

Abstract
Objective:
Gasdermin D (GSDMD) is the key executioner of the inflammatory cell death mechanism pyroptosis. Recent reports have also implicated GSDMD in other mechanisms of cell death, including apoptosis, necroptosis, and NETosis. Given the role of dysregulated cell death in autoimmune syndromes such as systemic lupus erythematosus (SLE), we hypothesized that GSDMD plays pathogenic roles by promoting inflammatory cell death leading to increased generation of nuclear autoantigens and autoantibodies.
Methods:
The imiquimod-induced model of SLE was tested in Gsdmd−/− (n=30) and wild-type (WT; n=34) C57BL/6 mice. At euthanasia, mice were examined for serum autoantibodies, immune complex deposition, organ inflammation, immune dysregulation and type I Interferon responses. A pristane-induced lung injury model in Gsdmd−/− (n=7) and WT (n=10) mice was used to confirm pulmonary phenotype. Regulation of various mechanisms of cell death was investigated in the mice.
Results:
Unexpectedly, Gsdmd−/− mice developed enhanced mortality, more severe renal and pulmonary inflammation, and exacerbated autoantibody production in response to imiquimod. Pulmonary involvement was also more severe in the pristane model in the absence of GSDMD. Lack of GSDMD was associated with increased circulating nuclear autoantigens (p<0.01), anti-dsDNA autoantibodies (p<0.01), tissue immune complex deposition (p<0.05), expansion of myeloid cell subsets (p<0.05), and enhanced B cell activation and plasma cell differentiation (p=0.001). In the absence of GSDMD, enhanced autoantigen generation was associated with increased local induction of cell death in vivo.
Conclusion:
GSDMD negatively regulates autoantigen generation and immune dysregulation in response to tissue injury and may play previously unappreciated protective roles in systemic autoimmunity.
Introduction
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune syndrome with pleiotropic clinical manifestations and profound dysregulation of innate and adaptive immune responses [1–5]. Standard of care treatment for SLE involves fairly nonspecific immunosuppression and there is a need for more targeted approaches [6, 7]. SLE is characterized by the formation of immune responses directed at nuclear autoantigens such as nucleic acids and histones. Dysregulation in mechanisms of cell death and impaired clearance of dead cells have been implicated in the pathogenesis of lupus. Indeed, enhanced apoptosis and impaired clearance of apoptotic material are operational in murine and human lupus [8]. Furthermore, enhanced formation of neutrophil extracellular traps (NETs) and impaired clearance of these structures have recently been described in the pathogenesis of SLE and its associated organ and vascular damage [9–12]. Dysregulation in various mechanisms of cell death has been proposed to play a role in the generation of immune complexes and activation of the type I Interferon (IFN) pathway characteristic of SLE, with downstream pathogenic effects [13–16].
The molecule gasdermin D (GSDMD) has been described as the key executioner of pyroptosis, a proinflammatory form of programmed cell death that occurs most frequently following infections with intracellular pathogens and is linked to inflammasome activation [17–20]. After activation through cleavage by caspases, the GSDMD N-terminal domain can oligomerize to form membrane pores and promote the release of intracellular contents including the proinflammatory cytokines IL-1β and IL-18, cytokines that generate an inflammasome-associated inflammatory response [21–23]. In addition to pyroptosis, GSDMD was recently implicated in the formation of NETs in the context of microbial inflammation [24–26] and in apoptosis, through the formation of mitochondrial pores and subsequent release of cytochrome c from the mitochondria [27, 28]. Therefore, modulation of GSDMD may have important roles in dysregulated mechanisms of cell death in SLE and other autoimmune diseases.
To test this hypothesis, we used a Toll-like receptor-7 (TLR-7)-induced model of SLE (the imiquimod-induced SLE model), to assess the role of GSDMD in clinical phenotype, immune dysregulation, and organ damage. This model was chosen because it replicates many features of human SLE in the context of many mouse strains and without the complex murine genetic confounders ,and because it has a relatively quick onset [29]. We hypothesized that deletion of Gsdmd would lead to a protective effect in clinical phenotype, through inhibition of inflammatory mechanisms of cell death that would decrease autoantigen generation, immune complex formation, inflammatory responses and organ damage. Instead, we discovered an unexpected protective role of GSDMD in lupus-like autoimmunity, through modulation of cell death and tissue damage.
Methods
For details on immunological and clinical assessment of mice, please see supplemental files.
Mice:
Wild-type C57BL/6 (WT) mice were purchased from The Jackson Laboratory. Breeding pairs of Gsdmd−/− mice on B6 background were a generous gift from Dr. Russel Vance at UC-Berkeley. Mice were bred under specific pathogen-free conditions and all treatments and experiments were done in accordance with NIH guidelines under the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) protocol numbers #A016–05-26 and #A019–05-03.
Imiquimod-induced lupus model:
Mice (8–10 week-old males and females) were treated with 5% imiquimod cream (Fougera) epicutaneously, 3 times a week for 5 weeks as previously described, to induce a lupus phenotype [30–32]. All in-vivo and in-vitro experiments were matched for age and sex between groups.
Pristane-induced acute inflammatory lung model:
Mice (8–10 week old males and females) were given a single intraperitoneal injection of 0.5 mL pristane (Sigma) to induce acute lung inflammation/hemorrhage as previously described [33, 34]. Mice were euthanized one week later to quantify lung pathology.
Statistical analysis:
Data were plotted using GraphPad Prism. Appropriate statistical tests were performed as described in figure legends. For endothelium vasorelaxation assays, two-way ANOVA with Tukey’s correction for multiple comparisons was used. For survival curve analysis, Kaplan-Meier survival analysis was used. For all other analysis, Mann-Whitney tests were performed to determine significance. Statistical significance levels are indicated as *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Results
GSDMD modulates clinical manifestations of murine lupus.
Topical application of the TLR-7 agonist imiquimod promotes a lupus phenotype characterized by systemic inflammation, splenomegaly, myeloid expansion, innate and adaptive immune dysregulation, enhanced type I IFN responses, renal immune complex deposition and inflammation, and vasculopathy [30]. For every clinical and immunologic measurement assessed we observed that, in the absence of imiquimod exposure, Gsdmd−/− mice and WT B6 mice did not show any significant differences. However, following exposure to the TLR-7 agonist, Gsdmd−/− treated mice displayed significantly larger spleens, lower body weights, and higher spleen/body weight ratios compared to imiquimod-treated WT mice (Figure 1A–C). Furthermore, compared to imiquimod-treated WT mice, imiquimod-treated Gsdmd−/− mice experienced significantly elevated death rates (Figure 1D). As previously reported, imiquimod treatment resulted in synthesis of anti-dsDNA autoantibodies, a hallmark of SLE [30–32]. Compared to WT mice, Gsdmd−/− mice exposed to imiquimod displayed significantly higher levels of anti-dsDNA autoantibodies, while total IgG was no different from WT mice (Figure 1E–F). In addition, imiquimod-treated Gsdmd−/− mice developed a significantly enhanced systemic proinflammatory burden with peripheral leukocytosis and anemia, when compared to treated-WT mice (Figure S1A–C) in the absence of significant differences in circulating proinflammatory cytokines (Figure S2). Overall, the absence of GSDMD was associated with enhanced mortality, systemic inflammation and autoantibody production in the context of TLR-7-dependent lupus.
