

Abstract
Excess of glucocorticoids, either due to disease or iatrogenic, increases bone resorption and decreases bone formation and is a leading cause of osteoporosis and bone fractures worldwide. Improved therapeutic strategies are sorely needed. We investigated whether activating Wnt/β-catenin signaling protects against the skeletal actions of glucocorticoids, using female mice lacking the Wnt/β-catenin antagonist and bone formation inhibitor Sost. Glucocorticoids decreased the mass, deteriorated the microarchitecture, and reduced the structural and material strength of bone in wild-type (WT), but not in Sost−/− mice. The high bone mass exhibited by Sost−/− mice is due to increased bone formation with unchanged resorption. However, unexpectedly, preservation of bone mass and strength in Sost−/− mice was due to prevention of glucocorticoid-induced bone resorption and not to restoration of bone formation. In WT mice, glucocorticoids increased the expression of Sost and the number of sclerostin-positive osteocytes, and altered the molecular signature of the Wnt/β-catenin pathway by decreasing the expression of genes associated with both anti-catabolism, including osteoprotegerin (OPG), and anabolism/survival, such as cyclin D1. In contrast in Sost−/− mice, glucocorticoids did not decrease OPG but still reduced cyclin D1. Thus, in the context of glucocorticoid excess, activation of Wnt/β-catenin signaling by Sost/sclerostin deficiency sustains bone integrity by opposing bone catabolism despite markedly reduced bone formation and increased apoptosis. This crosstalk between glucocorticoids and Wnt/β-catenin signaling could be exploited therapeutically to halt resorption and bone loss induced by glucocorticoids and to inhibit the exaggerated bone formation in diseases of unwanted hyperactivation of Wnt/β-catenin signaling. © 2016 American Society for Bone and Mineral Research.
Keywords: CORTICOSTEROIDS, OSTEOPOROSIS, WNT/β-CATENIN/LRPS, GENETIC ANIMAL MODELS, MOLECULAR PATHWAYS - REMODELING
Introduction
Excess of glucocorticoids, either endogenously or iatrogenically induced, leads to loss of bone and is one of the leading causes of increased bone fragility worldwide.(1) Glucocorticoids, produced and released by the adrenal glands in response to biological stress, regulate numerous physiological processes in a wide range of tissues.(2,3) Exaggerated production of glucocorticoids is found in patients with pituitary tumors producing adrenocorticotropin hormone (ACTH) (Cushing’s disease), or exhibiting Cushing’s syndrome due to ectopic ACTH production or autonomous adrenal production of cortisol. Further, glucocorticoid action progressively increases with age due to elevated hormonal adrenal secretion as well as conversion of inactive to active metabolites catalyzed by 11β-hydroxysteroid-dehydrogenase 1 in target tissues. In addition, glucocorticoids are extensively used for the treatment of immune and inflammatory conditions including rheumatoid arthritis, for the management of organ transplantation, and as components of chemotherapy regimens for hematological cancers including multiple myeloma. Both endogenous and pharmacologic increase in glucocorticoid levels are associated with severe adverse side effects manifested in several tissues and organs, in particular the skeleton.(4–8) Prolonged treatment with glucocorticoids leads to a dramatic loss of bone mineral and strength in cortical and cancellous bone, and increases atraumatic fractures in approximately 30% to 50% of patients.(9–14) Excess glucocorticoids also causes muscle weakness with the consequent loss of body balance and increased falls, which in turn contribute to elevate the risk of bone fractures.(8) It is estimated that 4.4 million individuals in the United States and the United Kingdom are chronically treated with oral glucocorticoids,(8,15) and the use of these agents extends worldwide.
Human and animal studies demonstrate that the mechanism of glucocorticoid-induced osteoporosis involves increased bone resorption, decreased bone formation, and increased apoptosis of osteocytes and osteoblasts, which together contribute to bone weakness and elevated fracture risk.(10–14,16–20) There is a rapid, early bone loss ascribed to both prolongation of lifespan of preexisting osteoclast and increased generation of new osteoclasts.(16,21,22) This initial phase is followed by a less pronounced chronic bone loss associated with reduction of osteoblasts and osteoclasts and low bone remodeling, due to reduced synthetic activity of osteoblasts through downregulation of critical genes, including osteocalcin (OCN), increased osteoblast apoptosis, and reduced generation of osteoclasts resulting from the loss of supporting cells.(1,10,12)
The Wnt/β-catenin pathway has a critical role in the control of bone acquisition and maintenance. Wnt/β-catenin signaling is activated by ligands of the Wnt family that bind to frizzled receptors and co-receptors, and also by downregulation of antagonists, including Dkk1 and Sost/sclerostin.(23) Human mutations responsible for high bone mass conditions, including gain-of-function of the LRP5 Wnt co-receptor (OMIM# 603506.0013), loss of expression of the Sost/sclerostin inhibitor in Van Buchem disease (OMIM# 239100) and sclerosteosis type 1 (OMIM# 269500), and loss-of-function of the sclerostin chaperone LRP4 in sclerosteosis type 2 (OMIM# 614305), demonstrate that activation of the Wnt/β-catenin pathway is linked to increased bone formation and bone gain.(24–26) In addition, genetic manipulation of β-catenin, the mediator of the canonical Wnt pathway, affects bone resorption due to regulation of the expression of osteoprotegerin (OPG), the decoy receptor for receptor activator of nuclear factor kappa B ligand (RANKL), demonstrating that this pathway is also linked to resorption.(27,28) Therefore, the Wnt/β-catenin signaling cascade can regulate bone mass by both bone anabolic and anti-catabolic mechanisms. Further, activation of Wnt/β-catenin signaling promotes osteoblast and osteocyte survival.(29) Based on these pieces of evidence, we investigated whether activation of Wnt/β-catenin signaling in female mice lacking Sost/sclerostin opposes glucocorticoid-induced loss of bone mineral and strength, and examined the cellular and molecular mechanisms involved.
