

Abstract
Molecular-assisted precision oncology gained tremendous ground with high-throughput next-generation sequencing (NGS), supported by robust bioinformatics. The quest for genomics-based cancer medicine set the foundations for improved patient stratification, while unveiling a wide array of neoantigens for immunotherapy. Upfront pre-clinical and clinical studies have successfully used tumor-specific peptides in vaccines with minimal off-target effects. However, the low mutational burden presented by many lesions challenges the generalization of these solutions, requiring the diversification of neoantigen sources. Oncoproteogenomics utilizing customized databases for protein annotation by mass spectrometry (MS) is a powerful tool toward this end. Expanding the concept toward exploring proteoforms originated from post-translational modifications (PTMs) will be decisive to improve molecular subtyping and provide potentially targetable functional nodes with increased cancer specificity. Walking through the path of systems biology, we highlight that alterations in protein glycosylation at the cell surface not only have functional impact on cancer progression and dissemination but also originate unique molecular fingerprints for targeted therapeutics. Moreover, we discuss the outstanding challenges required to accommodate glycoproteomics in oncoproteogenomics platforms. We envisage that such rationale may flag a rather neglected research field, generating novel paradigms for precision oncology and immunotherapy.
Keywords: Glycoproteogenomics, Oncoproteogenomics, Cancer neoantigens, Glycosylation, Precision oncology
Neoantigen-based cancer vaccines: status and milestones for clinical translation
Targeted therapeutics, alone or in combination with chemo and radiotherapy, have constituted a crucial milestone in the management of cancer patients, being particularly important for those at advanced stage facing limited therapeutic options [1], [2]. Over the last ten years, a plethora of antibodies has been developed targeting proteins overexpressed by cancer cells, which play key roles in relevant oncogenic pathways and tumor vasculature development [3], [4]. A few antibody-based treatments have already been introduced in clinical practice and many more are undergoing the late phase of clinical trials [5], [6]. Cancer antibodies have also been shown to inhibit cancer growth and spread by blocking key cellular processes and inducing antibody-dependent cellular cytotoxicity (ADCC), promoting cancer cell elimination [7]. The introduction of antibodies capable of inhibiting checkpoint molecules responsible by immune tolerance to cancer cells such as PD-1, PD-L1, and CTLA-4 has decisively boosted cancer immunotherapy, showing effective results, especially in combination with chemo and radiotherapy [8], [9].
Currently, many worldwide reference oncological centers provide comprehensive treatment options, which are elected according to the molecular features of the targeted lesions and adjusted to the spatio-temporal evolution over the course of disease management. However, the enormous potential of antibody-based targeted therapeutics has been, to some extent, limited by off-target toxicity and high tumor molecular heterogeneity [10]. On the other hand, a pre-existing immunogenic fingerprint translated by high mutational burden appears to be a pre-requisite for effective immunotherapies, limiting the generalization of immune checkpoint inhibitors [11].
The limitations inherent to antibody-based immunotherapy have prompted a quest for cancer neoantigens, i.e., peptides (segments of proteins) specifically found on the surface of cancer cells. Although classically associated to alterations in protein primary sequences due to non-synonymous mutations, it became widely accepted that these neoantigens may also arise from altered splicing mechanisms, gene fusions, endogenous retroelements, and other processes occurring at the genome and transcriptome levels [12], [13], [14]. Post-translational modifications (PTMs) may also decisively contribute to unique cancer molecular signatures, which can be explored to unleash immune responses against cancer cells [15], [16] (Figure 1). However, at early stages of neoantigen discovery, the immunotherapy field has neglected the potential of protein neoantigens due to tremendous molecular heterogeneity and limited potential for “one fits all” pharmacological solutions [17]. In recent years, the technological readiness of high-throughput molecular characterization platforms has challenged this concept. Next-generation DNA sequencing has allowed rapid tackle of the cancer genome for mutations that have been subsequently explored in vaccine formulations [18]. Moreover, mass spectrometry (MS) has been used to quantitatively characterize cancer neoantigens [19], [20], [21], and increasingly sophisticated bioinformatics tools are aiding in the real-time identification of most suited protein species to include in vaccine formulations [22]. Finally, lab-scale peptide synthesizers allow real-time production of small quantities of diverse molecules for vaccines under good manufacturing practice (GMP) conditions, providing an opportunity for vaccine production in loco [23]. Increasing numbers of improved vaccine delivery vehicles are also emerging, which are designed to boost immune responses against otherwise less immunogenic cancer neoantigens [24]. So far, pre-clinical studies in mice support the excellent therapeutic potential of whole genome-based multivalent cancer vaccines for melanomas and neuroblastomas [25], [26], [27], [28]. These solutions have remarkably reduced tumor burden and, in some cases, completely eradicated tumors in animal models, while generating an immunological memory capable of preventing metastasis [25], [26], [27], [28]. This has provided blueprints on how such approaches can be translated into clinical applications in humans. Namely, a phase I clinical trial is ongoing to evaluate the safety and effect of personalized neoantigen vaccines for pancreatic cancer based on next-generation sequencing (NGS) and major histocompatibility complex (MHC) affinity prediction algorithm (NCT03558945). The study hypothesizes that personalized neoantigen vaccines will be safe and capable of generating measurable neoantigen-specific CD4+ and CD8+ T cell responses. Moreover, combination therapies with immune checkpoint inhibitors have allowed to unleash previously compromised immune responses in pre-clinical trials [29], [30]. Such findings fostered phase I/II clinical trials focused on personalized cancer vaccines derived from mutated peptides in combination with nivolumab and ipilimumab, in patients with metastatic non-small cell lung cancer, microsatellite stable colorectal cancer, gastroesophageal adenocarcinoma, and metastatic urothelial cancer (NCT03953235 and NCT03639714). These initiatives illustrate the materialization of precision oncology and will, most likely, constitute the next cornerstone in cancer management.
Figure 1.

Neoantigens generated by modifications at DNA, RNA, and protein levelsas well as by PTMs
Cancer cells frequently express unique protein species that are not present in healthy tissues (neoantigens), holding tremendous potential for targeted therapies and immunotherapy. Neoantigens may derived primarily from alterations in genome but also in RNA processing and other events underlying protein synthesis. Protein maturation with PTMs, such as glycosylation and phosphorylation, adds a second layer of specificity, which is valuable toward more effective targeted therapeutics. Beyond genetic alterations, epigenetic regulation plays a key role in governing gene expression as well as processing, significantly contributing to the formation of a wide array of proteoforms either directly or indirectly modulating the expression of enzymes involved in PTMs of proteins. Me, methyl; Ac, acetyl; SLeA, sialyl-Lewis A; SLeX, sialyl-Lewis X; S3T, sialyl-3-T; STn, sialyl-Tn; GPI, glycosylphosphatidylinositol; PTM, post-translational modification.
While no longer considering blue-sky approaches, the efficiency of patient-tailored cancer neoantigen vaccines is still challenged by the low mutational frequency presented by many lesions [31]. More strikingly, comparative genomics has shown that, in some cases, metastases present a rather limited array of mutations in comparison to primary lesions [32]. As such, extending neoantigen discovery beyond peptide identification based on genomic sequencing stands as the next logical step. The emerging field of proteogenomics, exploring customized protein sequence databases by integrating genomics and transcriptomics data, provides a unique tool to interrogate the cancer proteome [33]. In fact, the integration of genomics and transcriptomics is crucial for protein identification, bringing proteomics one step closer to the exome. Accordingly, paired genomics, transcriptomics, and proteomics data of samples from the same tumors have demonstrated that the proteome contains novel information that cannot be discerned through genomic analysis alone [34].
An additional level of molecular complexity arises from the array of PTMs that decisively define protein biophysical and biochemical properties as well as functional roles. PTMs provide microenvironmental context and exponentially increase the number of protein species defined by the exome, leveraging a more complete view of tumor molecular heterogeneity and cancer biology [35]. Particularly, glycosylation is amongst the main PTMs of membrane proteins and it is well established that cancer cells present altered glycosylation patterns in comparison to corresponding healthy tissues [36], [37]. A significant number of studies have also consensually postulated an array of glycan modifications that appear of pancarcinoma nature [38], [39]. Moreover, many reports advocate that such molecular features are responsible for generating unique glycopeptide signatures at the cell surface [40]. Identifying these distinctive cancer-specific glycoproteoforms will provide bispecific (at the glycan and protein levels) molecular targets, with the potential to limit off-target effects.
The proof of concept regarding the potential of glycan-based proteogenomics has been highlighted in two recent studies, supporting the pursuit of more comprehensive research efforts [41], [42]. As such, this review focuses on illustrating the role of proteogenomics as a decisive tool for systems biology and, ultimately, precision oncology. Focus is set on the importance of addressing protein glycosylation and integrating glycomics and glycoproteomics into neoantigen discovery platforms, envisaging the generalization of cancer vaccines to tumors of distinct molecular natures.
Oncoproteogenomics toward neoantigen discovery
Cancer biomarker discovery has been mostly centered on the genome and transcriptome, with less, but growing emphasis, on the proteome. While the complexity of the tumor genomic background is being rapidly uncovered by large dimension sequencing studies [32], [43], [44], translation of its findings into resulting proteome remodeling remains poorly understood and explored. In fact, many reports disclosed a landscape of genetically relevant alterations and dysfunctional transcriptomes that reflect large numbers of non-synonymous single-nucleotide variants (SNVs), insertions and deletions (indels), alternative splicing variants, copy-number aberrations, and abnormal fusion genes [45]. However, the transcriptome frequently fails to mirror the proteome’s abundance, diversity [46], [47], and consequent functional impact on tumor initiation, progression, dissemination, and response to treatment. This has limited the easy translation of elegant transcriptome-derived patient stratification models into the everyday practice of most pathology laboratories. It has also delayed the identification of clinically relevant proteins for rational design of targeted therapeutics. The number of protein surrogates and/or targetable biomarkers for molecular-based patient stratification reaching the clinics remains scarce. The few successful examples include prostate-specific antigen (PSA) for detection [48], carcinoembryonic antigen (CEA) for patient management and prognosis [49], and CA125/mucin 16 (MUC16), which is a diagnostic marker and holds potential for targeted therapies [50], in prostate, colon, and ovarian cancer, respectively.
On the other side of the equation, the initial enthusiasm with oncoproteomics in the post-genomic era, which started 20 years ago, has been gradually losing ground. This is mostly due to protein identifications in conventional proteomics workflows relying on general protein databases that fail to reflect the uniqueness of the cancer transcriptome [51], limiting the breadth of identified biomarkers. Oncoproteogenomics has arisen from the pressing need to correct this discrepancy, enabling the accurate interpretation of large datasets generated by modern high-resolution mass spectrometers. Proteogenomics exploits sample-matched genomics and transcriptomics datasets to customize protein annotation, providing definitive proofs of protein translation [52]. The most used approaches for generating protein databases include whole-genome or exome sequencing, with emphasis on six-frame translation of whole genome sequences, which enables the identification of previously undiscovered exons and open reading frames [53]. Available exon annotations on the human genome may also be explored to generate junction sequences for all possible exons in a gene. DNA/RNA sequencing is also available to obtain SNV data and corresponding tumor-specific protein sequences [54].
When applied to cancer, proteogenomics is an exciting new concept, which attempts to detect tumor-specific changes in proteoforms that result from mutations or an altered transcription process, rather than solely focusing on improving proteome characterization (Figure 2). Over the past five years, these methodologies have become sufficiently mature to support the transition from proof of concept studies in cell lines and animal models to large-scale translational studies using human samples. Several examples show how oncoproteogenomics may complement and improve on the molecular subtyping and prognostication of breast [55], gastric [41], colorectal [46], and ovarian [56] tumors beyond previous genomics- and transcriptomics-based patterns. Most of these reports provide proteome-based networks highlighting functional protein nodules for targeted intervention. Furthermore, the depth of tumor proteogenomics profiling has led to the identification of breast cancer protein neoantigens that result from previously undescribed gene variants and non-coding gene regions, defying old paradigms and expanding our understanding of cancer molecular biology [20]. Such molecular signatures are possibly the consequence of significant cancer genome instability and arise as strong candidates for immunotherapy.
Figure 2.

Integrated multiomics data towarddiscovery of potential clinically relevant biomarkers and targeted therapeutics
Oncoproteogenomics concerns genomics and transcriptomics data from tumor samples, which is used for generating customized databases, to support protein annotation. The inclusion of glycomics information in the workflow allows more effective protein identification by MS/MS, including glycosylation site mapping, illustrating its value to gain insight on molecular information that cannot be achieved by the other omics toward precision oncology. Overall oncoproteogenomics is supported by several bioinformatics tools, which also contributes for identification of more suitable antigens toward cancer vaccines. WGS, whole-genome sequencing; SNV, single-nucleotide variant; indel, insertion and deletion; MS/MS, tandem mass spectrometry; HexNAc, N-acetylhexosamine; Hex, hexose.