Figure 1. GSDMD modulates clinical features of murine lupus.
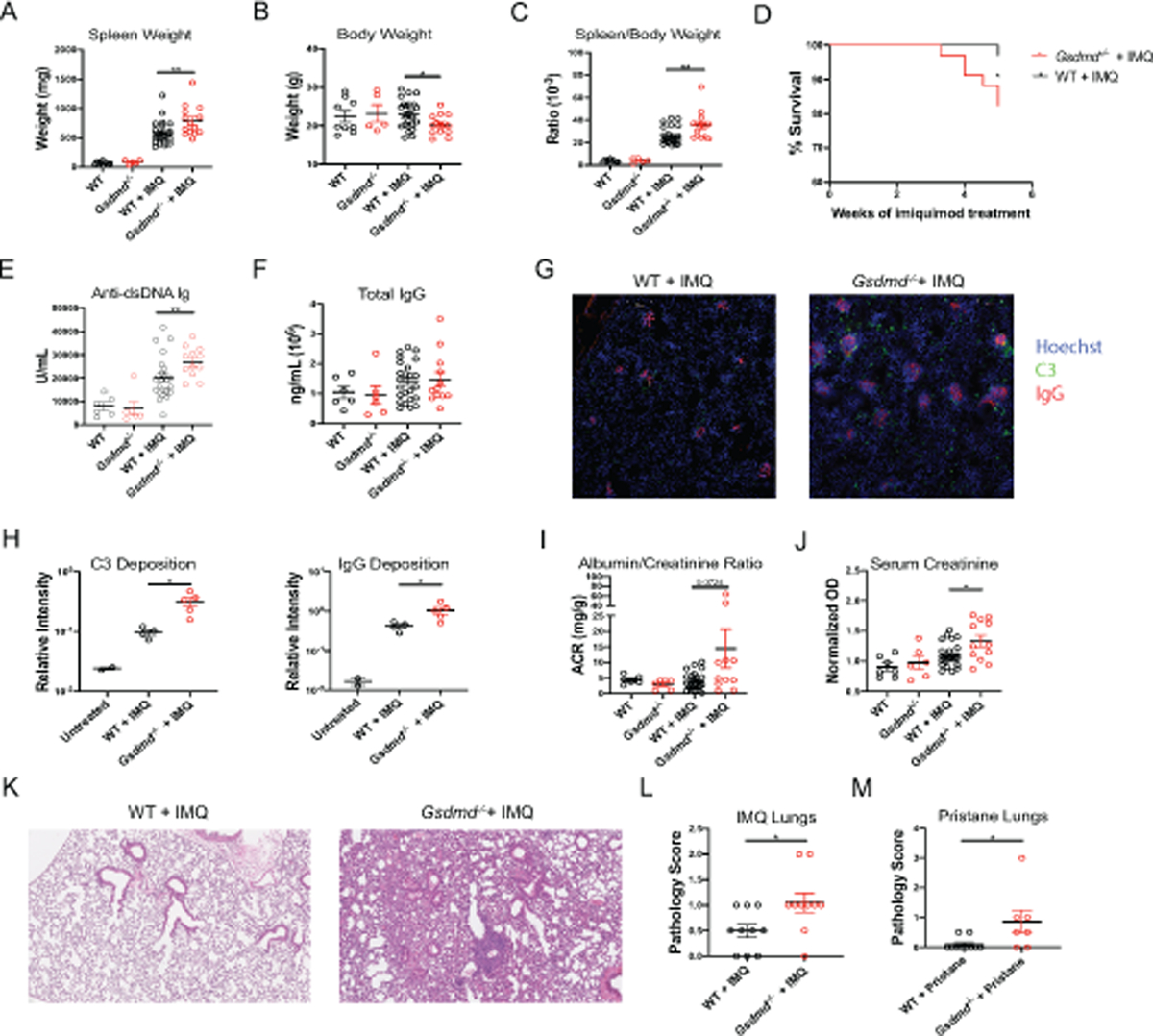
(A–C) Spleen weight, body weight, and spleen/body weight ratios of untreated and imiquimod-treated wild-type (WT) and Gsdmd−/− mice. (D) Survival curve of imiquimod-treated mice. (E, F) Serum anti-dsDNA and total IgG concentrations. (G–H) 10X images of frozen kidney sections stained for C3, IgG, and Hoescht and quantified using Fiji software. Images are representative of 4–5 mice. (I) Urine Albumin/Creatinine Ratio and (J) serum creatinine concentrations. (K–L) Formalin-fixed paraffin-embedded (FFPE) lung sections from imiquimod-treated mice were analyzed for bronchial and alveolar inflammation. (M) FFPE lung sections from pristane-treated mice were analyzed for overall severity. (A–C), n=9 WT, 5 Gsdmd−/−, 24 WT + IMQ, 12 Gsdmd−/− + IMQ. (D), n=34 WT + IMQ, 30 Gsdmd−/− + IMQ. (E, F, J) n=6 WT, 5 Gsdmd−/−, 24 WT + IMQ, 12 Gsdmd−/− + IMQ. (K-L) n=10 WT + IMQ, 10 Gsdmd−/− + IMQ. (M) n=10 WT + IMQ, 7 Gsdmd−/− + IMQ. Dots indicate individual mice. Results represent mean ± SEM. Statistics were calculated by non-parametric Mann-Whitney test for all except (D), where Kaplan-Meier survival analyses was performed; *p < 0.05, **p < 0.01.
The kidneys are often affected in SLE and lupus glomerulonephritis is one of the most prevalent causes of morbidity and mortality in this condition. As previously described, imiquimod induced complement C3 and IgG deposition in the glomeruli of WT mice. Consistent with an exacerbated autoimmune phenotype, Gsdmd−/− mice developed significantly more prominent glomerular immune complex deposition compared to WT mice (Figure 1G–H). While this specific mouse model of lupus is not characterized by nephrotic-range proteinuria or end stage renal disease, Gsdmd−/− mice had higher proteinuria and serum creatinine levels compared to imiquimod-treated WT mice (Figure 1I–J), indicating more impaired renal function.
Another common manifestation of SLE is lung and pleural inflammation. In more severe cases, diffuse alveolar hemorrhage (DAH) can be a life-threatening complication in SLE [35]. Previously unrecognized in the TLR-7 induced lupus model, we found by hematoxylin and eosin staining of lung formalin-fixed paraffin-embedded (FFPE) sections that topical skin imiquimod exposure induced lung inflammation, hemorrhage, and fibrosis. Furthermore, Gsdmd−/− lungs exhibited enhancement of pulmonary immune cell infiltration in response to imiquimod when compared to WT (Figure 1K–L).
Given an unexpected lung phenotype and the knockout mice being available on B6 background, we used an induced model of DAH that ensues rapidly and peaks approximately 2 weeks after administration of the hydrocarbon pristane to the peritoneum of B6 mice [33, 34]. Gsdmd−/− mice exposed to intraperitoneal pristane developed significantly accelerated and more severe lung hemorrhage, inflammation, and fibrosis when compared to WT mice exposed to pristane (Figure 1M).
Endothelial dysfunction and vasculopathy are hallmarks of murine and human lupus. Gsdmd−/− mice treated with imiquimod showed similar perturbations in endothelium-dependent vasorelaxation when compared to WT imiquimod-treated mice, suggesting that lack of GSDMD did not modulate lupus vasculopathy (Figure S3).