Materials and Methods
Mice and tissue procurement
Four-month-old female Sost−/− mice of C57BL/6 background generated by Li and colleagues(30) or wild-type (WT) littermate controls, 10 mice per group, were implanted with 90-day slow-release pellets delivering placebo, 1.4 mg/kg/day (GC1), or 2.1 mg/kg/day (GC2) prednisolone (Innovative Research of America, Sarasota, FL, USA) while under anesthesia. Previous studies showed that these doses reproduce in the mouse the hallmarks of glucocorticoid-induced osteoporosis and are equivalent to medium and high therapeutic glucocorticoid doses in humans.(31,32) Moreover, the C57BL/6 mouse is a reliable and reproducible model for glucocorticoid-induced skeletal deterioration, as demonstrated by consistent decreases in BMD, reduction in bone formation, and increased osteoblast/osteocyte apoptosis.(18,31) Mice were injected 10 and 3 days prior to euthanasia with 0.6% calcein and 1.0% Alizarin red solutions, respectively. Twenty-eight days after pellet implantation mice were euthanized. Bones and muscles were dissected. Bones were processed as indicated below in the BMD measurement and micro-computed tomography analysis, Bone histomorphometry and apoptosis analysis, and Immunohistochemistry sections. Wet weight of indicated muscles from both hind limbs was taken and averaged for each mouse. Analysis was performed in a blinded fashion.
BMD measurement and micro–computed tomography analysis
BMD of the total body excluding the head and the tail, the lumbar spine (L1–L6), and the femur, and lean body mass were measured by dual-energy X-ray absorptiometry (DXA) scanning by using a PIXImus II densitometer (GE Medical System, Lunar Division, Madison, WI, USA). Body weight and DXA measurements were performed 2 to 4 days before (initial) and 28 days (final) after pellet implantation.(33,34) Mice were randomized to the experimental groups based on spine BMD. Percent changes in body weight, lean body mass, and BMD were calculated with the following formula: [(final – initial)/initial] multiplied by 100.
For micro–computed tomography (μCT) analysis, L6 vertebrae were cleaned of soft tissue, fixed in 10% buffered formalin, and stored in 70% ethanol until scanned at 6-μm resolution (Skyscan 1172; SkyScan, Aartselaar, Belgium). Measurements were done 60 μm away from the growth plates as described.(35,36)
Serum biochemistry
Blood was collected 2 and 4 weeks after pellet implantation from the facial vein of 3-hour fasted mice. N-terminal propeptide of type I procollagen (P1NP), C-terminal telopeptides of type I collagen (CTX), and tartrate-resistant acid phosphatase form 5b (TRAP 5b) were measured using enzyme-linked immunosorbent assays (Immunodiagnostic Systems Inc., Boldon, UK).(34,37) Alkaline phosphatase was measured on a Randox Daytona analyzer (Randox Laboratories Limited, Crumlin, UK).
Quantitative PCR
Total RNA extraction and quantitative PCR (qPCR) were performed as reported.(38) Briefly, total RNA was extracted from lumbar vertebrae L5, femur, ex vivo cultured bones, using TRIzol (Invitrogen, Carlsbad, CA, USA). cDNA was synthesized by using a high-capacity cDNA reverse transcriptase kit (Applied Biosystems, Inc., Foster City, CA, USA). Gene expression was analyzed using primer probe sets from Applied Biosystems or from Roche Applied Science (Penzberg, Germany). Relative mRNA expression levels were normalized to the housekeeping gene ribosomal protein S2 (Rplp2) by using the ΔCt method.(36,38) Data are expressed as fold change, where the ratio between the gene of interest and the housekeeping gene for WT mice receiving placebo was considered as 1.
