

Abstract
Advances in molecular methods and the ability to share large population-based datasets are uncovering heterogeneity within diabetes types, and some commonalities between types. Within type 1 diabetes, endotypes have been discovered based on demographic (e.g. age at diagnosis, race/ethnicity), genetic, immunological, histopathological, metabolic and/or clinical course characteristics, with implications for disease prediction, prevention, diagnosis and treatment. In type 2 diabetes, the relative contributions of insulin resistance and beta cell dysfunction are heterogeneous and relate to demographics, genetics and clinical characteristics, with substantial interaction from environmental exposures. Investigators have proposed approaches that vary from simple to complex in combining these data to identify type 2 diabetes clusters relevant to prognosis and treatment. Advances in pharmacogenetics and pharmacodynamics are also improving treatment. Monogenic diabetes is a prime example of how understanding heterogeneity within diabetes types can lead to precision medicine, since phenotype and treatment are affected by which gene is mutated. Heterogeneity also blurs the classic distinctions between diabetes types, and has led to the definition of additional categories, such as latent autoimmune diabetes in adults, type 1.5 diabetes and ketosis-prone diabetes. Furthermore, monogenic diabetes shares many features with type 1 and type 2 diabetes, which make diagnosis difficult. These challenges to the current classification framework in adult and paediatric diabetes require new approaches. The ‘palette model’ and the ‘threshold hypothesis’ can be combined to help explain the heterogeneity within and between diabetes types. Leveraging such approaches for therapeutic benefit will be an important next step for precision medicine in diabetes.
Since the first remedies for ‘honey urine’ were described over 25 centuries ago, untangling the pathways that lead to what we call today diabetes has enabled better treatments. The current classification of diabetes recognises the major forms of type 1 diabetes, type 2 diabetes, gestational diabetes and other less common types, such as monogenic diabetes (including neonatal diabetes and MODY), diseases of the exocrine pancreas (e.g. cystic fibrosis-related diabetes and pancreatogenic [or type 3c] diabetes) or drug-induced diabetes (Table 1) [1]. Advances in molecular biology and genetic methods during the past decades have facilitated the discovery of a myriad of genetic associations that suggest diverse pathophysiology within each of those types, as well as some commonalities between types. The recent acceleration in the ability to share large population-based datasets has further increased the evidence for heterogeneity within and between diabetes types [2]. Integrating our understanding of genetics, immunology and metabolic abnormalities in each type of diabetes will boost the ability to use precision medicine to tailor treatments and improve health outcomes.
Keywords: Atypical, Classification, Diabetes, Endotype, Genetics, Heterogeneity, Palette, Precision medicine, Review, Threshold
Heterogeneity of diabetes within types and clinical consequences
Type 1 diabetes
Type 1 diabetes is diverse in its genetics, environmental influences, immunology, metabolic characteristics and clinical course. The heterogeneity of type 1 diabetes stems from its polygenic nature, with interactions of multiple genes with environmental factors. Recent advances in type 1 diabetes genetic knowledge and methods have facilitated genotype–phenotype correlations. Genetic risk scores (GRS), including HLA and non-HLA genes, can stratify the risk for development of islet autoimmunity, progression through preclinical stages, and diagnosis of type 1 diabetes [3].
The rapid secular rise in type 1 diabetes incidence (3–4% per year) suggests an environmental–genetic interaction that is initiating or accelerating the disease process. Research has demonstrated the influence of many environmental factors [4]. In a large prospective study of children with genetic risk for type 1 diabetes, prolonged infection with Enterovirus B was shown to increase risk of beta cell autoimmunity [5]. Further work has revealed interactive effects between specific gene profiles (CTLA-4 and HLA-DR-DQ4–8/8–4) and maternal gestational infections on risk and characteristics of beta cell autoimmunity [6].
In addition to the number, level and affinity of islet autoantibodies, the type of autoantibody at the initial seroconversion also imparts a varying risk of progression to clinical disease. The appearance of insulin autoantibodies (IAA) as the first autoantibodes, as opposed to GAD65 autoantibodies (GADA), is marked by a rapid earlier peak incidence, and faster progression to multiple autoantibodies and then diabetes [7].
Age is a marker of heterogeneity in type 1 diabetes (reviewed in [8]). Younger age is associated with higher risk and rate of progression through the stages of the disease, as well as distinct histological characteristics, immunological patterns and genetic influences. The underlying mechanisms for these differences, once known, will allow targeting of interventions to specific age groups and stages of preclinical disease.
The rate of beta cell function decline during the first year after diagnosis is highly variable, ranging from 0% to 58% [9]. Older age, lower HbA1c and higher BMI at diagnosis predict slower loss of C-peptide [9]. Beyond clinical characteristics, ~26% of variability in beta cell function persistence post diagnosis is explained by genetic factors, including the HLA region (although not the DR3/DR4 genotype that confers the highest risk of type 1 diabetes), PTPN22, INS region, and rs559047 (a Iong non-coding RNA [lncRNA] in chromosome 1 that is not associated with type 1 diabetes risk) [10]. Therefore, although many loci simultaneously determine type 1 diabetes risk, lower C-peptide levels and younger age of onset, this is not always the case. In the Chinese population, higher Chinese-specific type 1 diabetes GRS correlates with lower C-peptide [11], highlighting important ethnic differences in type 1diabetes genetics. Type 2 diabetes-associated loci (e.g. JAZF1 and TCF7L2) also influence C-peptide persistence in type 1 diabetes [10].
In addition, clinical heterogeneity in type 1 diabetes is reflected by differences in the risk of complications, which is related to a wide array of factors, from hyperglycaemia and longer diabetes duration, to race, ethnicity and socioeconomic factors. Higher residual beta cell function confers lower risk of microalbuminuria and retinopathy [12]. Recently, the SEARCH for Diabetes in Youth study showed different patterns of subclinical macrovascular complications according to phenotypes of different combinations of weight and glucose levels in blood [13]. A number of studies have shown influence on complications by candidate genes such as the VEGF gene for retinopathy, the ADIPOQ gene for coronary artery disease and the ELMO1 gene for nephropathy, although replication remains controversial (reviewed in [14]). The Genetics of Kidneys in Diabetes (GoKinD) study found four additional loci for nephropathy (near FRMD3, CHN2, CARS1 and an intergenic region of chromosome 13q), all with borderline significance but some evidence of replication in other studies.