Overall, absence of GSDMD worsened clinical and immunologic features of murine SLE including systemic, renal and lung inflammation, and enhanced autoantibody production.
GSDMD modulates expansion and activation of distinct subsets of myeloid cells and plasmacytoid dendritic cells.
We previously reported a significant expansion of the myeloid cell compartment in blood, spleen, and bone marrow of imiquimod-treated WT mice [31, 32]. Supporting these previous findings, WT mice exposed to imiquimod displayed splenic expansion of proinflammatory monocytes (CD11b+ Ly-6C+ Ly-6G−), neutrophils (CD11b+ Ly-6G+), conventional dendritic cells (DCs; CD11b+ CD11c+), and plasmacytoid DCs (pDCs; CD11c− PDCA-1+ B220+) (Figure 2). Compared to the WT mice, imiquimod-treated Gsdmd−/−mice had significantly higher numbers of splenic neutrophils and pro-inflammatory monocytes, but comparable numbers of conventional myeloid DCs (Figure 2A–E). Imiquimod-treated Gsdmd−/− mice splenic DCs displayed lower MHC class II expression but higher CD80 expression than imiquimod-treated WT (Figure 2F–G). Overall, these results indicated that lack of GSDMD led to enhanced myeloid cell expansion upon induction of lupus-like disease.
Figure 2. Gsdmd−/− mice display myeloid cell expansion and activation.
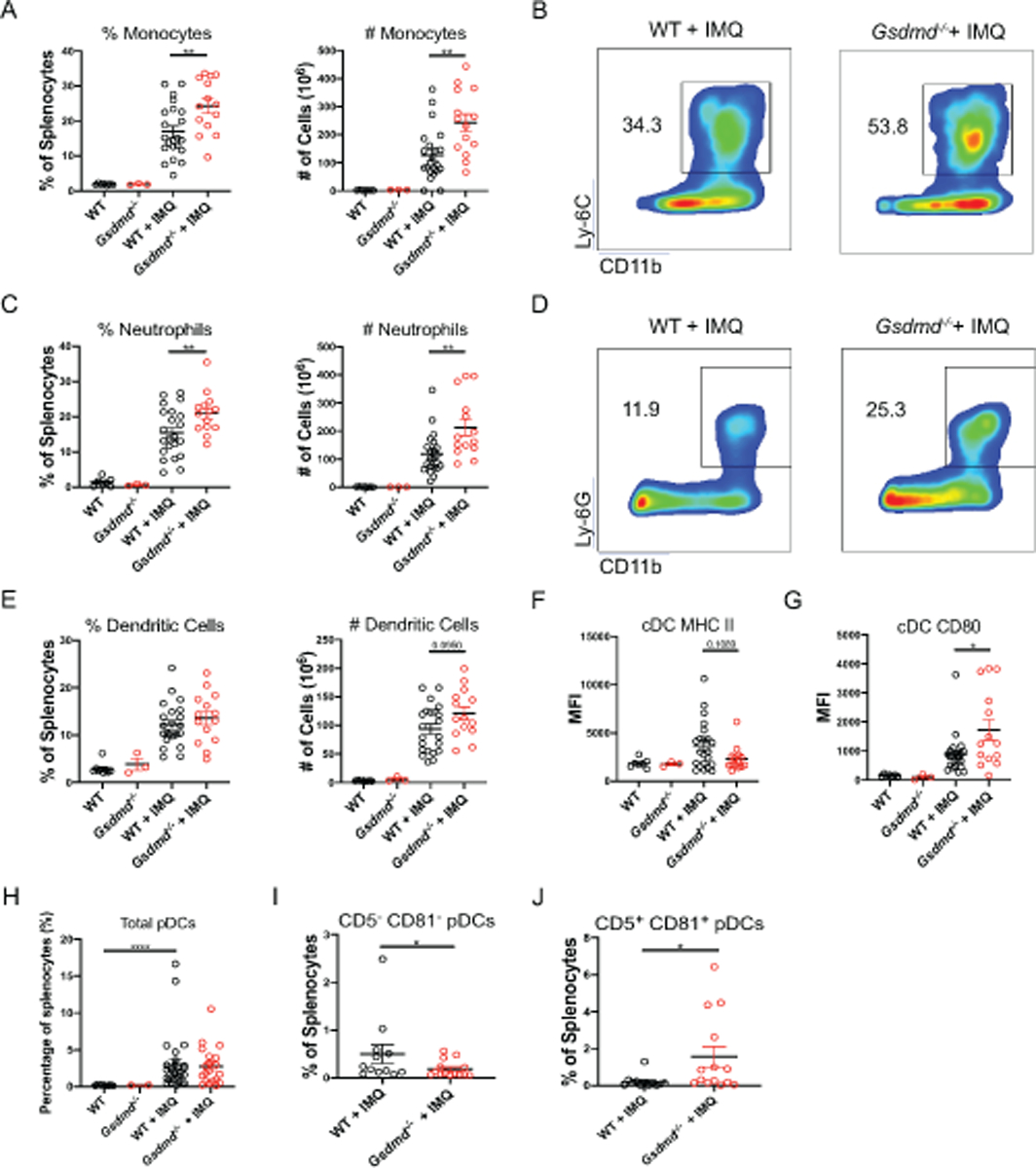
Splenocytes from untreated and imiquimod-treated WT and Gsdmd−/− mice were analyzed by flow cytometry to quantify (A, B) proinflammatory monocytes, (C, D) neutrophils, (E) conventional dendritic cells (cDC), cDC MHC II, and CD80 expression, (H–J) ‘classical’ and ‘nonclassical’ plasmacytoid DCs. (A–G) n=7 WT, 3 Gsdmd−/−, 24 WT + IMQ, 12 Gsdmd−/− + IMQ), (H–J) n=12 WT + IMQ, 14 Gsdmd−/− + IMQ. Dots indicate individual mice. Results represent mean ± SEM. Statistics were calculated by non-parametric Mann-Whitney test; *p < 0.05, **p < 0.01.
pDCs have been proposed to play critical roles in lupus through synthesis of type I IFNs [30]. While we confirmed significant expansion of splenic pDCs in imiquimod-treated mice, there were no differences between Gsdmd−/− and WT animals (Figure 2H). An additional biological homeostatic role for GSDMD was recently described and related to limiting cytosolic DNA surveillance. This mechanism involved the activation of GSDMD by the AIM2 inflammasome that suppressed cGAS-driven type I IFN responses to cytosolic DNA [36]. We considered that a mechanism by which Gsdmd−/− mice displayed worsening clinical phenotype could be related to a decrease in their ability to repress type I IFN responses. However, we observed lower splenic mRNA levels of interferon-stimulated genes (ISGs) in imiquimod-treated Gsdmd−/− mice compared to their WT counterparts (Figure S4A). Furthermore, kidneys and lungs from imiquimod-treated Gsdmd−/− expressed comparable levels of ISGs to imiquimod-treated WT mice (Figure S4B–C). These results suggested that the exacerbation of autoimmune responses in Gsdmd−/− following exposure to imiquimod was not due to upregulation of type I IFN responses.
