

Abstract
Toll‐like receptor 2 and 4 (TLR2, TLR4) signaling is implicated in atherosclerotic plaque formation. The two‐stage master regulator Virtual Inference of Protein‐activity by Enriched Regulon (VIPER) analysis of macrophage TLR2 and TLR4 signature genes integrated with coexpression network genes derived from 371 patient‐derived carotid specimens identifies activated RNA polymerase II transcriptional coactivator p15 (SUB1/Sub1, PC4) as a master regulon in the atherogenic TLR response. It is found that TLR2 and TLR4 signaling is proinflammatory and proatherosclerotic in chow‐fed apolipoprotein E‐deficient (ApoE −/−) mice. Through transgenic myeloid‐specific Sub1 knockout in ApoE −/− mice, it is discovered that these proatherosclerotic effects of TLR2 and TLR4 signaling are mediated by Sub1. Sub1 knockout in macrophages enhances anti‐inflammatory M2 macrophage polarization and cholesterol efflux. Irradiated low density lipoprotein receptor‐deficient (Ldlr −/−) mice transplanted with Sub1 −/− murine bone marrow display reduced atherosclerosis. Promoter analysis reveals Sub1‐dependent activation of interferon regulatory factor 1 (Irf1) transcription in a casein kinase 2 (Ck2)‐dependent manner, and Sub1‐knockout macrophages display decreased Irf1 expression. Artificial Irf1 overexpression in Sub1‐knockout macrophages enhances proinflammatory M1 skewing and lowers cholesterol clearance. In conclusion, the TLR master regulon Sub1, and its downstream effect on the transcription factor Irf1, promotes a proinflammatory M1 macrophage phenotype and enhances atherosclerotic burden in vivo.
Keywords: atherosclerosis; PC4; SUB1; TLR2; TLR4, Toll‐like receptor
The Toll‐like receptor (TLR) master regulon RNA polymerase II transcriptional coactivator p15 (Sub1), and its downstream effect on the transcription factor interferon regulatory factor 1 (Irf1), promotes a proinflammatory M1 macrophage phenotype and enhances atherosclerotic burden in mice in vivo. The findings suggest that targeting the SUB1/IRF1 axis may be an effective strategy toward combating proatherogenic, TLR‐induced M1 polarization.
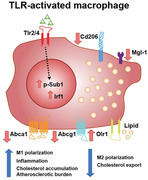
1. Introduction
Atherosclerosis is the leading cause of death and disability in developed countries and can precipitate a number of ischemic conditions, including coronary artery disease (CAD), carotid artery stenosis, ischemic stroke, peripheral arterial disease, and renal artery obstruction.[ 1 ] Atherosclerosis is a disease caused by plaque build‐up within the cardiovascular system that results in a thickening of arterial walls, a decrease in arterial wall elasticity, and a narrowing of the arterial lumen. The progression of atherosclerotic plaque development is defined by vascular wall damage, subendothelial accumulation of oxidized low‐density lipoprotein (oxLDL), proliferation of fibrous connective tissue, and elevated recruitment of macrophages.[ 2 ]
The interaction between subendothelial oxLDL and macrophages is central to atherogenesis, as oxLDL induces macrophage inflammation and pathogenic foam cell differentiation in the vascular intima.[ 3 ] This oxLDL‐induced macrophage activation is influenced by Toll‐like receptors (TLRs), a large family of membrane‐spanning, noncatalytic receptors that are displayed on the macrophage surface.[ 4 ] Specifically, two TLRs—TLR2 and TLR4—have been linked to atherosclerotic plaque development. Both TLR2 and TLR4 contribute to foam cell accumulation within apolipoprotein E‐deficient (ApoE −/−) murine aortic plaques, with TLR4 showing a more profound effect than TLR2.[ 5 ] Moreover, TLR4 expression is upregulated within human atherosclerotic lesions and cultured macrophages exposed to oxLDL.[ 6 ] Additionally, acute myocardial infarction patients display elevated TLR2 and TLR4 expression in circulating monocytes as well as elevated TLR4 expression in ruptured plaque macrophages.[ 7 ] Therefore, macrophage TLR2 and TLR4 are considered potential therapeutic targets for atherosclerosis, and TLR2‐ and TLR4‐targeting agents are currently in preclinical development.[ 8 ]
Although macrophage TLR2 and TLR4 have well‐established roles in atherosclerotic plaque development, their downstream effectors and their roles in atherogenesis are not clearly understood. Here, we first sought to identify master regulator transcription factors (TFs) that determine the transcriptional response to atherogenic TLR2 and TLR4 stimuli. A two‐stage master regulator Virtual Inference of Protein‐activity by Enriched Regulon (VIPER) analysis was implemented to overlay macrophage TLR transcription signatures with six coexpression networks derived from 371 patient‐derived carotid specimens to identify intersection among genes within the macrophage TLR transcription signatures and the associated master regulons in carotid atherosclerotic plaques. This VIPER analysis revealed that the transcription factor activated RNA polymerase II transcriptional coactivator p15 (SUB1/Sub1, PC4) is a novel master regulator of TLR2 and TLR4 signaling in carotid plaque macrophages. Based on this in silico evidence, we sought to explore what impact macrophage Sub1 activity has on the development of murine atherosclerosis. We discovered that the proatherosclerotic effects of TLR2 and TLR4 signaling in chow‐fed ApoE −/− mice are mediated by Sub1. We also found that Sub1‐knockout macrophages display an anti‐inflammatory M2 polarized phenotype and enhanced cholesterol efflux. Moreover, irradiated low density lipoprotein receptor‐deficient (Ldlr −/−) mice transplanted with Sub1 −/− bone marrow showed decreased atherosclerosis. The inflammatory phenotype of Sub1‐deficient macrophages is restored by artificial overexpression of proinflammatory interferon regulatory factor 1 (Irf1), thereby lowering their cholesterol efflux ability. The present study thus identifies Sub1 as a master regulon of the atherogenic TLR response in macrophages.
2. Results
2.1. Identification of Master Regulator TFs in the Macrophage TLR Response
TLR activation induces a restructuring of downstream signaling pathways, producing a pattern unique to TLR activity that enables identification of master regulator TFs that regulate this transcriptional program. By analyzing differences between the gene expression profiles of Pam3CSK4 (Pam)‐treated and lipopolysaccharide (LPS)‐treated versus nontreated bone marrow‐derived macrophages (BMDMs) (File S1, Supporting Information), we were able to extract the macrophage transcriptomic signatures associated with TLR2 and TLR4 activation, respectively. The differentially expressed genes (DEGs) identified in the BMDMs were mapped to orthologous human genes, and DEGs with no human counterpart were eliminated from further analysis. TLR2 activation was associated with 808 DEGs (418 upregulated and 390 downregulated genes; Figure 1A), while TLR4 activation was associated with 1246 DEGs (683 upregulated and 563 downregulated genes; Figure 1B).
Figure 1.
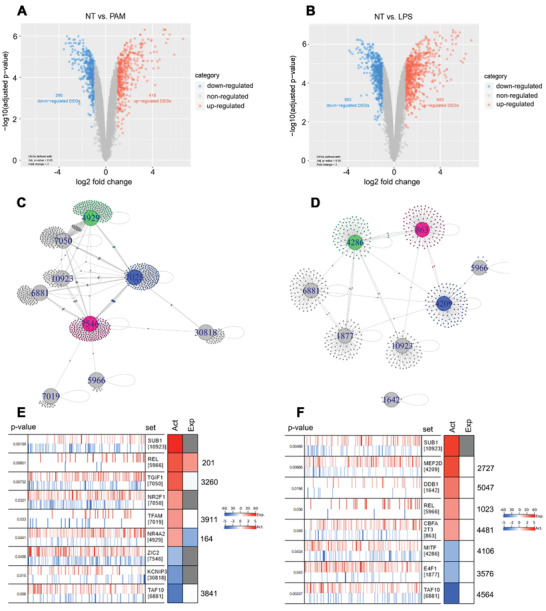
Identification of master transcription factors in the macrophage TLR2 and TLR4 responses. Differentially expressed genes (DEGs) identified between A) Pam‐treated (TLR2) and B) LPS‐treated (TLR4) versus nontreated bone marrow‐derived macrophages (BMDMs) represented by colored dots in a volcano plot; criteria for DEGs were: i) log2 fold‐change >1 and ii) p < 0.05; dots denote all transcripts detected by the microarray. Network diagrams of the master transcription factors (TFs) (large nodes labeled by TF's gene ID) and predicted targets (small dots) generated by Virtual Inference of Protein‐activity by Enriched Regulon (VIPER) analysis (p < 0.01) for C) the TLR2 network and D) the TLR4 network. Community structure predicted by fastgreedy.community module with edges denoting regulation of predicted targets by master TFs. VIPER evaluation of the master TFs for E) the TLR2 network and F) the TLR4 network. Right side: RTN and partial correlation analyses for repressed (blue) and activated (red) master TFs within coexpression networks reverse engineered and projected (vertical lines) to the TLR transcriptional signatures by msVIPER; x‐axis denotes the TLR signature sorted from most downregulated to most upregulated in Pam‐treated (TLR2) or LPS‐treated (TLR4) versus nontreated BMDMs as determined by the t‐statistic value from limma. Left side: The Act column of heatmap denotes activity, and the Exp column denotes value of differential expression within the TLR2 or TLR4 signature. Act signifies differential protein activity score in Pam‐treated (TLR2) or LPS‐treated (TLR4) versus nontreated BMDMs as predicted by the aREA module in VIPER, which analyzed coexpressed gene expression with TFs; red denotes increased activity, and blue denotes decreased activity post TLR2‐ or TLR4‐induction. Exp signifies differential transcript levels in Pam‐treated (TLR2) or LPS‐treated (TLR4) versus nontreated BMDMs as determined by the t‐statistic value from limma; red denotes upregulation, and blue denotes downregulation post TLR2‐ or TLR4‐induction.
Next, we generated six coexpression networks from the six transcriptomic datasets derived from human carotid plaques and normal carotid tissue samples (n = 371) to compare to the TLR signatures in order to identify master regulator TFs that regulate the macrophage TLR transcriptional signatures in atherosclerosis. The four Affymetrix datasets (GSE21545, GSE24495, GSE43292, and GSE28829) were preprocessed before being combined to prevent possible bias, i.e., quality control (Figure S1, Supporting Information), normalization, and correction for batch effects of specimen subgroups (Figure S2, Supporting Information). The two non‐Affymetrix datasets (GSE13922 and GSE1000927) were not subject to preprocessing. Variation in sample sizes was accounted for by implementing a shrinkage estimate of partial correlations[ 9 ] across TFs and their gene targets within every dataset; each edge within a network related to a significant partial correlation. The summary statistics for the six coexpression networks are provided in File S2 in the Supporting Information. We confirmed that there was concordance across all six datasets in the identified regulons, i.e., groups of genes regulated as a unit, generally by the same TF. p‐values were transformed to Z‐scores, and the six coexpression networks were combined into one network; edges from bigger studies were given more weight by applying Stouffer's method,[ 10 ] which assigns a sample size‐dependent weighted significance to each edge. The resulting combined network possessed edges that were coherent across every dataset.
