protein biosynthesis in addition to other metabolic pathways. The top three enriched KEGG pathways were ribosome biogenesis, TCA cycle, and aminoacyl-tRNA biosynthesis (Fig. 2D and supplemental Table S5). Taken together, both GO annotation and KEGG pathway analyses suggested that the substrates related to protein translation and metabolism were extensively regulated by acetylation in S. coelicolor.
Functional Interaction Networks of Acetylated Substrates in S. coelicolor
Protein–protein interactions occur widely in all organisms and are important in implementing and regulating cellular processes. However, how the acetylated protein interactions affect the regulation of S. coelicolor physiology and development is poorly understood. The interaction network of acetylated proteins was generated based on the STRING database and visualized using Cytoscape (36). A total of 462 acetylated proteins were identified as nodes and connected to each other (Fig. 3A and supplemental Table S6). Functional category analysis for these proteins demonstrated several highly interconnected clusters such as ribosome biogenesis, carbon metabolism, protein folding, and aminoacyl-tRNA biosynthesis (Fig. 3). Consistent with the GO and KEGG analysis results (Fig. 2), most acetylated proteins were enriched in protein translation and metabolic pathways in general. The top two identified clusters were ribosome-associated proteins (Fig. 3B) and carbon metabolism (Fig. 3C). The strong interactions among these acetylated pathway components suggested that acetylation may facilitate S. coelicolor to adapt to the changing soil environments.
Fig. 3.

Protein–protein interaction networks of acetylated proteins in Streptomyces coelicolor. Interaction networks of acetylated proteins were analyzed using the MCODE plug-in toolkit in the Cytoscape software. A, interaction network of all acetylated proteins. Detailed cluster information of proteins in ribosomes (B), carbon metabolism (C), metabolism in diverse environments (D), protein folding (E), and tRNA aminoacylation in translation (F) is shown. The respective MCODE score and protein–protein interaction score were listed in supplemental Table S6.
Sequence Motifs of Acetylation in S. coelicolor
The preferences for the amino acid motifs surrounding acetylation sites were observed in both prokaryotes and eukaryotes (4, 42, 43, 44). Here, the Motif-X program was used to search for overrepresented sequence patterns among all identified acetylated peptides. The preferred conserved consensus motifs with amino acid sequences from the −10 to +10 flanking region of the acetylated lysine were extracted (supplemental Table S7). As shown in Figure 4A, eight conserved sequences surrounding Kac sites were detected in the S. coelicolor acetylome. These motifs included ##Kac#F##, ##I#Kac##, ##Kac#H##, ##KacH##, ##FKac##, ##V#Kac##, ##KacL##, and ##LKac## (# indicates a random amino acid residue). The amino acids F, H, and L were frequently found in these motifs. Among the acetylation motifs identified here, motifs ##I#Kac##, ##Kac#F##, ##Kac#H##, ##KacH##, ##FKac##, and ##V#Kac## were mainly present as substrates in fungi (45), and the motifs ##KacH##, ##FKac##, and ##LKac## have also been reported as representatives in rice (44) and bacteria (46, 47). These results supported the notion that the preferred sequences for lysine acetylation are conserved among different organisms, implying a similar kinetics of acetylation.
Fig. 4.

Analysis of acylation motifs in Streptomyces coelicolor.A, sequence logo representation of overall acetylation motifs identified using Motif-X software. The motifs consisted of 20 residues (ten amino acids upstream and the other ten downstream to the site) surrounding the acetylated lysine. B, the under-represented and over-represented amino acids flanking the acetylated sites were shown using a heat map generated from the R-package script. C, the specific acetylation motifs by comparing acetylome and succinylome data. D, the specific succinylation motifs by comparing acetylome and succinylome data. The representatives of these motifs were analyzed by Motif-X.
The other two software, iceLogo and R program, were also used to analyze the acetylation motifs, and similar results were obtained. The heat map produced using the R program analysis not only showed the same overrepresented flanking sequences as in Motif-X program but also displayed that arginine (R) and lysine (K) tend to be absent at the −1 and +1 positions around Kac, suggesting that they are unpopular residues in the substrate sequences near Kac (Fig. 4B). iceLogo displayed similar results as the other software for both overrepresented and underrepresented flanking sequences of the acetylated lysine residues (supplemental Fig. S3).