Despite several examples of considerable technology readiness, when facing clinical translation, some outstanding challenges persist for oncoproteogenomics. Perhaps the most pressing difficulty relates to the fact that databases of putative protein sequences derived from genomics experiments are significantly larger than those explored in conventional proteomics, which dangerously increases the probability of false positives during protein annotation. Moreover, the incorporation of large genomics datasets into proteomics poses a significant computational challenge, translated by long processing times and frequently high false discovery rates [57]. This is further aggravated by the low relative abundance of peptide sequences derived from genetic abnormalities over protein species arising from genomics predictions. Alternatively, libraries inferred directly from RNA-seq, expressed sequence tag (EST), and cDNA bring us one step closer to the exome, while significantly reducing the amount of generated data in comparison to whole-genome sequencing [58]. Furthermore, transcriptome-generated databases allow the proteomic identification of RNA editing products, splice junctions, and fusion proteins. Nevertheless, oncoproteogenomics continues to be a fast-evolving field, and more detailed information on the opportunities and limitations of analytical and bioinformatics tools to assist database customization and accommodating genomics information can be found in recent reviews [59], [60]. Another key aspect is the capacity to translate proteogenomics data into relevant targets for immunotherapy. This aspect has been elegantly tackled by several computational pipelines to elect peptides with higher binding affinity to MHC-I molecules [61], [62], [63]. Moreover, neural networks exploiting annotated data on the binding kinetics of known peptides are being used to refine these models to estimate cancer neoantigen affinity to a wide array of different MHC classes and haplotypes, providing means for a more educated election of suitable immunogens [64], [65], [66]. However, even if a peptide has strong MHC binding prediction, this may be ineffective if upstream processing such as proteolysis prevents the actual loading of that peptide. Accordingly, several software are now available to aid prediction of proteasome specificity and protease cleavage sites, including NetChop20S, NetChopCterm, and ProteaSMM for MHC class I antigens, and PepCleaveCD4 and MHC-II-NP for MHC class II antigens, offering a second level of predicted peptide quality control [67], [68], [69], [70]. The notion that many predicted neoantigens may never constitute proteolysis products capable of being presented by MHC has again prompted proteomics. MHC molecules and associated peptides are currently being isolated from different cancers for tandem mass spectrometry (MS/MS) identification, generating key data for training machine learning algorithms to improve neoantigen prediction [71]. The thorough accomplishment of such goals will be decisive for translating oncoproteogenomics into clinically useful targets. Finally, critical challenges persist concerning the capacity of oncoproteogenomics to infer on relevant functional mechanisms adopted by cancer systems at multiple biological scales, ultimately pinpointing key biological network nodes for intervention [72]. A plethora of reports shows that this goal requires a comprehensive understanding of PTMs [39], [73]. PTMs are key for defining and regulating protein functions, degradation pathways, and even cellular locations, and many times play an essential role in the signaling pathways that define cell fate [72]. PTMs, such as phosphorylation, methylation, and glycosylation, are pivotal in the rapid modulation of protein functions in response to microenvironmental cues, providing potential links between metabolic alterations and protein activity [74]. Therefore, incorporating PTMs into oncoproteogenomics setups will bring us one step closer to systems biology settings, even though at the expenses of a new set of interdisciplinary analytical challenges.
PTMs are often of a transient nature, but exponentially increase the number of proteoforms, raising problems for accurate protein identification. Such dynamics are often accompanied by a complex and non-templated molecular organization, as is the case of glycosylation. As a result, more than half of the spectral information generated by MS/MS experiments usually remains unassigned due to the presence of PTMs, as elegantly demonstrated by Chick and his colleagues [75]. The accommodation of these subtleties requires an adaptation of conventional proteomics protocols, namely the incorporation of pre-enrichment methods and different separation techniques, inclusion of a diversified array of proteases and other enzymes prior to MS analysis, diversification of MS ionization and fragmentation methods, and dedicated bioinformatics analysis [76]. In addition, the physicochemical alterations resulting from PTMs may decisively interfere with MS-based protein identification at different levels. It is widely known that acid groups and other hydrophilic species are detrimental to ionization and might reduce the identification of modified peptides by electrospray analysis, which is used by the bulk of modern mass spectrometers [77]. Nevertheless, this effect may be significantly attenuated by the acidic buffers generally used in proteomics experiments to produce positive charged species at lower pH. It may also be compensated by the very high sensitivity and resolution of modern spectrometers, particularly when coupled to liquid chromatography (LC) operated in nano-settings. Another critical aspect is PTM site annotation. Site assignment is particularly challenging because MS/MS experiments involving modified peptides often originate fragment ions that are not accurately identifiable by the bioinformatics tools used in routine proteomics workflows. In fact, conventional proteomics search algorithms can dynamically accommodate several anticipated modifications but fail to identify peptides carrying unknown alterations [75]. And even the identification of peptides carrying expectable modifications may not be straightforward. Common database search algorithms face difficulties to score with high-confidence complex fragment ion spectra quite often containing information on PTM neutral loss or non-canonical fragmentation pathways [78]. This difficulty is generally overcome by resorting to the combination of different MS/MS fragmentation strategies [79], [80]. Nevertheless, recent advances in bioinformatics tools have generated reliable analytical platforms capable of supporting large dimension studies at this level. A key example is the generalization of the Byonic software, which has been used as a more reliable tool for glycoprotein and glycopeptide annotations [81], [82].
Despite such challenges, there are already some demonstrations upholding the decisive role of PTM monitoring for patient stratification. Most studies have explored phosphorylation to demonstrate its decisive role for pinpointing relevant disease-associated molecular pathways and how this information may be comprehensively fit into clinical models to improve patient management [56], [83], [84]. Histone acetylation has also been explored to gain insight on endometrial carcinogenesis, foreseeing new therapeutic approaches [85]. Emerging reports now point out the need to include protein glycosylation to gain insight on different levels of molecular information that cannot be deciphered by genomics, transcriptomics, and proteomics alone [37], [39]. Previous studies have shown the significance of glycosylation for patient stratification and identification of novel biomarkers and therapeutic targets [41], [42]. As example, altered sialylation and fucosylation have been used to improve the predictive value of PSA [86], [87], [88]. Targeting sialyl-Tn (STn) glycoforms of plasminogen in the serum of patients holds potential for non-invasive clinical diagnosis of individuals with gastric precancerous lesions [89]. Also, MUC16-STn was identified as an independent predictive biomarker of decreased response to chemotherapy in bladder cancer, whereas MUC16 and STn alone were not [90]. Furthermore, by greatly extending the number of identifiable cancer-specific proteoforms, PTMs will retrieve unforeseen cancer neoantigens to support cancer vaccine development based on a more comprehensive approach.
Cancer glycosylation for precise cancer targeting
Glycosylation is the most abundant and structurally complex PTM of membrane-anchored and secreted proteins. Despite presenting a non-templated structural nature, it is subjected to strict cellular regulation, rapidly responding to physiological alterations and distinct pathological contexts. As such, glycan heterogeneity reflects the harmonized activity of nucleotide sugar transporters, glycosyltransferases, and glycosidases in the endoplasmic reticulum (ER) and Golgi apparatus (GA) [36]. Altered glycogene expression, loss of cellular homeostasis, including metabolic shifts impacting in nucleotide sugar availability, and alterations in subcellular localization of glycosyltransferases and glycosidases are amongst the main events driving changes in glycan chains in cancer cells [36], [91]. On the other hand, loss-of-function mutations in glycogenes are not frequent and have therefore little impact on the establishment of this malignant glycophenotype [92]. The main classes of cell surface glycans are O-N-acetylgalactosamine (O-GalNAc) glycans, set up in the GA through the attachment of a GalNAc to serine (Ser) or threonine (Thr), and N-glycans, initiated in the ER with the addition of an oligosaccharide moiety to an asparagine (Asn) residue in a peptide consensus sequence of Asn-X-Ser/Thr (X corresponding to any amino acid except proline) [36]. Glycans are further processed throughout the secretory pathways according to the cellular repertoire of glycosyltransferases, glycosidases, and substrates. This may comprehend branching and/or elongation of core structures, sialylation, and termination with Lewis blood group-related antigens or ABO(H) blood group determinants, amongst other epitopes. Sialic acids may be further modified by O-acetylation, and galactose (Gal) and GlcNAc by sulfation, greatly expanding the complexity of the glycome [36]. Mature glycans may still experience structural remodeling by extracellular glycosyltransferases and glycosidases freely circulating in the plasma or carried by platelets, further increasing the breadth of its structural dynamics [93]. It is likely that this may also occur at the surface of cancer cells in solid tumors, which has not yet been demonstrated.
Glycans are crucial in early steps of protein biosynthesis and maturation, ensuring quality control of protein folding and contributing to define protein conformation and functional roles. Alterations in the composition of glycan chains and the distribution and the density of glycosites in proteins are common in cancer [39]. Glycomics studies of different nature and involving a wide number of distinct types of cancer have consistently demonstrated that more aggressive cancer cells increase the degree of protein sialylation and N-glycan branching [36], [91]. Concomitantly, glycoproteins may lose the capacity to present elongated O-glycans, thus acquiring simple cell glycophenotypes translated by the accumulation of short glycoforms such as the Tn, STn, and T antigens [94], [95], [96]. These events induce major phenotypic alterations toward enhanced cell motility and cell invasion and activate relevant oncogenic pathways, namely the EGFR and ErbB2 receptors [95], [96], [97], [98], [99]. The expression of truncated glycans further impacts the interactions of cancer cells with the immune system. Examples include STn antigen recognition by Siglec-15 on macrophages and dendritic cells, and sialyl-T (ST) antigen recognition by Siglec-9 on neutrophils and Siglec-7 on natural killer cells [100]. These interactions trigger an array of inhibitory signals on antigen-presenting cells, ultimately precluding T cell-mediated responses against tumor cells, as demonstrated by recent studies [101], [102]. It is likely that a deeper understanding of functional interactions between glycans and glycoconjugates with immune system intermediates may constitute the next cornerstone for immune checkpoint inhibition and, potentially, a more rational design of cancer vaccines. Cancer cells also often overexpress sialylated Lewis antigens such as sialyl-Lewis A (SLeA) and X (SLeX). These are terminal epitopes of glycan chains in proteins and glycolipids that play a decisive role in cancer cell hematogenous dissemination by interacting with E-selectin on activated endothelial cells [103]. Moreover, they promote cancer cell adhesion to platelets through P-selectin and lymphocytes through L-selectin, which are critical aspects to support survival in circulation and evade immune responses [104]. There are many other examples of how glycans impact key cancer hallmarks, as elegantly highlighted by recent reviews [39], [105].
There are numerous studies associating STn, SLeA, and other glycoforms with worst prognosis and metastatic spread [106], [107]. Notably, STn and SLeA glycans are rarely expressed by healthy tissues, where they are confined to cells specialized in secretion across the lumen of the gastrointestinal and respiratory tracts as well as in secreted mucins, acting as a protective barrier against pathogens and contributing to immunological homeostasis [37], [108]. These glycans are also not expressed in blood cells in circulation, standing as potential biomarkers [109], [110]. This has prompted their exploitation with relative success for targeted therapeutics and immunotherapy in pre-clinical and clinical settings, with emphasis on chimeric antigen receptor T (CAR-T) cells and cancer vaccines, as reviewed recently [111], [112]. Namely, CAR-Ts designed against cancer cells expressing the Tn and STn antigens hold potential toward this objective, given their restricted expression pattern in healthy human tissues [111], [113]. On the other hand, glycan-based vaccines, while capable of inducing high antibody titters, have not been able to induce effective and long-lasting cellular immunity against tumors [114], [115].
To this date, the establishment of immunization strategies capable of overcoming the low immunogenicity and immunosuppressive nature of cancer-associated glycans remains an outstanding challenge. However, the concept of neoantigens does not apply to glycans alone, [116] with the only known exception coming from the incorporation of N-glycolylneuraminic acid (Neu5Gc; sialic acid) from dietary sources such as red meat, which induces the formation of xenoantigens in human glycan chains [117]. Neoantigen signatures may be obtained from glycopeptides, granting bispecificity both via the glycan and the peptide chain [118], [119]. Supporting this hypothesis, glycoarray-based studies have identified autoantibodies against distinct mucin variable tandem repeat glycoproteoforms in the serum of cancer patients [120], [121]. Autoantibodies showed cancer-specific recognition of targeted glycans, regardless of disease-associated variations in glycan density and distribution in peptide chains, supporting the existence of molecular fingerprints of disease that should be explored for cancer targeting. Accordingly, understanding the repertoire of glycans stands out as a decisive step for zooming in on the cancer glycoproteome. We have recently explored this approach to demonstrate the presence of unusual glycoproteins at the cell surface, such as the case of nucleolin (NCL) [122]. NCL is mostly confined to the nucleus of healthy cells, but it may migrate to the cell surface where it appears glycosylated in different types of solid tumors [122], [123], [124], [125], [126] (Figure 3). These are striking observations since NCL presents neither hydrophobic transmembrane domains nor a targeting signal to the cell membrane that could justify glycosylation [127]. The mechanisms governing this protein mislocalization remain to be fully elucidated; nevertheless, it becomes clear that this cancer-specific alteration presents enormous potential for precise targeting. Many similar examples have been reported in the literature, which set an important novel paradigm for biomarker research and can be comprehensively explored in the future [128], [129], [130].