A current area of active debate is the definition of endotypes within type 1 diabetes that may have implications for treatment [15]. This concept is supported by the association between specific HLA alleles and the first autoantibody to appear in at-risk children [7]. A distinct histological phenotype associates with more aggressive insulitis, increased presence of B cells, altered insulin processing and onset under age 7 [16]. A rapid growth in the ability of ‘omics’ and bioinformatics to generate and integrate data may help advance identification of type 1 diabetes endotypes.
The heterogeneity of type 1 diabetes has implications for prediction, prevention, diagnosis and treatment. Beyond the risk associated with islet autoimmunity, predictive models can be optimised for specific background genetic risk, preclinical stage of the disease and age, among other characteristics [17]. The success of immunotherapies to prevent beta cell loss before and after diagnosis has been limited by heterogenous responses to these drugs. While age and immunological profiles seem to influence response, a better understanding of the factors involved in type 1 diabetes heterogeneity and different endotypes [15] will allow us to apply precision medicine and tailor therapy to the specific pathophysiological processes underlying each person’s diabetes (Fig. 1).
Fig 1.
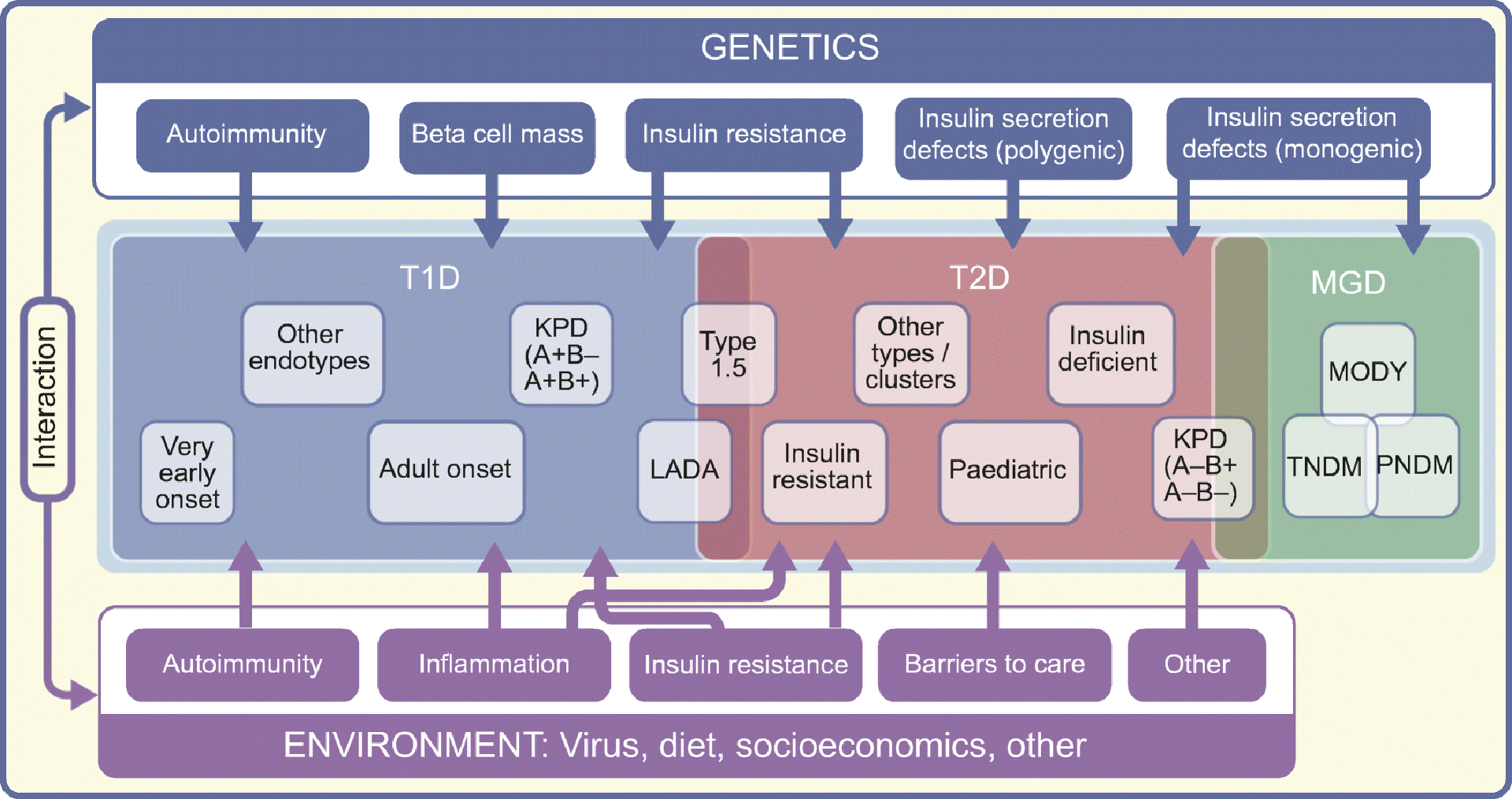
Illustration of heterogeneity within and between diabetes types. Genetic and environmental factors, and their interaction, influence multiple mechanisms (e.g. autoimmunity, reduced beta cell mass, insulin secretion defects, inflammation, barriers to health care) that may contribute in variable degree to the development and progression of diabetes in each individual. This individual variability leads to heterogeneity within diabetes types (e.g. very early onset type 1 diabetes, LADA) and between diabetes types (e.g. KPD, LADA type 1.5 diabetes). Therapeutic leverage of pathophysiological heterogeneity will allow advancement of precision medicine in diabetes. T1D, type 1 diabetes; T2D, type 2 diabetes; MGD, monogenic diabetes.
Type 2 diabetes
Key pathophysiological aspects in type 2 diabetes stem from environmental factors but notably require genetic susceptibility, are complexly interwoven and may even be self-reinforcing [18]. Many type 2 diabetes genes are related to obesity, several to insulin resistance, but most are linked to the beta cell [19].