VIPER analysis was then implemented to overlay the macrophage TLR transcription signatures with the coexpression network to identify intersection among genes within the signatures and the associated master regulons.[ 11 ] The VIPER analysis identified enrichment of TLR2 transcription genes in nine master regulator TFs (in order of most activated to most suppressed): SUB1 (NCBI Gene ID: 10923), the NF‐kB subunit Avian Reticuloendotheliosis Proto‐Oncogene (REL) (ID: 5966), TGFB Induced Factor Homeobox 1 (TGIF1) (ID: 7050), Nuclear Receptor Subfamily 2 Group F Member 1 (NR2F1) (ID: 7025), Transcription Factor A, Mitochondrial (TFAM) (ID: 7019), Nuclear Receptor Subfamily 4 Group A Member 2 (NR4A2) (ID: 4929), Zic Family Member 2 (ZIC2) (ID: 7546), Potassium Voltage‐Gated Channel Interacting Protein 3 (KCNIP3) (ID: 30818), and TATA‐Box Binding Protein Associated Factor 10 (TAF10) (ID: 6881)) (Figure 1C,E). Likewise, VIPER analysis identified enrichment of TLR4 transcription genes in eight master regulator TFs (in order of most activated to most suppressed): SUB1 (ID: 10923), Myocyte Enhancer Factor 2D (MEF2D) (ID: 4209), Damage Specific DNA Binding Protein 1 (DDB1) (ID: 1642), REL (ID: 5966), CBFA2/RUNX1 Partner Transcriptional Co‐Repressor 3 (CBFA2T3) (ID: 863), Melanocyte Inducing Transcription Factor (MITF) (ID: 4286), E4F Transcription Factor 1 (E4F1) (ID: 1877), and TAF10 (ID: 6881) (Figure 1D,F). Analyzing the structure of the TLR2 gene network, we found that the SUB1, TGIF1, NR2F1, NR4A2, ZIC2, and TAF10 regulons formed the network core, while the KCNIP3, REL, and TFAM regulons were more peripheral players (Figure 1C,E). Similarly, in the TLR4 gene network, SUB1, MEF2D, CBFA2T3, MITF, E4F1, and TAF10 regulons formed the network core, while the REL and DDB1 regulons were more peripheral players (Figure 1D,F). Gene set enrichment analysis (GSEA) was applied to the fastgreedy.community‐identified gene communities within the TLR2 gene network (n = 7 communities) and TLR4 gene network (n = 7 communities) to reveal the biological relevance of the pathways regulated by the master regulons (File S3, Supporting Information). Notably, the REL‐associated communities in both networks showed Gene Ontology molecular function (GO MF) enrichment for organic cyclic compound binding (GO MF: 0097159), heterocyclic compound binding (GO MF: 1901363), nucleic acid binding (GO MF: 0003676), and RNA polymerase II distal enhancer sequence‐specific binding (GO MF: 0003705).
Notably, there were three master regulons common to both TLR signatures: SUB1, REL, and TAF10. Among these three, the role of REL has already been well‐characterized in atherogenesis.[ 12 ] However, to our knowledge, SUB1 and TAF10 are novel master regulator TFs that have not been previously linked to atherosclerosis. Considering SUB1’s strong activation status and its more central position in both TLR networks, we chose to pursue further investigation on the role of macrophage SUB1 in atherosclerosis.
2.2. TLR2 and TLR4 Signaling Activates M1‐Skewing Sub1 in Murine Macrophages In Vitro and Atherosclerosis In Vivo
To understand the role of macrophage Sub1 in TLR‐induced atherogenesis, we generated Sub1 flox/flox (WT), myeloid‐specific hemizygous Lysozyme M (LysM)Cre/−/Sub1 flox/wt (HEMI) mice, and myeloid‐specific Sub1 KO mice (Figure S3, Supporting Information) and maintained BMDM cultures from WT, HEMI, and Sub1 KO mice in vitro. Real‐time quantitative PCR analysis (qPCR) revealed Sub1 mRNA downregulation in HEMI mice and Sub1 KO mice, with Sub1 KO mice showing negligible levels of Sub1 mRNA expression (Figure 2A). In contrast to mRNA levels, we observed similar Sub1 protein levels in WT and HEMI BMDMs (Figure 2B), suggesting haplosufficiency of Sub1 (i.e., one WT allele is sufficient for normal protein expression). For all further experiments, both the WT and HEMI phenotypes were chosen as control groups to control for any LysM Cre transgene effects.
Figure 2.
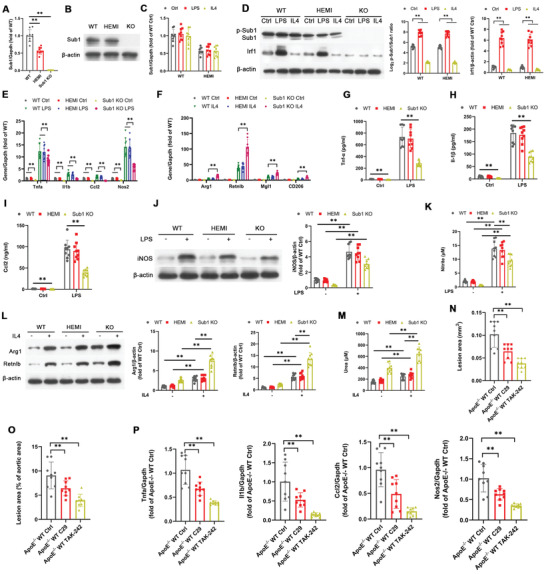
TLR2 and TLR4 signaling activates M1‐skewing Sub1 in murine macrophages in vitro and atherosclerosis in vivo. A–M) Bone marrow‐derived macrophages (BMDMs) isolated from Sub1 flox/flox (wild‐type, WT), LysM Cre/−/Sub1 flox/wt (hemizygous, HEMI), and LysM Cre/−/Sub1 flox/flox (knockout, KO) mice were stimulated with vehicle (Ctrl), Pam (100 ng mL−1, 8 h), LPS (100 ng mL−1, 8 h), or IL4 (5 ng mL−1, 8 h). A) qPCR of Sub1 mRNA levels. B) Immunoblotting of Sub1 protein levels. C) qPCR of Sub1 mRNA expression. D) Immunoblotting of Sub1, p‐Sub1, and Sub1's downstream target Irf1. Densitometric quantification of the p‐Sub1/Sub1 ratio and Irf1 protein expression. E) qPCR of M1 marker genes. F) qPCR of M2 marker genes. ELISA of G) Tnf‐α, H) Il‐1β, and I) Ccl2 secretion. J) Immunoblotting of iNOS protein expression and K) nitrite‐based iNOS activity. L) Immunoblotting of Arg1 and Retnlb protein expression and M) urea‐based Arg1 activity. N–P) ApoE −/−; Sub1 flox/flox (ApoE −/− WT) mice were fed a chow diet and administered vehicle (Ctrl), C29 (50 mg kg−1), or TAK‐242 (3 mg kg−1) by daily intraperitoneal (i.p.) injection for 14 weeks. n = 9 mice per group. N) Quantification of aortic root lesion areas based on 8–12 10 µm sections per mouse (30 µm apart). Scale bar = 200 µm. O) Quantification of total lesion areas in en face aortas. Scale bar = 1 cm. P) qPCR of M1 marker genes in isolated aortic root plaque macrophages. Data reported as means ± SDs. In vivo experiments: n = 9 mice per group. In vitro experiments: n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (A,O–P: one‐way ANOVA with Fisher's LSD; C–M: two‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group or n = 9 mice per group).
Macrophage phenotypes are polarized along a continuum from a proinflammatory M1 phenotype to an anti‐inflammatory, prorepair M2 phenotype.[ 13 ] The TLR4 agonist LPS, and to a lesser degree the TLR2 agonist Pam, drive M1 polarization; conversely, the cytokine IL‐4 drives M2 polarization.[ 14 ] As M1 macrophages promote atherogenesis while M2 macrophages promote atheroprotection,[ 15 ] we applied these factors to drive BMDM polarization in vitro. Consistent with our in silico TLR analysis (Figure 1E), mRNA levels of Sub1 in primary BMDMs were not changed by Pam or LPS exposure (Figure 2C). However, the p‐Sub1/Sub1 ratio (i.e., an indicator of casein kinase II (CkII, Ck2)‐mediated Sub1 phosphorylation)[ 16 ] and protein expression of Sub1's downstream target Irf1 were upregulated by Pam or LPS; the opposite effect was observed with the M2‐polarizing IL‐4 (Figure 2D). Decreased mRNA levels of the proinflammatory M1 markers Tumor Necrosis Factor Alpha (Tnfa, Tnf‐α), Interleukin 1 Beta (Il1b, Il‐1β), C‐C Motif Chemokine Ligand 2 (Ccl2, Ccl‐2), and Nitric Oxide Synthase 2 (Nos2, iNOS)[ 17 ] were observed under vehicle, Pam, and LPS conditions in Sub1 KO BMDMs (Figure 2E). However, higher mRNA levels of the anti‐inflammatory M2 markers Arginase 1 (Arg1, Arg1), Resistin Like Beta (Retnlb, Retnlb), Macrophage Galactose‐Type Lectin 1 (Mgl1, Mgl1), and CD206 Molecule (CD206, CD206)[ 17 ] were observed under vehicle and IL‐4 conditions in Sub1 KO BMDMs (Figure 2F). Secretion of the inflammatory markers Tnf‐α, Il‐1β, and Ccl‐2 was decreased in Sub1 KO BMDMs under vehicle, Pam, and LPS conditions (Figure 2G–I). Sub1 KO decreased iNOS protein expression and activity under vehicle, Pam, and LPS conditions (Figure 2J,K) but increased Arg1 and Retnlb protein expression as well as Arg1 activity under vehicle and IL‐4 conditions (Figure 2L,M). The above evidence confirms that TLR2 and TLR4 signaling activate Sub1 in murine macrophages, which skews them toward a proinflammatory M1 phenotype.
To validate the results of our findings in vivo, we conducted a series of experiments using pharmacological TLR inhibition or agonism in a chow‐fed ApoE −/− murine model of atherosclerosis. We administered the TLR2 inhibitor C29, the TLR4 inhibitor TAK‐242 (CLI‐095, resatorvid), or vehicle control to ApoE −/− mice fed a chow diet for 14 weeks. No significant differences in body weight or lipid profiles were observed between the three cohorts (Figure S4A–E, Supporting Information). Consistent with findings in chow‐fed ApoE −/− Tlr2 −/− and ApoE −/− Tlr4 −/− mice,[ 5 ] C29 or TAK‐242 reduced aortic atherosclerotic burden (Figure 2N,O) and downregulated the proinflammatory M1 markers Tnfa, Il1b, Ccl2, and Nos2 in macrophages isolated from aortic root plaques (Figure 2P). These findings confirm that TLR2 and TLR4 signaling are proinflammatory and proatherosclerotic in chow‐fed ApoE −/− mice.
2.3. Macrophage Sub1 Enhances TLR Signaling‐Induced Atherosclerosis in Chow‐Fed ApoE −/− Mice
To determine the role of Sub1 in TLR signaling‐induced atherosclerosis, we applied myeloid‐specific Sub1 KO in the chow‐fed ApoE −/− murine model of atherosclerosis. We administered vehicle control, the TLR2 agonist Pam, or the TLR4 agonist LPS to ApoE −/−; Sub1 flox/flox mice (ApoE −/− WT), ApoE −/−; LysM Cre/−/Sub1 flox/wt mice (ApoE −/− HEMI), and ApoE −/−; LysM Cre/−/Sub1 flox/flox mice (ApoE, Sub1 KO) fed a chow diet for 14 weeks. No significant differences in body weight were observed between the cohorts (Figure S5A, Supporting Information). Significant changes in lipid profiles were observed in LPS‐treated mice but not Pam‐treated mice; the LPS‐induced changes in total cholesterol, low‐density lipoprotein cholesterol (LDL‐C), and high‐density lipoprotein cholesterol (HDL‐C) were rescued by ApoE, Sub1 KO (Figure S5B–E, Supporting Information). Pam or LPS enhanced aortic atherosclerotic burden and reduced lesion collagen deposition (as detected by Masson's trichrome stain) from ApoE −/− WT and HEMI mice, effects abrogated in ApoE, Sub1 KO mice (Figure 3A–C). Pam or LPS enhanced proinflammatory M1 marker expression in plaque macrophages from ApoE −/− WT and HEMI mice, effects abrogated in ApoE, Sub1 KO mice (Figure 3D). As atherosclerotic progression is increased by macrophage proliferation within the atherosclerotic lesion,[ 18 ] we analyzed macrophage cell proliferation and apoptosis in vitro and in vivo. LPS reduced, while Sub1 KO had no significant impact, on macrophage proliferation in vitro or in atherosclerotic lesions in vivo by Ki67 immunofluorescence (Figure 3E,F). However, in vitro terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) assays showed that Pam or LPS enhanced apoptosis levels in macrophages in vitro or in atherosclerotic lesions in vivo, which were abrogated by Sub1 KO (Figure 3G,H). Correspondingly, Pam or LPS enhanced cleaved caspase‐3 staining in atherosclerotic lesions in vivo, effects abrogated by Sub1 KO (Figure 3I). Pam or LPS also enhanced iNOS+ M1 macrophage and decreased CD206+ M2 macrophage content in ApoE −/− WT and HEMI lesions, effects abrogated in ApoE, Sub1 KO lesions (Figure 3J). These combined findings indicate that the proatherosclerotic effects of TLR2 and TLR4 signaling in chow‐fed ApoE −/− mice are mediated by Sub1.
