

Abstract
Many DNA replication and DNA repair enzymes have been found to carry [4Fe4S] clusters. The major leading strand polymerase, DNA polymerase ε (Pol ε) from Saccharomyces cerevisiae, was recently reported to have a [4Fe4S] cluster located within the catalytic domain of the largest subunit, Pol2. Here the redox characteristics of the [4Fe4S] cluster in the context of that domain, Pol2CORE, are explored using DNA electrochemistry, and the effects of oxidation and rereduction on polymerase activity are examined. The exonuclease deficient variant D290A/E292A, Pol2COREexo–, was used to limit DNA degradation. While no redox signal is apparent for Pol2COREexo– on DNA-modified electrodes, a large cathodic signal centered at −140 mV vs NHE is observed after bulk oxidation. A double cysteine to serine mutant (C665S/C668S) of Pol2COREexo–, which lacks the [4Fe4S] cluster, shows no similar redox signal upon oxidation. Significantly, protein oxidation yields a sharp decrease in polymerization, while rereduction restores activity almost to the level of untreated enzyme. Moreover, the addition of reduced EndoIII, a bacterial DNA repair enzyme containing [4Fe4S]2+, to oxidized Pol2COREexo– bound to its DNA substrate also significantly restores polymerase activity. In contrast, parallel experiments with EndoIIIY82A, a variant of EndoIII, defective in DNA charge transport (CT), does not show restoration of activity of Pol2COREexo–. We propose a model in which EndoIII bound to the DNA duplex may shuttle electrons through DNA to the DNA-bound oxidized Pol2COREexo– via DNA CT and that this DNA CT signaling offers a means to modulate the redox state and replication by Pol ε.
Introduction
All organisms require genome replication with a high degree of fidelity and have evolved complex molecular machinery to accomplish the task.1 Eukaryotic DNA replication is carried out by B-family polymerases, and it is initiated by DNA primase and DNA polymerase (Pol) α, followed by elongation of the leading and lagging strands by Pol ε and Pol δ, respectively.2 Pol ε and Pol δ are highly processive multisubunit enzymes composed of a catalytic subunit and regulatory subunits.2,3 The catalytic domain of Pol ε and Pol δ exhibits both polymerase and 3′-5′ exonuclease (proofreading) activities.2−5
Pol ε is the largest of the replicative polymerases in Saccharomyces cerevisiae, and it is composed of four subunits, Pol2 (256 kDa), Dpb2 (79 kDa), Dpb3 (23 kDa), and Dpb4 (22 kDa).5 The catalytic subunit, Pol2, is a flexible two-lobed structure that contains the N-terminal domain (NTD) in lobe 1 and the C-terminal domain (CDT) in lobe 2. The NTD catalyzes DNA polymerization and proofreading nuclease activity, whereas the CTD is noncatalytic. The intrinsically high processivity of Pol ε arises in part from two insertions (∼100 residues each) in the NTD of Pol2 that envelop the nascent DNA double strand.5,6 Pol ε processivity is also modestly increased through interactions with proliferating cellular nuclear antigen (PCNA) and the two accessory subunits, Dpb3 and Dpb4.3,5 In contrast, the smaller enzyme Pol δ (3 subunits, 220 kDa total) does not have a domain that encircles the nascent DNA strand and only becomes highly processive when bound to the PCNA replication clamp.2,6 The functions of the three accessory subunits in Pol ε include mediating interactions between the polymerase and the DNA duplex and/or other biological molecules.5−7
An important feature of eukaryotic DNA replication and repair enzymes, including polymerases, is the presence of highly conserved cubane [4Fe4S] clusters.5,8−11 Iron–sulfur clusters are common redox cofactors found in or near the active sites of enzymes in all forms of life.11 These metal cofactors appear to be ubiquitous in DNA and RNA processing enzymes,10 with the most recent example being the RNA-dependent RNA polymerase of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).10a Eukaryotic Pol ε and Pol δ also contain a [4Fe4S] cluster within their catalytic subunit.4,5 Initial literature reports suggested that the role of the [4Fe4S] cluster of DNA processing enzymes is primarily structural in nature. However, structural requirements can be addressed using alternative methods, such as simple Zn2+ cations, that would obviate the need for a metabolically expensive [4Fe4S] cofactor.8 Traditionally, the roles of [4Fe4S] clusters in biology have focused on electron transfer.8−11 As with other cluster-containing DNA processing enzymes, the [4Fe4S] of Pol ε does not directly catalyze redox transformations during its enzymatic activity.4,5 Recent studies have shown instead that redox-active [4Fe4S] cofactors in DNA processing enzymes offer a means to modulate DNA binding and therefore have strong implications in DNA replication.8,12
The redox potentials of [4Fe4S] clusters in DNA repair enzymes shift to a physiologically relevant range (∼90 mV vs NHE) when bound to the DNA polyanion; it is this shift in potential that leads to a redox switch for binding DNA.8 In addition, electrons can migrate rapidly through duplexed DNA, allowing for long-range charge transport (CT).13 DNA-mediated redox signaling has been found between DNA-bound enzymes with [4Fe4S] cluster oxidation states of 2+ and 3+.8 Studies have demonstrated that DNA repair proteins may take advantage of DNA CT to scan the genome efficiently and identify lesions, mismatches, or other perturbations. Redox-active [4Fe4S] clusters are also utilized for substrate handoff in yeast and human primase through DNA CT.12 For yeast Pol δ, the redox-active [4Fe4S] cluster provides a means to modulate polymerase activity reversibly; DNA CT from guanine radicals, generated under conditions of oxidative stress, can lead to oxidation of the [4Fe4S] cluster in Pol δ, inhibiting replication, but rereduction of the cluster restores replication activity.12 While DNA CT appears to play an important role in the activity of various polymerases, no studies on DNA synthesis by Pol ε have been reported within the context of DNA CT to/from the enzyme-bound [4Fe4S] cluster.