A specific pDC subset that is phenotypically and functionally distinct from the classic type-I IFN producing pDC was recently described and associated with lymphocyte activation and differentiation rather that type I IFN production [37]. We found that Gsdmd−/− imiquimod-treated mice had significantly lower numbers of the ‘conventional’ type I-IFN-producing pDCs (CD5− CD81−; Figure 2I) but significant expansion of the “lymphocyte-activating” pDCs (CD5+ CD81+; Figure 2J).
Recent reports described that, in mouse neutrophils, caspase-11 or proteases such as neutrophil elastase can cleave GSDMD and subsequently induce the release of NETs [25, 26]. However, in the TLR-7 induced model of lupus, Gsdmd−/− bone marrow neutrophils were still capable of forming NETs, both spontaneously and in response to stimulation with the calcium ionophore A23187 (Figure S5). These observations indicated that lack of GSDMD in a murine model of lupus did not significantly impact the formation of NETs (Fig S5). Overall, GSDMD modulated the myeloid compartment and the activation and expansion of specific proinflammatory myeloid cell and pDC subsets, without significantly altering type I IFN responses or NET formation.
GDSMD modulates B cell differentiation and activation in murine lupus.
Given that Gsdmd−/− mice synthesize higher levels of specific autoantibodies but had no differences in total IgG production (Figure 1E–F), we further analyzed their B cell phenotype. Consistent with previous reports [30–32], imiquimod-treated mice developed a marked reduction of splenic naïve (IgD+ IgM+) and marginal zone (CD21+ CD23−) B cells. Gsdmd−/− treated mice had a more significant reduction of total, naïve, and MZ B cells compared to their WT counterparts (Figure 3A–D). While there was a reduction in immature B cell subsets, Gsdmd−/− CD19+ B cells were more activated (based on expression of CD40, CD44, CD80, CD86, and CD62L; Figure 3E–J) than WT mice, following imiquimod treatment. Furthermore, Gsdmd−/− mice developed significant expansion of splenic plasma cells after treatment with imiquimod, as compared to WT mice (Figure 3K–L). In contrast, various T cell subsets and their activation markers displayed no significant differences between imiquimod-treated Gsdmd−/− and WT mice (Figure S6). Specifically, there were no differences in distribution of CD4+ and CD8+ subsets, including naïve (CD44− CD62L+), central memory (CD44+ CD62L+), effector memory (CD44+ CD62L−) CD4+ T cells and Foxp3+ CD4+ T regulatory cells between Gsdmd−/− and WT mice (Figure S6). These results suggested that the expansion of activated B cells in the absence of GSDMD may have occurred independently of changes in T cell subsets.
Figure 3. Gsdmd−/− mice display enhanced B cell differentiation and activation.
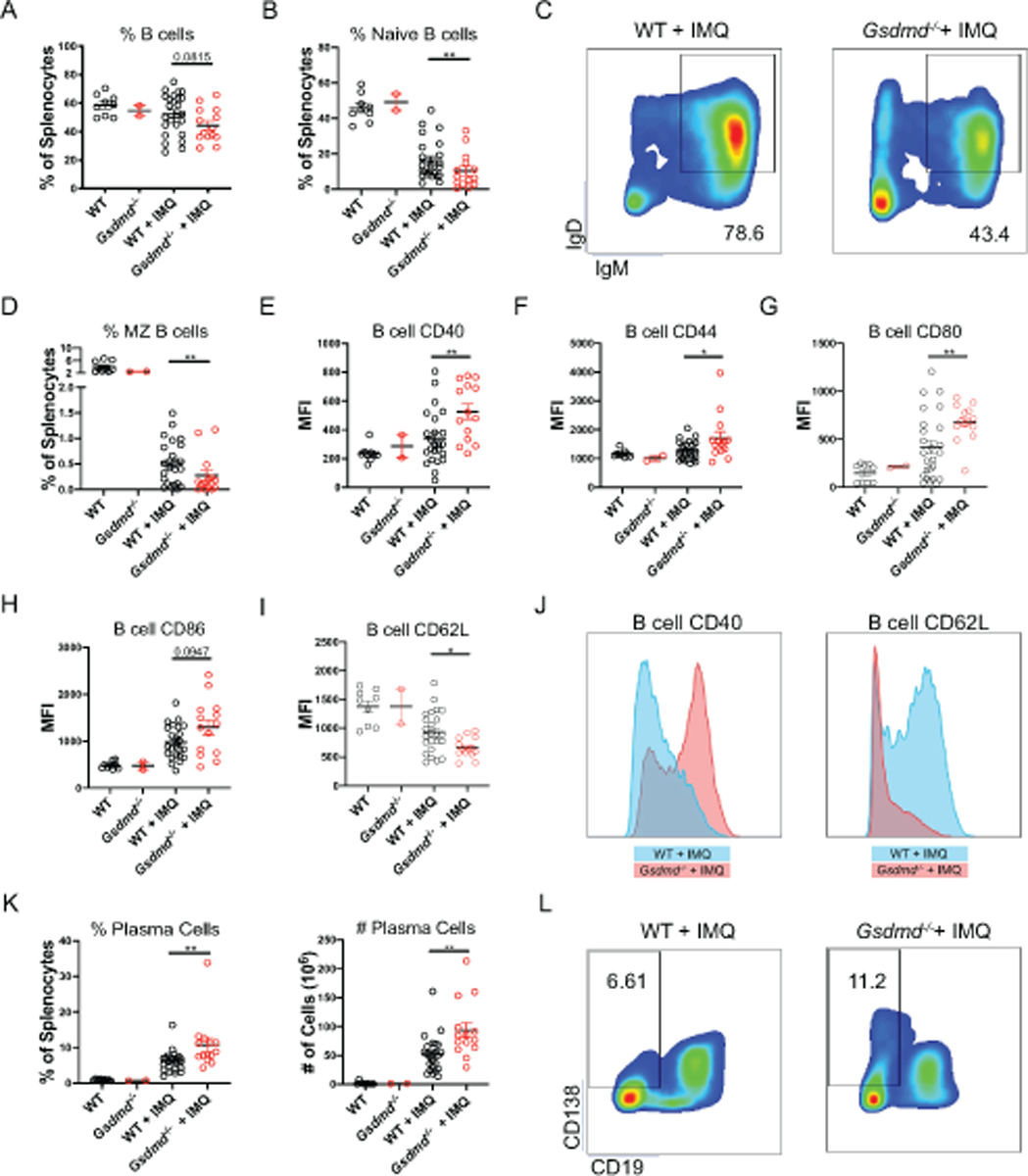
Spleens from untreated and imiquimod-treated WT and Gsdmd−/− mice were harvested and splenocyte subpopulations quantified by flow cytometry. (A–D) Dot plots of total splenic B cells, naïve B cells, marginal zone (MZ) B cells. (E–I) Dot plots of activation markers CD40, CD44, CD80, CD86, and CD62L expression on CD19+ B cells. (J–K) Representative histograms of CD19+ B cell expression of CD40 and CD62L. (K–L) Dot plots of splenic plasma cells. For all experiments: n=9 WT, 2 Gsdmd−/−, 24 WT + IMQ, 14 Gsdmd−/− + IMQ. Dots indicate individual mice. Results represent mean ± SEM. Statistics were calculated by non-parametric Mann-Whitney test; *p < 0.05, **p < 0.01.