The acetylation motifs showed completely different patterns compared with those of succinylation (Fig. 4). Particularly, the acetylation motifs displayed opposite preferences for the residues arginine (R) and lysine (K), which were the representatives in succinylation motifs near Ksuc (22). The specific and overlapped patterns of the modified motifs were then analyzed. As shown in Figure 4, C and D, the specific motifs mimicked the full modification spectrums in acetylation and succinylation, respectively. Also, no obvious overlapped motifs were identified between acetylation and succinylation, indicating that bimodification on the same lysine residue was likely from the stochastic events and with no specificity. Collectively, these results supported the notion that acetylation and succinylation target different lysine residues in the proteome, and moreover, stochastic bimodifications on some lysine residues were also inferred.
The Crosstalk Between Acetylation and Succinylation in S. coelicolor
A previous study has shown that pathways involved in protein biosynthesis and carbon metabolism were enriched by succinylation in S. coelicolor (22). In the current study, the identified acetylation substrates were also closely related to protein translation and central metabolic processes (Figs. 3 and 4). Key questions are raised that whether there is any crosstalk between lysine acetylation and succinylation and, if so, how it is regulated by the different sirtuins. To address these questions, quantitative acetylome and succinylome analyses were performed by knocking out either the desuccinylase ScCobB2 or the presumed deacetylase ScCobB1 (30) (Fig. 5A). The bacterial cells were cultured and harvested using the same conditions as shown in Figure 2A. In total, 3981 sites in 1522 proteins and 5982 sites in 1510 proteins were quantified in acetylome and succinylome, respectively (supplemental Table S8). The combined results revealed that 2511 sites were both acetylated and succinylated, accounting for 63.1% and 50.0% of the identified sites in acetylome and succinylome, respectively (Fig. 5B and supplemental Table S9). The bimodified proteins (1140) including those with acetylation and succinylation at the same sites and different sites in one protein, accounts for 74.9% and 75.5% of all the identified acetylome and succinylome proteins, respectively, suggesting extensive crosstalk between lysine acetylation and succinylation in S. coelicolor (Fig. 5C and supplemental Table S9).
Fig. 5.

Quantitative acylome analyses after knocking out SccobB1 or SccobB2.A, schematic representation of the workflow used in quantitative acylome analyses. The quantitative acetylome and quantitative succinylome analyses were performed following the similar procedures (see Experimental Procedures section). Only the quantitative acetylome was shown here to simplify. B, the intersection of acetylated and succinylated sites in Streptomyces coelicolor proteomes. C, the intersection of acetylated and succinylated proteins in S. coelicolor proteomes (the bimodified proteins including those with acetylation and succinylation at separate sites in one protein). D, the summary number of the differentially modified sites or proteins in quantitative acylomes. E, scatter plot showed the details of changes in succinylated peptide intensities between the S. coelicolor wildtype and ΔSccobB2 cells (left) and between the wildtype and ΔSccobB1 cells (right). F, scatter plot showed the details of changes in acetylated peptide intensities between the S. coelicolor wildtype and ΔSccobB2 cells (left) and between the wildtype and ΔSccobB1 cells (right). When p < 0.05, the change of modification level >2 was deemed as upregulated significantly (brown dots), and the change modification level <0.5 was deemed as downregulated significantly (cyan dots).
After deletion of SccobB2, the quantitative analyses showed that more sites (611 versus 541) were upregulated in succinylation, consistent with the previous notion that ScCobB2 is a global desuccinylase (22) (Fig. 5, D and E). On the other hand, when SccobB1 was deleted, more sites (506 versus 433) were downregulated in succinylation (Fig. 5, D and E), indicating that some lysine sites modified by succinylation might also be targeted by ScCobB1. As we know, no desuccinylase but a deacetylase activity was reported for ScCobB1 previously (30). The quantitative acetylome results showed that more sites (341 versus 249) were downregulated in acetylation after deletion of SccobB2 (Fig. 5, D and F), suggesting some lysine sites modified by acetylation could also be targeted by ScCobB2. However, less upregulated acetylation sites (246 versus 363) were observed in ΔSccobB1 (Fig. 5, D and F). This result implied other deacetylase(s) besides ScCobB1 may exist and play a leading role in S. coelicolor. Nevertheless, these observations from the quantitative analyses revealed a possible competitive relationship between acetylation and succinylation targeting upon the same lysine sites.