Here we report on the redox chemistry of the [4Fe4S] cluster in yeast Pol ε bound to DNA and its effects on polymerase activity. We examined the DNA electrochemistry of Pol2COREexo–, an exonuclease-deficient truncation of the catalytically active subunit of Pol ε.5a Pol2CORE is a polymerase- and exonuclease-active truncation of Pol2 (residues 1–1228) corresponding to the NTD. In addition, Pol2CORE is known to contain the [4Fe4S] cluster within a cysteine-rich domain “CysX”, composed of residues C665, C668, C667, and C763.5a We examined the exonuclease deficient (exo–) D290, E292A mutant of Pol2CORE, to limit DNA degradation by the polymerase.5a
Results and Discussion
DNA Electrochemistry
Cyclic voltammetry (CV) experiments were performed using DNA-modified gold electrodes to characterize the DNA-bound redox potentials of Pol2COREexo– (Figure 1 and Figures S1–S3). DNA-modified electrodes are an exceptional tool for the clean one-electron oxidation and/or reduction of [4Fe4S]-containing enzymes bound to DNA using DNA CT. By use of this system, electrons can be shuttled from the Au electrodes through the duplexed DNA, oxidizing or reducing the [4Fe4S] cluster of Pol2COREexo– depending on the applied potential (Eappl; Figure 1 top panel).
Figure 1.
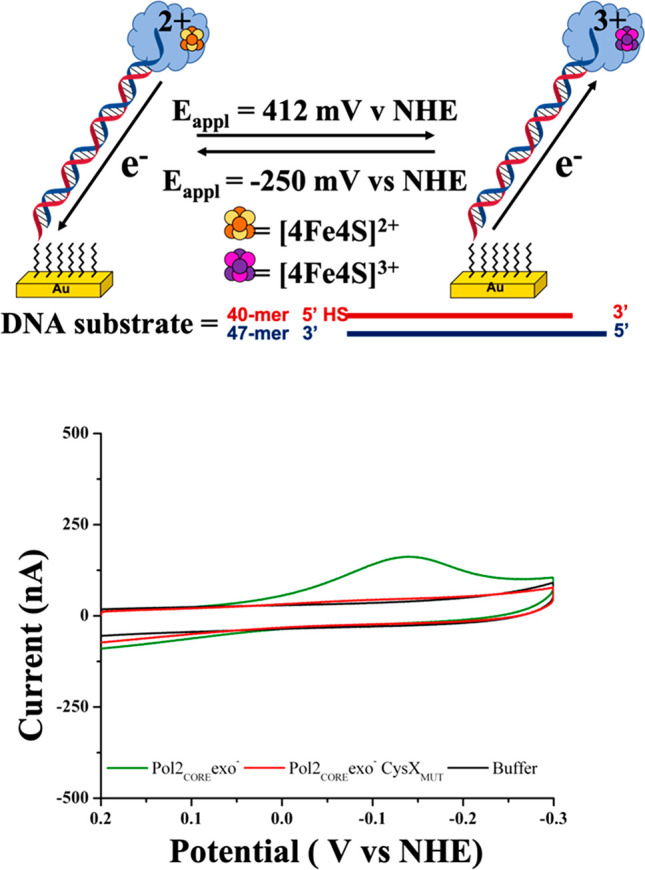
(Top panel) Electrochemical oxidation of Pol2COREexo– using DNA-modified electrodes. DNA substrate is attached to gold (Au) surface through a 5′ alkanethiol group. Complementary matched DNA strand is slightly longer, yielding a 7-nt overhang that serves as the natural binding substrate for Pol2COREexo–. (Bottom panel) CV scans of electrochemically oxidized Pol2COREexo– (5 μM) exhibit a large cathodic CV signal centered around −140 V vs NHE (green trace), but no signal is observed when Pol2COREexo–CysXMUT (red trace) or buffer (5 mM NaH2PO4, 50 mM NaCl, pH 7.0; black trace) is electrochemically oxidized. Potential applied (Eappl) for bulk oxidation is 412 mV vs NHE for 500 s. CV scan rate = 100 mV s–1. Pol2COREexo– protein sample concentrations used for cyclic voltammetry experiments are 5 μM (ε[4Fe4S]410 = 17 000 M–1 cm–1).
CV scans of electrochemically unaltered Pol2COREexo– (5 μM) samples did not show detectable cathodic or anodic signals on the DNA-modified electrodes (Figure S1). This result contrasts with CV scans on yeast Pol δ bound tightly with PCNA, where the cluster is redox-active on the DNA-modified electrode and exhibits a reversible electrochemical signal with a midpoint potential of 113 ± 5 mV vs NHE.12a It was originally expected that a similar reversible electrochemical signal might be observed for Pol2COREexo–, since Pol ε bears similarities to Pol δ, and Pol2COREexo– retains strong DNA-binding affinity.12a
Bulk electrolysis experiments were then performed to generate oxidized Pol2COREexo– (Figure 1 and Figure S1). A large cathodic signal centered at −140 mV vs NHE was observed for Pol2COREexo– after bulk oxidation (500 s, Eappl = 412 mV vs NHE; Figure 1 bottom panel, green trace). These findings resemble those obtained using human and yeast DNA primase, where electrochemically unaltered protein exhibits no signal, but the application of positive potentials (412 or 512 mV vs NHE) produces a large cathodic signal centered at −140 mV vs NHE.12b,12c
Our DNA electrochemical studies included a double cysteine to serine mutant (C665S/C668S; CysXMUT) of Pol2COREexo– (Figure 1 and Figure S2). As mentioned above, the [4Fe4S] cluster of Pol2 is in a cysteine rich domain called CysX within Pol2CORE. Studies have shown that Pol2COREexo–CysXMUT does not bind a [4Fe4S] cluster and its polymerase activity is severely compromised.5a,5d Haploid yeast cells expressing Pol ε CysXMUT have been demonstrated to be inviable.5a CV scans of Pol2COREexo–CysXMUT using DNA-modified electrodes do not show a significant cathodic signal, even after bulk oxidation (500 s, Eappl = 412 mV vs NHE; Figure 1 bottom panel, red trace). The absence of redox activity of Pol2COREexo–CysXMUT can thus be explained by the absence of the [4Fe4S] cluster. Bulk reduction (500 s, Eappl = −250 mV vs NHE) of protein and buffer samples also did not yield significant cathodic or anodic signals (data not shown).
Effects of Redox State on Polymerase Activity
These CV results prompted us to investigate the effect of oxidation of the [4Fe4S] cluster in Pol2COREexo– on its polymerase activity. A primer extension assay was utilized5a where a prewarmed solution containing dNTPs was mixed with a Pol2COREexo– solution containing a DNA template with a primer that could be elongated. The DNA substrate was composed of a 5′ fluorescein-labeled DNA primer (20-mer) annealed to a complementary template strand (50-mer). The conditions of the primer-extension assay were optimized to determine the concentration range (0.1–5 nM) of Pol2COREexo– that produced detectable, consistent, and reliable results. Primer extension products were separated using 20% denaturing urea polyacrylamide gel electrophoresis, imaged using a Typhoon scanner, and analyzed using ImageQuant software. These results are summarized in Figures 2, S4, and S5.