Given the differences in B cell phenotype observed in treated Gsdmd−/− mice compared to WT, we assessed if GSDMD was detected in lymphocytes and found that this protein is indeed expressed in WT B6 splenic B and T lymphocytes (Figure S7A–B). Furthermore, GSDMD protein levels increased following B cell activation with LPS, anti-IgM, or imiquimod stimulation in vitro, but not during T cell activation using anti-CD3/CD28 beads (Figure S7A–B). We then isolated splenic B cells from Gsdmd−/− and WT mice and quantified activation markers and proliferation rates in response to a variety of in vitro stimuli. Despite the expression of GSDMD on B cells and its upregulation with activation, we detected no significant differences on in vitro induction of activation and proliferation between WT and Gsdmd−/− B cells (Figure S8A–B). Overall, these results suggested that dysregulation of B cell responses in the absence of GSDMD were not the result of intrinsic B cell differences driven by GSDMD expression in these cells and instead may be driven by other mechanisms such as autoantigen generation or presentation.
GSDMD modulates specific proinflammatory cell death mechanisms and release of nuclear autoantigens in murine lupus.
To further characterize how the lack of GSDMD could promote enhanced immune dysregulation, immune complex deposition and organ damage in lupus, we quantified circulating levels of autoantigens. We observed significantly higher levels of circulating NET remnants and cell-free DNA (cfDNA) in imiquimod-treated Gsdmd−/− mice, when compared to WT-treated mice (Figure 4A–B). These results suggested that generation of higher levels of nuclear autoantigens may be implicated in autoantibody generation, immune complex deposition and tissue damage. As in vitro studies showed that neutrophils lacking GDSMD did not show significant differences in NET formation but displayed higher levels of circulating NET remnants and cfDNA were detected in circulation, we assessed whether GSDMD could modulate the clearance of death cells by macrophages and the clearance of NETs by serum nucleases. We quantified and compared the efficiency of WT and Gsdmd−/− peritoneal macrophages in phagocytosing apoptotic cells and found no significant differences (Figure 4C). Similarly, we observed no differences in the ability of nuclease-containing serum from imiquimod-treated WT and Gsdmd−/− mice to clear NETs. Again, WT and Gsdmd−/− NETs were no different in the extent of degradation (Figure S5). These results suggested that the increases in levels of circulating autoantigens in Gsdmd−/− mice were most likely driven by peripheral myeloid cell expansion and through differential modulation of certain types of cell death, rather than impaired clearance of dead cells or their products.
Figure 4. Gsdmd−/− mice display increased circulating autoantigens and enhanced release of HMGB1.
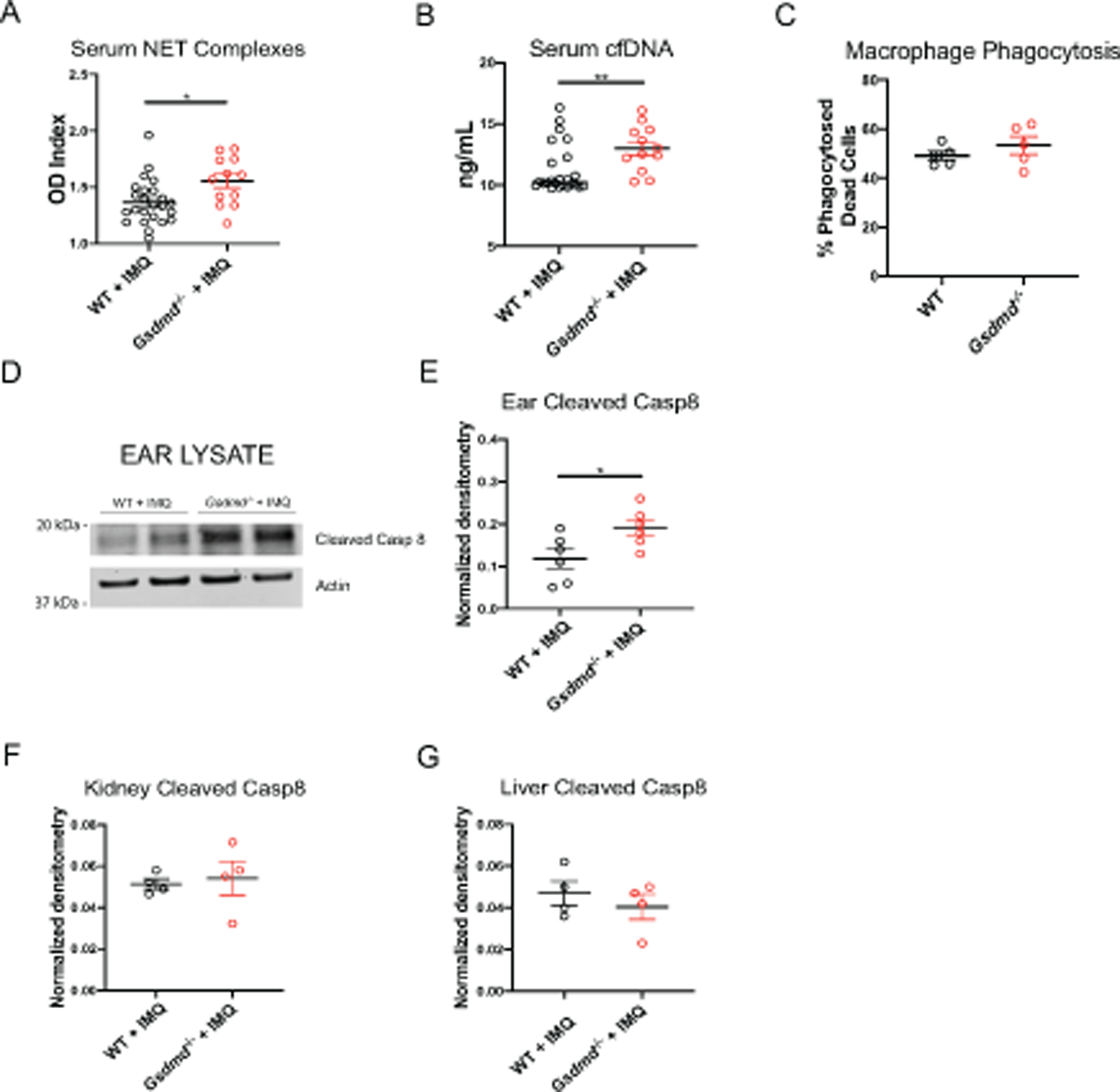
(A–B) Serum NET complexes and cell-free DNA (cfDNA) (n=24, WT + IMQ; n=12, Gsdmd−/− + IMQ). (C) Quantification of macrophage phagocytosis of apoptotic cells. (D) Immunoblot of cleaved caspase 8 in ear lysates of WT and Gsdmd−/− mice treated with imiquimod for 1 week. (E–G) Quantification of cleaved caspase 8 normalized to actin in ear, kidney, and liver lysates of WT and Gsdmd−/− mice treated with imiquimod for 1 week.