Type 2 diabetes occurs when acquired insulin resistance usually from obesity exists in the setting of innate and acquired inadequacy in beta cell function. However, the possible underlying mechanisms are multiple. Chronically high insulin demand triggers relative beta cell dysfunction. Adipocytes release inflammatory factors that may impair beta cells. Hypothalamic inflammation and appetite dysregulation may occur. Hypersecretion of islet amyloid polypeptide (IAPP) favours islet amyloidosis; hyperglycaemia promotes islet glucotoxicity, lipoglucotoxicity and beta cell apoptosis, which may explain observed moderate decreases in beta cell mass. There may be incretin hormone insufficiency/resistance as well as inappropriate hyperglucagonaemia [20]. Changes occur in the gut microbiome that alter released metabolites, potentially including inflammatory endotoxins, which has justified faecal transplant trials in type 2 diabetes, although with variable results [21]. Individual differences in the relative contribution of each of these pathways may underlie disease heterogeneity.
There are important racial/ethnic disparities in type 2 diabetes epidemiology, physiopathology, clinical course and response to treatment. Compared with non-Hispanic white people, rates of type 2 diabetes are greater in black, Asian and Hispanic people [22]. These differences are multifactorial and include biological differences, such as upregulated beta cell function and increased insulin resistance in African-Americans compared with whites, or greater risk of glucose metabolism abnormalities at lower levels BMI in south Asians compared with other groups [23]. These characteristics may add to and interact with socioeconomic aspects that are associated with race and ethnicity and influence key environmental exposures, such as diet, among others.
Age is a marker of heterogeneity in type 2 diabetes. Children develop insulin deficiency faster than adults, and their rate of chronic complications is also higher [24]. In addition, the social determinants of disease are likely to be stronger in paediatric than adult-onset type 2 diabetes. Finally, the shortage of drugs for type 2 diabetes that are approved for children complicates treatment and could impact outcomes in paediatrics.
Like diabetes itself, the long-term complications of diabetes result from the interplay of genetics and exposures. While hyperglycaemia exerts the strongest influence on complications, hypertension is also firmly linked with nephropathy and cardiovascular disease. Socioeconomic hurdles impact many aspects of diabetes diagnosis and treatment and thus contribute to a higher rate of complications. Several genes known to influence coronary artery disease in the general population also increase risk in individuals with type 2 diabetes (reviewed in [14]). Similarly to type 1 diabetes, although a number of plausible genes have been linked to microvascular complications[14], there are inconsistencies among studies, which may be related to the heterogeneity of type 2 diabetes.
Untangling the heterogeneity of type 2 diabetes may improve prediction of clinical outcomes and facilitate precision medicine. Novel biostatistical methodologies help leverage emerging information on the disease genetic architecture. Udler et al identified clusters of type 2 diabetes genetic loci involved in metabolic pathways, disease traits and clinical outcomes [25]. Ahlqvist et al used a data-driven cluster analysis to classify adult-onset diabetes into five subgroups with distinct phenotypes, risk of complications and genetic associations [26, 27]. For example, the insulin-deficient phenotype associates with retinopathy and neuropathy, while insulin-resistant individuals more often have nephropathy and macrovascular complications. Similarly, other authors have proposed the use of phenotypic and genetic information to identify diabetes subtypes [28]. An alternative approach aims to predict clinical outcomes of interest by using simple phenotypic measures (e.g. baseline renal function to predict diabetic nephropathy) [29].
Pharmacogenetics and pharmacodynamics may help explain variability in responses to type 2 diabetes treatment (reviewed in [30]). Variation at the TCF7L2 locus and possibly in genes that encode the sulfonylurea receptor (e.g. KCNJ11 and ABCC8), which also increase type 2 diabetes risk, impact the response to sulfonylureas (reviewed in [31]). The pharmacogenetics of metformin have been studied [31] but its cost-effectiveness and clinical applicability are not clear. Insulin-deficient diabetes responds poorly to drugs that increase insulin secretion (e.g. glucagon-like peptide 1 [GLP-1] receptor agonists). Individuals with type 2 diabetes that was predominantly insulin resistant seemed to respond worse to dipeptidyl peptidase 4 (DPP-4) inhibitor treatment. However, the complex interplay of insulin resistance and secretion, with relative contributions that are unclear and evolve with time, and the small effect size of type 2 diabetes-associated genetic variants, hinder the advancement of precision medicine. Current type 2 diabetes guidelines to intensify therapy after metformin are largely based on potential added benefits (e.g. weight reduction) or increased risk of side effects (e.g. hypoglycaemia).
Monogenic diabetes
Monogenic diabetes (neonatal diabetes or MODY) is a prime example of how understanding heterogeneity within diabetes types can lead to precision medicine. Clinical features and treatment are affected by the mutated gene and sometimes the specific variant [32].
Over 20 genetic causes of neonatal diabetes have been identified to date. While mutations in the potassium-channel genes KCNJ11 and ABCC8 [33] can cause both permanent (PNDM) or transient neonatal diabetes (TNDM), imprinting anomalies of chromosome 6q24 are the most common cause of TNDM. Genetic diagnosis of potassium-channel neonatal diabetes is extremely important because affected neonates can be treated with sulfonylurea tablets [34]. In KCNJ11 PNDM, the genetic mutations are correlated with the severity of the neurological abnormalities, which were thought to improve with sulfonylureas, although recent data casts doubts on this concept [35]. Mutations in key pancreas development transcription genes (e.g. PTF1A, GATA6 and PDX1) lead to pancreatic agenesis and PNDM.
The most common phenotype of monogenic diabetes is MODY, caused by mutations in any of at least 14 genes [36]. We have shown that the correct genetic diagnosis of MODY allows patients to receive the most effective treatment [37]. For example, sulfonylureas are effective as a treatment for MODY caused by mutations in HNF1A or HNF4A and, at low dose, ABCC8 genes [38]. GCK mutations cause stable fasting hyperglycaemia with a higher set point of glucose that does not require treatment except during pregnancy [39].
Other diabetes types
Pancreatogenic or type 3c diabetes results from exocrine pancreatic disease but the underlying mechanisms are an active research area. Other diabetes types include steroid-induced diabetes, gestational diabetes, congenital generalised lipodystrophy or Wolfram syndrome. In addition, rare and atypical cases of diabetes exist that cannot be accurately classified within the known diabetes types. Some of these cases may represent extreme phenotypes with mechanisms shared, although with milder severity, by more frequent diabetes types.