Figure 2.
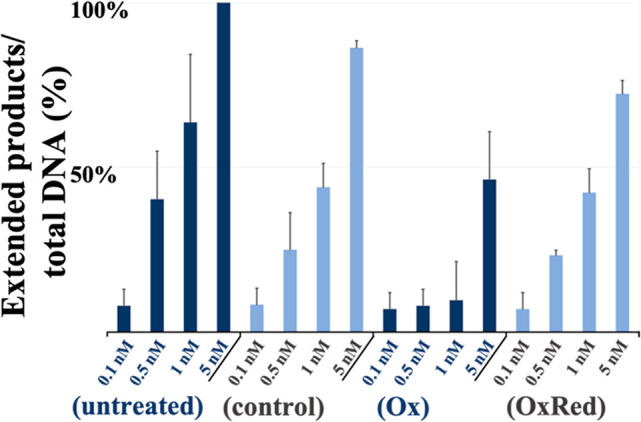
Bar graph summarizing averaged denaturing PAGE results of Pol2COREexo– primer extension assay upon [4Fe4S] cluster oxidation/reduction. (untreated) activity of untreated Pol2COREexo–: (control) activity of Pol2COREexo– after incubation on DNA modified electrodes without applied potential; (Ox) activity of Pol2COREexo– after bulk oxidation (600 s, Eappl = 412 mV vs NHE); (OxRed) activity of Pol2COREexo– after bulk oxidation (600 s, Eappl = 412 mV vs NHE) followed by bulk rereduction (600 s, Eappl = −250 mV vs NHE). The quantification of DNA products (as % of total) was obtained by dividing the signal of the total amount of extended products by the total amount of primer used (sum of all extended product signals and unextended primer signal). All experiments were carried out in triplicate; error bars indicate standard deviation.
Electrochemically untreated Pol2COREexo– yielded complete extension of the fluorescein-labeled DNA primer at a concentration of 5 nM. Pol2COREexo– samples with oxidized [4Fe4S] clusters (Ox) were prepared by performing bulk oxidation (600 s, Eappl = 412 mV vs NHE) on the DNA-modified electrodes. Protein concentrations (based on [4Fe4S]) had to be optimized (20 μL, 20 nM) since samples containing high concentrations of Pol2COREexo– (over 50 nM) or high-volume samples (over 25 μL) contained enough electrochemically unaltered protein (after bulk oxidation) to produce a strong product signal. In addition, we employed DNA-modified gold electrodes with large surface areas (A = 0.16 cm2), rather than our multiplexed chips, to decrease the amount of electrochemically unaltered protein remaining in solution. We estimate the yield of oxidized protein by taking the difference between the total charge obtained in the presence of Pol2COREexo– and the charge generated by electrolysis of the buffer alone giving >95% on average (Figure S4; see figure caption for calculation of bulk oxidation yield). Bulk electrolysis experiments were carried out in the absence of oxygen to prevent aerobic degradation of the clusters.12a Oxidized Pol2COREexo– samples showed a clear and dramatic decrease in replication when compared to Pol2COREexo– treated similarly on the electrodes but without oxidation (control; Figure 2 and Figure S5).
We then investigated whether a rereduction of the previously oxidized Pol2COREexo– samples would also influence polymerase activity. Samples of oxidized Pol2COREexo– were prepared as described above and then treated under bulk reduction conditions (600 s, Eappl = −250 mV vs NHE) on the same electrode (OxRed Pol2COREexo–). Primer extension assays of rereduced (OxRed) Pol2COREexo– revealed significant restoration of the polymerase activity (Figure 2). It should be noted that primer extension assays of Pol2COREexo– samples, subjected to incubation on the DNA-modified electrodes for equivalent time but without an applied potential (control), consistently show decreased levels of polymerization compared to untreated Pol2COREexo– (Figure 2 and Figure S5). Likely, the observed discrepancy is due to protein loss during the procedure on the electrode because of protein remaining bound to the DNA-modified electrode, as well as protein mechanically lost during removal from the electrode. These results suggest that oxidation of the [4Fe4S] of Pol2COREexo– results in the observed reversible inhibition of replication, resembling results previously obtained using Pol δ.12a Thus, the reversible oxidation and reduction of [4Fe4S] clusters in polymerases might provide a route through which polymerase activity is regulated.
It may be useful to consider how this inhibition of replication with oxidation may occur. As with other DNA-processing proteins containing [4Fe4S] clusters, the cluster is located far from the catalytic site in Pol2, and it is difficult to understand how cluster oxidation could affect catalysis. However, with other DNA-processing proteins, cluster oxidation was seen to increase binding to the DNA substrate significantly (>100×); in the case of highly processive polymerases, such tight binding could inhibit replication.
DNA CT Signaling between DNA-Bound Proteins
Next we asked whether the redox state of Pol2COREexo– and, as a result, its DNA polymerase activity could be changed through DNA CT with a DNA-binding repair enzyme containing a reduced [4Fe4S]2+ cluster. We employed endonuclease III (EndoIII), an established DNA CT-proficient base excision repair glycosylase from Escherichia coli.8,14 Could these two DNA-binding enzymes from different organisms function chemically as redox signaling partners to modulate DNA synthesis?
To accommodate Pol2COREexo– and EndoIII binding onto the same dsDNA, we increased the length of the primer/template DNA substrate. The footprint of each protein is ∼10 base pairs. The DNA substrate was composed of a 5′ fluorescein-tagged 40-mer primer (instead of the original 20-mer) which was annealed to a 50-mer complementary DNA strand, resulting in 40 nucleotides of double-stranded DNA. An identical set of experiments as the ones described for Figure 2 were carried out for Pol2COREexo– (untreated, control, Ox, and OxRed) using the longer primer:template DNA adduct (Figure 3). Ox and OxRed samples of Pol2COREexo– were prepared using DNA-modified electrodes before carrying out the primer extension assays. Results (untreated, control, Ox, and OxRed) using the 40:50 primer:template DNA substrate (Figure 3) follow similar patterns as extension assays using the 20:50 primer:template substrate. As expected, upon varying the primer length, we again observe a decrease in replication activity for the oxidized Pol2COREexo–(Ox) compared to both control and untreated samples. Similarly, we see that rereduction of oxidized Pol2COREexo– on the DNA electrode (OxRed) restores most of the activity.
Figure 3.