As GSDMD is essential for pyroptosis, this type of cell death could not represent a mechanism leading to enhanced generation of released nucleic acids in the KO mice [18–21]. Similarly, we did not find a difference in NET formation, as shown above (Figure S5). Given that in this lupus model imiquimod is applied to the skin, we hypothesized that the higher levels of circulating autoantigens in Gsdmd−/− treated mice might occur as a result of enhanced cell death in tissues. To investigate how GSDMD may regulate potential autoantigen release early on in the model, we treated the skin in the ears of mice with imiquimod for 1 week to assess cleavage of caspase 8, a molecule that is activated in multiple mechanisms of cell death [38]. Indeed, we observed significantly more cleaved caspase 8 in Gsdmd−/− skin in vivo compared to WT (Fig 4D–E). Furthermore, we found equal expression of active caspase 8 in organs not directly treated with imiquimod for 1 week, such as the kidneys and livers (Fig 4F–G). Overall, our data suggests that GSDMD plays a role in the initiation of murine lupus by restraining cell death at areas of insult as a result of direct TLR-7 agonist exposure.
To assess if overall enhancement of cell death could also be observed at later time-points in the affected organs such as lungs and kidney, we performed in situ TUNEL staining on FFPE sections from 5-week imiquimod-treated animals. Lung sections displayed higher levels of TUNEL positivity than kidney sections (Fig. S9A–B), suggesting enhanced cell death was triggered in pulmonary tissue in this model. There was increase in cell death in the lung tissues, but not in the kidneys, of Gsdmd−/−+ IMQ mice compared to WT + IMQ, although this did not reach statistical significance (Fig. S9A–B). While in situ TUNEL staining cannot reliably discriminate apoptotic from necrotic types of cell death [39], these results further support that lack of GSDMD exacerbates cell death in certain organs that are relevant to lupus pathology.
Given recent evidence describing a role for GSDMD in mitochondrial pore-forming ability in the context of apoptosis [27, 28], we further evaluated how the lack of this molecule may modify various types of cell death. Consistent with these reports, we found that GSDMD modulated cytochrome c release from the mitochondria on splenic lymphocytes in response to the proapoptotic stimulus staurosporine (STS; Fig 5A). Furthermore, lymphocytes lacking GSDMD appeared to be less efficient at undergoing apoptosis, as shown by in vitro TUNEL staining and visualization of DNA fragmentation (Fig 5B). Given the lower rate of apoptosis in Gsdmd−/− lymphocytes, we hypothesized that lack of GSDMD could instead modulate a form of necrotic cell death.
Figure 5. GSDMD restricts necrotic cell deaths in splenic lymphocytes.
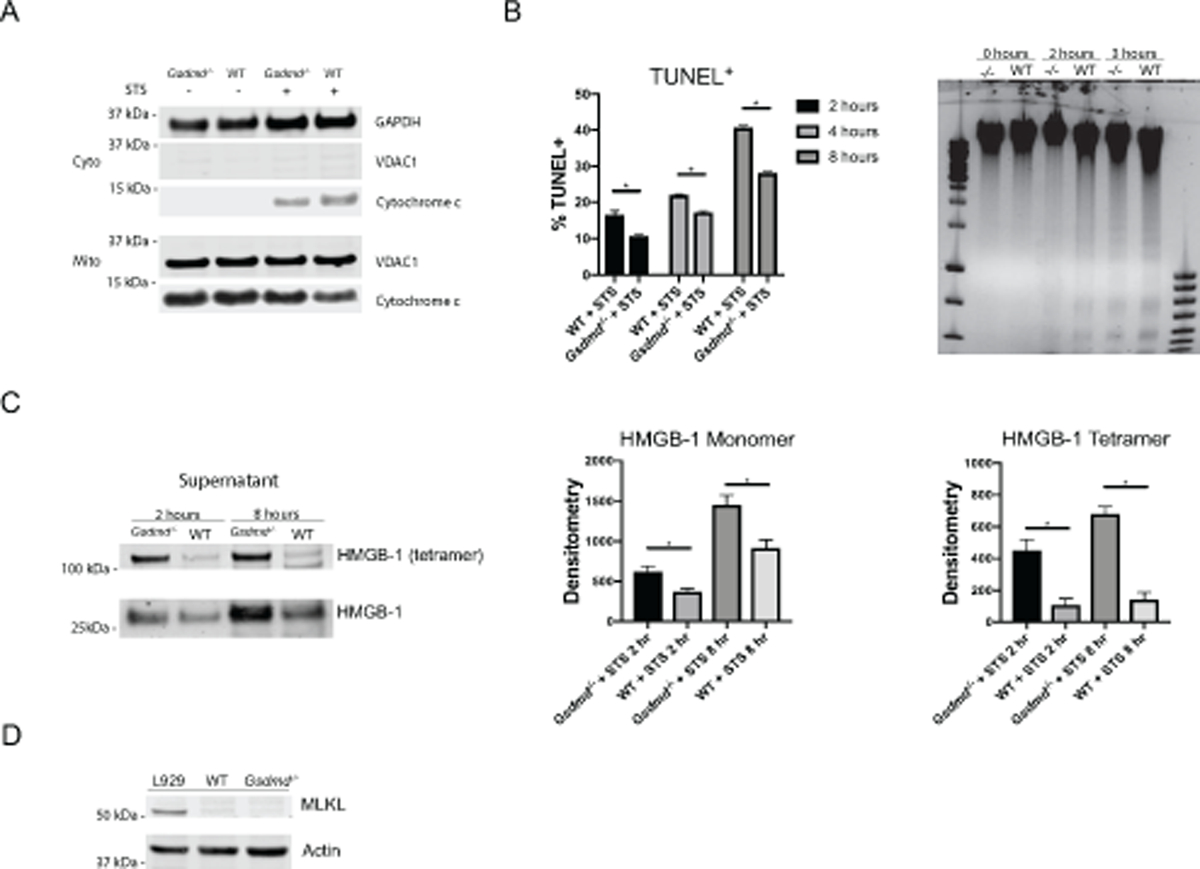
(A) Immunoblot of cytochrome c in mitochondrial and cytosolic fractions from lysates of splenic lymphocytes. (B) Scatter plot depicting TUNEL+ fractions in lymphocytes and agarose gel visualization of DNA fragmentation. (C) Immunoblot and quantification HMGB-1 release into supernatant fraction. (D) Immunoblot of MLKL in lymphocytes. L929 cells were used as positive controls. Images are representative of 3 independent experiments. *p<0.05.
High mobility group box 1 (HMGB1) is a chromatin protein involved in gene transcription, chromatin remodeling and organization of DNA [40]. HMGB1 can be released by cells and can bind to receptor for advanced glycation end-products (RAGE) and various TLRs promoting proinflammatory responses [41]. Key mechanisms by which HMGB1 is released from cells is through necrotic cell death or NET formation. Following stimulation with STS to induce apoptosis, Gsdmd−/− lymphocytes released significantly more extracellular monomeric and tetrameric HMGB-1 (Fig 5C). To investigate if this increased release of HMGB-1 could be due to programmed necrotic cell death such as necroptosis, we assessed expression of the total protein mixed lineage kinase domain like pseudokinase (MLKL) and its phosphorylation status. However, this possibility was ruled out given the lack of expression and thus lack of potential phosphorylation of MLKL in splenic lymphocytes (Fig 5D). Overall, in vitro results support that lack of GSDMD may promote enhanced autoantigen generation through modulation of necrotic types of cell death in immune cells.