Heterogeneity of diabetes between types
The classic phenotypic distinctions between type 2 diabetes (i.e. adult onset, obese) and type 1 diabetes (childhood onset, thin and white race) appear increasingly blurred. At the genetic level, both type 1 and type 2 diabetes have been positively associated with four loci—BMP8A, HLA region, CENPW and ASCC2—and show associations in opposite directions for a SNP in the BCAR1/CTRB1/CTRB2 region [10].
Improved analytical tools reveal that more type 1 diabetes appears in adulthood than in childhood, although it is lost in a sea of type 2 diabetes [40]. Almost 10% of predominantly white Europeans with adult-onset diabetes had evidence of islet autoimmunity [41]. Compared with paediatric type 1 diabetes, adult-onset classical type 1 diabetes often presents with higher C-peptide levels that decline more slowly [9], which attenuates the typical differences in beta cell function between type 1 diabetes and type 2 diabetes. Furthermore, although type 1 diabetes rates are 50% lower in blacks and Hispanics and 75% lower in Asians vs non-Hispanic whites [42], type 1 diabetes is not rare in any group.
Hawa et al [41] observed that most individuals with adult-onset autoimmune diabetes were insulin independent for an extended time post-diagnosis compared with people with classical type 1 diabetes. The term latent autoimmune diabetes in adults (LADA) was developed for these slow progressors [43]. While it is possible that false-positive islet autoantibody tests underlie some cases of type 2 diabetes being diagnosed as LADA, GADA positivity conferred a higher type 1 diabetes GRS and predicted incident diabetes in a population-based prospective study in adults [44]. LADA most likely is at the slowest extreme of the spectrum of variability in the rate of autoimmune beta cell loss observed in type 1 diabetes (Fig. 1). The clinical characteristics and genetic architecture of LADA [43] suggest an additional role for type 2 diabetes pathophysiological pathways. Insulin resistance induced by being obese or overweight (more frequent in LADA) or other factors could aggravate the imbalance between insulin secretion and requirements, thus triggering diabetes. Furthermore, chronic obesity upregulates proinflammatory innate and adaptive immune responses, producing a state of systemic inflammation possibly driven by antigens yet to be identified. Islet-specific T cell autoimmunity seems to increase the loss of beta cell function in obesity associated with diabetes (both type 1 and type 2) [45]. Further illustrating the heterogeneity between diabetes types, factors typically associated with type 2 diabetes (e.g. TCF7L2 variants, elevated BMI) may influence the progression of islet autoimmunity and development of diabetes [46].
In paediatrics, the clinical classification of diabetes can be challenging [47]. Greater obesity and ethic admixture may accelerate type 2 diabetes onset towards childhood. Although diabetic ketoacidosis (DKA) is more common in type 1 diabetes, hyperglycaemia-induced beta cell toxicity may promote DKA in up to 29% of individuals with type 2 diabetes. Therefore, C-peptide may be low at type 2 diabetes diagnosis and normal during the honeymoon phase of type 1 diabetes, confusing diabetes classification at onset. The terms ‘type 1.5 diabetes’, ‘double diabetes’ or ‘hybrid diabetes’ [48] have been used for obese adolescents with a clinical diagnosis of type 2 diabetes who test positive for autoantibodies. Although this nomenclature is not part of the classification framework used by the ADA [1] and other international guidelines, there is increasing interest in the notion that the immune system may be a pathogenic component of obesity and type 2 diabetes [49].