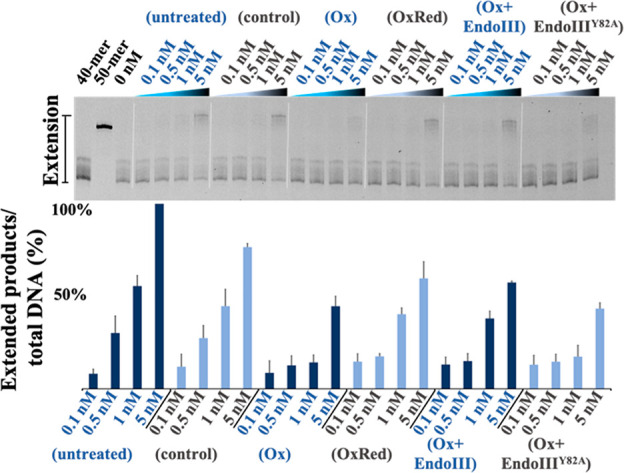
Pol2COREexo– primer extension assay results upon electrochemical oxidation/reduction using DNA-modified electrodes, and effect of DNA-mediated CT on the activity of oxidized Pol2COREexo. (Top panel) Representative example of quantitative Pol2COREexo– primer extension assay results on denaturing PAGE. (Bottom panel) Bar graph summarizing averaged results of Pol2COREexo– primer extension assay: (untreated) activity of untreated Pol2COREexo–; (control) activity of Pol2COREexo– after incubation with DNA modified electrodes without an applied potential; (Ox) activity of Pol2COREexo– after bulk oxidation (600 s, Eappl = 412 mV vs NHE); (OxRed) activity of Pol2COREexo– after bulk oxidation (600 s, Eappl = 412 mV vs NHE) followed by bulk rereduction (600 s, Eappl = −250 mV vs NHE). (Ox+EndoIII) is the activity of oxidized Pol2COREexo– after incubation with 50 nM EndoIII. (Ox+EndoIIIY82A) is the activity of oxidized Pol2COREexo– after incubation with 50 nM EndoIIIY82A. It should be noted that experiments with Ox, Ox+EndoIII, and Ox+EndoIIIY82A were performed using the same oxidized Pol2COREexo– sample. Bulk oxidation and bulk reduction were performed on 20 μL of 20 nM Pol2COREexo– using DNA-modified electrodes. All experiments were carried out in triplicate; error bars indicate standard deviation.
We then asked whether EndoIII could serve as the reductant bound to DNA. The oxidized Pol2COREexo– sample prepared (20 μL, 20 nM) was diluted with buffer (5 mM NaH2PO4, 50 mM NaCl, pH 7.0) to appropriate concentrations, combined with the DNA substrate, and then mixed with either oxidized Pol2COREexo– sample (Ox) or oxidized Pol2COREexo– sample and EndoIII (Ox+EndoIII) to reach the final concentrations (0.1–5 nM Pol2COREexo–); the solution was then incubated at ambient temperature before mixing with the prewarmed dNTPs to initiate the polymerase assay. The concentrations of EndoIII used were fixed at 50 nM (based on the concentration of [4Fe4S] cluster), 1 order of magnitude higher than the highest concentration of Pol2COREexo– used. The results (Figure 3) indicate that incubation of oxidized Pol2COREexo– with EndoIII restores polymerization to similar levels as those obtained when the protein is reduced on DNA-modified electrodes (compare Ox+EndoIII with OxRed, Figure 3). Still higher concentrations of EndoIII (250 nM) gave similar results (Figure S6). Overall, the activity of Ox+EndoIII is consistently slightly lower in activity than OxRed but higher than the oxidized sample. Again, we attribute these variations to the difficulty in removing oxidized protein from the electrode. As a control, we then included the variant EndoIIIY82A, which has similar enzymatic activity and binds to DNA with the same affinity as wt EndoIII but is deficient in carrying out DNA CT.8,12e,14de EndoIIIY82A was used following the same protocols as EndoIII (Ox+EndoIIIY82A). The results with the CT-deficient mutant show little if any increase in activity relative to oxidized Pol2COREexo–. These results taken together thus support the ability of the EndoIII protein to interact with Pol ε through DNA CT, yielding a restoration of polymerase activity upon cluster reduction to the 2+ form.
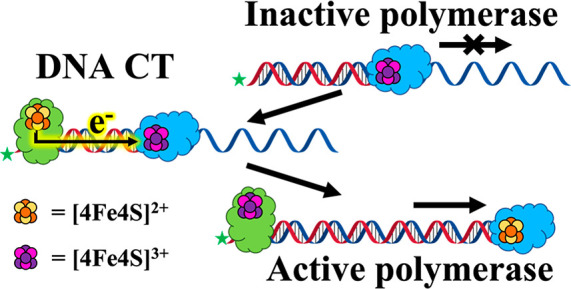
A model is presented in Figure 4, where Pol2COREexo–, the exonuclease deficient and truncated catalytic subunit of Pol ε, shows normal polymerase activity when presented with dNTPs and a DNA-primer extension substrate (Figure 4a). Bulk oxidation of Pol2COREexo– using DNA-modified electrodes results in a decrease in polymerase activity with fewer replication products (b → a′, Figure 4). Bulk reduction of the oxidized Pol2COREexo– sample using DNA-modified electrodes can restore replication (b → c → a, Figure 4). But also addition of excess EndoIII similarly restores the polymerase activity through DNA-mediated redox signaling (b → a′ → d, Figure 4). In contrast, addition of EndoIIIY82A, deficient in DNA CT, shows no restoration of polymerase activity; here DNA CT between proteins is not available and Pol2COREexo– remains inactive (b → a′ → e, Figure 4). Redox control of Pol ε would thus provide interesting opportunities and is intriguing to consider, but the in vivo mechanism and possible partners still require further investigation.
Figure 4.