To investigate the functional categories of the differentially modified proteins, Clusters of Orthologous Groups analysis was performed for the upregulated and downregulated proteins in both the quantitative acetylome and succinylome (supplemental Fig. S4). The quantitative succinylome results showed that the most upregulated proteins in ΔSccobB2 were involved in gene transcription and protein translation, whereas the most downregulated proteins in ΔSccobB1 were involved in cell metabolism. These results indicated that distinct proteins were regulated by ScCobB1 and ScCobB2, in agreement with the results of the motifs analysis shown in Figure 4. The quantitative acetylome results (supplemental Fig. S4) showed that the most downregulated proteins were involved in cell metabolism and signal transduction both in ΔSccobB1 and ΔSccobB2, implying ScCobB1 may not be the primary deacetylase and a coordination of ScCobB1 and ScCobB2 in the regulation of S. coelicolor acetylation to some extent.
To better understand the role of the PTM crosstalk, we mapped the bimodified proteins onto the KEGG pathways. The results showed that multiple pathways, particularly those involved in ribosome biogenesis and central metabolism, were enriched by bimodified proteins (Fig. 6 and supplemental Table S10). In detail, 42 of 53 ribosomal proteins were bimodified by acetylation or succinylation. RpoA/RpoB/RpoC, EF-Tu/EF-Ts, and FtsY/Ffh were the top ribosomal complexes whose subunits were both acetylated and succinylated (Fig. 6A and supplemental Fig. S5A). Nearly all the enzymes (85.7%) in central metabolism pathways were bimodified. Remarkably, enzymes in the TCA cycle were 100% bimodified, including α-ketoglutarate dehydrogenase components (SucAB), succinyl-CoA synthetase complexes (SucCD), and fumarate hydratase (Fig. 6B). In addition, all the enzymes from the glycolysis/gluconeogenesis pathway were identified as bimodified as well (Fig. 6C). It is worth noting that glyceraldehyde-3-phosphate dehydrogenase (GapA), which is a rate-limit enzyme in the glycolysis pathway, was extensively acetylated and succinylated (Fig. 6C and supplemental Fig. S5B). In GapA, 15 lysine sites were acetylated and 20 lysine sites were succinylated including 14 lysine sites were bimodified, and most of them were on the conserved enzymatic activity domains (5). The modified sites in response to SccobB1 or SccobB2 deletion were also quantified and mapped onto the KEGG pathways (Fig. 6 and supplemental Table S10), providing a bunch of candidates that were highly regulated by acetylation and succinylation. RpoA/RpoB/RpoC, GapA, AceE, SucAB, and fumarate hydratase had the most upregulated succinylation sites in ΔSccobB2, whereas AceE, citrate synthase, and SucA had the most upregulated acetylation sites in ΔSccobB1 (Fig. 6). Collectively, 91.9% of the sites were identified as acetylation and succinylation in the central metabolic pathway proteins, far higher than the average level of the bimodification sites (Fig. 5B), implying that more nonenzymatic reactions might be occurring in these pathways. Taken together, our analyses indicated extensive crosstalk between acetylation and succinylation both on the same sites and same proteins involved in the metabolic pathways.
Fig. 6.