Figure 3.
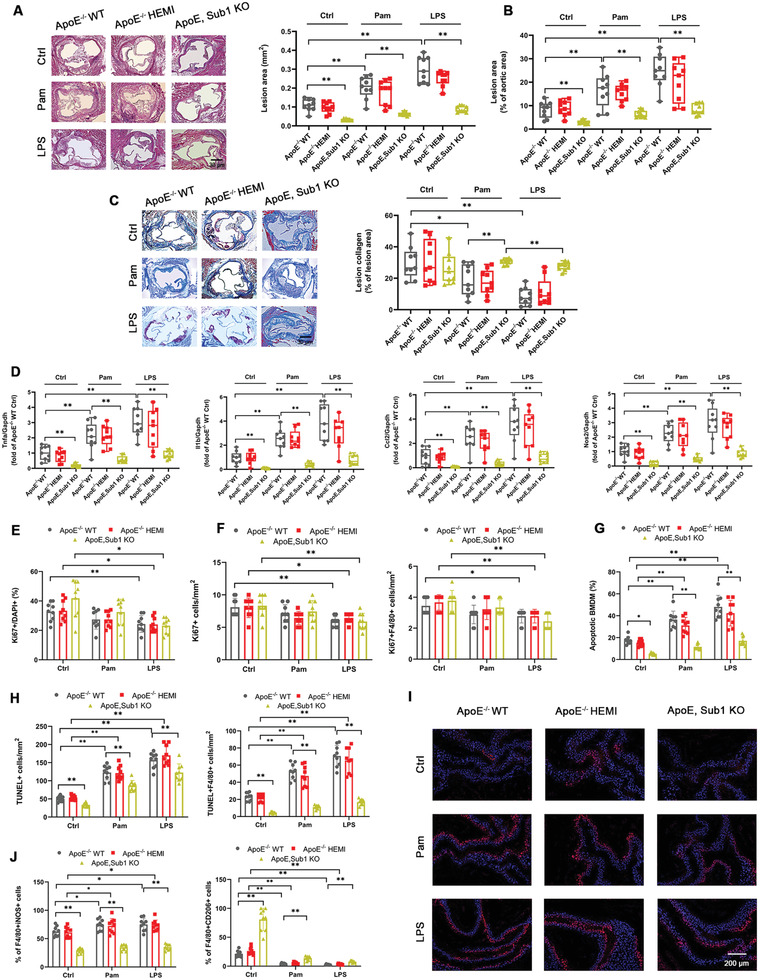
Macrophage Sub1 enhances TLR signaling‐induced atherosclerosis in chow‐fed ApoE −/− mice. ApoE −/−; Sub1 flox/flox (ApoE −/− WT), ApoE −/−; LysM Cre/−/Sub1 flox/wt (ApoE −/− HEMI), and ApoE −/−; LysM Cre/−/Sub1 flox/flox (ApoE, Sub1 KO) mice were fed a chow diet and administered vehicle (Ctrl), Pam (15 µg), or LPS (50 µg) by weekly intraperitoneal (i.p.) injection for 14 weeks. A) Representative H&E staining images showing lesion areas in aortic root sections. Quantification of aortic root lesion areas based on 8–12 10 µm sections per mouse (30 µm apart). Scale bar = 200 µm. B) Quantification of total lesion areas in en face aortas. C) Representative Masson's trichrome staining images and quantification of aortic root lesion collagen by Zeiss Axiovision software. Scale bar = 200 µm. D) qPCR of M1 marker genes in isolated aortic root plaque macrophages. E) In vitro bone marrow‐derived macrophages (BMDMs) proliferation under vehicle (Ctrl), Pam (100 ng mL−1), or LPS (100 ng mL−1) for 8 h and F) in vivo F4/80+ macrophage proliferation in aortic root lesions examined using anti‐Ki67 immunofluorescence. Scale bar = 100 µm. G) In vitro BMDM apoptosis levels under vehicle (Ctrl), Pam (100 ng mL−1), or LPS (100 ng mL−1) for 8 h assessed by TUNEL staining. Cell morphology analyzed by differential interference contrast (DIC) and nuclear staining by DAPI. Apoptotic cell percentage expressed as ratio of TUNEL+/DAPI+. Scale bar = 100 µm. n = 9 fields per group. H,I) In vivo F4/80+ macrophage apoptosis in aortic root lesions examined by TUNEL and cleaved caspase‐3 staining. Scale bar = 100 µm. J) Immunofluorescent staining analysis of M1 macrophages (iNOS+/F4/80+) and M2 macrophages (CD206+/F4/80+) in serial aortic root lesion sections. Data reported as means ± SDs. In vivo experiments: n = 9 mice per group. In vitro experiments: n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (two‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group or n = 9 mice per group).
2.4. Sub1 Mediates TLR Signaling‐Induced M1 Skewing in ApoE −/− Murine Macrophages
We conducted a series of in vitro experiments utilizing Pam, LPS, and IL‐4 stimulation in BMDMs isolated from ApoE −/− WT, ApoE −/− HEMI, and ApoE, Sub1 KO mice. Similar to WT BMDMs, the p‐Sub1/Sub1 ratio and protein expression of Sub1's downstream target Irf1 were upregulated by Pam or LPS; the opposite effect was observed with the M2‐polarizing cytokine IL‐4 (Figure 4A). Pam or LPS upregulated the proinflammatory M1 markers Tnfa, Il1b, Ccl2, and Nos2, effects abrogated in ApoE, Sub1 KO BMDMs (Figure 4B). The IL‐4‐induced anti‐inflammatory M2 markers Arg1, Retnlb, Mgl1, and CD206 were upregulated in ApoE, Sub1 KO BMDMs (Figure 4C). Pam or LPS upregulated secretion of the inflammatory markers Tnf‐α, Il‐1β, and Ccl‐2, effects abrogated in ApoE, Sub1 KO BMDMs (Figure 4D–F). Moreover, Pam or LPS upregulated iNOS protein expression and activity, effects abrogated in ApoE, Sub1 KO BMDMs (Figure 4G,H). IL‐4‐induced Arg1 and Retnlb protein expression as well as Arg1 activity were upregulated in ApoE, Sub1 KO BMDMs (Figure 4I,J). This evidence reveals that proinflammatory M1 skewing by TLR2 and TLR4 signaling in ApoE −/− macrophages is mediated by Sub1.
Figure 4.
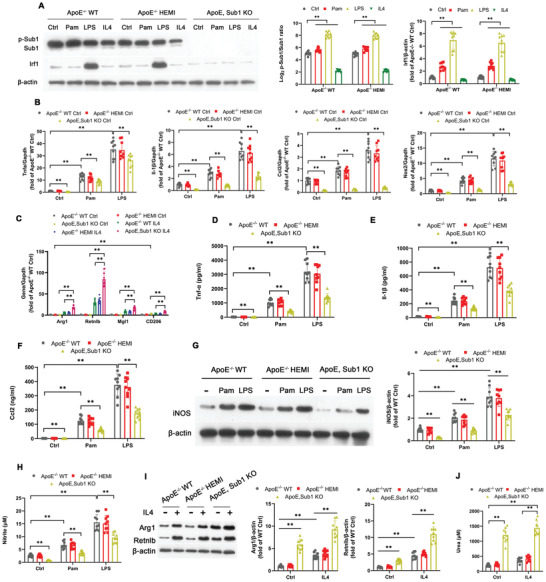
Sub1 mediates TLR signaling‐induced M1 skewing in ApoE −/− murine macrophages. Bone marrow‐derived macrophages (BMDMs) from ApoE −/−; Sub1 flox/flox (ApoE −/− WT), ApoE −/−; LysM Cre/−/Sub1 flox/wt (ApoE −/− HEMI), and ApoE −/−; LysM Cre/−/Sub1 flox/flox (ApoE, Sub1 KO) mice were treated with vehicle (Ctrl), Pam (100 ng mL−1), LPS (100 ng mL−1), or IL4 (5 ng mL−1) for 8 h. A) Immunoblotting of Sub1, p‐Sub1, and Sub1's downstream target Irf1. Densitometric quantification of the p‐Sub1/Sub1 ratio and Irf1 protein expression. B) qPCR of M1 marker genes. C) qPCR of M2 marker genes. ELISA of D) Tnf‐α, E) Il‐1β, and F) Ccl2 secretion. G) Immunoblotting of iNOS protein expression and H) nitrite‐based iNOS activity. I) Immunoblotting of Arg1 and Retnlb protein expression and J) urea‐based Arg1 activity. Data reported as means ± SDs. n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (two‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group).
2.5. oxLDL Exposure Activates Sub1, Which Reduces Cholesterol Efflux from Murine Macrophages
TLRs participate in the interaction between oxLDL and macrophages, thereby promoting oxLDL‐induced macrophage activation.[ 3 , 4 ] To understand the role of macrophage Sub1 in oxLDL‐induced macrophage activation, we conducted a series of in vitro experiments in WT, HEMI, and Sub1 KO BMDMs. We observed Sub1 activation in response to oxLDL exposure (Figure 5A). After oxLDL exposure, we observed reduced lipid accumulation in Sub1 KO BMDMs compared to WT and HEMI controls (Figure 5B). Moreover, lower [3H]‐cholesterol uptake (Figure 5C) as well as higher ApoE‐dependent autocrine cholesterol efflux (Ctrl; Figure 5C) and cholesterol efflux to Apolipoprotein A1 (ApoA1) and HDL‐C (Figure 5D) were observed in Sub1 KO BMDMs. ApoE mRNA levels were also upregulated in Sub1 KO BMDMs (Figure 5E). Consistent with the increased cholesterol efflux activity of Sub1 KO BMDMs, Sub1 KO BMDMs displayed upregulation of the cholesterol transporters ATP‐Binding Cassette Sub‐Family G Member 1 (Abcg1) and ATP‐Binding Cassette Subfamily A Member 1 (Abca1) coupled with downregulation of oxLDL Receptor 1 (Olr1) (Figure 5F).
Figure 5.