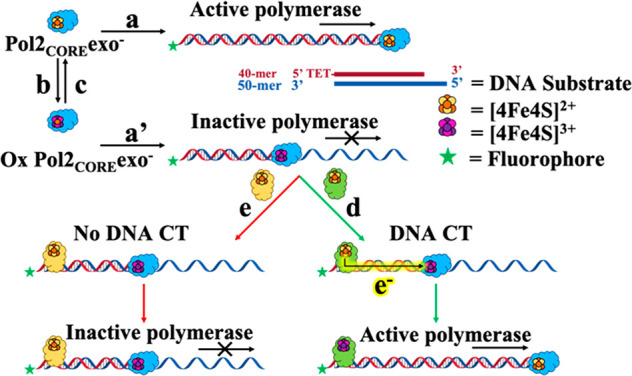
Model for DNA-mediated redox signaling between DNA processing enzymes Pol2COREexo– and EndoIII. (a) Pol2COREexo– in the presence of a DNA substrate and dNTPs has normal polymerase activity. (b) Bulk oxidation using DNA-modified electrodes produces oxidized Pol2COREexo– (a′). Oxidized Pol2COREexo– shows diminished polymerization in the presence of DNA substrate and dNTPs. (c) Rereduction of oxidized Pol2COREexo– by bulk electrolysis using DNA-modified electrodes restores Pol2COREexo– polymerization activity. (d) EndoIII is CT proficient and can reduce oxidized Pol2COREexo– through DNA-mediated redox signaling. (e) EndoIIIY82A is CT-deficient and cannot reduce DNA-bound and oxidized Pol2COREexo–; therefore the oxidized Pol2COREexo– remains inactive.
Conclusion
Taken together, the results reported here illustrate that the [4Fe4S] cluster of Pol2COREexo– is redox-active when the protein is bound to DNA and that the cluster oxidation state affects the polymerase activity reversibly; oxidation of the cluster inhibits DNA synthesis. Significantly, DNA-mediated CT may occur between a repair protein, also containing a redox-active [4Fe4S], and Pol2COREexo– to restore the polymerase activity of Pol ε. Redox signaling through DNA CT thus has the potential to rapidly modulate replication by Pol ε.
Experimental Section
General Methods
All reagents were obtained from commercial sources and used as received unless stated otherwise. Water used to prepare buffer solutions was purified on a Milli-Q Reference Ultrapure Water Purification System. Electrochemistry experiments used a standard three-electrode cell composed of multiplexed Au chip or a continuous Au(111) surface (0.16 cm2) bearing DNA modification as the working electrode, an Ag/AgCl reference electrode in 3 M NaCl (BASInc.), and a 1 mm diameter Pt wire (Lesker) as the counter electrode.12,13 Potentials were converted from Ag/AgCl to NHE by adding 212 mV to the potentials measured by the Ag/AgCl; this conversion accounted for both ambient temperature and the use of 3 M NaCl for reference storage.12a To prevent cluster degradation in the presence of O2, all electrochemical manipulations and polymerase activity assays involving oxidized or reduced Pol2COREexo– samples were carried out under strict anaerobic conditions in vinyl chambers (glove bags) kept at atmospheres of 2–4% H2 in argon or N2 (≤1 ppm of O2) with Pd scrubbing towers (Coy Laboratories). Buffers were degassed by bubbling argon for a minimum of 2 h and stored under anaerobic conditions. UV–vis data were acquired using a Cary 100 Bio (Agilent) spectrophotometer. SDS–PAGE gel images were acquired using a Typhoon FLA 9000 (GE), and the resulting images were analyzed using ImageQuant software.
Protein Purifications
1xFLAG-tagged Pol2COREexo– and Pol2COREexo– CysXMUT were expressed in yeast and initial purification via M2 resin as previously described for full-length Pol ε.5a . 1 mM DTT (instead of TCEP) was added to the elution fractions, which were then concentrated on a 50 kDa cutoff filter (Amicon) and loaded onto a Superose 12 PC 3.2/30 column (GEHealthcare) equilibrated with 25 mM HEPES, pH 7.6, 10% glycerol, 300 mM NaAc, 1 mM DTT, and 0.005% NP-40. E. coli EndoIII and EndoIIIY82A were expressed and purified according to previously published protocols.15a
General DNA Preparation
All oligonucleotides were purchased from Integrated DNA Technologies (IDT) and purified by reverse-phase high performance liquid chromatography using a C-18 column (Agilent). Masses of purified oligonucleotides were confirmed by MALDI-TOF/TOF mass spectrometry (Bruker AutoFlex) using MALDIAnalyzer ionization. Oligonucleotides containing thiol modifications were obtained from IDT in their disulfide form and were chemically reduced using 50-fold excess of tris(2-carboxyethyl)phosphine HCl and purified using Micro BioSpin columns pre-equilibrated with DNA buffer (50 mM NaCl, 5 mM NaH2PO4, pH 7.0) prior to annealing. DNA strands were annealed (1:1, 50 μM oligonucleotides in 100 μL) for 5 min at 90 °C followed by cooling to ambient temperature over 90 min in argon-sparged DNA buffer (50 mM NaCl, 5 mM NaH2PO4, pH 7.0). Annealed DNA samples were kept at −20 °C, used within 1 week, and thawed immediately before use.
DNA Electrochemistry
DNA-modified electrodes12−15 were prepared by deprotecting and purifying a 40-mer ssDNA bearing a 5′-thiol modification and annealing it to a complementary well-matched 47-mer ssDNA. Annealing of these two strands yields a 7-nucleotide overhang that may serve as a natural binding site for Pol2COREexo–. The resulting double stranded DNA (dsDNA) substrate was incubated overnight on a set of multiplexed gold electrodes to produce self-assembled low-density DNA monolayers, with surface coverage of about 15–20 pmol cm–2 with respect to dsDNA (determined by established protocols16). The electrode surfaces were then washed, passivated using β-mercaptohexanol, and rinsed again using degassed DNA buffer (5 mM NaH2PO4, 50 mM NaCl, pH 7.0). The electrode subassembly was brought into the anaerobic chamber and connected to the potentiostat equipped with a multiplexer (CH Instruments). Pol2COREexo– samples, originally stored at −80 °C, were then brought into the anaerobic chamber and thawed immediately before the start of electrochemical characterizations. The concentrations of Pol2COREexo– samples used for electrochemical characterizations were first adjusted to 5 μM based on [4Fe4S] cluster absorbance (ε[4Fe4S]410 = 17 000 M–1 cm–1). Cyclic voltammetry (CV; 100 mV/s scan rate) and square wave voltammetry (SQWV; 15 Hz frequency, 25 mV amplitude) scans were performed on 25 μL samples of buffer (5 mM NaH2PO4, 50 mM NaCl, pH 7.0), Pol2COREexo–, or Pol2COREexo– CysXMUT each using one separate quadrant of the multiplexed electrode subassembly.
DNA Substrates for Electrochemistry
40-mer with 5′ thiol modification: 5′ HS-GTG CTG CAA CGT GTC TGC GCG CTG AGT GCA CGC AAC TCG C 3′.