The enriched bimodified proteins are involved in protein translation and carbon metabolism pathways. The bimodified (acetylated and succinylated) proteins enriched in ribosomes (A), glycolysis/gluconeogenesis (B), pyruvate metabolism (C), and tricarboxylic acid (TCA) cycle (D) were mapped onto the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The acetylation-only sites are shown as red dots, succinylation-only sites are shown as blue dots, and bimodified sites are shown as brown dots. The modified sites in response to knocking out of SccobB1 or SccobB2 were quantified and mapped onto the KEGG pathways. The acetylation sites regulated by ΔSccobB1 and the succinylation sites regulated by ΔSccobB2 are shown in red and blue arrows, respectively. ACO, aconitate hydratase; AceE, pyruvate dehydrogenase E1 component; AceF, pyruvate dehydrogenase E2 component (dihydrolipoamide acetyltransferase); AcsA, acetyl-CoA synthetase; Adh, alcohol dehydrogenase; Aldh, aldehyde dehydrogenase; CS, citrate synthase; Eno, phosphopyruvate hydratase; Fba, fructose-bisphosphate aldolase; FH, fumarate hydratase; GapA, glyceraldehyde-3-phosphate dehydrogenase; Glk, glucokinase; GpmA, phosphoglycerate mutase; IDH, isocitrate dehydrogenase; Lpd, dihydrolipoamide dehydrogenase; MDH, malate dehydrogenase; N-resize, upregulated; PfkA, 6-phosphofructokinase; Pgi, glucose-6-phosphate isomerase; Pgk, phosphoglycerate kinase; Pyk, pyruvate kinase; S-resize, downregulated; SDH, succinate dehydrogenase; SucA, 2-oxoglutarate dehydrogenase E1 component; SucB, 2-oxoglutarate dehydrogenase E2 component; SucC, acetyl-CoA synthetase subunit beta; SucD, acetyl-CoA synthetase subunit alpha.
Deacylation by Sirtuins In vitro
To further explore the interplay between acetylation and succinylation, in vitro tests were performed by treating these sites with different sirtuins. Five lysine-containing peptides were randomly selected from metabolic pathways showing both acetylation and succinylation (supplemental Table S1). These peptides and their corresponding acetylated and succinylated products were synthesized and tested with ScCobB1 or ScCobB2 treatment in vitro, using the E. coli CobBL treatment as the positive control. As shown in Figures 7 and S6, all the succinylated peptides were desuccinylated by ScCobB2, whereas all the acetylated peptides except SucC-K45ac were deacetylated by ScCobB1, demonstrating evident crosstalk by different sirtuins. As expected, ScCobB2 displayed a specific desuccinylase activity (Fig. 7B), and CobBL displayed both deacetylase and desuccinylase activities (Fig. 7C). However, ScCobB1 was not shown as a specific deacetylase, and it also displayed weak desuccinylase activity for some peptides (Fig. 7A). In summary, these results confirmed the regulatory crosstalk of PTMs in S. coelicolor. The deacylation by ScCobB1 and ScCobB2 may work together in elaborately regulating cellular biological processes (e.g., cell metabolism).
Fig. 7.

Deacylation analysis using ScCobB1 and ScCobB2 in vitro. The regulatory crosstalk between deacetylation and desuccinylation was tested by treating the same synthetic peptides with sirtuins. The assays were performed as explained in the Experimental procedures section, using NAD+ as the cofactor. The assays without NAD+ in the mixtures were used as the negative controls (see supplemental Fig. S5 for more details). A, the modified peptides were incubated with ScCobB1. Relatively, low deacetylase and desuccinylase activities were observed. B, the modified peptides were incubated with ScCobB2. A specific desuccinylase activity was observed. C, the bifunctional CobBL was used as a positive control that showed both the deacetylase and desuccinylase activities.
Discussion
In the current study, we reported the first proteome-wide analysis of lysine acetylation and quantitative proteomic analyses including both acetylation and succinylation in S. coelicolor. Bioinformatics analysis showed that the acetylated proteins were involved in a broad range of cellular functions ranging from protein translation to cellular metabolic pathways (Fig. 2). Comparative analysis revealed that the acetylation and succinylation highly overlapped in proteins as well as in some lysine loci in S. coelicolor proteomes (Fig. 4, Fig. 5, Fig. 6). Also, distinct motif patterns of acetylation and succinylation were suggested in this study (Fig. 4), supporting the notion that the two PTMs targeted different lysine residues (25). The stochastic modifications on the same lysine residues might be due to the nonenzymatic reactions by either succinyl-CoA or acetyl-CoA (or acetyl phosphate) (10, 23). In addition, our in vitro experiments suggested possible regulatory crosstalk by sirtuins, which indicated a coordination between acetylation and succinylation to regulate S. coelicolor physiology. However, more analyses are still needed to uncover the physiological significance underlining the stochastic modifications as well as the competitive relationship between the sirtuins.