Figure 3.
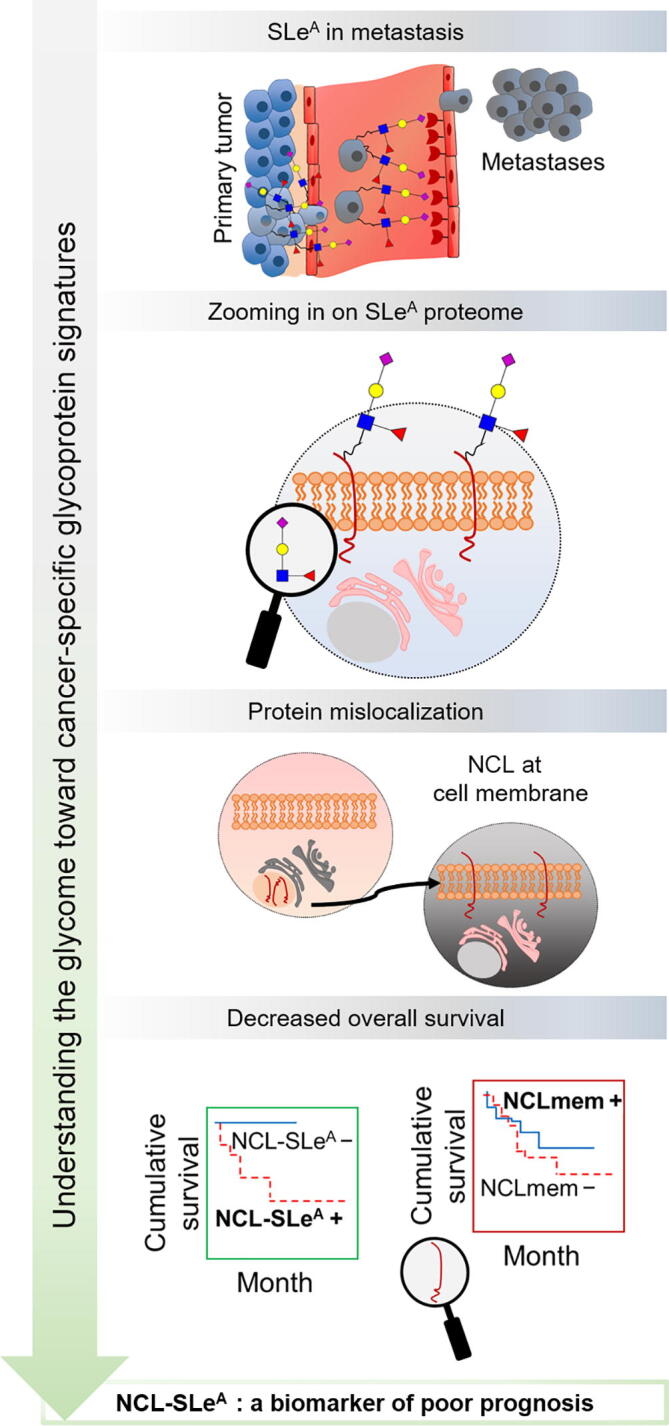
Zooming in on the sugar coating of cancer cells has provided the identification of glycoproteins holding potential for targeted therapies
SLeA, a terminal glycan epitope of glycolipids and membrane glycoproteins, is commonly overexpressed in gastric tumors and is an important ligand of E-selectin, playing a key role on E-selectin-mediated disease dissemination. Exploring the SLeA proteome allows to increase specificity and overcome some expression associated with non-malignant conditions. NCL-SLeA, a well-known mislocalized protein in cancer, has emerged as a top-ranked targetable glycoprotein at cell membrane in gastric cancer and a potential E-selectin ligand. Moreover, only the protein glycoform was associated with decreased overall survival, showing the added value of glycosylation for biomarker discovery. NCL, nucleolin; NCL-SLeA, nucleolin-sialyl-Lewis A glycoform; NCLmem, nucleolin expressed at the cell membrane.
When taking into account the relevance of integrating glycan information into protein surrogates, there are several examples on how considering cancer-associated glycoforms of classical biomarkers has improved their clinical value (PSA, MUC16, CEA, and CD133) [86], [90], [131], [132], [133]. Moreover, focusing on glycans on HER2, PD-1, and PD-L1 has allowed to provide important footprints for patient stratification, better predict treatment response, and improve the efficacy of therapeutic antibodies [102], [134], [135], [136], [137], [138], [139]. In summary, glycosylation provides functional and microenvironmental contexts to cell membrane proteins, and targeting glycosylated moieties may lead to previously unforeseen glycoproteomics signatures at the cell surface, paving the way toward disease-specific glycoproteoforms.
Glycomics and glycoproteomics: concepts, strategies, and challenges facing biomarker discovery
The comprehensive combination of glycomics and glycoproteomics will be crucial toward the identification of novel cancer biomarkers of clinical value, with emphasis on unforeseen cancer-specific signatures reflecting molecular alterations at the genome, transcriptome, proteome, and metabolome levels. Glycomics addresses the repertoire of glycans in a biological system and is a critical milestone for identification of glyconeoantigens. MS is by far the most widely used analytical tool in a field that has been progressing fast, backed by the increasing sensitivity and resolution of mass spectrometers [140], [141]. Moreover, important steps have been made toward protocol standardization [142], automatization [143], and bioinformatics [144], setting the foundations for carbohydrate metrology. However, despite intense research and robust proofs of concept, the implementation of MS-based glycome signatures in clinical practice has yet to be established and poses significant analytical, clinical, and regulatory difficulties, namely the lack of well-defined glycan standards as well as biased reproducibility resulting from a wide array of MS architectures. Notably, lectin microarrays of variable architectures have been proposed as high-throughput alternative technologies, which may be particularly important in the context of liquid biopsies, but of limited use for glycoantigen discovery [145]. Facing mature technology, the field should now push toward a comprehensive, large-scale interrogation of the human glycome. This will decisively prompt our understanding of glycan diversity in health and disease, including the foundations to assertively tackle the glycoproteome.
Glycomics
Glycan analysis poses a significant analytical challenge due to its non-templated and structural heterogeneity and frequent co-existence of isomeric/isobaric structures in the same sample [146], [147]. MS remains the gold standard technique, but support from nuclear magnetic resonance (NMR), complementary enzymatic methods, and immunoassays may be required for more detailed structural elucidation. Typical protocols initiate with the selective release of glycans from glycoproteins by either enzymatic or chemical methods. N-glycans are isolated by peptide-N-glycosidase F (PNGase F) or PNGase A digestion, whereas O-GalNAc glycans are obtained by reductive β-elimination through chemical methods [148].
Focusing on N-glycans, PNGase F digestion has the advantage of tagging substituted Asn residues in protein chains, facilitating downstream glycosite identification by glycoproteomics [149]. This can be achieved in proteins in solutions, immobilized in polyacrylamide gels, and even in tumor sections [150], [151], [152]. However, the immobilization of purified glycoproteins or protein mixtures on polyvinylidene fluoride (PVDF) membranes prior to enzymatic and chemical treatments significantly facilitates glycan release from minute amounts of biological material (nano-femtomole) [153] (Figure 4A). Notably, PNGase F works well for glycoproteins, whereas PNGase A is more recommended for deglycosylation of glycopeptides [154], [155]. Moreover, PNGase A differs from PNGase F in the fact that it is able to cleave N-linked glycans with or without α(1,3)-linked core fucose residues, whereas PNGase F is incapable of cleaving glycans showing this particular structural feature [156]. Glycans may then be directly analyzed by MS in their native form, providing an overview on the main classes of available structures. Alternatively, they may be resolved by LC into isomeric structures, as advocated by glycomics consensus guidelines [157]. The upfront identification of regioisomers may be accomplished by LC–MS/MS using different types of columns [hydrophilic interaction (HILIC) [158], zwitterionic interaction, porous graphitized carbon (PGC) [159], or C4-C18 reverse phase [160]]. However, complementary sample treatment with sialidases and other specific exoglycosidases may be necessary for more detailed structural characterization of complex stereoisomers. A promising approach to overcome this limitation is to include ion mobility spectrometry (IMS) as an additional gas-phase separation dimension, which will probably constitute routine in a near future [161]. Glycans may be derivatized prior to analysis to facilitate chromatography and improve ionization properties. The most popular, well-established, and widely applied derivatization method is permethylation, allowing to address both N- and O-glycans at an omics scale. Permethylation stabilizes labile sugars such as fucose and sialic acids, preventing fucose migration and enabling detection by soft ionization methods, such as matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) [162], [163]. Moreover, it renders glycans more hydrophobic, thus facilitating separation by conventional C18 reverse phase LC columns and positive ionization by both MALDI and ESI, and improving sensitivity. Another popular derivatization approach for N-glycans comprehends reductive amination, which resorts to labels such as 2-aminobenzamide or 2-aminobenzoic acid, allowing fluorescence detection and quantification [164]. The introduction of fluorescent tags has also permitted the construction of MS-independent platforms for rapid N-glycomics, crucial for the generalization of glycomics analysis at a wider scale. Other less explored approaches for quantitative analysis of N-glycans include isobaric tags, such as aminoxyTMT [165] and QUANTITY [166]. These tags can be conjugated with the reducing end of N-glycans, allowing their quantification by LC–MS using report ions. Currently, underivatized glycan analysis has been gaining ground with the introduction of PGC–LC columns that allow a good separation of isomeric structures. Moreover, important steps have been made toward standardization, with the recent establishment of PGC–LC–MS N-glycan retention libraries and elution mapping resources [167]. While the field evolves toward less time-consuming PGC platforms, permethylation still remains by far the most used derivatization method for glycan analysis at the micromolar scale. In addition, product ion spectra of permethylated glycans are greatly informative by providing ions derived from both glycosidic linkages and cross-ring fragmentations [168], [169], and several libraries exist for the interpretation of LC chromatograms [160], [170].
Figure 4.

Glycomics and glycoproteomics conventional workflows for the identification of potential targetable glycobiomarkers
A. Immobilization of extracted glycoproteins from biological material on PVDF membrane allows consecutive release of N-glycans and O-glycans by PNGase F digestion and reductive β-elimination, respectively. This protocol comprises analysis of the released and reduced glycans by PGC–nanoLC–ESI–MS/MS. B. An achievement for the field of (glyco)proteomics in the last years was the introduction of MALDI Imaging, which adds spatial information to molecular specificity provided by MS. The main steps of a MALDI-MSI protocol for protein and N-glycan analyses are very similar. After removal of paraffin in FFPE tissues, PNGase F or trypsin is applied on the tissue section, to release N-glycans from glycoproteins or obtain digested peptides, followed by application of MALDI matrix. Besides the average mass spectrum obtained for each tissue section, the distribution of the most abundant glycans or peptides can be visualized individually across the tissue. Moreover, identification of N-glycans and/or peptides can be confirmed by MS/MS on tissue extracts. For the sequential analysis of peptides on the same tissue section used previously for N-glycan analysis, an intermediate washing step to remove MALDI matrix, N-glycans, and PNGase F is necessary in addition to the antigen retrieval step. C. The information generated by glycomics experiments is pivotal for guiding glycoproteomics by employing a glycan-targeted proteomics approach. Therefore, this includes an initial enrichment of extracted cell membrane proteins for the glycan of interest, followed by separation of glycoproteins according to the molecular weight and analysis by nanoLC–MS/MS using a bottom-up approach. Combined bioinformatics tools are employed toward curated data, through comparison with transcriptomics data, protein expression data from healthy and tumor tissues, and evaluation of subcellular location. PVDF, polyvinylidene fluoride; PNGase F, peptide-N-glycosidase F; EIC, extracted ion chromatogram; PGC, porous graphitized carbon; LC, liquid chromatography; ESI, electrospray ionization; FFPE, formalin-fixed paraffin-embedded; α-CHCA, α-cyano-4-hydroxycinnamic acid; MALDI-MSI, matrix-assisted laser desorption/ionization mass spectrometry imaging; PNA, peanut agglutinin; VVA, Vicia villosa agglutinin; NeuAse, neuraminidase.