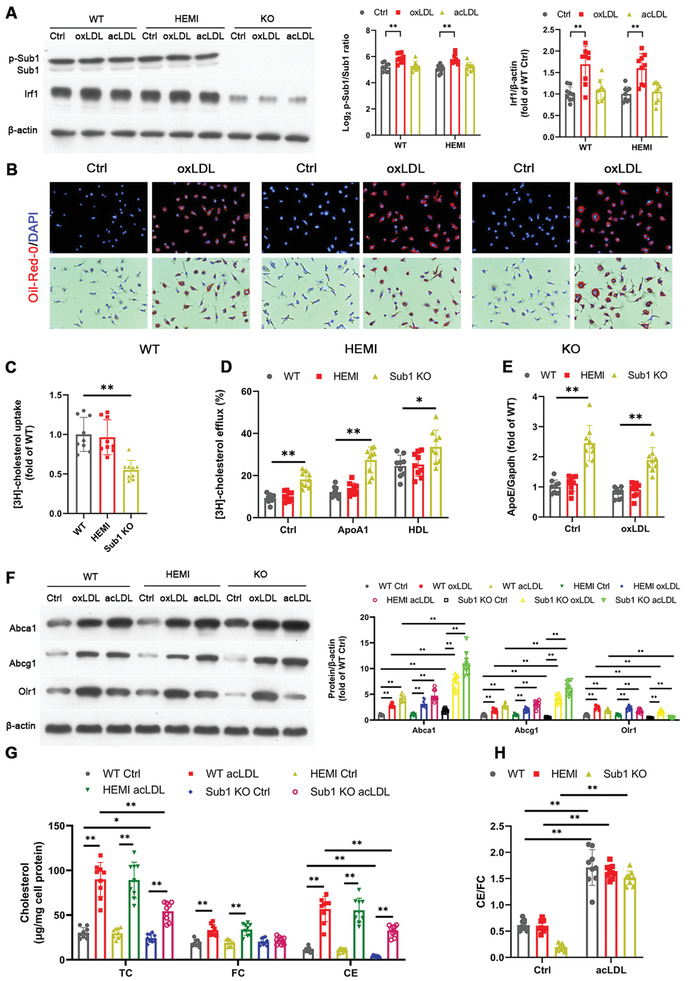
Macrophage Sub1 reduces cholesterol efflux from murine macrophages. The following experiments employed bone marrow‐derived macrophages (BMDMs) from Sub1 flox/flox (wild‐type, WT), LysM Cre/−/Sub1 flox/wt (hemizygous, HEMI), and LysM Cre/−/Sub1 flox/flox (knockout, KO) mice. A) Immunoblotting of Sub1, p‐Sub1, and Sub1's downstream target Irf1 following exposure to vehicle (Ctrl) or modified LDLs (30 µg mL−1 oxLDL or 50 µg mL−1 acLDL, 24 h). Densitometric quantification of the p‐Sub1/Sub1 ratio and Irf1 protein expression. B) Representative images of neutral lipid accumulation upon oxLDL incubation (30 µg mL−1, 24 h). Scale bar, 100 µm. C) Cholesterol uptake after exposure to [3H] cholesterol‐labeled acLDL (50 µg mL−1) for 30 min. All values normalized to total protein levels and expressed as fold of WT. D) Cholesterol efflux after treatment with [3H] cholesterol for 48 h. E) qPCR of ApoE mRNA expression. F) Immunoblotting of Abca1, Abcg1, and Olr1 following exposure to Ctrl or modified LDLs (30 µg mL−1 oxLDL or 50 µg mL−1 acLDL, 24 h). G) Accumulation of total cholesterol (TC), free cholesterol (FC), and esterified cholesterol (CE), and H) the CE/FC ratio in BMDMs following acLDL exposure (50 µg mL−1, 24 h). Data reported as means ± SDs. n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (A,E–H: two‐way ANOVA with Fisher's LSD; C,D: one‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group).
Increases in free cholesterol accumulation within macrophages lead to an inflammatory phenotype and an increased likelihood of foam cell formation.[ 19 ] Under acetylated LDL (acLDL)‐treated conditions, accumulation of total cholesterol (TC), cholesterol ester (CE), and free cholesterol (FC) all decreased in Sub1 KO BMDMs (Figure 5G). Nevertheless, the ratio of esterified/free cholesterol (CE/FC) did not significantly change in Sub1 KO BMDMs, indicating no significant effect on cholesterol mobilization and storage (Figure 5H). The above results indicate that Sub1 KO lowers macrophage cholesterol accumulation under acLDL‐treated conditions.
Mice on high‐fat diets (HFDs) typically show elevated serum LDL‐C levels due to the inflammatory response of liver macrophages;[ 20 ] therefore, we hypothesized that myeloid Sub1 KO in HFD‐fed mice would lower serum LDL‐C levels. Indeed, HFD‐fed Sub1 KO mice showed lower serum LDL‐C levels relative to their WT and HEMI counterparts (Figure S6, Supporting Information). The above results indicate that Sub1 KO in macrophages lowers circulating LDL‐C levels under HFD conditions.
2.6. Transplantation of Sub1 Knockout Macrophages Reduces Western Diet‐Induced Atherosclerosis in Ldlr −/− Mice
The consumption of a Western diet (i.e., high cholesterol and saturated fatty acid content) contributes to enhanced circulating cholesterol levels, oxLDL‐induced macrophage activation, and pathogenic foam cell differentiation.[ 21 ] To analyze the role of Sub1‐deficient macrophages in Western diet‐induced atherosclerosis in vivo, we examined the effect of transplanting transgenic bone marrow into a Western diet‐fed Ldlr −/− murine model of atherosclerosis. Irradiated Ldlr −/− mice transplanted with either Sub1 flox/flox bone marrow (WT→Ldlr −/−), LysM Cre/−/Sub1 flox/wt bone marrow (HEMI→Ldlr −/−), LysM Cre/−/Sub1 flox/flox bone marrow (Sub1 KO→Ldlr −/−), or LysM Cre/−/Sub1 flox/flox; Signal Transducer And Activator Of Transcription 6 (Stat6)−/− bone marrow (Sub1, Stat6 KO→Ldlr −/−) were fed a Western diet for 12 weeks. As Stat6 signaling is a key mediator of M2 macrophage polarization,[ 22 ] we used Sub1, Stat6 KO→Ldlr −/− mice to assess whether Sub1 KO's effects were primarily mediated through M2 polarization. Immunoblotting confirmed Sub1 and Stat6 knockdown in the appropriate bone marrow samples prior to transplantation (Figure S7, Supporting Information). There were no significant differences in body weight or lipid profiles in the four cohorts (Figure S8A–E, Supporting Information), probably due to the effects of Ldlr −/− phenotype in increasing LDL‐C levels.
Sub1 KO→Ldlr −/− aortic root atherosclerotic lesions were considerably smaller than the two control cohorts; notably, the addition of Stat6 KO abrogated the antiatherosclerotic effects of Sub1 KO (Figure 6A,B). Lesion collagen deposition (as detected by Masson's trichrome stain) was similar in the four cohorts (Figure 6C). Plaque macrophages from Sub1 KO→Ldlr −/− lesions displayed decreased proinflammatory M1 marker expression, effects abrogated by Stat6 KO (Figure 6D). By Ki67 immunofluorescence, there were no significant differences in macrophage proliferation in vitro or in atherosclerotic lesions in vivo among the four cohorts (Figure 6E,F). However, in vitro TUNEL assay showed decreased apoptosis levels in Sub1 KO macrophages under basal conditions and oxLDL challenge, which were abrogated by Stat6 KO (Figure 6G). Moreover, in vivo TUNEL assay and cleaved caspase‐3 staining detected lower apoptosis levels in Sub1 KO→Ldlr −/− lesions, effects abrogated in Sub1, Stat6 KO→Ldlr −/− lesions (Figure 6H,I). In addition, Sub1 KO→Ldlr −/− atherosclerotic lesions displayed decreased proinflammatory iNOS+ M1 macrophage content coupled with increased anti‐inflammatory CD206+ M2 macrophage content, effects abrogated in Sub1, Stat6 KO→Ldlr −/− lesions (Figure 6J). These data indicate that macrophage Sub1 KO reduces atherosclerosis primarily through promoting anti‐inflammatory M2 polarization as opposed to affecting macrophage proliferation or apoptosis.
Figure 6.
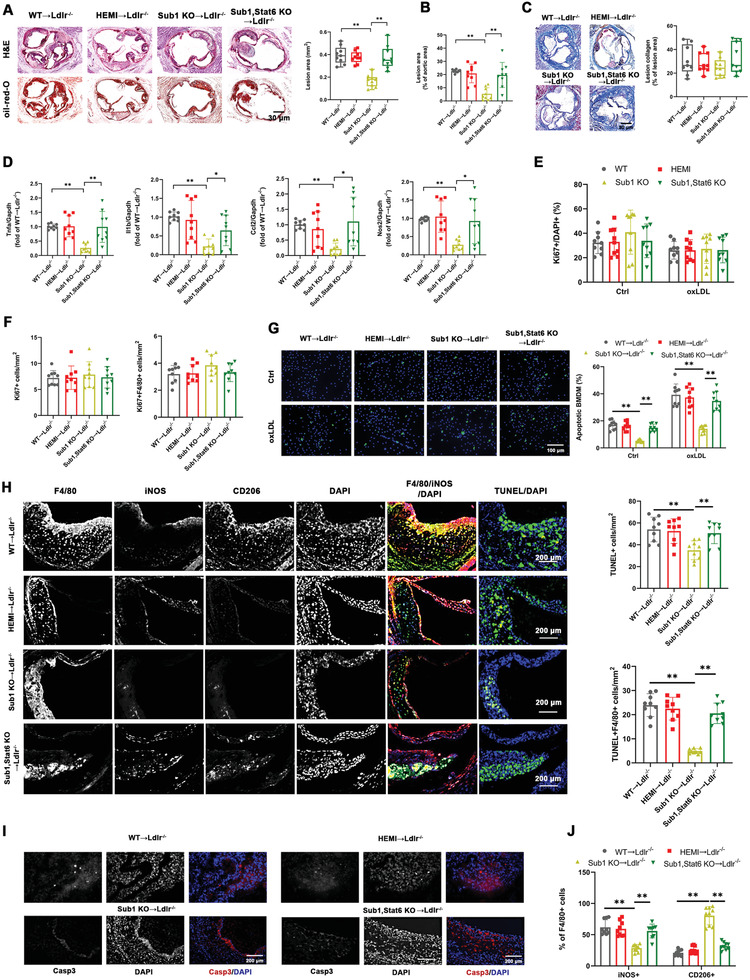
Transplantation of Sub1 knockout macrophages reduces Western diet‐induced atherosclerosis in Ldlr −/− recipient mice. Irradiated Ldlr −/− mice transplanted with bone marrow from Sub1 flox/flox mice (WT→Ldlr −/−), LysM Cre/−/Sub1 flox/wt mice (HEMI→Ldlr −/−), LysM Cre/−/Sub1 flox/flox mice (Sub1 KO→Ldlr −/−), or LysM Cre/−/Sub1 flox/flox; Stat6 −/− mice (Sub1, Stat6 KO→Ldlr −/−) were fed a Western diet for 12 weeks. A) Representative H&E and Oil Red O staining images showing lesion areas in aortic root sections. Scale bar = 200 µm. Quantification of aortic root lesion areas based on 8–12 10 µm sections per mouse (30 µm apart). B) Quantification of total lesion areas in en face aortas. C) Representative Masson's trichrome staining images and quantification of aortic root lesion collagen by Zeiss Axiovision software. Scale bar = 200 µm. D) qPCR of M1 marker genes in isolated aortic root plaque macrophages. E) In vitro bone marrow‐derived macrophages (BMDMs) proliferation under vehicle (Ctrl) or oxLDL (50 µg mL−1, 24 h) conditions and F) in vivo F4/80+ macrophage proliferation in aortic root lesions examined using anti‐Ki67 immunofluorescence. Scale bar = 100 µm. G) In vitro BMDM apoptosis levels under vehicle (Ctrl) or oxLDL (50 µg mL−1, 24 h) conditions assessed by TUNEL staining. Cell morphology analyzed by differential interference contrast (DIC) and nuclear staining by DAPI. Apoptotic cell percentage expressed as ratio of TUNEL+/DAPI+. Scale bar = 100 µm. n = 9 fields per group. H,I) In vivo F4/80+ macrophage apoptosis in aortic root lesions examined by TUNEL and cleaved caspase‐3 staining. Scale bar = 100 µm. J) Immunofluorescent staining analysis of M1 macrophages (iNOS+/F4/80+) and M2 macrophages (CD206+/F4/80+) in serial aortic root lesion sections. Data reported as means ± SDs. In vivo experiments: n = 9 mice per group. In vitro experiments: n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (A–D,F,H,J: one‐way ANOVA with Fisher's LSD; E,G: two‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group or n = 9 mice per group).