In the current study, we confirmed the desuccinylase activity of ScCobB2 both in vivo (Fig. 6) and in vitro (Fig. 7). However, because of the relatively weak deacetylase activity shown by ScCobB1 (Figs. 5D and 7), we still cannot exclude the possibility that another global deacetylase(s) is functional in S. coelicolor. Although the mechanism for the acylation crosstalk remains unclear, our systemic analysis provided a basis for further exploration of the roles of PTMs in cell metabolism as well as other pathways in S. coelicolor. Future directions could include investigating the dynamic homeostasis of the acylations in vivo and characterizing other possible deacetylases in S. coelicolor.
Data availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (48) with the dataset identifier PXD027205 and PXD019536. The results were created using MaxQuant with version 1.6.15.0. Alternatively, the dataset can be accessed in MS-Viewer (49) with the search key “zbzofokood” (quantitative succinylome) and “schqt1dyvv” (quantitative acetylome).
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We thank Jingjie PTM Biolab (Hangzhou, China) for help in analyzing the proteomic data.
Funding and additional information
This work was supported by the National Key R&D Program of China (2019YFA0904003 to W. Z.), Strategic Priority Research Program of the Chinese Academy of Sciences, China (XDB38020300 to W. Z.), National Natural Science Foundation of China (31921006 to G.-p. Z. and 82000273 to P. L.), Guangdong Basic and Applied Basic Research Foundation (2021A1515012511 to W. Z.), and China Postdoctoral Science Foundation (BX20200092 to P. L.).
Author contributions
P. L., G.-p. Z., and W. Z. methodology; H. Z., Y. Y., and J. W. formal analysis; H. Z. and Y. Y. investigation; F. L., Z. G., and S. Z. resources; W. Z. writing–original draft.
Footnotes
Present address for Hong Zhang: Department of Cell Biology and Molecular Genetics, University of Maryland, College Park, MD, USA.
Contributor Information
Peng Li, Email: lipeng4754@163.com.
Guo-ping Zhao, Email: gpzhao@sibs.ac.cn.
Wei Zhao, Email: wei.zhao1@siat.ac.cn.
Supplemental data
Supplemental Figures S1–S6
References
- 1.Walsh C.T., Garneau-Tsodikova S., Gatto G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z.A., Cole P.A. The chemical biology of reversible lysine post-translational modifications. Cell Chem. Biol. 2020;27:953–969. doi: 10.1016/j.chembiol.2020.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z.B., Zeng R., Xiong Y., Guan K.L., Zhao S. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C.F., Grishin N.V., Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., Zwaans B.M., Tishkoff D., Ho L., Lombard D., He T.C. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M., Peng C., Anderson K.A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G.R., Green M.F., Madsen A.S., Schmiesing J. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C., Falck J.R., Peng J., Gu W., Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M., Ding J., Czyz D., Hu R., Ye Z., He M. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinert B.T., Iesmantavicius V., Wagner S.A., Schölz C., Gummesson B., Beli P., Nyström T., Choudhary C. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell. 2013;51:265–272. doi: 10.1016/j.molcel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Starai V.J., Celic I., Cole R.N., Boeke J.D., Escalante-Semerena J.C. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 12.Starai V.J., Escalante-Semerena J.C. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J. Mol. Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Figlia G., Willnow P., Teleman A.A. Metabolites regulate cell signaling and growth via covalent modification of proteins. Dev. Cell. 2020;54:156–170. doi: 10.1016/j.devcel.2020.06.036. [DOI] [PubMed] [Google Scholar]