Analysis of O-glycans has been particularly challenging due to the lack of enzymatic approaches. In fact, until recently O-glycosidase was the only available enzyme to address this objective. However, this enzyme catalyzes the removal of core 1 and core 3 O-linked disaccharides from glycoproteins but does not act on more elongated O-glycans that are commonly found in human cell glycome [171]. Notably, O-proteases, such as StcE (acting on mucins) and OpeRATOR, have been recently introduced for studying O-glycopeptides [172], [173], holding true potential to improve sequence coverage, glycosite mapping, and glycoform analysis. Accordingly, O-glycomics analysis still relies on chemical methods that often degrade the protein backbone and thus significantly reduce the sensitivity of analysis. However, addition of a reducing agent in the most widely employed O-glycan release strategy, reductive β-elimination, prevents the glycan degradation resulting from “peeling reactions”. The limitation of lacking enzymatic approaches has been recently addressed by the introduction of a semi-quantitative method that exploits a mimetic of the Tn antigen (benzyl-GalNAc) as a scaffold to determine the structure of more extended glycan chains [174], [175]. Moreover, it increased sensitivity of analysis by 100–1000 folds when compared to chemical methods and identified a more complex repertoire of O-glycans [174]. However, while extremely elegant, this technique is limited to cell culture-based approaches, being unsuited to support studies in vivo or ex vivo. In addition, many new tools have been introduced in the recent years to overcome several limitations associated with the analysis and quantification of both N- and O-glycans. For example, solid-phase chemoenzymatic approaches have been proposed to improve N- and O-glycomics [176], [177], [178] and several chemical methods have been introduced for easy identification of different types of sialic acid linkages in glycan chains (α2,3, α2,6, α2,8, or α2,9) [179], which are decisive for defining functional traits. Overall, MS has paved the way for functional glycomics and glycobiomarker discovery. In this context, another major achievement was the introduction of MS imaging, which enables analysis in situ using fresh but also formalin-fixed embedded tissues with high sensitivity [180], [181]. The generalization of this approach confers spatial resolution to glycomics and glycoproteomics studies and paves the way for more robust and context-customized glycoproteomics (Figure 4B). Collectively, the field has reached the maturity to support large-scale multiomics studies.
Glycoproteomics
The bulk of genetic and epigenetic alterations in protein glycosylation pathways occurring in cancer cells are reflected on the cell surface [182], [183]. These include changes not only in the structure, length, and charge of glycan chains but also in the abundance and occupancy of glycosites in a protein, which decisively shape protein functions and concomitantly provide molecular signatures for targeted therapeutics and immunotherapy [39], [91], [184]. Providing information on the nature and abundance of glycosylated proteins, as well as on the distribution and composition of glycosites for a given biological milieu, has been the subject of glycoproteomics. Nevertheless, cell membrane glycoproteins constitute a small portion of the proteome [185]. A sample pre-enrichment for the glycans of interest, guided by a prior knowledge of the glycome, is often elected as the starting point to overcome this limitation [90], [122]. Samples may be pre-enriched for species of interest by physical methods, immunoprecipitation, and affinity chromatography targeting specific glycans with antibodies or lectins, prior to analysis by MS [80], [122], [186]. Digestions with glycosidases may also be introduced to generate desired glycostructures for lectin affinity chromatography prior to MS analysis. As a typical example, many studies addressing glycoproteins carrying short-chain STn antigens in cancer employ sialidases to generate the Tn antigen, enabling enrichment with the Vicia villosa agglutinin (VVA) lectin [90]. A similar rationale has been used to enrich samples for proteins carrying ST antigens using the peanut agglutinin (PNA) lectin [187] (Figure 4C). Building on these protocols, in the last decade, Vakhrushev et al. [80] developed an elegant strategy for high-throughput identification of O-GalNAc glycosites by exploiting genome editing technologies. This strategy is based on knockout of COSMC (C1GALT1-specific chaperone), which restricts O-glycosylation to the simplest Tn and STn antigens suitable for VVA enrichment [80], [186]. This simplification of cell glycosylation has allowed to overcome O-glycosylation heterogeneity, facilitating MS/MS-based glycopeptide identification, which significantly contributes to expanding current knowledge of human O-glycoproteome [80], [186].
MS coupled to LC using nano-flow stands as the gold standard technique for glycoprotein annotation from minute amounts of starting material [188]. The development of highly sensitive and accurate mass spectrometer analyzers enabled deep proteome mining, providing the identification and quantification of a vast array of proteins and peptides while simultaneously informing on their PTMs [141]. Currently, standard ionization methods for protein and PTM analyses involve MALDI and nano-ESI. Additionally, the employment of hybrid mass analyzers, such as quadrupole time-of-flight (QTOF), time-of-flight/time-of-flight (TOF/TOF), ion trap/orbitrap (IT/Orbitrap), and quadrupole/orbitrap (Q/Orbitrap), which allow MS/MS experiments, has contributed to increasing confidence in protein identification and accurate mapping of PTM sites [76]. Moreover, the new generation of mass spectrometers with a tribrid architecture, namely combining quadrupole, linear ion trap, and orbitrap mass analyzers, will enhance protein coverage, improve fragmentation, and provide more comprehensive identification and characterization of proteomes [189].
Protein and PTM identification by MS can be achieved by different strategies. The most widely used approach consists in a bottom-up analysis of peptides derived from the digestion of proteins with different proteases by nanoLC–MS/MS. Conventionally, C18 reverse phase columns have been shown to be sufficiently versatile for chromatography prior to MS [190]. However, HILIC chromatography enables good separation of glycopeptides from protein mixtures, as well as efficient separation between neutral and sialoglycopeptides, while allowing more efficient structural characterization, mainly in the presence of a neutral or zwitterionic stationary phase [191], [192]. Noteworthily, glycosylation often renders proteins less prone to proteases, limiting the success of these approaches [193]. As such, many studies combine different broad-spectrum proteolytic enzymes to increase protein coverage and the chances of glycopeptide identification. Alternatively, the middle-down approach enables the analysis of large peptides resulting from mild proteolysis [194]. Finally, top-down analysis contemplates the identification of intact protein mass by MS followed by direct ion dissociation in the gas phase [195]. Relative and absolute quantification of proteins and PTMs has been classically achieved by 2-DE, before MS-based approaches emerged [196]. MS-based methods involve stable isotopic metabolic labeling [stable isotope labeling by amino acids in cell culture (SILAC) and stable isotope labeling with amino acids in mammal (SILAM)] and post-metabolic labeling [isobaric tags for relative and absolute quantification (iTRAQ), tandem mass tag (TMT), and isotope-coded affinity tag (ICAT)] [197]. However, with the increased resolution and sensitivity of modern mass spectrometers, label-free quantification (LFQ) methods have been gaining ground. These methods generally include the computational analysis of MS ion intensity, spectral counting, chromatogram peak area determination, or targeted approaches, such as selected reaction monitoring (SRM) [197]. MS/MS, with different fragmentation methods, provides more structural information, such as glycosites. Collision-induced dissociation (CID) is the most used and easily available ion fragmentation methodology. It consists in the collision of selected molecular ions with an inert gas (argon, nitrogen, or helium), leading to the fragmentation of protonated amide linkages, while frequently favoring neutral loss of glycan moieties [198]. Although capable of providing significant information on peptide sequence, CID does not provide ideal diagnostic ion information for the identification of glycopeptides. Contrastingly, electron capture dissociation (ECD) induces preferential cleavage of peptide backbones at the N-Cα bond, preventing the glycan-associated neutral loss, and thus being a better approach for glycopeptide identification [199]. More recently, electron transfer dissociation (ETD) was developed, showing many similarities with ECD; however, it can be performed in ion trap mass spectrometers or even in QTOF type instruments [200]. Moreover, high-energy collision dissociation (HCD) was implemented essentially in orbitrap platforms. Although similar to CID, in HCD, fragmentation is carried at higher collision energies, ensuring accurate glycopeptide diagnosis through the generation of typical glycan oxonium ions [201]. Lastly, electron-transfer/higher-energy collision dissociation (EThcD) is a hybrid dissociation method, resulting from combination of ETD and HCD. It provides higher peptide sequence coverage by simultaneously providing HCD diagnostic glycan ions and ETD-derived peptide fragments with preserved information on modification sites, facilitating PTM site assignment [202]. Namely, it has shown promising results for glycoproteomics analysis by providing both glycan and peptide fragment spectral information through cleavage of amide and glycosidic linkages [203]. Regardless of the methodological approach selected for protein identification, there is still a gap between protein identification and biomarker discovery. In silico approaches have revealed more accurate assignments and identified relevant glycobiomarkers for clinical translation [204]. This strategy encompasses several bioinformatics tools to validate protein identification (SequestHT, Proteome Discoverer, and SwissProt database), glycosylation sites (NetNGlyc and NetOGlyc), biological functions (Panther, STRING, Cytoscape, and UniProtKB), and associations to histological data (Oncomine and The Human Protein Atlas) [204] (Figure 4C). Recently, a novel in silico prediction approach for mucin-type O-GalNAcylation termed ISOGlyP (https://isoglyp.utep.edu/) has also been introduced to assist the identification of potential glycoproteins [205]. Moreover, Byonic software has emerged as a powerful tool toward automated identification of glycopeptide mass spectra, at level of glycosite modification. It allows the use of a generic glycan list, composed by a wide array of N- and O-glycan structures, or a customized list for search and identification of modified peptides. Nevertheless, it still requires manual validation [81], [82]. In summary, given the large panoply of methodological approaches, it is now possible to personalize workflows toward more accurate access to cellular and tissue glycome and glycoproteome, ultimately facilitating clinical translation.
Glycoproteogenomics: a new concept in the crossroad between nucleic acids, proteins, and carbohydrates
The full characterization of the glycoproteome remains a daunting analytical enterprise whose success is directly linked to the level of understanding about the nature of proteoforms and glycoforms. The array of proteoforms available for glycosylation is dependent on the genome and the subsequent events that culminate in protein synthesis and maturation [35]. On the other hand, glycosylation is not a direct gene product, but rather the result of a highly regulated process mediated by a wide array of glycosyltransferases, glycosidases, chaperones, and sugar donors along protein secretory pathways [39]. As such, not a single omics can accurately predict glycoproteome, making it necessary to adapt conventional strategies to accommodate molecular information of distinct origins. This has been, to some extent, partially minimized by shaping conventional proteomics workflows to accommodate the presence of glycans. However, significant amounts of glycoproteomics data generated by nanoLC–MS/MS runs remain undeciphered due to the lack of genome-customized protein databases used for protein annotation.
Glycoproteogenomics is a rather novel concept that attempts to bridge genes, proteins, and PTMs to bring new awareness to protein functions and ultimately provide targetable protein nodes (Figure 5). To our knowledge, the concept was first explored by Rolland et al. [42] in 2017, by integrating N-glycoproteomics and transcriptome sequencing to identify relevant signaling networks and therapeutic targets in lymphomas. In the following year, Mun et al. [41] combined mRNA, protein, phosphorylation, and N-glycosylation data to identify different subtypes of diffuse gastric cancer in young populations. PTMs are particularly important for defining subtypes associated with immune- and invasion-related pathways. However, the term glycoproteogenomics was only formally introduced by Lin et al. [206] in a study concerning human serum α-2-HS-glycoprotein. The authors have demonstrated correlations between gene polymorphisms and changes in glycosylation for this protein [206]. A wide variability of proteoforms was also observed, suggesting need for quantitative profiling to foresee improved biomarkers for different disease states. Taken together, these studies elegantly highlight the pivotal role played by glycans in human biology and the importance of associating different omics for improved cancer biomarkers. However, it remains crucial to progress beyond incrementality toward more integrated and interconnected approaches.
Figure 5.

Onco-glycoproteogenomics towardsafer and more effective immunotherapy and precision oncology
The comprehensive combination of the different omics is crucial for discovery of unforeseen tumor unique molecular signatures. Moreover, it will allow a thorough understanding of microenvironmental and functional contexts of glycoproteoforms, toward the rational design of targeted therapeutics.
In a broader sense, we conceptualize that glycoproteogenomics addresses the protein species resulting from a wide array of events, including gene polymorphisms and mutations, differential transcription, and glycosylation occurring in biological milieus. These events generate a wide number of glycoproteoforms for the same protein, which require integrated analytical workflows for comprehensive characterization. Proteogenomics has led the way by providing the conceptual framework to accomplish this goal. Adapting current approaches to accommodate the structural diversity of glycans and the physicochemical subtleties introduced by glycosylation in proteins will be the next logical step toward glycoproteogenomics. We anticipate that genomics and glycomics-customized protein identification methods will be crucial for understanding the role of glycoproteins in cancer. The identification of unforeseen molecular signatures is plausible to pave the way for neoantigen discovery and novel therapeutics.
Concluding remarks
Novel sources of cancer neoantigens are urgently needed to address cold tumors, showing low mutational frequencies, lack of T cell infiltration, and poor response to current immune checkpoint inhibitors. Glycans are essential components of biological systems that hold enormous potential toward this end, since they are present at the cell surface and can be easily targeted by ligands of different natures, including lectins of the immune system. Nevertheless, they are often neglected in the context of biomarker research facing the enormous amount of information on the cancer genome, transcriptome, and proteome. The technological readiness of high-throughput genomics and proteomics platforms has not only prompted cancer neoantigen discovery but also led to the generation of effective cancer vaccines. Moreover, genomics, transcriptomics, and more recently, proteogenomics have improved the molecular subtyping of human tumors and currently constitute a paramount framework toward precision oncology. Nevertheless, zooming in on the glycoproteome of cancer cells backed by a profound understanding about the cancer glycome may provide yet unforeseen molecular signatures, constituting another crucial milestone toward systems biology. However, mapping of the glycoproteome cannot be achieved without bridging different sources of information. It becomes imperative to systematize procedures to bring together genomics and transcriptomics information, enabling the accurate customization of proteomics databases with the integration of cancer-specific gene polymorphisms and alternative splicing transcripts. It is also mandatory to integrate PTMs that decisively define molecular functions. In this sense, glycosylation holds tremendous potential, as it provides well-known glycan cancer signatures and confers functional and metabolic contexts to the membrane proteome. Furthermore, setting the aim in cancer glycoproteogenomics-derived peptide identification will provide bispecific neoantigens. This wish list comes with outstanding computational challenges, aiming at integrating and extracting information from multiple layers of data. Nevertheless, it will undoubtedly unveil novel and even unexpected molecular signatures, providing a more thorough understanding of biological systems, improved tumor classification capacity, and a more rational design of personalized cancer therapies.