47-mer (well matched): 5′ CTG TCG TGC GAG TTG CGT GCA CTC AGC GCG CAG ACA CGT TGC AGC AC 3′.
Primer Extension Assays
Immediately prior to activity assays, a sample of Pol2COREexo– (20 nM) was prepared and aliquoted (20 μL each). One aliquot was kept without manipulation (untreated), one was incubated in the DNA-modified electrodes without an applied potential (control), another one was electrochemically oxidized (Ox; 600 s, Eappl = 412 mV vs NHE), and a fourth was electrochemically oxidized and reduced (OxRed; 600 s, Eappl = 412 mV vs NHE followed by 600 s, Eappl = −250 mV vs NHE) using a continuous DNA-modified Au(111) surface electrode (A = 0.16 cm2) electrodes. Low sample volumes (20 μL) and continuous Au-electrode wafers were employed to increase the yield of bulk oxidized/reduced Pol2COREexo– and therefore decrease the amount of electrochemically unaltered protein. The time that Pol2COREexo– samples were incubated on DNA-modified electrodes was kept constant (1 h) across all samples for every experiment.
The enzymatic activities of Pol2COREexo– (untreated, control, Ox, and OxRed) were investigated using a primer extension assay adapted from an established protocol.5a Briefly, reaction mixture A (Mix A), containing untreated, control, Ox, or OxRed Pol2COREexo–, DNA substrate (20 nM; 20:50 primer:template), Tris-HCl (20 mM, pH 7.8), sodium acetate (40 mM), and bovine serum albumin (BSA, 0.1 mg mL–1), was prepared and kept on ice. Reaction mixture B (Mix B), containing Tris-HCl (20 mM, pH 7.8), magnesium acetate (16 mM), BSA (0.1 mg mL–1), and dNTP mix (0.2 mM each), was prepared, aliquoted, and preincubated for at least 20 min at 30 °C. The primer extension assay was initiated by addition of 10 μL of Mix A to 10 μL of prewarmed Mix B followed by incubation at 30 °C for 10 min. The activity assay was terminated by addition of 20 μL of stop solution (95% formamide, 20 mM EDTA, 0.1% bromophenol blue, 0.05% xylene cyanol). Primer extension products were separated on a 20% denaturing urea polyacrylamide gel at 90 W for 2.5 h and visualized by fluorescence imaging using a Typhoon FLA 9000.
Primer extension assays using EndoIII and EndoIIIY82A as the redox signaling partner were carried out as described above with the following modifications. Reaction mixture A′ (Mix A′) employed the use of a 40:50 primer:template DNA substrate (instead of 20:50) and contained EndoIII or EndoIIIY82A (50 nM). All other concentrations, volumes, and conditions were kept constant. Visual discrimination between starting primer and extended product required an increase in SDS–PAGE separation time from 2.5 to 3.5 h. It is important to note that activity assays Ox, Ox+EndoIII, and Ox+EndoIIIY82A were performed using the same electrochemically oxidized (600 s, Eappl = 412 mV vs NHE) Pol2COREexo– sample. Primer extension assay investigating two different concentrations of EndoIII and EndoIIIY82A (50 nM and 250 nM; Figure S6) were performed as described above. Also note that the same electrochemically oxidized (600 s, Eappl = 412 mV vs NHE) Pol2COREexo– sample was used for assays Ox, Ox+EndoIII, and Ox+EndoIIIY82A in both concentrations.
DNA Substrates for Primer Extension Assay
20-mer with 5′ tetrachlorofluorescein (TET) modification: 5′ TET-CGA GCC GTC TAC TCA ACT CA 3′.
40-mer with 5′ tetrachlorofluorescein (TET) modification: 5′ TET- CGA GCC GTC TAC TCA ACT CAT CCA GAA CAA CGT CAC TGA C 3′.
50-mer (well matched): 5′ CAG CTT GAT AGT CAG TGA CGT TGT TCT GGA TGA GTT GAG TAG ACG GCT CG 3′.
Acknowledgments
We are grateful to the Swedish Cancer Foundation (E.J.), Swedish Research Council (E.J.), Tryggers Stiftelse (E.J.), and NIH (Grant NIH GM126904 (J.K.B.)) for their financial support. We thank Prof. Jason Slinker for fabrication of multiplexed chips. We also thank Dr. Rebekah Silva and Dr. Adela Nano for help with protein assays, instrumentation, and valuable discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c07150.
CV data of Pol2COREexo–, Pol2COREexo–CysXMUT, and DNA buffer; bulk oxidation yield curve; SDS–PAGE primer extension assay image (20:50 primer:template); and primer extension assay comparing activity of Pol2COREexo– samples treated with EndoIII (50–250 nM) and EndoIIIY82A (50–250 nM) results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Ganai R.; Johansson E. DNA Replication-A Matter of Fidelity. Mol. Cell 2016, 62, 745–755. 10.1016/j.molcel.2016.05.003. [DOI] [PubMed] [Google Scholar]; b Kunkel T. A.; Bebenek K. DNA Replication Fidelity. Annu. Rev. Biochem. 2000, 69, 497–529. 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]; c Rivera-Mulia J. C.; Gilbert D. M. Replicating Large Genomes: Divide and Conquer. Mol. Cell 2016, 62, 756–765. 10.1016/j.molcel.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Burgers P. M. J.; Kunkel T. A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. 10.1146/annurev-biochem-061516-044709. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lujan S. A.; Williams J. S.; Kunkel T. A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. 10.1016/j.tcb.2016.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mondol T.; Stodola J. L.; Galletto R.; Burgers P. M. PCNA Accelerates the Nucleotide Incorporation Rate by DNA Polymerase Delta. Nucleic Acids Res. 2019, 47, 1977–1986. 10.1093/nar/gky1321. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee M. Y. W. T.; Wang X.; Zhang S.; Zhang Z.; Lee E. Y. C. Regulation and Modulation of Human DNA polymerase δ Activity and Function. Genes 2017, 8, 190. 