As macrophage infiltration into the myocardium can produce ventricular hypertrophy,[ 23 ] we investigated any possible effects of macrophage Sub1 KO on the murine myocardium. We noted no significant differences in the interventricular septum or ventricular myocardium among the four cohorts (Figure S9A, Supporting Information). We also observed no signs of aortic valve calcification by Alizarin Red staining among the four cohorts (Figure S9B, Supporting Information). We also found no significant differences in macrophage infiltration within the myocardium among the four cohorts (Figure S9C, Supporting Information).
2.7. Macrophage Sub1 Upregulates M1‐Skewing Irf1 Expression in a Ck2‐Dependent Manner
The proinflammatory transcription factor Irf1 is known to play a role in regulating several proteins involved in atherosclerosis‐related inflammation, such as Retinoic Acid‐Inducible Gene I (RIG‐I) and Interleukin 8 (Il‐8).[ 24 ] Sub1 is phosphorylated by the protein kinase Ck2, which is induced by LPS and is associated with proinflammatory downstream gene expression in macrophages.[ 25 ] Moreover, the combination of Sub1 and Ck2 is necessary for downstream promoter element (DPE)‐dependent transcription of Irf1.[ 26 ] Therefore, we hypothesized that Irf1 may be a key downstream intermediary for Sub1's effects on atherogenesis. Consistent with our immunoblotting findings (Figures 2D and 5A), we confirmed Irf1 mRNA downregulation in Sub1 KO BMDMs by qPCR (Figure 7A). For in vivo validation, we confirmed that C29 or TAK‐242 reduced Irf1 mRNA expression in macrophages isolated from chow‐fed ApoE −/− WT murine aortic root plaques (Figure S10A, Supporting Information). Conversely, Pam or LPS enhanced Irf1 mRNA expression in plaque macrophages from chow‐fed ApoE −/− WT and HEMI mice, effects abrogated in ApoE, Sub1 KO mice (Figure S10B, Supporting Information). Moreover, Irf1 mRNA expression was downregulated in Western diet‐fed Sub1 KO→Ldlr −/− aortic root plaque macrophages (Figure S10C, Supporting Information). ChIP studies with an anti‐Sub1 antibody confirmed that BMDMs possess Sub1 expression at the Irf1 promoter (Figure 7B). Moreover, ChIP studies with an anti‐trimethylated lysine 4 of histone 3 (H3K4me3) antibody, a marker of open chromatin structure, suggests reduced Irf1 promoter activity in Sub1 KO BMDMs (Figure 7B).
Figure 7.
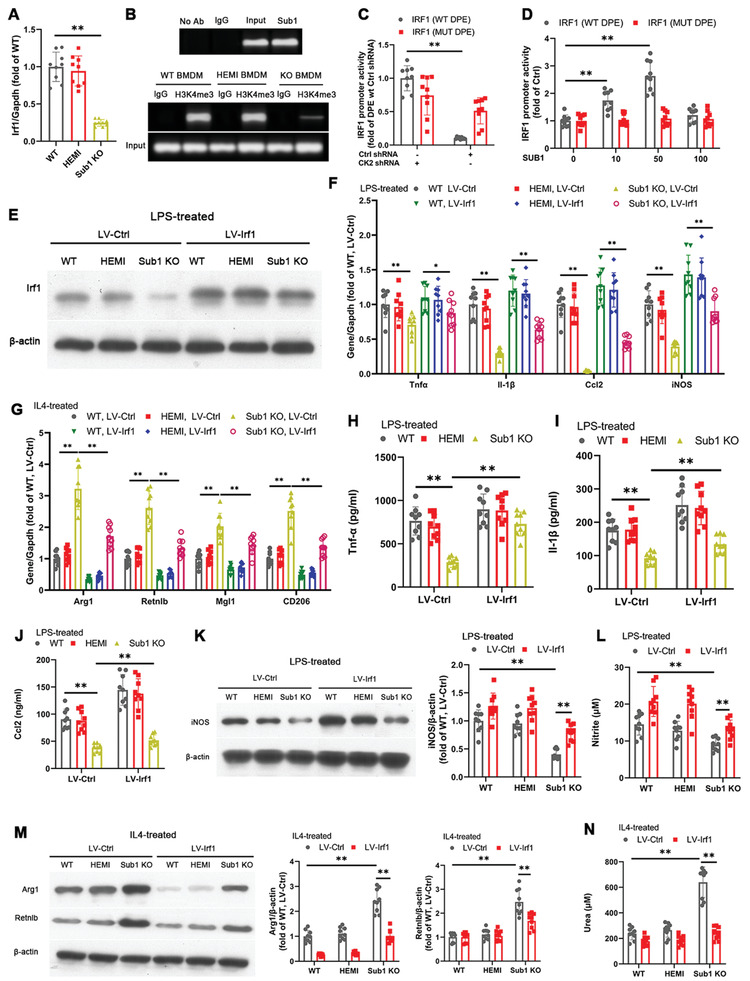
Macrophage Sub1 upregulates M1‐skewing Irf1 expression in a Ck2‐dependent manner. Unless otherwise stated, the following experiments employed Sub1 flox/flox (wild‐type, WT), LysM Cre/−/Sub1 flox/wt (hemizygous, HEMI), and LysM Cre/−/Sub1 flox/flox (knockout, KO) bone marrow‐derived macrophages (BMDMs). A) qPCR of Irf1 mRNA levels. B) Chromatin immunoprecipitation (ChIP)‐based detection of Sub1's interaction with the Irf1 promoter in WT BMDMs (top). Open confirmation of Irf1 promoter detected by H3K4me3 antibody ChIP in BMDMs (bottom). Human THP‐1 cells were transfected with the WT IRF1 promoter or mutant (MUT) IRF1 promoter alone or in combination with C) CK2 shRNA or scrambled control shRNA, or D) Lenti‐GIII‐CMV‐CK2‐HA or control vector. Renilla luciferase vector was cotransfected as normalization control. Luciferase reporter activity was detected 48 h post‐transfection. E–N) BMDMs were transfected with Lenti‐GIII‐CMV‐Irf1‐HA (LV‐Irf1) to enable stable Irf1 overexpression. Where indicated, BMDMs were stimulated with M1‐polarizing LPS (100 ng mL−1, 8 h) or M2‐polarizing IL4 (5 ng mL−1, 8 h). E) Immunoblotting of Irf1. F) qPCR of M1 marker genes under vehicle (Ctrl) or LPS conditions. G) qPCR of M2 marker genes under vehicle (Ctrl) or IL4 conditions. ELISA of H) Tnf‐α, I) Il‐1β, and J) Ccl2 secretion under vehicle (Ctrl) or LPS conditions. K) Immunoblotting of iNOS protein expression and L) nitrite‐based iNOS activity under vehicle (Ctrl) or LPS conditions. M) Immunoblotting of Arg1 and Retnlb protein expression and N) urea‐based Arg1 activity under vehicle (Ctrl) or IL4 conditions. Data reported as means ± SDs. n = 3 biological replicates × 3 technical replicates. *p < 0.05 and **p < 0.01 (A: one‐way ANOVA with Fisher's LSD; B–N) two‐way ANOVA with Fisher's LSD; comparing n = 3 in vitro biological replicates per group).
To ascertain whether Sub1's promotion of Irf1 expression is Ck2‐dependent, we first generated luciferase reporter constructs of human IRF1 with WT and mutant DPE binding sites. The WT DPE binding site construct showed decreased luciferase reporter activity with shRNA‐induced CK2 knockdown; however, no significant difference was observed with the mutant DPE binding site construct (Figure 7C). CK2 overexpression dose‐dependently increased luciferase reporter activity of the WT DPE binding site construct, but had no effect on the mutant DPE binding site construct (Figure 7D). This evidence indicates that SUB1 promotes IRF1 expression in a CK2‐dependent manner.
Seeing that Sub1 promotes Irf1 expression in BMDMs, we examined whether lentiviral Irf1 overexpression (Figure 7E) could rescue the effects of Sub1 KO on BMDM polarization. Irf1 overexpression in Sub1 KO BMDMs upregulated proinflammatory M1 marker expression under LPS conditions (Figure 7F) but downregulated anti‐inflammatory M2 marker expression under IL‐4 conditions (Figure 7G). Irf1 overexpression in Sub1 KO BMDMs also upregulated Tnf‐α, Il‐1β, and Ccl‐2 secretion under LPS conditions (Figure 7H–J). Moreover, Irf1 overexpression in Sub1 KO BMDMs enhanced iNOS protein expression and activity under LPS conditions (Figure 7K,L) but decreased Arg1 and Retnlb protein expression as well as Arg1 activity under IL‐4 conditions (Figure 7M,N). Irf1 overexpression reversed cholesterol handling in Sub1 KO BMDMs (Figure S11A,B, Supporting Information). Irf1 overexpression downregulated Abca1 and Abcg1 expression but upregulated Olr1 levels in cholesterol‐loaded Sub1 KO BMDMs (Figure S11C, Supporting Information). These results indicate that Sub1‐induced Irf1 activity is an important contributor to the heightened inflammatory response and dysfunctional cholesterol handling in macrophages.
3. Discussion
Inflammatory macrophage polarization increases the propensity of atherosclerosis.[ 15 ] However, the molecular mechanisms underlying this phenomenon are not clearly understood. Herein, we sought to identify master regulator TFs regulating the macrophage TLR transcriptional signature in atherosclerosis. We accomplished this in silico analysis in two stages; first we employed TLR2‐ and TLR4‐stimulated and control macrophages to identity the TLR2 and TLR4 transcriptional signatures in macrophages. Second, we used six publicly available whole transcriptome datasets on atherosclerosis patient‐derived carotid specimens to generate an integrated gene coexpression network. Superimposition of the TLR transcriptional signatures with the coexpression network identified three master regulons common to both TLR signatures: SUB1, the NF‐kB subunit REL, and TAF10. As a novel master regulon in atherosclerosis, we further investigated the role of macrophage Sub1 in atherosclerosis. Our follow‐up experiments revealed that the proatherosclerotic effects of TLR2 and TLR4 signaling in chow‐fed ApoE −/− mice are mediated by Sub1. Moreover, Sub1 KO macrophages show reduced atherogenic characteristics such as anti‐inflammatory M2 skewing and improved cholesterol handling. In irradiated Western diet‐fed Ldlr −/− mice, macrophage Sub1 KO reduced atherosclerotic burden. We also showed that expression of the proinflammatory transcription factor Irf1 was reduced upon Sub1 KO and artificially restoring Irf1 expression in Sub1 KO macrophages reversed Sub1 KO's positive effects on macrophage polarization and cholesterol handing. This combined evidence identifies Sub1 as a master regulon of the atherogenic TLR response in macrophages.