CRediT author statement
José Alexandre Ferreira: Conceptualization, Methodology, Software, Data curation, Writing - original draft, Supervision, Project administration, Funding acquisition. Marta Relvas-Santos: Methodology, Software, Data curation, Writing - original draft, Visualization. Andreia Peixoto: Writing - review & editing. André M.N. Silva: Software, Resources, Writing - review & editing, Supervision. Lúcio Lara Santos: Resources, Writing - review & editing, Supervision, Project administration, Funding acquisition. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
The authors wish to acknowledge the Portuguese Foundation for Science and Technology (FCT) for the PhD grants (Grant Nos. SFRH/BD/111242/2015 awarded to AP and SFRH/BD/146500/2019 awarded to MRS) and the FCT assistant researcher grant (Grant No. CEECIND/03186/2017 awarded to JAF). FCT is co-financed by European Social Fund (ESF) under Human Potential Operation Programme (POPH) from National Strategic Reference Framework (NSRF). The authors also acknowledge the FCT funding for CI-IPOP research unit (Grant No. PEst-OE/SAU/UI0776/201) and LAQV-REQUIMTE research unit (Grant No. UIDB/50006/2020), the Portuguese Oncology Institute of Porto Research Centre (Grant Nos. CI-IPOP-29-2020, CI-IPOP-58-2020, and CI-IPOP-Proj.70-bolsa2019-GPTE), and the PhD Program in Biomedical Sciences of ICBAS-University of Porto in Portugal. The authors also thank the “Early stage cancer treatment, driven by context of molecular imaging (ESTIMA)” framework (Grant No. NORTE-01-0145-FEDER-000027) and the IPO-Score (Grant No. DSAIPA/DS/0042/2018) for financial support. This work was financed by European Regional Development Fund (ERDF) through the COMPETE 2020 – Operational Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT / Ministry for Science, Technology and Higher Education (MCTES).
Handled by Song Liu
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China.
References
- 1.Bayat Mokhtari R., Homayouni T.S., Baluch N., Morgatskaya E., Kumar S., Das B. Combination therapy in combating cancer. Oncotarget. 2017;8:38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma R.A., Plummer R., Stock J.K., Greenhalgh T.A., Ataman O., Kelly S. Clinical development of new drug-radiotherapy combinations. Nat Rev Clin Oncol. 2016;13:627–642. doi: 10.1038/nrclinonc.2016.79. [DOI] [PubMed] [Google Scholar]
- 3.Yang L., Shi P., Zhao G., Xu J., Peng W., Zhang J. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8. doi: 10.1038/s41392-020-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azevedo R., Ferreira J.A., Peixoto A., Neves M., Sousa N., Lima A. Emerging antibody-based therapeutic strategies for bladder cancer: a systematic review. J Control Release. 2015;214:40–61. doi: 10.1016/j.jconrel.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Seebacher N.A., Stacy A.E., Porter G.M., Merlot A.M. Clinical development of targeted and immune based anti-cancer therapies. J Exp Clin Cancer Res. 2019;38:156. doi: 10.1186/s13046-019-1094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplon H., Reichert J.M. Antibodies to watch in 2019. MAbs. 2019;11:219–238. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zahavi D., AlDeghaither D., O’Connell A., Weiner L.M. Enhancing antibody-dependent cell-mediated cytotoxicity: a strategy for improving antibody-based immunotherapy. Antibody Therapeutics. 2018;1:7–12. doi: 10.1093/abt/tby002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidel J.A., Otsuka A., Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Afonso J., Santos L.L., Longatto-Filho A., Baltazar F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat Rev Urol. 2020;17:77–106. doi: 10.1038/s41585-019-0263-6. [DOI] [PubMed] [Google Scholar]
- 10.Rosenblum D., Joshi N., Tao W., Karp J.M., Peer D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun. 2018;9:1410. doi: 10.1038/s41467-018-03705-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J.Y., Kronbichler A., Eisenhut M., Hong S.H., van der Vliet H.J., Kang J. Tumor mutational burden and efficacy of immune checkpoint inhibitors: a systematic review and meta-analysis. Cancers (Basel) 2019;11:1798. doi: 10.3390/cancers11111798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahles A., Lehmann K.V., Toussaint N.C., Huser M., Stark S.G., Sachsenberg T. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell. 2018;34:211–224. doi: 10.1016/j.ccell.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W., Lee K.W., Srivastava R.M., Kuo F., Krishna C., Chowell D. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25:767–775. doi: 10.1038/s41591-019-0434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith C.C., Selitsky S.R., Chai S., Armistead P.M., Vincent B.G., Serody J.S. Alternative tumour-specific antigens. Nat Rev Cancer. 2019;19:465–478. doi: 10.1038/s41568-019-0162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mnatsakanyan R., Shema G., Basik M., Batist G., Borchers C.H., Sickmann A. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev Proteomics. 2018;15:515–535. doi: 10.1080/14789450.2018.1483340. [DOI] [PubMed] [Google Scholar]
- 16.RodrIguez E., Schetters S.T.T., van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol. 2018;18:204–211. doi: 10.1038/nri.2018.3. [DOI] [PubMed] [Google Scholar]
- 17.Wirth T.C., Kuhnel F. Neoantigen targeting-dawn of a new era in cancer immunotherapy? Front Immunol. 2017;8:1848. doi: 10.3389/fimmu.2017.01848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lancaster E.M., Jablons D., Kratz J.R. Applications of next-generation sequencing in neoantigen prediction and cancer vaccine development. Genet Test Mol Biomarkers. 2020;24:59–66. doi: 10.1089/gtmb.2018.0211. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q., Douglass J., Hwang M.S., Hsiue E.H., Mog B.J., Zhang M. Direct detection and quantification of neoantigens. Cancer Immunol Res. 2019;7:1748–1754. doi: 10.1158/2326-6066.CIR-19-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson H.J., Socciarelli F., Vacanti N.M., Haugen M.H., Zhu Y., Siavelis I. Breast cancer quantitative proteome and proteogenomic landscape. Nat Commun. 2019;10:1600. doi: 10.1038/s41467-019-09018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gubin M.M., Zhang X., Schuster H., Caron E., Ward J.P., Noguchi T. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhanda S.K., Mahajan S., Paul S., Yan Z., Kim H., Jespersen M.C. IEDB-AR: immune epitope database-analysis resource in 2019. Nucleic Acids Res. 2019;47:W502–W506. doi: 10.1093/nar/gkz452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truex N.L., Holden R.L., Wang B.Y., Chen P.G., Hanna S., Hu Z. Automated flow synthesis of tumor neoantigen peptides for personalized immunotherapy. Sci Rep. 2020;10:723. doi: 10.1038/s41598-019-56943-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheetz L., Park K.S., Li Q., Lowenstein P.R., Castro M.G., Schwendeman A. Engineering patient-specific cancer immunotherapies. Nat Biomed Eng. 2019;3:768–782. doi: 10.1038/s41551-019-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oberli M.A., Reichmuth A.M., Dorkin J.R., Mitchell M.J., Fenton O.S., Jaklenec A. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017;17:1326–1335. doi: 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali O.A., Emerich D., Dranoff G., Mooney D.J. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med. 2009;1:8ra19. doi: 10.1126/scitranslmed.3000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barker S.E., Grosse S.M., Siapati E.K., Kritz A., Kinnon C., Thrasher A.J. Immunotherapy for neuroblastoma using syngeneic fibroblasts transfected with IL-2 and IL-12. Br J Cancer. 2007;97:210–217. doi: 10.1038/sj.bjc.6603857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morandi F., Frassoni F., Ponzoni M., Brignole C. Novel immunotherapeutic approaches for neuroblastoma and malignant melanoma. J Immunol Res. 2018;2018:8097398. doi: 10.1155/2018/8097398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L., Wang Y., Miao L., Liu Q., Musetti S., Li J. Combination immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol Ther. 2018;26:45–55. doi: 10.1016/j.ymthe.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahtinen S., Feola S., Capasso C., Laustio N., Groeneveldt C., Ylosmaki E.O. Exploiting preexisting immunity to enhance oncolytic cancer immunotherapy. Cancer Res. 2020;80:2575–2585. doi: 10.1158/0008-5472.CAN-19-2062. [DOI] [PubMed] [Google Scholar]
- 31.Chu Y., Liu Q., Wei J., Liu B. Personalized cancer neoantigen vaccines come of age. Theranostics. 2018;8:4238–4246. doi: 10.7150/thno.24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Priestley P., Baber J., Lolkema M.P., Steeghs N., de Bruijn E., Shale C. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575:210–216. doi: 10.1038/s41586-019-1689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park H., Bae J., Kim H., Kim S., Kim H., Mun D.G. Compact variant-rich customized sequence database and a fast and sensitive database search for efficient proteogenomic analyses. Proteomics. 2014;14:2742–2749. doi: 10.1002/pmic.201400225. [DOI] [PubMed] [Google Scholar]
- 34.Zhang B., Whiteaker J.R., Hoofnagle A.N., Baird G.S., Rodland K.D., Paulovich A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat Rev Clin Oncol. 2019;16:256–268. doi: 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S. How many human proteoforms are there? Nat Chem Biol. 2018;14:206–214. doi: 10.1038/nchembio.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Azevedo R., Peixoto A., Gaiteiro C., Fernandes E., Neves M., Lima L. Over forty years of bladder cancer glycobiology: where do glycans stand facing precision oncology? Oncotarget. 2017;8:91734–91764. doi: 10.18632/oncotarget.19433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandes E., Sores J., Cotton S., Peixoto A., Ferreira D., Freitas R. Esophageal, gastric and colorectal cancers: looking beyond classical serological biomarkers towards glycoproteomics-assisted precision oncology. Theranostics. 2020;10:4903–4928. doi: 10.7150/thno.42480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kudelka M.R., Ju T., Heimburg-Molinaro J., Cummings R.D. Simple sugars to complex disease—mucin-type O-glycans in cancer. Adv Cancer Res. 2015;126:53–135. doi: 10.1016/bs.acr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peixoto A., Relvas-Santos M., Azevedo R., Santos L.L., Ferreira J.A. Protein glycosylation and tumor microenvironment alterations driving cancer hallmarks. Front Oncol. 2019;9:380. doi: 10.3389/fonc.2019.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bassani-Sternberg M., Coukos G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr Opin Immunol. 2016;41:9–17. doi: 10.1016/j.coi.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 41.Mun D.G., Bhin J., Kim S., Kim H., Jung J.H., Jung Y. Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell. 2019;35:111–124. doi: 10.1016/j.ccell.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Rolland D.C.M., Basrur V., Jeon Y.K., McNeil-Schwalm C., Fermin D., Conlon K.P. Functional proteogenomics reveals biomarkers and therapeutic targets in lymphomas. Proc Natl Acad Sci U S A. 2017;114:6581–6586. doi: 10.1073/pnas.1701263114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adalsteinsson V.A., Ha G., Freeman S.S., Choudhury A.D., Stover D.G., Parsons H.A. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun. 2017;8:1324. doi: 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.PCAWG Transcriptome Core Group, Calabrese C., Davidson N.R., Demircioglu D., Fonseca N.A., He Y. Genomic basis for RNA alterations in cancer. Nature. 2020;578:129–136. doi: 10.1038/s41586-020-1970-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barry M.J. Clinical practice. Prostate-specific-antigen testing for early diagnosis of prostate cancer. N Engl J Med. 2001;344:1373–1377. doi: 10.1056/NEJM200105033441806. [DOI] [PubMed] [Google Scholar]