Altogether, these data indicate a context-dependent role of GSDMD in cell death mechanisms. Specifically, GSDMD appeared important in restraining cell death in barrier tissues early on in the model, specifically at the site of TLR-7 agonist insult, as well as in lungs at later stages of the disease. Furthermore, Gsdmd−/− splenic lymphocytes were less efficient at undergoing apoptosis but, instead, died through a potentially more immunogenic mechanism resulting in significantly higher levels of HMGB-1 release. These results suggested that enhanced barrier cell death following imiquimod treatment, and activation of alternative mechanisms of proinflammatory necrotic cell death in immune cells may contribute to the enhanced generation of autoantigens, promoting the exacerbated lupus phenotype and immune dysregulation observed in the absence of GSDMD.
Discussion
In this study, we investigated the role of GSDMD in the development of systemic autoimmunity and tissue damage in murine lupus. We initially hypothesized that GSDMD would play a pathogenic role in SLE by promoting proinflammatory forms of cell death and systemic inflammation. This would in turn lead to enhanced release of intracellular autoantigens and generation of proinflammatory cytokines, all plausible mechanisms involved in the promotion of loss of immunologic tolerance and inflammation [18, 19, 21–23, 42]. Unexpectedly, we found that the absence of GSDMD in a model of TLR-7 induced lupus enhanced mortality and exacerbated lupus clinical phenotype, including kidney and pulmonary damage, systemic inflammation, and enhanced autoantigen and autoantibody production. This was associated with features of immune dysregulation such as myeloid cell expansion and B cell activation and differentiation. Furthermore, we observed that GSDMD deficiency contributed to enhanced cell death specific to the site of local TLR-7 agonist insult. To our knowledge, this is the first report to observe prominent pulmonary inflammation and immune cell infiltration within this specific animal model, and its exacerbation in the absence of GSMD. Furthermore, we confirmed this enhanced clinical pulmonary phenotype in the absence of GSDMD in the pristane model of DAH that is typically elicited in B6 background. Of note, this pristane model of DAH has previously been described to be driven by enhanced cell death as a direct result of pristane migration to the lungs [34], which further supports that dysregulated cell death in the absence of GSDMD may promote lung damage. This observation is also supported by finding evidence of increased cell death in the lungs of imiquimod-treated mice, which was exacerbated in the absence of GSDMD. Altogether, we found that GSDMD restricts systemic autoimmune phenotypes characteristic of SLE, at least in part, by controlling tissue-specific cell death.
Given that dysregulation in the type I IFN pathway and elevation of a type I IFN signature are characteristic of human and murine SLE, and the recently described role of GSDMD in controlling type I IFN signaling, we assessed whether lack of this molecule would enhance type I IFN pathways [15, 16, 36, 43, 44]. However, Gsdmd−/− mice did not exhibit enhanced type I IFN responses or expansion of pDCs in tissues, suggesting that the mechanisms exacerbating lupus autoimmunity were not linked to enhanced dysregulation in this group of cytokines. We cannot completely rule out that elevation of ISGs could have occurred in the KOs at earlier time points during the disease and were not detected at euthanasia. Instead, GSDMD deficiency associated with expansion of the myeloid compartment, including neutrophils and proinflammatory monocytes. In addition, we found evidence for enhanced B cell activation and expansion of plasma cells, suggesting that GSDMD plays an important role in limiting B cell activation and differentiation following exposure to TLR-7 agonists. It is possible that the expansion of the recently described ‘non-conventional’ pDCs in the absence of GSDMD plays a role in the enhanced maturation of B cells; however this remains to be systematically demonstrated [37]. We found that both murine T- and B- lymphocytes expressed GSDMD and that this molecule was upregulated during B cell activation. However, the effect of GSDMD on B cell activation and differentiation did not appear to be intrinsic but rather, may be triggered by activation of the myeloid cell compartment or other dysregulated cell types. While we did not observe any differences in T cell phenotype, we cannot rule out that the differential modulation of B cell responses in mice lacking GSDMD is not dependent of T cell abnormalities. Whether decreases in lymphocyte apoptosis with the absence of GSDMD could promote the persistence of autoreactive cells as a mechanism promoting autoimmunity should be considered, as we detected less TUNEL-positive lymphocytes in Gsdmd−/− mice. One significant difference between WT mice and Gsdmd−/− was that the latter displayed significant increases in circulating cell-free DNA, suggesting enhanced cell death or impaired clearance of death material. However, we saw no defects in dead cell clearance in Gsdmd−/− macrophages, suggesting enhanced cell death likely played a role. This was further supported by the observation of enhanced caspase 8 cleavage at the site of imiquimod insult (skin) during the initiation of the model. This observation suggests specific and stimulation-dependent roles of GSDMD in barrier cells, where they are known to be significantly expressed [45]. This is further supported by the observation of enhanced cell death in the lungs at later time-points, in association with increased lung inflammation, further supporting a specific and context-dependent regulatory role of GSDMD.
Within immune cells, we detected enhanced release of extracellular HMGB1 in Gsdmd−/− lymphocytes, despite decreases in in vitro apoptosis. This may indicate that lack of GSDMD may direct immune cells to die by necrotic cell death rather than apoptosis, promoting a more proinflammatory cell death in the context of TLR activation, even in the absence of pyroptotic cell death. This necrotic cell death did not appear to be necroptosis, as lymphocytes in this model did not express the proper machinery, MLKL, to undergo such a mechanism. It is nevertheless possible that other immune or non-immune cells preferentially died by necroptosis in the absence of GSDMD and future studies should further investigate this. Enhanced levels of overall and necrotic cell death in the absence of GSDMD may have not only led to the generation of elevated levels of autoantigens that could promote immune complex formation and direct tissue damage but could have also amplified several types of inflammatory responses in various tissues, such as what was observed in lungs and kidneys.
These observations provide important new insights into the multifaceted roles of GSDMD. A pathogenic role for GSDMD was described in experimental autoimmune encephalomyelitis (a mouse model of multiple sclerosis) and in familial Mediterranean fever, an autoinflammatory syndrome [46, 47]. In contrast, and somewhat similar to the findings in our study, GSDMD was recently described to have a protective role in noninfectious liver injury by preventing necroptosis and apoptosis in liver cells through modulation of caspase-8 cleavage [48]. Overall, this supports our hypothesis that GSDMD may act as a negative regulator of some forms of cell death at sites of tissue injury. Our observations that GSDMD may have a protective role in murine lupus autoimmunity support the concept that this molecule has complex roles in immune responses, with both protective and pathogenic effects that may be context dependent. Future studies should examine other genetically-prone or induced models of SLE to further support our observation, as well as examine the expression and modulation of GSDMD in the context of human lupus and autoimmune syndromes.
Despite previous reports that GSDMD was required for NET formation [25, 26], we found that Gsdmd−/− bone marrow (BM) neutrophils were able to form NETs both spontaneously and following in-vitro stimulation with a calcium ionophore. Furthermore, imiquimod-treated Gsdmd−/− mice had higher circulating NETs compared to imiquimod-treated WT mice. These observations could be explained by a display in similar ability to form NETs but increased circulating and tissue neutrophil numbers as a part of the myeloid expansion observed in Gsdmd−/− mice. Given the many pathways that lead to NET formation [49], our findings further reveal that GSDMD’s role in a variety of cell death mechanisms may remain context dependent. Given the description of GSDMD’s role in promoting apoptosis by other groups, it remains possible that GSDMD could serve a protective role in other models, such as cancer, as tumor cells behave differently than noncancerous cells [27, 28]. Our observations suggest an even more complex role for GSDMD in health and disease and suggest that therapies that could target GDSMD need to be carefully evaluated in specific disease states. Indeed, further understanding the putative immunoregulatory roles of GSDMD will be key before defining the conditions that may benefit from modulation of this complex molecule.