- 14.Colak G., Xie Z., Zhu A.Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y.H., Lin H., Zhao Y., Tan M. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics. 2013;12:3509–3520. doi: 10.1074/mcp.M113.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li P., Zhang H., Zhao G.P., Zhao W. Deacetylation enhances ParB-DNA interactions affecting chromosome segregation in Streptomyces coelicolor. Nucleic Acids Res. 2020;48:4902–4914. doi: 10.1093/nar/gkaa245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vazquez B.N., Thackray J.K., Serrano L. Sirtuins and DNA damage repair: SIRT7 comes to play. Nucleus. 2017;8:107–115. doi: 10.1080/19491034.2016.1264552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei W., Liu T., Li X., Wang R., Zhao W., Zhao G., Zhao S., Zhou Z. Lysine acetylation regulates the function of the global anaerobic transcription factor FnrL in Rhodobacter sphaeroides. Mol. Microbiol. 2017;104:278–293. doi: 10.1111/mmi.13627. [DOI] [PubMed] [Google Scholar]
- 18.Han X., Shen L., Wang Q., Cen X., Wang J., Wu M., Li P., Zhao W., Zhang Y., Zhao G. Cyclic AMP inhibits the activity and promotes the acetylation of acetyl-CoA synthetase through competitive binding to the ATP/AMP pocket. J. Biol. Chem. 2017;292:1374–1384. doi: 10.1074/jbc.M116.753640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong S.Y., Ng L.T., Ng L.F., Inoue T., Tolwinski N.S., Hagen T., Gruber J. The role of mitochondrial non-enzymatic protein acylation in ageing. PLoS One. 2016;11 doi: 10.1371/journal.pone.0168752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali I., Conrad R.J., Verdin E., Ott M. Lysine acetylation goes global: From epigenetics to metabolism and therapeutics. Chem. Rev. 2018;118:1216–1252. doi: 10.1021/acs.chemrev.7b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang G., Meyer J.G., Cai W., Softic S., Li M.E., Verdin E., Newgard C., Schilling B., Kahn C.R. Regulation of UCP1 and mitochondrial metabolism in Brown adipose tissue by reversible succinylation. Mol. Cell. 2019;74:844–857.e847. doi: 10.1016/j.molcel.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H., Li P., Ren S., Cheng Z., Zhao G., Zhao W. ScCobB2-mediated lysine desuccinylation regulates protein biosynthesis and carbon metabolism in Streptomyces coelicolor. Mol. Cell. Proteomics. 2019;18:2003–2017. doi: 10.1074/mcp.RA118.001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagner G.R., Payne R.M. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabari B.R., Zhang D., Allis C.D., Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cel. Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosono S., Tamura M., Suzuki S., Kawamura Y., Yoshida A., Nishiyama M., Yoshida M. Changes in the acetylome and succinylome of Bacillus subtilis in response to carbon source. PLoS One. 2015;10:e0131169. doi: 10.1371/journal.pone.0131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinert B.T., Iesmantavicius V., Moustafa T., Scholz C., Wagner S.A., Magnes C., Zechner R., Choudhary C. Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol. Syst. Biol. 2014;10:716. doi: 10.1002/msb.134766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou H., Finkemeier I., Guan W., Tossounian M.A., Wei B., Young D., Huang J., Messens J., Yang X., Zhu J., Wilson M.H., Shen W., Xie Y., Foyer C.H. Oxidative stress-triggered interactions between the succinyl- and acetyl-proteomes of rice leaves. Plant Cell Environ. 2018;41:1139–1153. doi: 10.1111/pce.13100. [DOI] [PubMed] [Google Scholar]
- 28.Gaviard C., Broutin I., Cosette P., Dé E., Jouenne T., Hardouin J. Lysine succinylation and acetylation in Pseudomonas aeruginosa. J. Proteome Res. 2018;17:2449–2459. doi: 10.1021/acs.jproteome.8b00210. [DOI] [PubMed] [Google Scholar]
- 29.Pan J., Chen R., Li C., Li W., Ye Z. Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 2015;14:4309–4318. doi: 10.1021/acs.jproteome.5b00485. [DOI] [PubMed] [Google Scholar]
- 30.Mikulik K., Felsberg J., Kudrnacova E., Bezouskova S., Setinova D., Stodulkova E., Zidkova J., Zidek V. CobB1 deacetylase activity in Streptomyces coelicolor. Biochem. Cell Biol. 2012;90:179–187. doi: 10.1139/o11-086. [DOI] [PubMed] [Google Scholar]