The SEARCH for Diabetes in Youth study implemented the ADA classification framework [1] in youth with diabetes using the presence/absence of two aetiologic markers, namely, autoimmunity and insulin sensitivity, to define four groups [50]. Most youth fell into categories that align with traditional descriptions of type 1 diabetes (autoimmune/insulin sensitive; 54.5%) and type 2 diabetes (non-autoimmune/insulin resistant; 15.9%). Evidence indicated that the autoimmune/insulin-resistant group represented autoimmune type 1 diabetes with obesity but not a distinct aetiologic entity. The non-autoimmune/insulin sensitive group (~10%) was deemed to require additional testing to detect potential missed islet autoimmunity [51] or MODY mutations. These findings suggest that standard case definitions can be operationalised to classify type of diabetes in youth (Fig. 2).
Fig. 2.
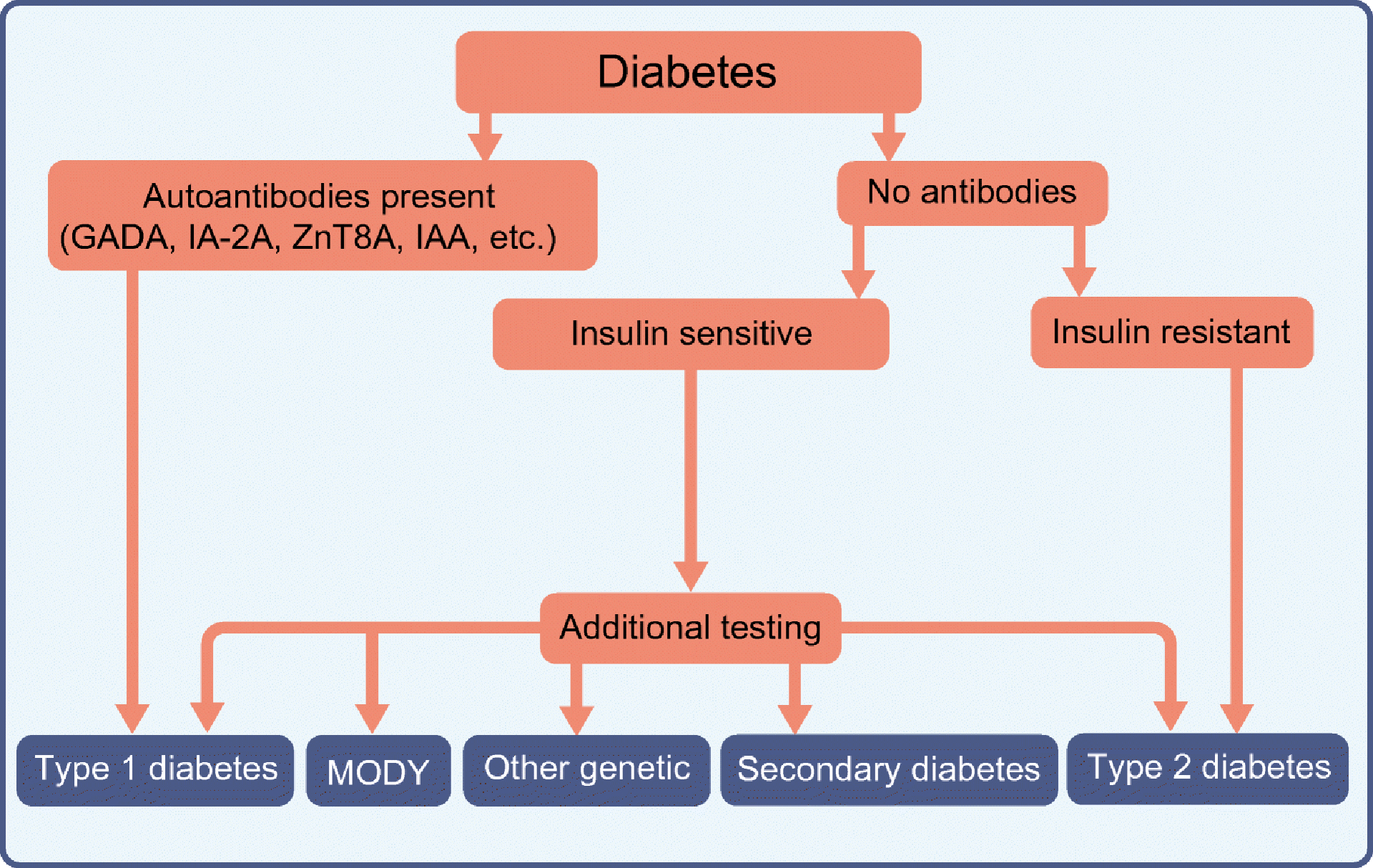
Algorithm for classification of paediatric diabetes based on the presence or absence of autoimmunity and insulin sensitivity. Secondary causes of diabetes include steroid-induced diabetes and cystic fibrosis-related diabetes. Other genetic causes of diabetes include lipodystrophy, mitochondrial diabetes, etc. IA-2A, insulin antigen-2 autoantibodies; IAA, insulin autoantibodies, ZnT8A, zinc transporter 8 autoantibodies. Modified from [58] with the permission of American Diabetes Association. © 2014 American Diabetes Association.
Monogenic diabetes shares many features with type 1 type 2 diabetes, which makes diagnosis of monogenic diabetes difficult. In a recent study of individuals with type 1 diabetes for 50 years or more, 27.5% had monogenic variants, 7.9% of which were classified as ‘likely pathogenic’, underscoring the importance of genetic testing for clinically diagnosed type 1 diabetes[52]. The key features that distinguish monogenic diabetes from type 1 diabetes are autoantibody negativity (although GADA have been reported in 1% of individuals with MODY individuals), the presence of C-peptide and, particularly for GCK MODY, lower HbA1c. The key distinguishing features between MODY and type 2 diabetes are lower age at diagnosis, lower BMI and autosomal dominant family history. A ‘MODY probability calculator’ can aid in the clinical decision to conduct genetic testing [53]. Pathophysiological mechanisms in monogenic diabetes may also underlie type 2 diabetes. For example, the E23K variant in KCNJ11, whose mutation causes PNDM, increases type 2 diabetes risk (Fig. 1). This observation supports future studies on atypical diabetes, as they may provide insight into more frequent types.
Ketosis-prone diabetes (KPD) represents another example of heterogeneity between diabetes types [54], with phenotypes distinct from both type 1 and type 2 diabetes (Fig. 1). This emerging syndrome, widespread in multi-ethnic populations around the world, comprises multiple diabetes forms with distinct aetiologies. The presence/absence of islet autoantibodies and C-peptide (‘AB’ classification system) among patients who present with DKA defines KPD subtypes. Three of these subtypes have a distinct pathophysiology (monogenic, immunological, metabolic) that differs from those of type 1 diabetes or type 2 diabetes, and have divergent natural histories [55].
Two different approaches may help explain the heterogeneity within and between diabetes types. The palette model proposes that multiple pathophysiological processes contributing to diabetes risk and progression, with variable relative importance in each individual, determine the phenotype and could be leveraged for therapy [56]. The threshold hypothesis contends that clinical diabetes develops when the combined influences of genetic and environmental diabetogenic factors exceeds a threshold [57]. These two models can be combined to understand the joint effect and interactions between multiple factors with variable contribution to diabetes risk in different individuals (Fig. 3). This perspective supports the search for endotypes (e.g. within type 1 diabetes) that elucidate heterogeneity within diabetes types. It also helps explain heterogeneity between diabetes types and downplays the need to classify diabetes types into distinct categories. Leveraging the knowledge on disease heterogeneity for therapeutic purposes will be an important next step for the advancement of precision medicine in diabetes.
Fig. 3.