- 49.Becerra A.Z., Probst C.P., Tejani M.A., Aquina C.T., Gonzalez M.G., Hensley B.J. Evaluating the prognostic role of elevated preoperative carcinoembryonic antigen levels in colon cancer patients: results from the National Cancer Database. Ann Surg Oncol. 2016;23:1554–1561. doi: 10.1245/s10434-015-5014-1. [DOI] [PubMed] [Google Scholar]
- 50.Felder M., Kapur A., Gonzalez-Bosquet J., Horibata S., Heintz J., Albrecht R. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi: 10.1186/1476-4598-13-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheynkman G.M., Shortreed M.R., Cesnik A.J., Smith L.M. Proteogenomics: integrating next-generation sequencing and mass spectrometry to characterize human proteomic variation. Annu Rev Anal Chem (Palo Alto Calif) 2016;9:521–545. doi: 10.1146/annurev-anchem-071015-041722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfaro J.A., Sinha A., Kislinger T., Boutros P.C. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nat Methods. 2014;11:1107–1113. doi: 10.1038/nmeth.3138. [DOI] [PubMed] [Google Scholar]
- 53.Ang M.Y., Low T.Y., Lee P.Y., Wan Mohamad Nazarie W.F., Guryev V., Jamal R. Proteogenomics: from next-generation sequencing (NGS) and mass spectrometry-based proteomics to precision medicine. Clin Chim Acta. 2019;498:38–46. doi: 10.1016/j.cca.2019.08.010. [DOI] [PubMed] [Google Scholar]
- 54.Reuter J.A., Spacek D.V., Pai R.K., Snyder M.P. Simul-seq: combined DNA and RNA sequencing for whole-genome and transcriptome profiling. Nat Methods. 2016;13:953–958. doi: 10.1038/nmeth.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E. Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nesvizhskii A.I. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11:1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lowe R., Shirley N., Bleackley M., Dolan S., Shafee T. Transcriptomics technologies. PLoS Comput Biol. 2017;13:e1005457. doi: 10.1371/journal.pcbi.1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pereira R., Oliveira J., Sousa M. Bioinformatics and computational tools for next-generation sequencing analysis in clinical genetics. J Clin Med. 2020;9:132. doi: 10.3390/jcm9010132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D'Souza M., Sulakhe D., Wang S., Xie B., Hashemifar S., Taylor A. Strategic integration of multiple bioinformatics resources for system level analysis of biological networks. Methods Mol Biol. 2017;1613:85–99. doi: 10.1007/978-1-4939-7027-8_5. [DOI] [PubMed] [Google Scholar]
- 61.Paul S., Sidney J., Sette A., Peters B. TepiTool: a pipeline for computational prediction of T cell epitope candidates. Curr Protoc Immunol. 2016;114:18.19.1–24. doi: 10.1002/cpim.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abella J.R., Antunes D.A., Clementi C., Kavraki L.E. APE-Gen: a fast method for generating ensembles of bound peptide-MHC conformations. Molecules. 2019;24:881. doi: 10.3390/molecules24050881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nielsen M., Lundegaard C., Blicher T., Lamberth K., Harndahl M., Justesen S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu J., Wang W., Zhang J., Zhou B., Zhao W., Su Z. DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front Immunol. 2019;10:2559. doi: 10.3389/fimmu.2019.02559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao T., Cheng L., Zang T., Hu Y. Peptide-major histocompatibility complex class I binding prediction based on deep learning with novel feature. Front Genet. 2019;10:1191. doi: 10.3389/fgene.2019.01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baldauf M.C., Gerke J.S., Kirschner A., Blaeschke F., Effenberger M., Schober K. Systematic identification of cancer-specific MHC-binding peptides with RAVEN. Oncoimmunology. 2018;7:e1481558. doi: 10.1080/2162402X.2018.1481558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nielsen M., Lundegaard C., Lund O., Kesmir C. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics. 2005;57:33–41. doi: 10.1007/s00251-005-0781-7. [DOI] [PubMed] [Google Scholar]
- 68.Tenzer S., Peters B., Bulik S., Schoor O., Lemmel C., Schatz M.M. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell Mol Life Sci. 2005;62:1025–1037. doi: 10.1007/s00018-005-4528-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoze E., Tsaban L., Maman Y., Louzoun Y. Predictor for the effect of amino acid composition on CD4+ T cell epitopes preprocessing. J Immunol Methods. 2013;391:163–173. doi: 10.1016/j.jim.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paul S., Karosiene E., Dhanda S.K., Jurtz V., Edwards L., Nielsen M. Determination of a predictive cleavage motif for eluted major histocompatibility complex class II ligands. Front Immunol. 2018;9:1795. doi: 10.3389/fimmu.2018.01795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Purcell A.W., Ramarathinam S.H., Ternette N. Mass spectrometry-based identification of MHC-bound peptides for immunopeptidomics. Nat Protoc. 2019;14:1687–1707. doi: 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- 72.Boja E.S., Rodriguez H. Proteogenomic convergence for understanding cancer pathways and networks. Clin Proteomics. 2014;11:22. doi: 10.1186/1559-0275-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vasaikar S., Huang C., Wang X., Petyuk V.A., Savage S.R., Wen B. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell. 2019;177:1035–1049. doi: 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Metallo C.M., Vander Heiden M.G. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chick J.M., Kolippakkam D., Nusinow D.P., Zhai B., Rad R., Huttlin E.L. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol. 2015;33:743–749. doi: 10.1038/nbt.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silva A.M.N., Vitorino R., Domingues M.R.M., Spickett C.M., Domingues P. Post-translational modifications and mass spectrometry detection. Free Radic Biol Med. 2013;65:925–941. doi: 10.1016/j.freeradbiomed.2013.08.184. [DOI] [PubMed] [Google Scholar]
- 77.Greer S.M., Cannon J.R., Brodbelt J.S. Improvement of shotgun proteomics in the negative mode by carbamylation of peptides and ultraviolet photodissociation mass spectrometry. Anal Chem. 2014;86:12285–12290. doi: 10.1021/ac5035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eng J.K., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 79.Nardiello D., Palermo C., Natale A., Quinto M., Centonze D. Strategies in protein sequencing and characterization: multi-enzyme digestion coupled with alternate CID/ETD tandem mass spectrometry. Anal Chim Acta. 2015;854:106–117. doi: 10.1016/j.aca.2014.10.053. [DOI] [PubMed] [Google Scholar]
- 80.Vakhrushev S.Y., Steentoft C., Vester-Christensen M.B., Bennett E.P., Clausen H., Levery S.B. Enhanced mass spectrometric mapping of the human GalNAc-type O-glycoproteome with SimpleCells. Mol Cell Proteomics. 2013;12:932–944. doi: 10.1074/mcp.O112.021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bern M., Kil Y.J., Becker C. Byonic: advanced peptide and protein identification software. Curr Protoc Bioinformatics. 2012;Chapter 13:Unit13.20. doi: 10.1002/0471250953.bi1320s40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee L.Y., Moh E.S., Parker B.L., Bern M., Packer N.H., Thaysen-Andersen M. Toward automated N-glycopeptide identification in glycoproteomics. J Proteome Res. 2016;15:3904–3915. doi: 10.1021/acs.jproteome.6b00438. [DOI] [PubMed] [Google Scholar]
- 83.Chen Y.J., Roumeliotis T.I., Chang Y.H., Chen C.T., Han C.L., Lin M.H. Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell. 2020;182:226–244. doi: 10.1016/j.cell.2020.06.012. [DOI] [PubMed] [Google Scholar]
- 84.Clark D.J., Dhanasekaran S.M., Petralia F., Pan J., Song X., Hu Y. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell. 2020;180:207. doi: 10.1016/j.cell.2019.12.026. [DOI] [PubMed] [Google Scholar]
- 85.Dou Y., Kawaler E.A., Cui Zhou D., Gritsenko M.A., Huang C., Blumenberg L. Proteogenomic characterization of endometrial carcinoma. Cell. 2020;180:729–748. doi: 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Llop E., Ferrer-Batalle M., Barrabes S., Guerrero P.E., Ramirez M., Saldova R. Improvement of prostate cancer diagnosis by detecting PSA glycosylation-specific changes. Theranostics. 2016;6:1190–1204. doi: 10.7150/thno.15226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gratacos-Mulleras A., Duran A., Asadi Shehni A., Ferrer-Batalle M., Ramirez M., Comet J. Characterisation of the main PSA glycoforms in aggressive prostate cancer. Sci Rep. 2020;10:18974. doi: 10.1038/s41598-020-75526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haga Y., Uemura M., Baba S., Inamura K., Takeuchi K., Nonomura N. Identification of multisialylated LacdiNAc structures as highly prostate cancer specific glycan signatures on PSA. Anal Chem. 2019;91:2247–2254. doi: 10.1021/acs.analchem.8b04829. [DOI] [PubMed] [Google Scholar]
- 89.Gomes C., Almeida A., Ferreira J.A., Silva L., Santos-Sousa H., Pinto-de-Sousa J. Glycoproteomic analysis of serum from patients with gastric precancerous lesions. J Proteome Res. 2013;12:1454–1466. doi: 10.1021/pr301112x. [DOI] [PubMed] [Google Scholar]
- 90.Cotton S., Azevedo R., Gaiteiro C., Ferreira D., Lima L., Peixoto A. Targeted O-glycoproteomics explored increased sialylation and identified MUC16 as a poor prognosis biomarker in advanced-stage bladder tumours. Mol Oncol. 2017;11:895–912. doi: 10.1002/1878-0261.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pinho S.S., Reis C.A. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 92.Hansen L., Lind-Thomsen A., Joshi H.J., Pedersen N.B., Have C.T., Kong Y. A glycogene mutation map for discovery of diseases of glycosylation. Glycobiology. 2015;25:211–224. doi: 10.1093/glycob/cwu104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee-Sundlov M.M., Ashline D.J., Hanneman A.J., Grozovsky R., Reinhold V.N., Hoffmeister K.M. Circulating blood and platelets supply glycosyltransferases that enable extrinsic extracellular glycosylation. Glycobiology. 2017;27:188–198. doi: 10.1093/glycob/cww108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Radhakrishnan P., Dabelsteen S., Madsen F.B., Francavilla C., Kopp K.L., Steentoft C. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci U S A. 2014;111:E4066–E4075. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Freitas D., Campos D., Gomes J., Pinto F., Macedo J.A., Matos R. O-glycans truncation modulates gastric cancer cell signaling and transcription leading to a more aggressive phenotype. EBioMedicine. 2019;40:349–362. doi: 10.1016/j.ebiom.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peixoto A., Fernandes E., Gaiteiro C., Lima L., Azevedo R., Soares J. Hypoxia enhances the malignant nature of bladder cancer cells and concomitantly antagonizes protein O-glycosylation extension. Oncotarget. 2016;7:63138–63157. doi: 10.18632/oncotarget.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin M.-C., Chien P.-H., Wu H.-Y., Chen S.-T., Juan H.-F., Lou P.-J. C1GALT1 predicts poor prognosis and is a potential therapeutic target in head and neck cancer. Oncogene. 2018;37:5780–5793. doi: 10.1038/s41388-018-0375-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tajadura-Ortega V., Gambardella G., Skinner A., Halim A., Van Coillie J., Schjoldager K.T.G. O-linked mucin-type glycosylation regulates the transcriptional programme downstream of EGFR. Glycobiology. 2020;31:200–210. doi: 10.1093/glycob/cwaa075. [DOI] [PubMed] [Google Scholar]