- 31.Huang D.W., Sherman B.T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones P., Binns D., Chang H.Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A.F., Sangrador-Vegas A., Scheremetjew M., Yong S.Y. InterProScan 5: Genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou M.F., Schwartz D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics. 2011 doi: 10.1002/0471250953.bi1315s35. Chapter 13, Unit 13.15-24. [DOI] [PubMed] [Google Scholar]
- 34.Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 35.Rajaram S., Oono Y. NeatMap--non-clustering heat map alternatives in R. BMC bioinformatics. 2010;11:45. doi: 10.1186/1471-2105-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., Jensen L.J. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su G., Morris J.H., Demchak B., Bader G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinformatics. 2014;47:11–24. doi: 10.1002/0471250953.bi0813s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bader G.D., Hogue C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bentley S.D., Chater K.F., Cerdeno-Tarraga A.M., Challis G.L., Thomson N.R., James K.D., Harris D.E., Quail M.A., Kieser H., Harper D., Bateman A., Brown S., Chandra G., Chen C.W., Collins M. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 40.Cao J., Wang T., Wang Q., Zheng X., Huang L. Functional insights into protein acetylation in the hyperthermophilic archaeon Sulfolobus islandicus. Mol. Cell. Proteomics. 2019;18:1572–1587. doi: 10.1074/mcp.RA119.001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y., Tong M., Bai X., Liu X., Cai X., Luo X., Zhang P., Cai W., Vallee I., Zhou Y., Liu M. Comprehensive proteomic analysis of lysine acetylation in the foodborne pathogen Trichinella spiralis. Front. Microbiol. 2017;8:2674. doi: 10.3389/fmicb.2017.02674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu F., Yang M., Wang X., Yang S., Gu J., Zhou J., Zhang X.E., Deng J., Ge F. Acetylome analysis reveals diverse functions of lysine acetylation in Mycobacterium tuberculosis. Mol. Cell. Proteomics. 2014;13:3352–3366. doi: 10.1074/mcp.M114.041962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H., Diao H., Wang C., Lin Y., Yu F., Lu H., Xu W., Li Z., Shi H., Zhao S., Zhou Y., Zhang Y. Acetylproteomic analysis reveals functional implications of lysine acetylation in human spermatozoa (sperm) Mol. Cell. Proteomics. 2015;14:1009–1023. doi: 10.1074/mcp.M114.041384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong Y., Peng X., Cheng Z., Liu W., Wang G.L. A comprehensive catalog of the lysine-acetylation targets in rice (Oryza sativa) based on proteomic analyses. J. Proteomics. 2016;138:20–29. doi: 10.1016/j.jprot.2016.01.019. [DOI] [PubMed] [Google Scholar]
- 45.Lv Y. Proteome-wide profiling of protein lysine acetylation in Aspergillus flavus. PLoS One. 2017;12:e0178603. doi: 10.1371/journal.pone.0178603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lv B., Yang Q., Li D., Liang W., Song L. Proteome-wide analysis of lysine acetylation in the plant pathogen Botrytis cinerea. Sci. Rep. 2016;6:29313. doi: 10.1038/srep29313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Wang F., Bao X., Fu L. Systematic analysis of lysine acetylome reveals potential functions of lysine acetylation in Shewanella baltica, the specific spoilage organism of aquatic products. J. Proteomics. 2019;205:103419. doi: 10.1016/j.jprot.2019.103419. [DOI] [PubMed] [Google Scholar]
- 48.Vizcaino J.A., Csordas A., Del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.W., Wang R., Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker P.R., Chalkley R.J. MS-viewer: A web-based spectral viewer for proteomics results. Mol. Cell. Proteomics. 2014;13:1392–1396. doi: 10.1074/mcp.O113.037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figures S1–S6
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (48) with the dataset identifier PXD027205 and PXD019536. The results were created using MaxQuant with version 1.6.15.0. Alternatively, the dataset can be accessed in MS-Viewer (49) with the search key “zbzofokood” (quantitative succinylome) and “schqt1dyvv” (quantitative acetylome).