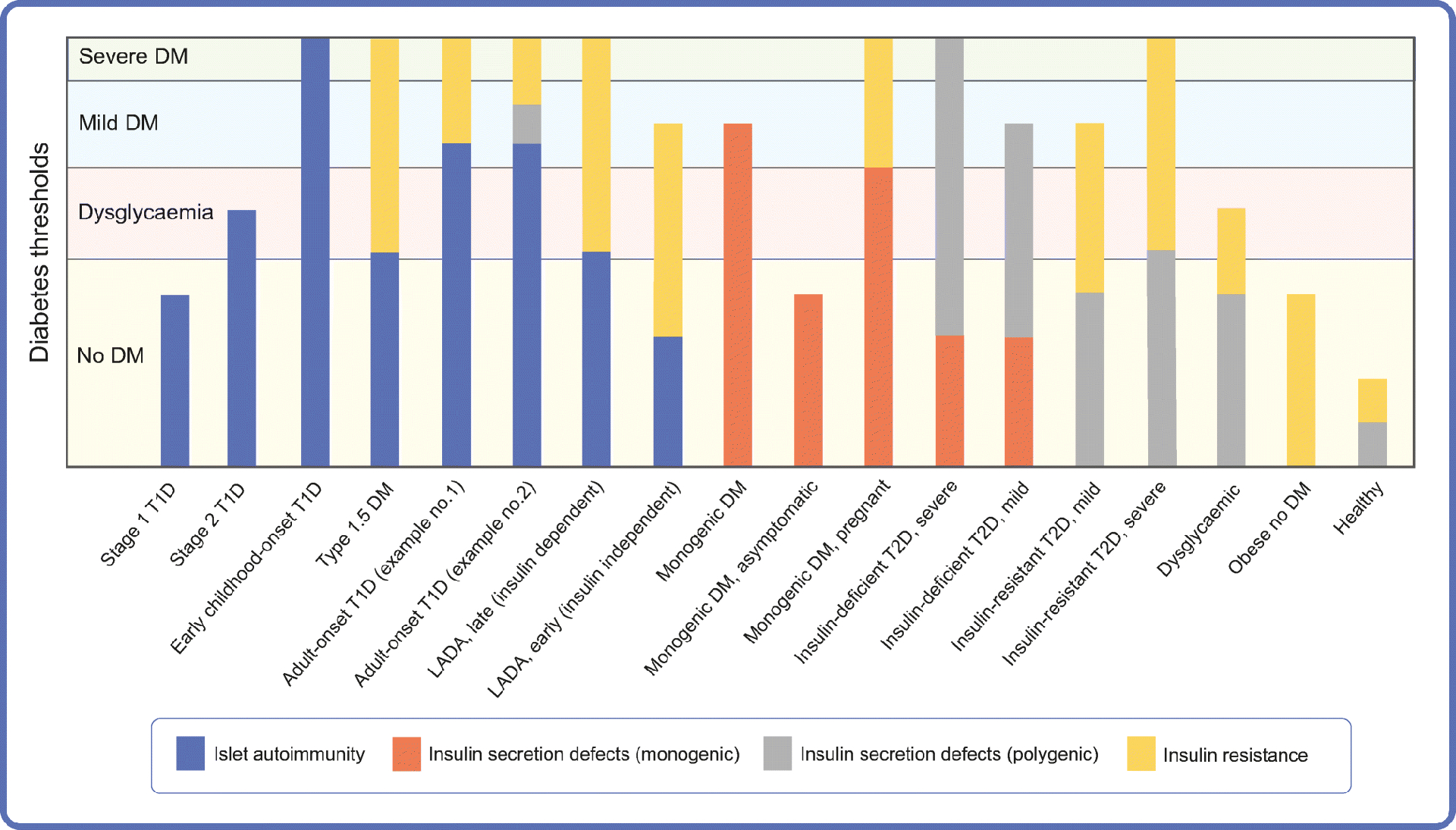
Model of diabetes pathophysiology that integrates the palette model [56] and the threshold hypothesis [57]. Multiple pathophysiological pathways (colours in a palette) can be at play in a given individual, as represented here (e.g. islet autoimmunity, monogenic insulin secretion defect, polygenic insulin secretion defect, insulin resistance), in addition to others not illustrated here (e.g. low beta cell mass due to monogenic mutation). The degrees of abnormality for each diabetogenic pathway in a given individual are combined to determine whether the blood glucose level crosses the threshold for dysglycaemia or diabetes (e.g. compare stages 1 and 2 of type 1 diabetes, or asymptomatic MODY and MODY with symptoms). The relative predominance of each diabetogenic pathway determines the clinical phenotype (e.g. compare insulin-resistant type 2 diabetes and insulin-deficient type 2 diabetes). Different pathophysiological processes may be involved in diabetes types currently considered a single entity (e.g. examples 1 and 2 of adult-onset type 1 diabetes). This perspective provides a framework to understand heterogeneity within and between diabetes types, address the challenges of diabetes classification into distinct categories, and may be leveraged for personalised treatment. The x-axis presents some examples of possible palettes in different individuals or over time in a given individual. DM, diabetes; T1D, type 1 diabetes; T2D, type 2 diabetes; LADA, latent autoimmune diabetes in adults.
Table 1.
Diabetes classification according to the ADA [1]
| Type | Description |
|---|---|
| Type 1 diabetes | Caused by autoimmune beta cell destruction, usually leading to absolute insulin deficiency |
| Type 2 diabetes | Caused by progressive loss of adequate insulin secretion, frequently with insulin resistance |
| Gestational diabetes | Diagnosed in the second or third trimesters, without overt diabetes prior to gestation |
| Monogenic diabetes | There are two subgroups: MODY, and neonatal diabetes |
| Disease of the exocrine pancreas | Caused by pancreatitis, cystic fibrosis, and other disorders of the pancreas. |
| Chemical or drug-induced | Caused by glucocorticoids, HIV/AIDS drugs, organ transplantation drugs |
Funding
Work in the authors’ laboratories is supported NIH 1 R01 DK121843-01 (MJR), R01 DK101411 (AB). WAH is supported by the University of Washington Diabetes Research Center (5P30 DK017047).