- 99.Ferreira I.G., Pucci M., Venturi G., Malagolini N., Chiricolo M., Dall'Olio F. Glycosylation as a main regulator of growth and death factor receptors signaling. Int J Mol Sci. 2018;19:580. doi: 10.3390/ijms19020580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rodrigues E., Macauley M.S. Hypersialylation in cancer: modulation of inflammation and therapeutic opportunities. Cancers (Basel) 2018;10:207. doi: 10.3390/cancers10060207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perdicchio M., Cornelissen L.A., Streng-Ouwehand I., Engels S., Verstege M.I., Boon L. Tumor sialylation impedes T cell mediated anti-tumor responses while promoting tumor associated-regulatory T cells. Oncotarget. 2016;7:8771–8782. doi: 10.18632/oncotarget.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perdicchio M., Ilarregui J.M., Verstege M.I., Cornelissen L.A., Schetters S.T., Engels S. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci U S A. 2016;113:3329–3334. doi: 10.1073/pnas.1507706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trinchera M., Aronica A., Dall'Olio F. Selectin ligands sialyl-Lewis a and sialyl-Lewis x in gastrointestinal cancers. Biology (Basel) 2017;6:16. doi: 10.3390/biology6010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Borsig L. Selectins in cancer immunity. Glycobiology. 2018;28:648–655. doi: 10.1093/glycob/cwx105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vajaria B.N., Patel P.S. Glycosylation: a hallmark of cancer? Glycoconj J. 2017;34:147–156. doi: 10.1007/s10719-016-9755-2. [DOI] [PubMed] [Google Scholar]
- 106.Hauselmann I., Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol. 2014;4:28. doi: 10.3389/fonc.2014.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oliveira-Ferrer L., Legler K., Milde-Langosch K. Role of protein glycosylation in cancer metastasis. Semin Cancer Biol. 2017;44:141–152. doi: 10.1016/j.semcancer.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 108.Ferreira J.A., Magalhaes A., Gomes J., Peixoto A., Gaiteiro C., Fernandes E. Protein glycosylation in gastric and colorectal cancers: toward cancer detection and targeted therapeutics. Cancer Lett. 2017;387:32–45. doi: 10.1016/j.canlet.2016.01.044. [DOI] [PubMed] [Google Scholar]
- 109.Neves M., Azevedo R., Lima L., Oliveira M.I., Peixoto A., Ferreira D. Exploring sialyl-Tn expression in microfluidic-isolated circulating tumour cells: a novel biomarker and an analytical tool for precision oncology applications. Nat Biotechnol. 2019;49:77–87. doi: 10.1016/j.nbt.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 110.Furukawa Y., Tara M., Ohmori K., Kannagi R. Variant type of sialyl Lewis X antigen expressed on adult T cell leukemia cells is associated with skin involvement. Cancer Res. 1994;54:6533–6538. [PubMed] [Google Scholar]
- 111.Posey A.D., Jr., Schwab R.D., Boesteanu A.C., Steentoft C., Mandel U., Engels B. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–1454. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steentoft C., Migliorini D., King T.R., Mandel U., June C.H., Posey A.D., Jr. Glycan-directed CAR-T cells. Glycobiology. 2018;28:656–669. doi: 10.1093/glycob/cwy008. [DOI] [PubMed] [Google Scholar]
- 113.Loureiro L.R., Feldmann A., Bergmann R., Koristka S., Berndt N., Arndt C. Development of a novel target module redirecting UniCAR T cells to sialyl Tn-expressing tumor cells. Blood Cancer J. 2018;8:81. doi: 10.1038/s41408-018-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mettu R., Chen C.-Y., Wu C.-Y. Synthetic carbohydrate-based vaccines: challenges and opportunities. J Biomed Sci. 2020;27:9. doi: 10.1186/s12929-019-0591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nishat S., Andreana P.R. Entirely carbohydrate-based vaccines: an emerging field for specific and selective immune responses. Vaccines (Basel) 2016;4:19. doi: 10.3390/vaccines4020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reily C., Stewart T.J., Renfrow M.B., Novak J. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–366. doi: 10.1038/s41581-019-0129-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yehuda S., Padler-Karavani V. Glycosylated biotherapeutics: immunological effects of N-glycolylneuraminic acid. Front Immunol. 2020;11:21. doi: 10.3389/fimmu.2020.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Malaker S.A., Penny S.A., Steadman L.G., Myers P.T., Loke J.C., Raghavan M. Identification of glycopeptides as posttranslationally modified neoantigens in leukemia. Cancer Immunol Res. 2017;5:376–384. doi: 10.1158/2326-6066.CIR-16-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sharma P., Marada V., Cai Q., Kizerwetter M., He Y., Wolf S.P. Structure-guided engineering of the affinity and specificity of CARs against Tn-glycopeptides. Proc Natl Acad Sci U S A. 2020;117:15148–15159. doi: 10.1073/pnas.1920662117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Blixt O., Bueti D., Burford B., Allen D., Julien S., Hollingsworth M. Autoantibodies to aberrantly glycosylated MUC1 in early stage breast cancer are associated with a better prognosis. Breast Cancer Res. 2011;13:R25. doi: 10.1186/bcr2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wandall H.H., Blixt O., Tarp M.A., Pedersen J.W., Bennett E.P., Mandel U. Cancer biomarkers defined by autoantibody signatures to aberrant O-glycopeptide epitopes. Cancer Res. 2010;70:1306–1313. doi: 10.1158/0008-5472.CAN-09-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fernandes E., Freitas R., Ferreira D., Soares J., Azevedo R., Gaiteiro C. Nucleolin-Sle A glycoforms as E-selectin ligands and potentially targetable biomarkers at the cell surface of gastric cancer cells. Cancers (Basel) 2020;12:861. doi: 10.3390/cancers12040861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fujiki H., Watanabe T., Suganuma M. Cell-surface nucleolin acts as a central mediator for carcinogenic, anti-carcinogenic, and disease-related ligands. J Cancer Res Clin Oncol. 2014;140:689–699. doi: 10.1007/s00432-014-1587-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Losfeld M.E., Khoury D.E., Mariot P., Carpentier M., Krust B., Briand J.P. The cell surface expressed nucleolin is a glycoprotein that triggers calcium entry into mammalian cells. Exp Cell Res. 2009;315:357–369. doi: 10.1016/j.yexcr.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 125.Losfeld M.E., Leroy A., Coddeville B., Carpentier M., Mazurier J., Legrand D. N-glycosylation influences the structure and self-association abilities of recombinant nucleolin. FEBS J. 2011;278:2552–2564. doi: 10.1111/j.1742-4658.2011.08180.x. [DOI] [PubMed] [Google Scholar]
- 126.Dorota H.Ł., Sylwia K.K., Weronika D., Anna L. The lectin-binding pattern of nucleolin and its interaction with endogenous galectin-3. Cell Mol Biol Lett. 2014;19:461–482. doi: 10.2478/s11658-014-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hovanessian A.G., Puvion-Dutilleul F., Nisole S., Svab J., Perret E., Deng J.S. The cell-surface-expressed nucleolin is associated with the actin cytoskeleton. Exp Cell Res. 2000;261:312–328. doi: 10.1006/excr.2000.5071. [DOI] [PubMed] [Google Scholar]
- 128.Monferran S., Paupert J., Dauvillier S., Salles B., Muller C. The membrane form of the DNA repair protein Ku interacts at the cell surface with metalloproteinase 9. EMBO J. 2004;23:3758–3768. doi: 10.1038/sj.emboj.7600403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sajic T., Ciuffa R., Lemos V., Xu P., Leone V., Li C. A new class of protein biomarkers based on subcellular distribution: application to a mouse liver cancer model. Sci Rep. 2019;9:6913. doi: 10.1038/s41598-019-43091-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lee K., Byun K., Hong W., Chuang H.Y., Pack C.G., Bayarsaikhan E. Proteome-wide discovery of mislocated proteins in cancer. Genome Res. 2013;23:1283–1294. doi: 10.1101/gr.155499.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hsiao C.J., Tzai T.S., Chen C.H., Yang W.H., Chen C.H. Analysis of urinary prostate-specific antigen glycoforms in samples of prostate cancer and benign prostate hyperplasia. Dis Markers. 2016;2016:8915809. doi: 10.1155/2016/8915809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao Q., Zhan T., Deng Z., Li Q., Liu Y., Yang S. Glycan analysis of colorectal cancer samples reveals stage-dependent changes in CEA glycosylation patterns. Clin Proteomics. 2018;15:9. doi: 10.1186/s12014-018-9182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mak A.B., Blakely K.M., Williams R.A., Penttila P.A., Shukalyuk A.I., Osman K.T. CD133 protein N-glycosylation processing contributes to cell surface recognition of the primitive cell marker AC133 epitope. J Biol Chem. 2011;286:41046–41056. doi: 10.1074/jbc.M111.261545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Peiris D., Spector A.F., Lomax-Browne H., Azimi T., Ramesh B., Loizidou M. Cellular glycosylation affects Herceptin binding and sensitivity of breast cancer cells to doxorubicin and growth factors. Sci Rep. 2017;7:43006. doi: 10.1038/srep43006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dong Y., Li W., Gu Z., Xing R., Ma Y., Zhang Q. Inhibition of HER2-positive breast cancer growth by blocking the HER2 signaling pathway with HER2-glycan-imprinted nanoparticles. Angew Chem Int Ed Engl. 2019;58:10621–10625. doi: 10.1002/anie.201904860. [DOI] [PubMed] [Google Scholar]
- 136.Hsu J.M., Li C.W., Lai Y.J., Hung M.C. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018;78:6349–6353. doi: 10.1158/0008-5472.CAN-18-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang M., Wang J., Wang R., Jiao S., Wang S., Zhang J. Identification of a monoclonal antibody that targets PD-1 in a manner requiring PD-1 Asn58 glycosylation. Commun Biol. 2019;2:392. doi: 10.1038/s42003-019-0642-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee H.H., Wang Y.N., Xia W., Chen C.H., Rau K.M., Ye L. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell. 2019;36:168–178. doi: 10.1016/j.ccell.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen D., Tan S., Zhang H., Wang H., He W., Shi R. The FG loop of PD-1 serves as a “hotspot” for therapeutic monoclonal antibodies in tumor immune checkpoint therapy. iScience. 2019;14:113–124. doi: 10.1016/j.isci.2019.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Everest-Dass A.V., Moh E.S.X., Ashwood C., Shathili A.M.M., Packer N.H. Human disease glycomics: technology advances enabling protein glycosylation analysis - part 1. Expert Rev Proteomics. 2018;15:165–182. doi: 10.1080/14789450.2018.1421946. [DOI] [PubMed] [Google Scholar]
- 141.Eliuk S., Makarov A. Evolution of orbitrap mass spectrometry instrumentation. Annu Rev Anal Chem (Palo Alto Calif) 2015;8:61–80. doi: 10.1146/annurev-anchem-071114-040325. [DOI] [PubMed] [Google Scholar]
- 142.Kolarich D., Rapp E., Struwe W.B., Haslam S.M., Zaia J., McBride R. The minimum information required for a glycomics experiment (MIRAGE) project: improving the standards for reporting mass-spectrometry-based glycoanalytic data. Mol Cell Proteomics. 2013;12:991–995. doi: 10.1074/mcp.O112.026492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walsh I., Nguyen-Khuong T., Wongtrakul-Kish K., Tay S.J., Chew D., Jose T. GlycanAnalyzer: software for automated interpretation of N-glycan profiles after exoglycosidase digestions. Bioinformatics. 2019;35:688–690. doi: 10.1093/bioinformatics/bty681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rojas-Macias M.A., Mariethoz J., Andersson P., Jin C., Venkatakrishnan V., Aoki N.P. Towards a standardized bioinformatics infrastructure for N- and O-glycomics. Nat Commun. 2019;10:3275. doi: 10.1038/s41467-019-11131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shang Y., Zeng Y., Zeng Y. Integrated microfluidic lectin barcode platform for high-performance focused glycomic profiling. Sci Rep. 2016;6:20297. doi: 10.1038/srep20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mulloy B, Dell A, Stanley P, J HP. Structural analysis of glycans. In: Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, et al. editors. Essentials of glycobiology. New York: Cold Spring Harbor; 2015, p. 639–52.