Macrophage accumulation within the arterial wall is a prominent feature in atherosclerotic plaques.[ 2 ] Exposure to various stimuli within the plaque microenvironment (e.g., TLR ligands, cytokines, oxidized lipids, etc.) can skew the polarization of plaque macrophages toward a proinflammatory M1 phenotype (classically activated by lipopolysaccharide and/or IFN‐ɣ exposure) and away from an anti‐inflammatory M2 phenotype (alternatively activated by IL‐4 or IL‐13 exposure).[ 13 ] It is well‐recognized that skewing favoring M1 over M2 polarization is a key element in atherosclerotic progression;[ 15 ] M1 macrophages secrete proinflammatory cytokines and promote atherogenesis and plaque vulnerability while M2 macrophages secrete anti‐inflammatory cytokines and promote atheroprotection.[ 15 ] Consistent with this inflammatory model of atherogenesis, we observed that TLR2 and TLR4 signaling are proinflammatory and proatherosclerotic in chow‐fed ApoE −/− mice and that these proatherosclerotic effects are mediated by Sub1. Moreover, we found that Sub1 KO increased M2 marker expression (i.e., CD206, Arg1, Mgl1, and Retnlb)[ 17 ] and decreased M1 marker expression (i.e., Tnfα, Il‐1β, Ccl2, and iNOS)[ 17 ] in macrophages under their respective priming conditions. These findings indicate that Sub1 supports skewing of the M1/M2 polarization balance toward a proinflammatory, proatherogenic M1 state. Recognizing that Sub1 is a master regulon of TLR signaling in macrophages, these results are consistent with TLR2 or TLR4 signaling activation also skewing macrophages toward the M1 phenotype.[ 27 ]
In addition to secreting inflammatory mediators, plaque macrophages can absorb oxLDL to form atherogenic foam cells or, contrarily, efflux cholesterol through reverse cholesterol transport to reverse plaque progression.[ 28 ] Although oxLDL exposure does not skew macrophages toward either M1 or M2,[ 29 ] macrophage polarization can adversely affect their cholesterol handling. For instance, the M1 macrophage‐secreted cytokine Il‐1β lowers ApoE‐mediated cholesterol efflux and cholesterol transporter expression,[ 30 ] leading to foam cell formation. Here, we observed that Sub1 KO macrophages showed decreased proinflammatory cytokine secretion coupled with enhanced ApoE‐mediated cholesterol efflux and cholesterol transporter expression under cholesterol‐loading conditions. In vivo, Sub1 KO→Ldlr −/− atherosclerotic plaques displayed decreased size, enhanced M2 macrophage content, and decreased M1 macrophage content, all of which were abrogated by KO of the M2‐polarization transcription factor Stat6. Therefore, anti‐inflammatory M2 skewing contributes to the observed reduction in atherosclerotic burden in Sub1 KO→Ldlr −/− mice.[ 3 ] Validating these findings, macrophage Sub1 KO on an ApoE −/− background also reduced atherosclerotic burden and promoted M2 skewing. These results support Sub1's role in promoting a proinflammatory, proatherogenic environment via M1 skewing.
The phagocytosis of apoptotic cells by macrophages, a process termed efferocytosis, also plays a key role in controlling plaque inflammation and atherosclerotic progression.[ 31 ] Efferocytosis reduces the release of danger‐associated molecular pattern molecules (DAMPs) by necrotic cells and also suppresses proinflammatory responses in macrophages.[ 31 ] Macrophage polarization can also adversely affect the proper efferocytosis within plaques, with M2 macrophages displaying enhanced efferocytosis abilities relative to M1 macrophages.[ 32 ] Here, we observed lower apoptotic indices in Sub1 KO→Ldlr −/− plaques, which were abrogated by Stat6 KO. Thus, these lower plaque apoptosis levels may be attributed to Sub1 KO‐mediated increases in plaque M2 macrophage content. These results also support Sub1’s role in promoting a proinflammatory, proatherogenic environment via M1 skewing.
The family of interferon regulatory factors (IRFs) play a critical role in monocyte lineage development, monocyte‐to‐macrophage differentiation, and M1/M2 polarization.[ 33 ] Specifically, Irf1 (as well as Irf5 and Irf8) are associated with M1 polarization, while Irf3 and Irf4 are associated with M2 polarization.[ 33 ] More recent work has revealed that LPS/TLR4‐induced, HDL‐C‐regulated DEGs display a strong enrichment for NFκB p65‐ and Irf1‐dependent genes, and HDL‐C's proinflammatory effects in macrophages require NFκB p65 and are partly dependent on Irf1.[ 34 ] Consistent with this evidence, our in silico analysis found that the NF‐kB subunit REL and the upstream effector of IRF1—SUB1—are master regulons of TLR signaling in macrophages and that SUB1, as an upstream effector of IRF1, is associated with M1 polarization. As we show that Irf1 overexpression is able to rescue the effects of Sub1 KO in macrophages, our study is the first to suggest that the Sub1/Irf1 axis may be a critically active pathway in proatherogenic, TLR‐induced M1 polarization within plaque macrophages.
There are a number of limitations to our findings. First, atherosclerotic plaques derived from different vascular compartments or those of varying stages or stability can significantly differ in terms of macrophage polarization.[ 35 ] Therefore, the evidence from heterogeneous carotid specimens presented here cannot be generalized to all atherosclerotic plaques. Second, the data accompanying the GEO datasets employed in our bioinformatics analysis did not include important factors such as treatment history, patient survival, and mutational burden. Third, for the carotid specimens whose transcriptomic profiles were included in our bioinformatics analysis, we could not obtain the associated tissue samples for histological analysis to verify the conclusions drawn by our bench studies. Fourth, we did not fully analyze the effects of Irf1 in the context of Sub1 KO using in vivo models. Future research should pursue this line of enquiry.
4. Conclusion
In conclusion, this study demonstrates how in silico identification of disease‐associated master regulons can be a useful method to pinpoint key master TFs. We also demonstrate the role of the TLR master regulon Sub1, and its downstream effect on the transcription factor Irf1, in promoting proinflammatory M1 polarization and atherosclerosis. Our findings suggest that targeting the SUB1/IRF1 axis may be an effective strategy toward combating proatherogenic, TLR‐induced M1 polarization.
5. Experimental Section
Experimental Design
The transcriptional master regulator technique, a method combining transcription signatures with coexpression analysis,[ 36 ] was applied to pinpoint master regulator TFs. The goals were accomplished in three stages, which are outlined below: i) extraction of TLR transcription signatures for TLR2 and TLR4 from whole transcriptome data, ii) determination of master regulator TFs from the two TLR signatures, and iii) follow‐up in vitro and in vivo studies on the novel master regulon SUB1. Please refer to the Supporting Information for the scripts and coding employed in steps (i) and (ii).
Extraction of TLR Transcription Signatures from Whole Transcriptome Data
Pam is a bacterial lipopeptide mimic that stimulates TLR2 signaling, while Gram‐negative bacterial LPS stimulates TLR4 signaling.[ 37 ] Microarray data from whole transcriptome analyses of i) Pam‐treated versus nontreated mouse BMDMs cultures at 8 h of exposure (for the TLR2 analysis) and ii) LPS‐treated versus nontreated BMDMs cultures at 8 h of exposure (for the TLR4 analysis) were extracted from the Gene Expression Omnibus (GEO) database (accession number: GSE89988). This microarray data was derived using an Illumina TotalPrep RNA Amplification Kit (Ambion, Grand Island, NY) and a MouseWG‐6 v2.0 R2 Expression BeadChip (Illumina, San Diego, CA) that contains 45 281 probes. The 8 h timepoint was chosen as steady‐state transcript levels peak around the 6 h mark during the murine macrophage response to TLR stimuli.[ 38 ] The Bioconductor Limma package in R was used to identify DEGs in i) Pam‐treated versus nontreated BMDMs and ii) LPS‐treated versus nontreated BMDMs. BioMart (Ensembl) was used to map murine Ensembl IDs to human Ensembl IDs.
Determination of Master Regulator TFs from Integrating the TLR Transcription Signatures and Atherosclerosis Datasets
Herein, the transcriptional master regulator technique was applied, which first required the construction of a coexpression network in atherosclerosis, followed by identification of the master regulator TFs that generate the TLR2 and TLR4 signatures. First, six independent transcriptome datasets comprising 371 patient‐derived carotid specimens were retrieved from the GEO database: GSE43292 (n = 32 plaques and 32 normal tissue; GPL6244 Human Gene 1.0 ST Array platform, Affymetrix, Santa Clara, CA), GSE24495 (n = 113 plaques; GPL10687 Rosetta/Merck Human RSTA 1.0 Array platform, Affymetrix), GSE21545 (n = 126 plaques; GPL570 Human Genome U133 Plus 2.0 Array platform, Affymetrix), GSE28829 (n = 16 advanced plaques; GPL570 Human Genome U133 Plus 2.0 Array platform, Affymetrix), GSE13922 (n = 11 plaques; GPL6255 humanRef‐8 v2.0 Expression BeadChip platform, Illumina), and GSE100927 (n = 29 plaques and 12 normal tissue; GPL17077 Agilent‐039494 SurePrint G3 Human GE v2 8x60K Microarray platform, Agilent Technologies, Palo Alto, CA). The whole transcriptome data were preprocessed prior to analysis. First, Affy and affyPLM (Bioconductor) were used to perform quality control (QC) on the Affymetrix datasets.[ 39 ] Briefly, affyPLM fits the probe‐level robust regressions (probe‐level model, PLM) to produce probe‐set summaries. The relative log expression (RLE) and normalized unscaled standard error (NUSE) values were derived from the PLM in order to quantitatively assess array quality. RLE values were computed for each probe‐set in the array by calculating the ratio of the expression of a particular probe‐set and the median expression of each probe‐set across all arrays. Since most probes were not expected to change across arrays, the distribution of the log‐ratios should be centered around zero. The NUSE values represent the individual probe standard error (SE) from the PLM fit. The SE values were normalized at the probe‐level across arrays so that the distribution of SE values across arrays should be centered around unity. Then, robust multiarray averaging (RMA) was used to normalize microarray data from the Affymetrix datasets.[ 40 ] Due to the presence of multiple platforms, ComBat was then used to eliminate batch effects within the samples.[ 41 ]
Using the processed data, a coexpression analysis followed by master regulon identification was performed for the TLR2 and TLR4 gene networks. Briefly, the Bioconductor RTN package in R was employed for transcriptional network inference and analysis.[ 36 ] Coexpression analysis was executed with the corpcor partial correlation package (CRAN R project) using the pcor.shrink tool in default settings.[ 9 ] The fdrtool package (CRAN R project) was utilized to estimate the false discovery rate (FDR) and statistical significance of every partial correlation.[ 36 ] VIPER analysis based on multiple‐sample gene expression signatures (the Bioconductor msVIPER module) was used to identify the master regulons in each gene network.[ 42 ] The igraph fastgreedy.community module (CRAN R project) was used to determine community structure for the TLR2 and TLR4 gene networks.[ 43 ]
Gene Set Enrichment Analysis for the Gene Communities
GSEA was performed on the gene communities identified within the TLR2 and TLR4 gene networks. The GSEA was performed against multiple ontologies: GO terms (including molecular function (MF), cellular component (CC), and biological process (BP)), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome (REAC), Transfac (TF), miRTarBase (MIRNA), CORUM protein complexes (CORUM), Human Phenotype Ontology (HP), Human Protein Atlas (HPA), and Online Mendelian Inheritance in Man (OMIM).
Animal Models
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the Second Affiliated Hospital of Chongqing Medical University (no. 2019653). Wild‐type C57BL/6, Ldlr −/−, ApoE −/−, and Stat6 −/− mice on a C57BL6 genetic background were purchased from the Jackson Laboratory (Bar Harbor, ME). First, mice possessing a floxed allele of Sub1 (Sub1 flox/flox; herein termed WT) were created and then these were crossed with LysM Cre mice expressing Cre‐recombinase in the myeloid lineage to produce LysM Cre/−/Sub1 flox/flox (herein termed Sub1 KO) mice and myeloid‐specific hemizygous LysM Cre/−/Sub1 flox/wt (herein termed HEMI) mice.[ 44 ] Then, Sub1, Stat6 KO mice and ApoE, Sub1 KO mice were created by crossbreeding Sub1 KO mice with Stat6 −/− or ApoE −/− mice, respectively. Myeloid lineage‐specific knockout and germline transmission were confirmed by PCR using the following genotyping primers: Lox gt forward, 5′‐GTG CTC CTT AAG TGT TGC AG‐3′; Lox gt reverse, 5′‐CCC TTC ATG TAA GTA TTC TC‐3′; Frt gt forward, 5′‐GAC TCT TGG ACA GCC AAG CTC‐3′; and Frt gt reverse, 5′‐TTT CAT GAT GCC TGG CCT TTC.