Abbreviations:
- GADA
GAD65 autoantibodies
- GRS
Genetic risk score
- IAA
Insulin autoantibodies
- KPD
Ketosis-prone diabetes
- LADA
Latent autoimmune diabetes in adults
- PNDM
Permanent neonatal diabetes
- TNDM
Transient neonatal diabetes
References
- [1].ADA (2020) 2. Classification and Diagnosis of Diabetes:. Diabetes Care 43(Suppl 1): S14–S31. 10.2337/dc20-S002 [DOI] [PubMed] [Google Scholar]
- [2].Bowman P, Flanagan SE, Hattersley AT (2018) Future roadmaps for precision medicine applied to diabetes: rising to the challenge of heterogeneity. J Diabetes Res 2018: 3061620. 10.1155/2018/3061620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Redondo MJ, Geyer S, Steck AK, et al. (2018) A type 1 diabetes genetic risk score predicts progression of islet autoimmunity and development of type 1 diabetes in individuals at risk. Diabetes Care 41(9): 1887–1894. 10.2337/dc18-0087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rewers M, Ludvigsson J (2016) Environmental risk factors for type 1 diabetes. Lancet 387(10035): 2340–2348. 10.1016/S0140-6736(16)30507-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vehik K, Lynch KF, Wong MC, et al. (2019) Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat Med 25(12): 1865–1872. 10.1038/s41591-019-0667-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lynch KF, Lee HS, Torn C, et al. (2018) Gestational respiratory infections interacting with offspring HLA and CTLA-4 modifies incident beta-cell autoantibodies. J Autoimmun 86: 93–103. 10.1016/j.jaut.2017.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Krischer JP, Liu X, Vehik K, et al. (2019) Predicting islet cell autoimmunity and type 1 diabetes: an 8-year TEDDY study progress report. Diabetes Care 42(6): 1051–1060. 10.2337/dc18-2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Leete P, Mallone R, Richardson SJ, Sosenko JM, Redondo MJ, Evans-Molina C (2018) The effect of age on the progression and severity of type 1 diabetes: potential effects on disease mechanisms. Current Diabetes Reports 18(11): 115. 10.1007/s11892-018-1083-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hao W, Gitelman S, DiMeglio LA, Boulware D, Greenbaum CJ, Type 1 Diabetes TrialNet Study Group (2016) Fall in C-peptide during first 4 years from diagnosis of type 1 diabetes: variable relation to age, HbA1c, and insulin dose. Diabetes Care 39(10): 1664–1670. 10.2337/dc16-0360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McKeigue PM, Spiliopoulou A, McGurnaghan S, et al. (2019) Persistent C-peptide secretion in type 1 diabetes and its relationship to the genetic architecture of diabetes. BMC Medicine 17(1): 165. 10.1186/s12916-019-1392-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhu M, Xu K, Chen Y, et al. (2019) Identification of novel T1D risk loci and their association with age and islet function at diagnosis in autoantibody-positive T1D individuals: based on a two-stage genome-wide association study. Diabetes Care 42(8): 1414–1421. 10.2337/dc18-2023 [DOI] [PubMed] [Google Scholar]
- [12].Lachin JM, McGee P, Palmer JP, Group DER (2014) Impact of C-peptide preservation on metabolic and clinical outcomes in the Diabetes Control and Complications Trial. Diabetes 63(2): 739–748. 10.2337/db13-0881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kahkoska AR, Nguyen CT, Adair LA, et al. (2019) Longitudinal phenotypes of type 1 diabetes in youth based on weight and glycemia and their association with complications. The Journal of clinical endocrinology and metabolism 104(12): 6003–6016. 10.1210/jc.2019-00734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dahlström E, Sandholm N (2017) Progress in defining the genetic basis of diabetic complications. Curr Diab Rep 17(9): 80. 10.1007/s11892-017-0906-z [DOI] [PubMed] [Google Scholar]
- [15].Battaglia M, Ahmed S, Anderson MS, et al. (2020) Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care 43(1): 5–12. 10.2337/dc19-0880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Leete P, Willcox A, Krogvold L, et al. (2016) Differential Insulitic Profiles Determine the Extent of beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes 65(5): 1362–1369. 10.2337/db15-1615 [DOI] [PubMed] [Google Scholar]
- [17].Michels A, Zhang L, Khadra A, Kushner JA, Redondo MJ, Pietropaolo M (2015) Prediction and prevention of type 1 diabetes: update on success of prediction and struggles at prevention. Pediatric Diabetes 16(7): 465–484. 10.1111/pedi.12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].DeFronzo RA, Ferrannini E, Groop L, et al. (2015) Type 2 diabetes mellitus. Nature Reviews Disease Primers 1: 15019. 10.1038/nrdp.2015.19 [DOI] [PubMed] [Google Scholar]
- [19].Flannick J, Mercader JM, Fuchsberger C, et al. (2019) Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570(7759): 71–76. 10.1038/s41586-019-1231-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pearson ER (2019) Type 2 diabetes: a multifaceted disease. Diabetologia 62(7): 1107–1112. 10.1007/s00125-019-4909-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Aron-Wisnewsky J, Clément K, Nieuwdorp M (2019) Fecal microbiota transplantation: a future therapeutic option for obesity/diabetes? Curr Diab Rep 19(8): 51. 10.1007/s11892-019-1180-z [DOI] [PubMed] [Google Scholar]
- [22].Rodríguez JE, Campbell KM (2017) Racial and ethnic disparities in prevalence and care of patients with type 2 diabetes. Clin Diabetes 35(1): 66–70. 10.2337/cd15-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hills AP, Arena R, Khunti K, et al. (2018) Epidemiology and determinants of type 2 diabetes in south Asia. Lancet Diabetes Endocrinol 6(12): 966–978. 10.1016/S2213-8587(18)30204-3 [DOI] [PubMed] [Google Scholar]
- [24].Nadeau KJ, Anderson BJ, Berg EG, et al. (2016) Youth-Onset Type 2 Diabetes Consensus Report: current status, challenges, and priorities. Diabetes Care 39(9): 1635–1642. 10.2337/dc16-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Udler MS, Kim J, von Grotthuss M, et al. (2018) Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med 15(9): e1002654. 10.1371/journal.pmed.1002654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ahlqvist E, Storm P, Käräjämäki A, et al. (2018) Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol 6(5): 361–369. 10.1016/S2213-8587(18)30051-2 [DOI] [PubMed] [Google Scholar]
- [27].Ahlqvist E, Tuomi T, Groop L (2019) Clusters provide a better holistic view of type 2 diabetes than simple clinical features. Lancet Diabetes Endocrinol 7(9): 668–669. 10.1016/S2213-8587(19)30257-8 [DOI] [PubMed] [Google Scholar]
- [28].Li L, Cheng WY, Glicksberg BS, et al. (2015) Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci Transl Med 7(311): 311ra174. 10.1126/scitranslmed.aaa9364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dennis JM, Shields BM, Henley WE, Jones AG, Hattersley AT (2019) Disease progression and treatment response in data-driven subgroups of type 2 diabetes compared with models based on simple clinical features: an analysis using clinical trial data. Lancet Diabetes Endocrinol 7(6): 442–451. 10.1016/S2213-8587(19)30087-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gloyn AL, Drucker DJ (2018) Precision medicine in the management of type 2 diabetes. Lancet Diabetes Endocrinol 6(11): 891–900. 10.1016/S2213-8587(18)30052-4 [DOI] [PubMed] [Google Scholar]