- 147.Narimatsu Y., Joshi H.J., Nason R., Van Coillie J., Karlsson R., Sun L. An atlas of human glycosylation pathways enables display of the human glycome by gene engineered cells. Mol Cell. 2019;75:394–407. doi: 10.1016/j.molcel.2019.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Morelle W., Michalski J.C. Analysis of protein glycosylation by mass spectrometry. Nat Protoc. 2007;2:1585–1602. doi: 10.1038/nprot.2007.227. [DOI] [PubMed] [Google Scholar]
- 149.Rudd P., Karlsson N.G., Khoo K.H., Packer N.H. In: Essentials of glycobiology. Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., editors. Cold Spring Harbor; New York: 2015. Glycomics and glycoproteomics; pp. 653–666. [Google Scholar]
- 150.Plummer T.H., Jr., Elder J.H., Alexander S., Phelan A.W., Tarentino A.L. Demonstration of peptide: N-glycosidase F activity in endo-beta-N-acetylglucosaminidase F preparations. J Biol Chem. 1984;259:10700–10704. [PubMed] [Google Scholar]
- 151.Kuster B., Wheeler S.F., Hunter A.P., Dwek R.A., Harvey D.J. Sequencing of N-linked oligosaccharides directly from protein gels: in-gel deglycosylation followed by matrix-assisted laser desorption/ionization mass spectrometry and normal-phase high-performance liquid chromatography. Anal Biochem. 1997;250:82–101. doi: 10.1006/abio.1997.2199. [DOI] [PubMed] [Google Scholar]
- 152.Powers T.W., Jones E.E., Betesh L.R., Romano P.R., Gao P., Copland J.A. Matrix assisted laser desorption ionization imaging mass spectrometry workflow for spatial profiling analysis of N-linked glycan expression in tissues. Anal Chem. 2013;85:9799–9806. doi: 10.1021/ac402108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jensen P.H., Karlsson N.G., Kolarich D., Packer N.H. Structural analysis of N- and O-glycans released from glycoproteins. Nat Protoc. 2012;7:1299–1310. doi: 10.1038/nprot.2012.063. [DOI] [PubMed] [Google Scholar]
- 154.Tarentino A.L., Plummer T.H., Jr Oligosaccharide accessibility to peptide: N-glycosidase as promoted by protein-unfolding reagents. J Biol Chem. 1982;257:10776–10780. [PubMed] [Google Scholar]
- 155.Tarentino A.L., Trimble R.B., Plummer T.H., Jr Enzymatic approaches for studying the structure, synthesis, and processing of glycoproteins. Methods Cell Biol. 1989;32:111–139. doi: 10.1016/s0091-679x(08)61169-3. [DOI] [PubMed] [Google Scholar]
- 156.Tretter V., Altmann F., Marz L. Peptide-N4-(N-acetyl-beta-glucosaminyl)asparagine amidase F cannot release glycans with fucose attached alpha 1→3 to the asparagine-linked N-acetylglucosamine residue. Eur J Biochem. 1991;199:647–652. doi: 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- 157.Campbell M.P., Abrahams J.L., Rapp E., Struwe W.B., Costello C.E., Novotny M. The minimum information required for a glycomics experiment (MIRAGE) project: LC guidelines. Glycobiology. 2019;29:349–354. doi: 10.1093/glycob/cwz009. [DOI] [PubMed] [Google Scholar]
- 158.Wuhrer M., de Boer A.R., Deelder A.M. Structural glycomics using hydrophilic interaction chromatography (HILIC) with mass spectrometry. Mass Spectrom Rev. 2009;28:192–206. doi: 10.1002/mas.20195. [DOI] [PubMed] [Google Scholar]
- 159.Kolarich D., Windwarder M., Alagesan K., Altmann F. Isomer-specific analysis of released N-glycans by LC-ESI MS/MS with porous graphitized carbon. Methods Mol Biol. 2015;1321:427–435. doi: 10.1007/978-1-4939-2760-9_29. [DOI] [PubMed] [Google Scholar]
- 160.Hu Y., Shihab T., Zhou S., Wooding K., Mechref Y. LC-MS/MS of permethylated N-glycans derived from model and human blood serum glycoproteins. Electrophoresis. 2016;37:1498–1505. doi: 10.1002/elps.201500560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fernandez-Lima F., Kaplan D.A., Suetering J., Park M.A. Gas-phase separation using a trapped ion mobility spectrometer. Int J Ion Mobil Spectrom. 2011;14:93–98. doi: 10.1007/s12127-011-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhou S., Dong X., Veillon L., Huang Y., Mechref Y. LC-MS/MS analysis of permethylated N-glycans facilitating isomeric characterization. Anal Bioanal Chem. 2017;409:453–466. doi: 10.1007/s00216-016-9996-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wuhrer M., Koeleman C.A., Hokke C.H., Deelder A.M. Mass spectrometry of proton adducts of fucosylated N-glycans: fucose transfer between antennae gives rise to misleading fragments. Rapid Commun Mass Spectrom. 2006;20:1747–1754. doi: 10.1002/rcm.2509. [DOI] [PubMed] [Google Scholar]
- 164.Ruhaak L.R., Zauner G., Huhn C., Bruggink C., Deelder A.M., Wuhrer M. Glycan labeling strategies and their use in identification and quantification. Anal Bioanal Chem. 2010;397:3457–3481. doi: 10.1007/s00216-010-3532-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hahne H., Neubert P., Kuhn K., Etienne C., Bomgarden R., Rogers J.C. Carbonyl-reactive tandem mass tags for the proteome-wide quantification of N-linked glycans. Anal Chem. 2012;84:3716–3724. doi: 10.1021/ac300197c. [DOI] [PubMed] [Google Scholar]
- 166.Yang S., Wang M., Chen L., Yin B., Song G., Turko I.V. QUANTITY: an isobaric tag for quantitative glycomics. Sci Rep. 2015;5:17585. doi: 10.1038/srep17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Abrahams J.L., Campbell M.P., Packer N.H. Building a PGC-LC-MS N-glycan retention library and elution mapping resource. Glycoconj J. 2018;35:15–29. doi: 10.1007/s10719-017-9793-4. [DOI] [PubMed] [Google Scholar]
- 168.Zhao C., Xie B., Chan S.Y., Costello C.E., O'Connor P.B. Collisionally activated dissociation and electron capture dissociation provide complementary structural information for branched permethylated oligosaccharides. J Am Soc Mass Spectrom. 2008;19:138–150. doi: 10.1016/j.jasms.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 169.Lemoine J., Fournet B., Despeyroux D., Jennings K.R., Rosenberg R., de Hoffmann E. Collision-induced dissociation of alkali metal cationized and permethylated oligosaccharides: influence of the collision energy and of the collision gas for the assignment of linkage position. J Am Soc Mass Spectrom. 1993;4:197–203. doi: 10.1016/1044-0305(93)85081-8. [DOI] [PubMed] [Google Scholar]
- 170.Zhou S., Hu Y., Mechref Y. High-temperature LC-MS/MS of permethylated glycans derived from glycoproteins. Electrophoresis. 2016;37:1506–1513. doi: 10.1002/elps.201500568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Koutsioulis D., Landry D., Guthrie E.P. Novel endo-alpha-N-acetylgalactosaminidases with broader substrate specificity. Glycobiology. 2008;18:799–805. doi: 10.1093/glycob/cwn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Malaker S.A., Pedram K., Ferracane M.J., Bensing B.A., Krishnan V., Pett C. The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci U S A. 2019;116:7278–7287. doi: 10.1073/pnas.1813020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yang S., Onigman P., Wu W.W., Sjogren J., Nyhlen H., Shen R.F. Deciphering protein O-glycosylation: solid-phase chemoenzymatic cleavage and enrichment. Anal Chem. 2018;90:8261–8269. doi: 10.1021/acs.analchem.8b01834. [DOI] [PubMed] [Google Scholar]
- 174.Kudelka M.R., Antonopoulos A., Wang Y., Duong D.M., Song X., Seyfried N.T. Cellular O-Glycome Reporter/Amplification to explore O-glycans of living cells. Nat Methods. 2016;13:81–86. doi: 10.1038/nmeth.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kudelka M.R., Nairn A.V., Sardar M.Y., Sun X., Chaikof E.L., Ju T. Isotopic labeling with cellular O-glycome reporter/amplification (ICORA) for comparative O-glycomics of cultured cells. Glycobiology. 2018;28:214–222. doi: 10.1093/glycob/cwy005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang S., Hu Y., Sokoll L., Zhang H. Simultaneous quantification of N- and O-glycans using a solid-phase method. Nat Protoc. 2017;12:1229–1244. doi: 10.1038/nprot.2017.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Adamczyk B., Stockmann H., O'Flaherty R., Karlsson N.G., Rudd P.M. High-throughput analysis of the plasma N-glycome by UHPLC. Methods Mol Biol. 2017;1503:97–108. doi: 10.1007/978-1-4939-6493-2_8. [DOI] [PubMed] [Google Scholar]
- 178.Zhang T., Madunic K., Holst S., Zhang J., Jin C., Ten Dijke P. Development of a 96-well plate sample preparation method for integrated N- and O-glycomics using porous graphitized carbon liquid chromatography-mass spectrometry. Mol Omics. 2020;16:355–363. doi: 10.1039/c9mo00180h. [DOI] [PubMed] [Google Scholar]
- 179.de Haan N., Yang S., Cipollo J., Wuhrer M. Glycomics studies using sialic acid derivatization and mass spectrometry. Nat Rev Chem. 2020;4:229–242. doi: 10.1038/s41570-020-0174-3. [DOI] [PubMed] [Google Scholar]
- 180.Powers T.W., Holst S., Wuhrer M., Mehta A.S., Drake R.R. Two-dimensional N-glycan distribution mapping of hepatocellular carcinoma tissues by MALDI-imaging mass spectrometry. Biomolecules. 2015;5:2554–2572. doi: 10.3390/biom5042554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Heijs B., Holst S., Briaire-de Bruijn I.H., van Pelt G.W., de Ru A.H., van Veelen P.A. Multimodal mass spectrometry imaging of N-glycans and proteins from the same tissue section. Anal Chem. 2016;88:7745–7753. doi: 10.1021/acs.analchem.6b01739. [DOI] [PubMed] [Google Scholar]
- 182.Ju T., Lanneau G.S., Gautam T., Wang Y., Xia B., Stowell S.R. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 2008;68:1636–1646. doi: 10.1158/0008-5472.CAN-07-2345. [DOI] [PubMed] [Google Scholar]
- 183.Dall'Olio F., Trinchera M. Epigenetic bases of aberrant glycosylation in cancer. Int J Mol Sci. 2017;18:998. doi: 10.3390/ijms18050998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shental-Bechor D., Levy Y. Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc Natl Acad Sci U S A. 2008;105:8256–8261. doi: 10.1073/pnas.0801340105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Apweiler R., Hermjakob H., Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 186.Steentoft C., Vakhrushev S.Y., Vester-Christensen M.B., Schjoldager K.T., Kong Y., Bennett E.P. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- 187.Campos D., Freitas D., Gomes J., Magalhaes A., Steentoft C., Gomes C. Probing the O-glycoproteome of gastric cancer cell lines for biomarker discovery. Mol Cell Proteomics. 2015;14:1616–1629. doi: 10.1074/mcp.M114.046862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wuhrer M., Koeleman C.A., Hokke C.H., Deelder A.M. Protein glycosylation analyzed by normal-phase nano-liquid chromatography–mass spectrometry of glycopeptides. Anal Chem. 2005;77:886–894. doi: 10.1021/ac048619x. [DOI] [PubMed] [Google Scholar]
- 189.Senko M.W., Remes P.M., Canterbury J.D., Mathur R., Song Q., Eliuk S.M. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal Chem. 2013;85:11710–11714. doi: 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- 190.Thaysen-Andersen M., Packer N.H. Advances in LC-MS/MS-based glycoproteomics: getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim Biophys Acta. 2014;1844:1437–1452. doi: 10.1016/j.bbapap.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 191.Jensen P.H., Mysling S., Hojrup P., Jensen O.N. Glycopeptide enrichment for MALDI-TOF mass spectrometry analysis by hydrophilic interaction liquid chromatography solid phase extraction (HILIC SPE) Methods Mol Biol. 2013;951:131–144. doi: 10.1007/978-1-62703-146-2_10. [DOI] [PubMed] [Google Scholar]
- 192.Palmisano G., Lendal S.E., Engholm-Keller K., Leth-Larsen R., Parker B.L., Larsen M.R. Selective enrichment of sialic acid-containing glycopeptides using titanium dioxide chromatography with analysis by HILIC and mass spectrometry. Nat Protoc. 2010;5:1974–1982. doi: 10.1038/nprot.2010.167. [DOI] [PubMed] [Google Scholar]
- 193.Goettig P. Effects of glycosylation on the enzymatic activity and mechanisms of proteases. Int J Mol Sci. 2016;17:1969. doi: 10.3390/ijms17121969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pandeswari P.B., Sabareesh V. Middle-down approach: a choice to sequence and characterize proteins/proteomes by mass spectrometry. RSC Adv. 2019;9:313–344. doi: 10.1039/c8ra07200k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Toby T.K., Fornelli L., Kelleher N.L. Progress in top-down proteomics and the analysis of proteoforms. Annu Rev Anal Chem (Palo Alto Calif) 2016;9:499–519. doi: 10.1146/annurev-anchem-071015-041550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ning F., Wu X., Wang W. Exploiting the potential of 2DE in proteomics analyses. Expert Rev Proteomics. 2016;13:901–903. doi: 10.1080/14789450.2016.1230498. [DOI] [PubMed] [Google Scholar]
- 197.Ankney J.A., Muneer A., Chen X. Relative and absolute quantitation in mass spectrometry-based proteomics. Annu Rev Anal Chem (Palo Alto Calif) 2018;11:49–77. doi: 10.1146/annurev-anchem-061516-045357. [DOI] [PubMed] [Google Scholar]
- 198.Kolli V., Dodds E.D. Energy-resolved collision-induced dissociation pathways of model N-linked glycopeptides: implications for capturing glycan connectivity and peptide sequence in a single experiment. Analyst. 2014;139:2144–2153. doi: 10.1039/c3an02342g. [DOI] [PubMed] [Google Scholar]
- 199.Mormann M., Paulsen H., Peter-Katalinić J. Electron capture dissociation of O-glycosylated peptides: radical site-induced fragmentation of glycosidic bonds. Eur J Mass Spectrom (Chichester) 2005;11:497–511. doi: 10.1255/ejms.738. [DOI] [PubMed] [Google Scholar]
- 200.Kim M.S., Pandey A. Electron transfer dissociation mass spectrometry in proteomics. Proteomics. 2012;12:530–542. doi: 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Scott N.E., Parker B.L., Connolly A.M., Paulech J., Edwards A.V., Crossett B. Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M000031-MCP201. M000031-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Frese C.K., Altelaar A.F., van den Toorn H., Nolting D., Griep-Raming J., Heck A.J. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal Chem. 2012;84:9668–9673. doi: 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- 203.Yu Q., Wang B., Chen Z., Urabe G., Glover M.S., Shi X. Electron-transfer/higher-energy collision dissociation (EThcD)-enabled intact glycopeptide/glycoproteome characterization. J Am Soc Mass Spectrom. 2017;28:1751–1764. doi: 10.1007/s13361-017-1701-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Azevedo R., Silva A.M.N., Reis C.A., Santos L.L., Ferreira J.A. In silico approaches for unveiling novel glycobiomarkers in cancer. J Proteomics. 2018;171:95–106. doi: 10.1016/j.jprot.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 205.Mohl J.E., Gerken T.A., Leung M.Y. ISOGlyP: de novo prediction of isoform specific mucin-type O-glycosylation. Glycobiology. 2021;31:168–172. doi: 10.1093/glycob/cwaa067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Lin Y.H., Zhu J., Meijer S., Franc V., Heck A.J.R. Glycoproteogenomics: a frequent gene polymorphism affects the glycosylation pattern of the human serum fetuin/alpha-2-HS-glycoprotein. Mol Cell Proteomics. 2019;18:1479–1490. doi: 10.1074/mcp.RA119.001411. [DOI] [PMC free article] [PubMed] [Google Scholar]