10.3390/genes8070190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jain R.; Rice W. J.; Malik R.; Johnson R. E.; Prakash L.; Prakash S.; Ubarretxena-Belandia I.; Aggarwal A. K. Cryo-EM Structure and Dynamics of Eukaryotic DNA Polymerase δ Holoenzyme. Nat. Struct. Mol. Biol. 2019, 26, 955–962. 10.1038/s41594-019-0305-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lancey C.; Tehseen M.; Raducanu V. S.; Rashid F.; Merino N.; Ragan T. J.; Savva C. G.; Zaher M. S.; Shirbini A.; Blanco F. J.; Hamdan S. M.; De Biasio A. Structure of the Processive Human Pol δ Holoenzyme. Nat. Commun. 2020, 11, 1109. 10.1038/s41467-020-14898-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a ter Beek J.; Parkash V.; Bylund G. O.; Osterman P.; Sauer-Eriksson A. E.; Johansson E. Structural Evidence for an Essential Fe-S Cluster in the Catalytic Core Domain of DNA Polymerase. Nucleic Acids Res. 2019, 47, 5712–5722. 10.1093/nar/gkz248. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yuan Z.; Georgescu R.; Schauer G. D.; O’Donnell M. E.; Li H. Structure of the Polymerase ε Holoenzyme and Atomic Model of the Leading Strand Replisome. Nat. Commun. 2020, 11, 3165. 10.1038/s41467-020-16910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hogg M.; Osterman P.; Bylund G. O.; Ganai R. A.; Lundstrom E. B.; Sauer-Eriksson A. E.; Johansson E. Structural Basis for Processive DNA Synthesis by Yeast DNA Polymerase. Nat. Struct. Mol. Biol. 2014, 21, 49–55. 10.1038/nsmb.2712. [DOI] [PubMed] [Google Scholar]; d Jain R.; Vanamee E. S.; Dzikovski B. G.; Buku A.; Johnson R. E.; Prakash L.; Prakash S.; Aggarwal A. K. An Iron-Sulfur Cluster in the Polymerase Domain of Yeast DNA Polymerase δ. J. Mol. Biol. 2014, 426, 301–308. 10.1016/j.jmb.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Asturias F. J.; Cheung I. K.; Sabouri N.; Chilkova O.; Wepplo D.; Johansson E. Structure of Saccharomyces cerevisiae DNA Polymerase Epsilon by Cryo-Electron Microscopy. Nat. Struct. Mol. Biol. 2006, 13, 35–43. 10.1038/nsmb1040. [DOI] [PubMed] [Google Scholar]; f Chilkova O.; Jonsson B.-H.; Johansson E. The Quaternary Structure of DNA Polymerase ϵ from Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 14082–14086. 10.1074/jbc.M211818200. [DOI] [PubMed] [Google Scholar]
- Chilkova O.; Stenlund P.; Isoz I.; Stith C. M.; Grabowski P.; Lundstrom E. B.; Burgers P. M.; Johansson E. The Eukaryotic Leading and Lagging Strand DNA Polymerases are Loaded onto Primer-Ends via Separate Mechanisms but Have Comparable Processivity in the Presence of PCNA. Nucleic Acids Res. 2007, 35, 6588–6597. 10.1093/nar/gkm741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Goswami P.; Abid Ali F.; Douglas M. E.; Locke J.; Purkiss A.; Janska A.; Eickhoff P.; Early A.; Nans A.; Cheung A. M. C.; Diffley J. F. X.; Costa A. Structure of DNA-CMG-Pol Epsilon Elucidates the Roles of the Non-Catalytic Polymerase Modules in the Eukaryotic Replisome. Nat. Commun. 2018, 9, 5061. 10.1038/s41467-018-07417-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b He H.; Li Y.; Dong Q.; Chang A.-Y.; Gao F.; Chi Z.; Su M.; Zhang F.; Ban Y.; Martienssen R.; Chen Y.-H.; Li F. Coordinated Regulation of Heterochromatin Inheritance by Dpb3-Dpb4 Complex. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 12524–12529. 10.1073/pnas.1712961114. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Aksenova A.; Volkov K.; Maceluch J.; Pursell Z. F.; Rogozin I. B.; Kunkel T. A.; Pavlov Y. I.; Johansson E. Mismatch Repair-Independent Increase in Spontaneous Mutagenesis in Yeast Lacking Non-Essential Subunits of DNA Polymerase I. PLoS Genet. 2010, 6, e1001209 10.1371/journal.pgen.1001209. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tsubota T.; Maki S.; Kubota H.; Sugino A.; Maki H. Double-Stranded DNA Binding Properties of Saccharomyces cerevisiae DNA Polymerase ε and of the Dpb3p-Dpb4p Subassembly. Gene Cells 2003, 8, 873–888. 10.1046/j.1365-2443.2003.00683.x. [DOI] [PubMed] [Google Scholar]
- Barton J. K.; O’Brien E.; Silva R. B. M. Redox Chemistry in the Genome: Emergence of the [4Fe4S] Cofactor in Repair and Replication. Annu. Rev. Biochem. 2019, 88, 163–190. 10.1146/annurev-biochem-013118-110644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Netz D. J. A.; Stith C. M.; Stumpfig M.; Kopf G.; Vogel D.; Genau H. M.; Stodola J. L.; Lill R.; Burgers P. M. J.; Pierik A. J. Eukaryotic DNA Polymerases Require an Iron-Sulfur Cluster for the Formation of Active Complexes. Nat. Chem. Biol. 2012, 8, 125–132. 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Klinge S.; Hirst J.; Maman J. D.; Krude T.; Pellegrini L. An Iron-Sulfur Domain of the Eukaryotic Primase is Essential for RNA Primer Synthesis. Nat. Struct. Mol. Biol. 2007, 14, 875–877. 10.1038/nsmb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Weiner B. E.; Huang H.; Dattilo B. M.; Nilges M. J.; Fanning E.; Chazin W. J. An Iron-Sulfur Cluster in the C-Terminal Domain of the p58 Subunit of Human DNA Primase. J. Biol. Chem. 2007, 282, 33444–33451. 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- a Maio N.; Lafont B. A. P.; Sil D.; Li Y.; Bollinger J. M. Jr.; Krebs C.; Pierson T. C.; Linehan W. M.; Rouault T. A. Fe-S Cofactors in the SARS-CoV-2 RNA-Dependent RNA Polymerase are Potential Antiviral Targets. Science 2021, 373, 236–241. 10.1126/science.abi5224. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Girbig M.; Misiaszek A. D.; Vorlander M. K.; Lafita A.; Grotsch H.; Baudin F.; Bateman A.; Muller C. W. Cryo_EM Structures of Human RNA Polymerase III in its Unbound and Transcribing States. Nat. Struct. Mol. Biol. 2021, 28, 210–219. 10.1038/s41594-020-00555-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shimberg G. D.; Michalek J. L.; Oluyadi A. A.; Rodrigues A. V.; Zucconi B. E.; Neu H. M.; Ghosh S.; Sureschandra K.; Wilson G. M.; Stemmler T. L.; Michel S. L. J. Cleavage and Polyadenylation Specificity Factor 30: An RNA-Binding Zinc Finger Protein with an Unexpected 2Fe-2S Cluster. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 4700–4705. 10.1073/pnas.1517620113. [DOI] [PMC free article] [PubMed] [Google Scholar]; d White M. F.; Dillingham M. S. Iron-Sulphur Clusters in Nucleic Acid Processing Enzymes. Curr. Opin. Struct. Biol. 2012, 22, 94–100. 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]; e Lukianova O. A.; David S. S. A Role for Iron-Sulfur Clusters in DNA Repair. Curr. Opin. Chem. Biol. 2005, 9, 145–151. 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]; f Baranovskiy A. G.; Siebler H. M.; Pavlov Y. I.; Tahirov T. H. Iron-Sulfur Clusters in DNA Polymerases and Primases of Eukaryotes. Methods Enzymol. 2018, 599, 1–20. 10.1016/bs.mie.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rees D. C.; Howard J. B. The Interface Between the Biological and Inorganic Worlds: Iron-Sulfur Metalloclusters. Science 2003, 300, 929–931. 10.1126/science.1083075. [DOI] [PubMed] [Google Scholar]; b Beinert H.; Holm R. H.; Munch E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]; c Johnson D. C.; Dean D. R.; Smith A. D.; Johnson M. K. Structure, Function, and Formation of Biological Iron-Sulfur Clusters. Annu. Rev. Biochem. 2005, 74, 247–281. 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- a Bartels P. L.; Stodola J. L.; Burgers P. M. J.; Barton J. K. A Redox Role for the [4Fe4S] Cluster of Yeast DNA Polymerase δ. J. Am. Chem. Soc. 2017, 139, 18339–18348. 10.1021/jacs.7b10284. [DOI] [PMC free article] [PubMed] [Google Scholar]; b O’Brien E.; Holt M. E.; Thompson M. K.; Salay L. E.; Ehlinger A. C.; Chazin W. J.; Barton J. K. The [4Fe4S] Cluster of Human DNA Primase Functions as a Redox Switch Using DNA Charge Transport. Science 2017, 355, eaag1789. 10.1126/science.aag1789. [DOI] [PMC free article] [PubMed] [Google Scholar]; c O’Brien E.; Salay L. E.; Epum E. A.; Friedman K. L.; Chazin W. J.; Barton J. K. Yeast Require Redox Switching in DNA Primase. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 13186–13191. 10.1073/pnas.1810715115. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tse E. C. M.; Zwang T. J.; Barton J. K. The Oxidation State of [4Fe4S] Clusters Modulates the DNA-Binding Affinity of DNA Repair Proteins. J. Am. Chem. Soc. 2017, 139, 12784–12792. 10.1021/jacs.7b07230. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Grodick M. A.; Segal H. M.; Zwang T. J.; Barton J. K. DNA-Mediated Signaling by Proteins with 4Fe-4S Clusters is Necessary for Genomic Integrity. J. Am. Chem. Soc. 2014, 136, 6470–6478. 10.1021/ja501973c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tse E. C. M.; Zwang T. J.; Bedoya S.; Barton J. K. Effective Distance for DNA-Mediated Charge Transport Between DNA Repair Proteins. ACS Cent. Sci. 2019, 5, 65–72. 10.1021/acscentsci.8b00566. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zwang T. J.; Tse E. C. M.; Barton J. K. Sensing DNA Through DNA Charge Transport. ACS Chem. Biol. 2018, 13, 1799–1809. 10.1021/acschembio.8b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ekanger L. A.; Oyala P. H.; Moradian A.; Sweredoski M. J.; Barton J. K. Nitric Oxide Modulates Endonuclease III Redox Activity by a 800 mV Negative Shift upon [Fe4S4] Cluster Nitrosylation. J. Am. Chem. Soc. 2018, 140, 11800–11810. 10.1021/jacs.8b07362. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ha Y.; Arnold A. R.; Nuñez N. N.; Bartels P. L.; Zhou A.; David S. S.; Barton J. K.; Hedman B.; Hodgson K. O.; Solomon E. Sulfur K-Edge XAS Studies of the Effect of DNA Binding on the [Fe4S4] Site in EndoIII and MutY. J. Am. Chem. Soc. 2017, 139, 11434–11442. 10.1021/jacs.7b03966. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Romano C. A.; Sontz P. A.; Barton J. K. Mutants of the Base Excision Repair Glycosylase Endonuclease III: DNA Charge Transport as a First Step in Lesion Detection. Biochemistry 2011, 50, 6133–6145. 10.1021/bi2003179. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gorodetsky A. A.; Boal A. K.; Barton J. K. Direct Electrochemistry of Endonuclease III in the Presence and Absence of DNA. J. Am. Chem. Soc. 2006, 128, 12082–12083. 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- a Pheeney C. G.; Arnold A. R.; Grodick M. A.; Barton J. K. Multiplexed Electrochemistry of DNA-Bound Metalloproteins. J. Am. Chem. Soc. 2013, 135, 11869–11878. 10.1021/ja4041779. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Slinker J. D.; Muren N. B.; Gorodetsky A. A.; Barton J. K. Multiplexed DNA-Modified Electrodes. J. Am. Chem. Soc. 2010, 132, 2769–2774. 10.1021/ja909915m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yu H.-Z.; Luo C.-Y.; Sankar C. G.; Sen D. Voltammetric Procedure for Examining DNA-Modified Surfaces: Quantitation, Cationic Binding Activity, and Electron-Transfer Kinetics. Anal. Chem. 2003, 75, 3902–3907. 10.1021/ac034318w. [DOI] [PubMed] [Google Scholar]; b Boon E. M.; Salas J. E.; Barton J. K. An Electrical Probe of Protein-DNA Interactions on DNA Modified Surfaces. Nat. Biotechnol. 2002, 20, 282–286. 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]; c Kelley S. O.; Barton J. K.; Jackson N. M.; Hill M. G. Electrochemistry of Methylene Blue Bound to a DNA-modified Electrode. Bioconjugate Chem. 1997, 8, 31–37. 10.1021/bc960070o. [DOI] [PubMed] [Google Scholar]; d Boon E. M.; Barton J. K.; Bhagat V.; Nersissian M.; Wang W.; Hill M. G. Reduction of Ferricyanide by Methylene Blue at a DNA-Modified Rotating-Disk Electrode. Langmuir 2003, 19, 9255–9259. 10.1021/la030266u. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.