- [31].Florez JC (2017) Pharmacogenetics in type 2 diabetes: precision medicine or discovery tool? Diabetologia 60(5): 800–807. 10.1007/s00125-017-4227-1 [DOI] [PubMed] [Google Scholar]
- [32].Shields BM, Shepherd M, Hudson M, et al. (2017) Population-based assessment of a biomarker-based screening pathway to aid diagnosis of monogenic diabetes in young-onset patients. Diabetes Care 40(8): 1017–1025. 10.2337/dc17-0224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].De Franco E, Flanagan SE, Houghton JA, et al. (2015) The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 386(9997): 957–963. 10.1016/S0140-6736(15)60098-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pearson ER, Flechtner I, Njolstad PR, et al. (2006) Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. The New England Journal Of Medicine 355(5): 467–477. 10.1056/NEJMoa061759 [DOI] [PubMed] [Google Scholar]
- [35].Svalastoga P, Sulen Å, Fehn JR, et al. (2020) Intellectual Disability in KATP channel neonatal diabetes. Diabetes Care 43(3): 526–533. 10.2337/dc19-1013 [DOI] [PubMed] [Google Scholar]
- [36].Shepherd M, Shields B, Hammersley S, et al. (2016) Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care 39(11): 1879–1888. 10.2337/dc16-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Carlsson A, Shepherd M, Ellard S, et al. (2020) Absence of islet autoantibodies and modestly raised glucose values at diabetes diagnosis should lead to testing for MODY: lessons from a 5-year pediatric Swedish national cohort study. Diabetes Care 43(1): 82–89. 10.2337/dc19-0747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Reilly F, Sanchez-Lechuga B, Clinton S, et al. (2019) Phenotype, genotype and glycaemic variability in people with activating mutations in the ABCC8 gene: response to appropriate therapy. Diabet Med 37(5):879–884. 10.1111/dme.14145 [DOI] [PubMed] [Google Scholar]
- [39].Dickens LT, Naylor RN (2018) Clinical management of women with monogenic diabetes during pregnancy. Curr Diab Rep 18(3): 12. 10.1007/s11892-018-0982-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT (2018) Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross-sectional, genetically stratified survival analysis from UK Biobank. The lancet Diabetes & endocrinology 6(2): 122–129. 10.1016/S2213-8587(17)30362-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hawa MI, Kolb H, Schloot N, et al. (2013) Adult-onset autoimmune diabetes in Europe is prevalent with a broad clinical phenotype: Action LADA 7. Diabetes Care 36(4): 908–913. 10.2337/dc12-0931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Spanakis EK, Golden SH (2013) Race/ethnic difference in diabetes and diabetic complications. Current Diabetes Reports 13(6): 814–823. 10.1007/s11892-013-0421-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cousminer DL, Ahlqvist E, Mishra R, et al. (2018) First genome-wide association study of latent autoimmune diabetes in adults reveals novel insights linking immune and metabolic diabetes. Diabetes Care 41(11): 2396–2403. 10.2337/dc18-1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Rolandsson O, Hampe CS, Sharp SJ, et al. (2020) Autoimmunity plays a role in the onset of diabetes after 40 years of age. Diabetologia 63(2): 266–277. 10.1007/s00125-019-05016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Brooks-Worrell BM, Boyko EJ, Palmer JP (2014) Impact of islet autoimmunity on the progressive β-cell functional decline in type 2 diabetes. Diabetes Care 37(12): 3286–3293. 10.2337/dc14-0961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Redondo MJ, Evans-Molina C, Steck AK, Atkinson MA, Sosenko J (2019) The influence of type 2 diabetes-associated factors on type 1 diabetes. Diabetes Care 42(8): 1357–1364. 10.2337/dc19-0102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dabelea D, Hamman R, Knowler W (2017) Chapter 15: Diabetes in youth. In: Cowie CC, Casagrande SS, Menke A et al. (eds) Diabetes in America, 3rdedition. NIH, Bethesda. Available fromwww.niddk.nih.gov/about-niddk/strategic-plans-reports/diabetes-in-america-3rd-edition#suggest. Accessed 18 June 2020 [Google Scholar]
- [48].Libman IM, Becker DJ (2003) Coexistence of type 1 and type 2 diabetes mellitus: “double” diabetes? Pediatric diabetes 4(2): 110–113. 10.1034/j.1399-5448.2003.00012.x [DOI] [PubMed] [Google Scholar]
- [49].Brooks-Worrell BM, Palmer JP (2019) Setting the stage for islet autoimmunity in type 2 diabetes: obesity-associated chronic systemic inflammation and endoplasmic reticulum (ER) stress. Diabetes Care 42(12): 2338–2346. 10.2337/dc19-0475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dabelea D, Pihoker C, Talton JW, et al. (2011) Etiological approach to characterization of diabetes type: the SEARCH for Diabetes in Youth Study. Diabetes Care 34(7): 1628–1633. 10.2337/dc10-2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brooks-Worrell B, Narla R, Palmer JP (2013) Islet autoimmunity in phenotypic type 2 diabetes patients. Diabetes Obes Metab 15 Suppl 3: 137–140. 10.1111/dom.12167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Yu MG, Keenan HA, Shah HS, et al. (2019) Residual β cell function and monogenic variants in long-duration type 1 diabetes patients. J Clin Invest 129(8): 3252–3263. 10.1172/JCI127397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shields BM, McDonald TJ, Ellard S, Campbell MJ, Hyde C, Hattersley AT (2012) The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 55(5): 1265–1272. 10.1007/s00125-011-2418-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Balasubramanyam A, Nalini R, Hampe CS, Maldonado M (2008) Syndromes of ketosis-prone diabetes mellitus. Endocrine Reviews 29(3): 292–302. 10.1210/er.2007-0026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Balasubramanyam A, Garza G, Rodriguez L, et al. (2006) Accuracy and predictive value of classification schemes for ketosis-prone diabetes. Diabetes care 29(12): 2575–2579. 10.2337/dc06-0749 [DOI] [PubMed] [Google Scholar]
- [56].McCarthy MI (2017) Painting a new picture of personalised medicine for diabetes. Diabetologia 60(5): 793–799. 10.1007/s00125-017-4210-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wasserfall C, Nead K, Mathews C, Atkinson MA (2011) The threshold hypothesis: solving the equation of nurture vs nature in type 1 diabetes. Diabetologia 54(9): 2232–2236. 10.1007/s00125-011-2244-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hamman RF, Bell RA, Dabelea D et al. (2014) The SEARCH for Diabetes in Youth Study: rationale, findings, and future directions. Diabetes Care 37(12):3336–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]