Supplementary Material
Acknowledgments:
We thank Wanxia Tsai from the NIAMS Office of Science and Technology, the NIAMS Flow Cytometry Section (James Simone, Kevin Tinsley and Jeffrey Lay), and the NIH Department of Laboratory Medicine for technical support. Furthermore, we thank Dr. Vance Russell at UC-Berkeley for the generous gift of Gsdmd−/− breeding pairs.
Funding:
This work was supported by the NIH/NIAMS IRP (ZIAAR041199).
References
- 1.Tiffin N, Adeyemo A, and Okpechi I, A diverse array of genetic factors contribute to the pathogenesis of systemic lupus erythematosus. Orphanet J Rare Dis, 2013. 8: p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman A and Isenberg DA, Systemic lupus erythematosus. N Engl J Med, 2008. 358(9): p. 929–39. [DOI] [PubMed] [Google Scholar]
- 3.Tsokos GC, Systemic lupus erythematosus. N Engl J Med, 2011. 365(22): p. 2110–21. [DOI] [PubMed] [Google Scholar]
- 4.Rivas-Larrauri F and Yamazaki-Nakashimada MA, Systemic lupus erythematosus: Is it one disease? Reumatol Clin, 2016. 12(5): p. 274–81. [DOI] [PubMed] [Google Scholar]
- 5.Tselios K and Urowitz MB, Cardiovascular and Pulmonary Manifestations of Systemic Lupus Erythematosus. Curr Rheumatol Rev, 2017. 13(3): p. 206–218. [DOI] [PubMed] [Google Scholar]
- 6.Kasturi S and Sammaritano LR, Corticosteroids in Lupus. Rheum Dis Clin North Am, 2016. 42(1): p. 47–62, viii. [DOI] [PubMed] [Google Scholar]
- 7.Durcan L and Petri M, Why targeted therapies are necessary for systemic lupus erythematosus. Lupus, 2016. 25(10): p. 1070–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mistry P and Kaplan MJ, Cell death in the pathogenesis of systemic lupus erythematosus and lupus nephritis. Clin Immunol, 2017. 185: p. 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jimenez-Alcazar M, et al. , Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science, 2017. 358(6367): p. 1202–1206. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan MJ and Radic M, Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol, 2012. 189(6): p. 2689–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villanueva E, et al. , Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol, 2011. 187(1): p. 538–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hakkim A, et al. , Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proceedings of the National Academy of Sciences of the United States of America, 2010. 107(21): p. 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan MJ, Role of neutrophils in systemic autoimmune diseases. Arthritis Res Ther, 2013. 15(5): p. 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith CK and Kaplan MJ, The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol, 2015. 27(5): p. 448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crow MK, Type I interferon in the pathogenesis of lupus. J Immunol, 2014. 192(12): p. 5459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett L, et al. , Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med, 2003. 197(6): p. 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kayagaki N, et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature, 2015. 526(7575): p. 666–71. [DOI] [PubMed] [Google Scholar]
- 18.He WT, et al. , Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res, 2015. 25(12): p. 1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruhl S and Broz P, The gasdermin-D pore: Executor of pyroptotic cell death. Oncotarget, 2016. 7(36): p. 57481–57482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi J, et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature, 2015. 526(7575): p. 660–5. [DOI] [PubMed] [Google Scholar]
- 21.Qi X, Formation of membrane pores by gasdermin-N causes pyroptosis. Sci China Life Sci, 2016. 59(10): p. 1071–1073. [DOI] [PubMed] [Google Scholar]
- 22.Evavold CL, et al. , The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity, 2018. 48(1): p. 35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuriakose T and Kanneganti TD, Gasdermin D Flashes an Exit Signal for IL-1. Immunity, 2018. 48(1): p. 1–3. [DOI] [PubMed] [Google Scholar]
- 24.Kambara H, et al. , Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep, 2018. 22(11): p. 2924–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen KW, et al. , Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol, 2018. 3(26). [DOI] [PubMed] [Google Scholar]
- 26.Sollberger G, et al. , Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol, 2018. 3(26). [DOI] [PubMed] [Google Scholar]
- 27.Rogers C, et al. , Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun, 2019. 10(1): p. 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuchiya K, et al. , Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun, 2019. 10(1): p. 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richard ML and Gilkeson G, Mouse models of lupus: what they tell us and what they don’t. Lupus Sci Med, 2018. 5(1): p. e000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yokogawa M, et al. , Epicutaneous application of toll-like receptor 7 agonists leads to systemic autoimmunity in wild-type mice: a new model of systemic Lupus erythematosus. Arthritis Rheumatol, 2014. 66(3): p. 694–706. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, et al. , Accelerated model of lupus autoimmunity and vasculopathy driven by toll-like receptor 7/9 imbalance. Lupus Sci Med, 2018. 5(1): p. e000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, et al. , Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight, 2018. 3(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barker TT, et al. , Pathogenic role of B cells in the development of diffuse alveolar hemorrhage induced by pristane. Lab Invest, 2011. 91(10): p. 1540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuang H, et al. , Pathogenesis of Diffuse Alveolar Hemorrhage in Murine Lupus. Arthritis Rheumatol, 2017. 69(6): p. 1280–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hariri LP, et al. , Acute fibrinous and organizing pneumonia in systemic lupus erythematosus: a case report and review of the literature. Pathol Int, 2010. 60(11): p. 755–9. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee I, et al. , Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity, 2018. 49(3): p. 413–426 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, et al. , A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc Natl Acad Sci U S A, 2017. 114(8): p. 1988–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tummers B and Green DR, Caspase-8: regulating life and death. Immunol Rev, 2017. 277(1): p. 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grasl-Kraupp B, et al. , In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology, 1995. 21(5): p. 1465–8. [DOI] [PubMed] [Google Scholar]
- 40.Stros M, HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta, 2010. 1799(1–2): p. 101–13. [DOI] [PubMed] [Google Scholar]
- 41.Sims GP, et al. , HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol, 2010. 28: p. 367–88. [DOI] [PubMed] [Google Scholar]
- 42.Rathkey JK, et al. , Live-cell visualization of gasdermin D-driven pyroptotic cell death. J Biol Chem, 2017. 292(35): p. 14649–14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wenzel J, et al. , Enhanced type I interferon signaling and recruitment of chemokine receptor CXCR3-expressing lymphocytes into the skin following treatment with the TLR7-agonist imiquimod. J Cutan Pathol, 2005. 32(4): p. 257–62. [DOI] [PubMed] [Google Scholar]
- 44.Andrade D, et al. , Interferon-alpha and angiogenic dysregulation in pregnant lupus patients who develop preeclampsia. Arthritis Rheumatol, 2015. 67(4): p. 977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei-Leston AC, Murphy AG, and Maloy KJ, Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front Immunol, 2017. 8: p. 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, et al. , Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J Exp Med, 2019. [DOI] [PMC free article] [PubMed]
- 47.Kanneganti A, et al. , GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J Exp Med, 2018. 215(6): p. 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang C, et al. , Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis, 2019. 10(7): p. 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kenny EF, et al. , Diverse stimuli engage different neutrophil extracellular trap pathways. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.