Unless otherwise specified, mice were provided standard rodent chow (44.2% carbohydrates, 6.2% fat, and 18.6% crude protein; T.2018, Harlan, Indianapolis, IN) and tap water ad libitum. The various experimental regimens were initiated at 8 weeks of age. For the TLR inhibitor and agonist studies, male ApoE −/− mice were intraperitoneally (i.p.) administered vehicle (20% DMSO in phosphate‐buffered saline (PBS), Ctrl), C29 (50 mg kg−1 daily) (BioVision, Milpitas, CA),[ 45 ] TAK‐242 (3 mg kg−1 daily) (Sigma, St. Louis, MO),[ 46 ] Pam (15 µg (0.01 µmol) weekly) (Sigma),[ 47 ] or LPS (50 µg (0.01 µmol) weekly) (Sigma) for 14 weeks.[ 48 ] For lipid metabolic studies, male WT, HEMI, and Sub1 KO mice were fed standard rodent chow or a HFD (60% fat, TD06414, Harlan) for 20 weeks.
For the bone marrow transplantation model, only male Ldlr −/− donor and male Ldlr −/− recipients were used. Five to seven days before bone marrow transplantation, donor mice and Ldlr −/− recipient mice were provided with antibiotic‐supplemented water. On the day of donor cell injection, recipient mice were exposed to two equal doses of radiation (4.5 Gy for 1.4 min per dose) separated by a 3 h interval. The Ldlr −/− recipient mice received approximately six million bone marrow cells in the retro‐orbital sinus. Animals were fed on regular chow for six weeks after transplantation and, thereafter, with a standardized Western diet (21% kcal from fat and 0.2% cholesterol, TD88137, Harlan) for 12 weeks. To confirm engraftment, recipient animals were anesthetized and perfused with 4% PBS in formaldehyde after drawing blood. The blood was subjected to genotyping for Cre transgene.
Serum Lipid Profiling
Serum levels of total cholesterol, LDL‐C, HDL‐C, and triglycerides were assayed with a test kit (01218LH, Beijing Leadman Biochemistry, Beijing, China) using a Siemens AD‐VIA‐2400 biochemical analyzer (Erlangen, Germany).
Analysis of Murine Aortic Atherosclerosis
Whole ApoE −/− mice aortas were analyzed by en face Oil Red O staining. For M1 marker analysis, CD14+ macrophages were isolated from aortic root plaques using anti‐CD14 magnetic beads (Invitrogen, Carlsbad, CA) as previously described[ 49 ] and subjected to qPCR analysis as described below. For aortic root lesion staining, frozen embedded sections of the aortic root starting just distal to the three cusp‐region of the aortic valve were collected from ApoE −/− mice and Ldlr −/− recipient mice. About 70 such sections were acquired, and every third section was subjected to immunohistochemical analysis. 10 µm thick lesion sections (30 µm apart) were stained using hematoxylin and eosin (H & E; PerkinElmer, Waltham, MA), Oil Red O (Sigma), and Masson's Trichome (Sigma), and captured using a RVL‐100 microscope (Echo Laboratories, San Diego, CA). Immunofluorescent staining was performed as described below.
Immunofluorescent Staining
For cultured BMDM staining, the primary antibodies were as follows: Ki67 (NB500‐170, Novus Biologicals, Littleton, CO) and Galectin‐3 (Mac2) (ab76245, Abcam, Cambridge, MA). For staining of aortic root sections, the following primary antibodies were used: EGF‐like module‐containing mucin‐like hormone receptor‐like 1 (F4/80) (PA5‐32399, Invitrogen), iNOS (ab3523, Abcam), CD206 (ab64693, Abcam), cleaved caspase‐3 (9661, Cell Signaling Technology (CST), Danvers, MA), CD68 (ab125212, Abcam), α‐actinin (ab137346, Abcam), CD3 (ab16669, Abcam), and Irf1 (8478, CST). The goat antirabbit secondary antibodies Alexa Fluor 647 (red; ab150115, Abcam) and Alexa Fluor 488 (green; ab150077, Abcam) were employed for immunofluorescent staining. A TUNEL Assay Kit (G3250, Promega, Madison, WI) was used to evaluate apoptosis levels in cultured BMDMs and aortic root sections. Cultured BMDMs and aortic root sections were also subjected to 4′,6‐diamidino‐2‐phenylindole (DAPI, Sigma) staining to identify nuclei. Images were captured using a RVL‐100 microscope.
Murine BMDM Culture
Murine BMDMs were differentiated from bone marrow cells through the use of L929‐conditioned medium. L929‐conditioned medium causes the bone marrow monocyte/macrophage progenitor cells to proliferate and differentiate into mature BMDMs.[ 50 ] Briefly, the L929‐conditioned medium was prepared as follows: L929 murine fibroblasts (ATCC, Manassas, VA) were collected after seven days of culture, centrifuged at 2000 × g for 5 min, and the supernatant was filtered using a 0.45 µm low protein binding filter. Bone marrow cells from murine femurs and tibias were obtained by flushing the marrow tissue with Dulbecco's Modified Eagle's Medium (DMEM; GE Hyclone, Pittsburgh, PA), centrifuging at 1400 × g for 5 min, and resuspension. Bone marrow cells were differentiated into mature BMDMs in DMEM supplemented with 20% fetal bovine serum (FBS), 15% L929‐conditioned medium, penicillin/streptomycin, and l‐glutamine on nontissue culture treated plates (BD Falcon, Franklin Lakes, NJ) for six days, with medium changed at day in vitro (DIV) 4.
Mature BMDMs were exposed to PBS vehicle control, Pam (100 ng mL−1), LPS (100 ng mL−1), or IL‐4 (5 ng mL−1, Sigma) for 8 h prior to analysis. Enzyme‐linked immunosorbent assay (ELISA) was used to measure Tnf‐α, Il‐1β, and Ccl‐2 secreted into the media. For modified LDL experiments, the BMDMs were exposed to 30 µg mL−1 oxLDL (Yiyuan Biotechnology, Guangzhou, China) or 50 µg mL−1 acLDL (Yiyuan Biotechnology) for 24 h prior to analysis.
Cholesterol Accumulation, Uptake, Efflux, and Content Assays
BMDMs were oxLDL‐exposed, stained, and visualized for cholesterol accumulation. Briefly, coverslip‐grown BMDMs were treated with oxLDL (30 µg mL−1) for 24 h, fixed in 4% paraformaldehyde (in PBS) after washing, washed with PBS followed by 60% isopropanol and incubated with 0.3% Oil Red O (in 60% isopropanol) for 1 h, and washed two times in PBS and again once with 60% isopropanol. DAPI was used to stain the BMDMs. Images were captured using a RVL‐100 microscope.
The BMDM cholesterol uptake assay was performed as described previously.[ 51 ] Briefly, acLDL was preincubated in 1 µCi mL−1 [3H]‐cholesterol (PerkinElmer) for 16 h at 37 °C. BMDMs were then treated with 50 µg mL−1 [3H]‐acLDL for 30 min. Cells were washed and treated with room‐temperature 0.5 N NaOH to lyse cells. From each condition, radioactivity aliquots were measured and standardized to BMDM protein obtained from simultaneous culture in different dishes.
The BMDM cholesterol efflux assay was performed as described elsewhere.[ 51 ] For efflux, BMDMs were treated with 1 µCi mL−1 [3H]‐cholesterol (PerkinElmer) for 48 h, PBS‐washed, and incubated with serum‐free media (Ctrl) or media containing HDL‐C (50 µg mL−1, Yiyuan Biotechnology) or ApoA1 (30 µg mL−1, Yiyuan Biotechnology). Four hours later, radioactivity aliquots (150 µL) were measured and standardized‐to‐total cell lysates.
BMDM cholesterol content was measured using the Cholesterol/Cholesteryl Ester Quantification Colorimetric Kit (K603‐100, Biovision, Mountain View, CA) after treating with acLDL (50 µg mL−1) for 24 h. CE was calculated by subtracting FC from TC.
qPCR Analysis
RNA isolation from BMDMs was performed using the RNeasy Mini Kit (74104, Qiagen, Hilden, Germany). An Agilent Bioanalyzer was used to check RNA concentration, integrity, and purity. Routine qPCR was performed using Gapdh as an endogenous housekeeping control. Primers used for qPCR are provided in Table S1 in the Supporting Information.
Immunoblotting
Routine procedures were followed for protein extraction and immunoblotting using β‐actin as a loading control.[ 52 ] The primary antibodies were as follows: β‐actin (ab8227, Abcam), p‐Sub1/Sub1 (G‐20, Santa Cruz Biotechnology), iNOS (ab3523, Abcam), Arg1 (ab124917, Abcam), Retnlb (ab11429, Abcam), Abca1 (NB400‐105, Novus Biologicals), Abcg1 (NB400‐132, Novus Biologicals), Olr1 (ab60178, Abcam), and Irf1 (8478, CST). To quantify the band intensities, the films were scanned using a Scanmaker 1000XL (Microtek Lab, Santa Fe Springs, CA). The resulting images were analyzed using an Image Pro Plus v6.1 Analyzer (Media Cybernetics, Rockville, MD).
iNOS and Arg1 Enzymatic Activity Assays
For assaying iNOS activity, BMDM supernatants were mixed at a 1:1 ratio with Griess reagent (Sigma). Absorbance levels were then measured at 543 nm with a SpectraMax 190 (Molecular Devices, Sunnyvale, CA). The nitrite concentration was calculated with a sodium nitrite 22 standard. For assaying Arg1 activity in BMDMs, the Quantichrome Urea Assay Kit (Bioassay Systems, Hayward, CA) was used.
Chromatin Immunoprecipitation (ChIP)
ChIP was carried out using standard protocols as previously described.[ 53 ] The following primary antibodies were used: IgG (ab171870, Abcam), Irf1 (8478, CST), Sub1 (NB100‐59775, Novus Biologicals), and H3K4me3 (ab8580, Abcam).
Vector Constructs
Human CK2 shRNA and scrambled control shRNA in pGIPz lentiviral plasmids were obtained from OpenBiosystems (Thermo Scientific, Huntsville, AL). Control lentiviral vector and lentiviral vectors expressing human CK2 (Lenti‐GIII‐CMV‐CK2‐HA) and murine Irf1 (Lenti‐GIII‐CMV‐Irf1‐HA) were obtained from Applied Biological Materials (Richmond, BC, Canada). Human IRF1 promoter fragments (−1312 to +50) containing either the WT DPE site that enables SUB1 binding (AGACGTG) or a mutant (MUT) DPE site that does not enable SUB1 binding (CTCATGT) were constructed as previously described[ 26 ] and cloned into an IDTSmart vector (Integrated DNA Technologies, Coralville, IA). The WT and MUT fragments were then subcloned into a pGL3‐basic luciferase reporter plasmid (Promega) to form pGL3‐basic/WT IRF1 promoter or pGL3‐basic/MUT IRF1 promoter constructs, respectively. Insert orientations were determined by restriction enzyme digestion and constructs were fully sequenced. Lipofectamine 2000 (Invitrogen) was employed to transfect plasmids according to the kit's instructions.
Luciferase Reporter Assays
Immortalized human THP‐1 cells were transfected with pGL3‐basic/WT IRF1 promoter or pGL3‐basic/mutant IRF1 promoter (750 ng) alone or in combination with CK2 shRNA or scrambled control shRNA using Lipofectamine 2000. In the second experiment, THP‐1 cells were transfected with pGL3‐basic/WT IRF1 promoter or pGL3‐basic/mutant IRF1 promoter (750 ng) alone or in combination with Lenti‐GIII‐CMV‐CK2‐HA or control vector. For both experiments, Renilla luciferase control vector (20 ng pRL‐TK, Promega) was cotransfected as control. Luciferase reporter activity was detected 48 h post‐transfection with the Dual Luciferase Reporter Assay System (Promega) via normalization to Renilla luciferase activity.
Statistical Analysis
Data were presented as means ± standard deviations (SDs) unless stated otherwise. All in vitro experiments consisted of n = 3 biological replicates × 3 technical replicates per experimental group and were represented as 9 individual data points. All in vivo studies consisted of n = 9 mice per cohort. Statistical tests were performed on data from independent biological replicates (n = 3 biological replicates per in vitro experimental group; n = 9 mice per in vivo cohort). No data were excluded from the statistical analysis. Normal distributions were validated with the Kolmogorov–Smirnov test. For comparisons between two groups, a two‐tailed unpaired Student's t‐test was applied. For comparisons among three or more groups, one‐way ANOVA or two‐way ANOVA followed by Fisher's least significant difference (LSD) post‐hoc testing were applied as indicated. A p‐value of less than 0.05 deemed a significance threshold for all analyses. Analyses were performed using SAS Enterprise Guide (v.4.3; SAS Institute) and R (v.3.0.2; R Foundation for Statistical Computing, Vienna, Austria).
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Y.Q.C., J.L.D., and R.Z.H. conceived and designed the study. R.Z.H., J.L.D., Z.C.H., X.R.C., Y.C., H.R.L., H.Z., Y.Y.L., L.W.L., Y.X.F., Y.W., W.H.S., Z.R.K., Y.Q.C., and N.D.M. performed the experimental procedures. R.Z.H., Y.C., J.L.D., N.D.M., and H.Z. analyzed the data. R.Z.H. and N.D.M. drafted the manuscript.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant number 81960095). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Medical research projects of the Chongqing Science and Technology Commission and Chongqing Health Committee (2020FYYX047) and Kuanren Talents Program of the Second Affiliated Hospital of Chongqing Medical University.
Huang R., Hu Z., Chen X., Cao Y., Li H., Zhang H., Li Y., Liang L., Feng Y., Wang Y., Su W., Kong Z., Melgiri N., Jiang L., Li X., Du J., Chen Y., The Transcription Factor SUB1 Is a Master Regulator of the Macrophage TLR Response in Atherosclerosis. Adv. Sci. 2021, 8, 2004162. 10.1002/advs.202004162
Contributor Information
Jianlin Du, Email: jianlindunev@cqmu.edu.cn.
Yunqing Chen, Email: paulchen@cqmu.edu.cn.
Data Availability Statement
Research data are not shared.
References
- 1.a) Newsom L. C., Ann. Pharmacother. 2015, 49, 484; [DOI] [PubMed] [Google Scholar]; b) Conti C. R., Cardiovasc. Innovations Appl. 2019, 3, 279. [Google Scholar]
- 2. Mori H., Finn A. V., Kolodgie F. D., Davis H. R., Joner M., Virmani R., Physiological Assessment of Coronary Stenoses and the Microcirculation, Springer, Berlin: 2017, p. 21. [Google Scholar]
- 3. Moore K. J., Tabas I., Cell 2011, 145, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tall A. R., Yvan‐Charvet L., Nat. Rev. Immunol. 2015, 15, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Higashimori M., Tatro J. B., Moore K. J., Mendelsohn M. E., Galper J. B., Beasley D., Arterioscler., Thromb., Vasc. Biol. 2011, 31, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu X. H., Shah P. K., Faure E., Equils O., Thomas L., Fishbein M. C., Luthringer D., Xu X.‐P., Rajavashisth T. B., Yano J., Circulation 2001, 104, 3103. [DOI] [PubMed] [Google Scholar]
- 7. Ishikawa Y., Satoh M., Itoh T., Minami Y., Takahashi Y., Akamura M., Clin. Sci. 2008, 115, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu C., Ding X., Zhou C., Ye P., Sun Y., Wu J., Zhang A., Huang X., Ren L., Wang K., Sci. Rep. 2017, 7, 15799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schäfer J., Strimmer K., Stat. Appl. Genet. Mol. Biol. 2005, 4, 32. [DOI] [PubMed] [Google Scholar]
- 10. Langfelder P., Mischel P. S., Horvath S., PloS one 2013, 8, e61505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailey P., Chang D. K., Nones K., Johns A. L., Patch A.‐M., Gingras M.‐C., Miller D. K., Christ A. N., Bruxner T. J., Quinn M. C., Nature 2016, 531, 47.26909576 [Google Scholar]
- 12. Ayllón B. T., Souilhol C., Oakley F., Evans P., Heart 2019, 105, A148. [Google Scholar]
- 13. Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., Gordon S., Hamilton J. A., Ivashkiv L. B., Lawrence T., Immunity 2014, 41, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yunna C., Mengru H., Lei W., Weidong C., Eur. J. Pharmacol. 2020, 877, 173090. [DOI] [PubMed] [Google Scholar]
- 15. Wynn T. A., Chawla A., Pollard J. W., Nature 2013, 496, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jonker H. R., Wechselberger R. W., Pinkse M., Kaptein R., Folkers G. E., FEBS J. 2006, 273, 1430. [DOI] [PubMed] [Google Scholar]
- 17. Colin S., Chinetti‐Gbaguidi G., Staels B., Immunol. Rev. 2014, 262, 153. [DOI] [PubMed] [Google Scholar]
- 18. Robbins C. S., Hilgendorf I., Weber G. F., Theurl I., Iwamoto Y., Figueiredo J.‐L., Gorbatov R., Sukhova G. K., Gerhardt L. M., Smyth D., Nat. Med. 2013, 19, 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y., Schwabe R. F., DeVries‐Seimon T., Yao P. M., Gerbod‐Giannone M.‐C., Tall A. R., Davis R. J., Flavell R., Brenner D. A., Tabas I., J. Biol. Chem. 2005, 280, 21763. [DOI] [PubMed] [Google Scholar]
- 20. Malik P., Berisha S. Z., Santore J., Agatisa‐Boyle C., Brubaker G., Smith J. D., J. Lipid Res. 2011, 52, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lian Z., Perrard X.‐y. D., Peng X., Raya J. L., Hernandez A. A., Johnson C. G., Lagor W. R., Pownall H. J., Hoogeveen R. C., Simon S. I., Arterioscler., Thromb., Vasc. Biol. 2020, 40, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Geng T., Yan Y., Xu L., Cao M., Xu Y., Pu J., Yan J. C., Cell. Signalling 2020, 72, 109628; [DOI] [PubMed] [Google Scholar]; b) Yu T., Gan S., Zhu Q., Dai D., Li N., Wang H., Chen X., Hou D., Wang Y., Pan Q., Nat. Commun. 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miguel‐Carrasco J. L., Zambrano S., Blanca A. J., Mate A., Vázquez C. M., J. Inflammation 2010, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang F., Xia W., Liu F., Li J., Wang G., Gu J., Cardiovasc. Res. 2011, 93, 190. [DOI] [PubMed] [Google Scholar]
- 25. Singh N. N., Ramji D. P., J. Mol. Med. 2008, 86, 887. [DOI] [PubMed] [Google Scholar]
- 26. Lewis B. A., Sims R. J. III, Lane W. S., Reinberg D., Mol. Cell 2005, 18, 471. [DOI] [PubMed] [Google Scholar]
- 27. Da Silva T. A., Zorzetto‐Fernandes A. L., Cecílio N. T., Sardinha‐Silva A., Fernandes F. F., Roque‐Barreira M. C., Sci. Rep. 2017, 7, 7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cuchel M., Rader D. J., Circulation 2006, 113, 2548. [DOI] [PubMed] [Google Scholar]
- 29. Bisgaard L. S., Mogensen C. K., Rosendahl A., Cucak H., Nielsen L. B., Rasmussen S. E., Pedersen T. X., Sci. Rep. 2016, 6, 35234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen M., Li W., Wang N., Zhu Y., Wang X., Am. J. Physiol.: Cell Physiol. 2007, 292, C1493. [DOI] [PubMed] [Google Scholar]
- 31. Kojima Y., Weissman I. L., Leeper N. J., Circulation 2017, 135, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Korns D. R., Frasch S. C., Fernandez‐Boyanapalli R., Henson P. M., Bratton D. L., Front. Immunol. 2011, 2, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chistiakov D. A., Myasoedova V. A., Revin V. V., Orekhov A. N., Bobryshev Y. V., Immunobiology 2018, 223, 101. [DOI] [PubMed] [Google Scholar]
- 34. van der Vorst E. P., Theodorou K., Wu Y., Hoeksema M. A., Goossens P., Bursill C. A., Aliyev T., Huitema L. F., Tas S. W., Wolfs I. M., Cell Metab. 2017, 25, 197. [DOI] [PubMed] [Google Scholar]
- 35.a) Shaikh S., Brittenden J., Lahiri R., Brown P. A., Thies F., Wilson H. M., Eur. J. Vasc. Endovasc. Surg. 2012, 44, 491; [DOI] [PubMed] [Google Scholar]; b) Tabas I., Bornfeldt K. E., Circ. Res. 2016, 118, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castro M. A., de Santiago I., Campbell T. M., Vaughn C., Hickey T. E., Ross E., Tilley W. D., Markowetz F., Ponder B. A., Meyer K. B., Nat. Genet. 2016, 48, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin B., Dutta B., Fraser I. D., Cell Syst. 2017, 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmitz F., Mages J., Heit A., Lang R., Wagner H., Eur. J. Immunol. 2004, 34, 2863. [DOI] [PubMed] [Google Scholar]
- 39. Gautier L., Cope L., Bolstad B. M., Irizarry R. A., Bioinformatics 2004, 20, 307. [DOI] [PubMed] [Google Scholar]
- 40. Irizarry R. A., Hobbs B., Collin F., Beazer‐Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P., Biostatistics 2003, 4, 249. [DOI] [PubMed] [Google Scholar]
- 41. Johnson W. E., Li C., Rabinovic A., Biostatistics 2007, 8, 118. [DOI] [PubMed] [Google Scholar]
- 42. Alvarez M. J., Shen Y., Giorgi F. M., Lachmann A., Ding B. B., Ye B. H., Califano A., Nat. Genet. 2016, 48, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clauset A., Newman M. E., Moore C., Phys. Rev. E 2004, 70, 066111. [DOI] [PubMed] [Google Scholar]
- 44. Clausen B., Burkhardt C., Reith W., Renkawitz R., Förster I., Transgenic Res. 1999, 8, 265. [DOI] [PubMed] [Google Scholar]
- 45. Chen S., Lyu C., Zhou J., Huang S., Zhang Y., Liu G., Liu K., Chen D., Hu Y., Zhou L., Neurosci. Lett. 2018, 686, 33. [DOI] [PubMed] [Google Scholar]
- 46. Wang X. Q., Wan H. Q., Wei X. J., Zhang Y., Qu P., Mol. Med. Rep. 2016, 14, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mullick A. E., Tobias P. S., Curtiss L. K., J. Clin. Invest. 2005, 115, 3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ostos M. A., Recalde D., Zakin M. M., Scott‐Algara D., FEBS Lett. 2002, 519, 23. [DOI] [PubMed] [Google Scholar]
- 49. Maione A. S., Cipolletta E., Sorriento D., Borriello F., Soprano M., Rusciano M. R., D'Esposito V., Markabaoui A. K., De Palma G. D., Martino G., Atherosclerosis 2017, 256, 53. [DOI] [PubMed] [Google Scholar]
- 50. Weischenfeldt J., Porse B., Cold Spring Harbor Protoc. 2008, 2008, pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- 51. Ouimet M., Franklin V., Mak E., Liao X., Tabas I., Marcel Y. L., Cell Metab. 2011, 13, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ben J., Jiang B., Wang D., Liu Q., Zhang Y., Qi Y., Tong X., Chen L., Liu X., Zhang Y., Nat. Commun. 2019, 10, 1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Janzer A., Stamm K., Becker A., Zimmer A., Buettner R., Kirfel J., J. Biol. Chem. 2012, 287, 30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research data are not shared.