Bone histomorphometry and apoptosis analysis
L1 to L3 vertebrae were fixed in 10% neutral buffered formalin and embedded undecalcified in methyl methacrylate as described.(20) Dynamic histomorphometry measurements were performed in 7-μm unstained bone sections under epifluorescence microscopy. Histomorphometric analysis was performed using OsteoMeasure high-resolution digital video system (OsteoMetrics, Decatur, GA, USA) interfaced to an Olympus BX51 fluorescence microscope (Olympus America Inc., Center Valley, PA, USA).(39) Osteoclasts were quantified on lumbar vertebra L2 thin sections stained for tartrate-resistant acid phosphatase (TRAP) and counterstained with Toluidine Blue as published.(34,37) An osteoclast was defined as a TRAP-positive cell attached to the bone surface with more than one nucleus. Apoptosis of osteoblasts and osteocytes was detected by the transferase-mediated biotin-dUTP nick end-labeling (TUNEL) reaction (Fisher Scientific, Pittsburgh, PA, USA) in undecalcified vertebral bone sections counterstained with 2% methyl green, mounted on silane-coated glass slides (Scientific Device Laboratory, Inc., Des Plaines, IL, USA), as described.(20) The prevalence of apoptotic osteoblasts and osteocytes was calculated by enumerating the total number and TUNEL-positive cells exhibiting condense chromatin, nuclear fragmentation, or cell shrinkage.
Immunohistochemistry
The expression of sclerostin was visualized in paraffin-embedded femora from 5-month-old WT and Sost−/− mice 28 days after pellet implantation, as described.(35,40) Sclerostin was visualized in deparaffinized sections of decalcified femurs, treated with 3% H2O2 to inhibit endogenous peroxidase activity, blocked with serum, and then incubated with goat polyclonal anti-mouse sclerostin antibody (1:100; Abcam, Cambridge, MA, USA; catalog Ab63097).(34) Sections were then incubated with anti-goat biotinylated secondary antibody followed by avidin conjugated peroxidase (Vectastain Elite ABC Kit; Vector Laboratories, Burlingame, CA, USA; catalog PK-6105). Color was developed with a diaminobenzidine substrate chromogen system (DAKO, Carpinteria, CA, USA). Cells expressing the protein of interest are stained in brown, whereas negative cells are green-blue.
Mechanical testing
Mechanical properties of L6 vertebrae were determined by axial compression after removal of vertebral processes and the cranial and caudal endplates. Vertebral bodies were loaded at a rate of 0.5 mm/min until failure (100P225 Modular Test Machine) as described.(41,42) Structural properties were obtained from the load/displacement curves and material-level properties were calculated using published equations to normalize to vertebral height, cross-sectional area, and bone volume.(43)
Ex vivo cultures
Femoral and tibial long bones were harvested from WT and Sost−/− mice, and then maintained in α-MEM containing 10% FBS and 1% penicillin/streptomycin for 24 hours. Bones were treated with 1 μM dexamethasone or vehicle (ethanol) with or without an anti-sclerostin antibody(44) (10 μg/mL; Acris Antibodies, San Diego, CA, USA; catalog AP13235PU-N) or vehicle (PBS) for 6 hours. mRNA was isolated and gene expression was measured by qPCR, as indicated above in the Quantitative PCR section.
Mineralization assay
Primary osteoblastic cells were isolated from the neonatal calvarial bones of C57BL/6 mice, Sost−/−, or WT littermate control mice, and plated at 5000 cells/cm2 density in α-MEM medium with 10% FBS and 1% penicillin/streptomycin as described.(31,45) Osteogenic medium was used after cultures reached confluence consisting of 50 μg/mL ascorbic acid and 10 mM β-glycerophosphate and treated with 1 μM dexamethasone or the corresponding vehicle (ethanol). Medium was replaced every 2 to 3 days, and mineralization was visualized using the OsteoImage Mineralization Assay Kit (Lonza, Basel, Switzerland), and then quantified by microplate reader for 492/520 nm excitation/emission fluorescence.
Statistical analysis
Data are expressed as means ± standard deviations (SDs). Sample differences were evaluated using SigmaPlot 12.0 (Systat Software, Inc., San Jose, CA, USA). Data of the in vivo experiments were analyzed by two-way ANOVA using genotype and treatment as independent variables. When ANOVA detected a significant interaction between the variables or a significant main effect of genotype, a post hoc test was used to determine the significance of the effect of the treatment within each genotype. The only exception was the analysis of the mechanical testing data in which one-way ANOVA for each genotype using treatment as independent variable was used, because the marked differences between WT and Sost−/− mice in these parameters precluded the detection of the effect of glucocorticoids when two-way ANOVA was used. All pairwise multiple comparison procedures within one-way or two-way ANOVA tests using the Tukey method were utilized to detect genotype and/or treatment differences. Results of the ANOVA and post hoc test are presented in the Supporting Information. Data of the in vitro experiments were analyzed by two-tailed Student’s t test. Data of the ex vivo experiments, in which bones from each leg of the same mouse were treated with vehicle or dexamethasone, were analyzed by paired t test. Means were considered significantly different at p < 0.05.
Study approval
All animal procedures were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine, and animal care was carried out in accordance with institutional guidelines.
Results
Sost−/− mice are protected from bone loss and decreased bone strength induced by glucocorticoids, but not from the loss of muscle mass
Mice lacking the osteocyte-derived antagonist of Wnt/β-catenin signaling Sost/sclerostin (Sost−/− mice) displayed high bone mass and strength compared to WT littermate controls (Fig. 1), consistent with earlier reports.(30) Glucocorticoid administration at two different doses of prednisolone (GC1 = 1.4 or GC2 = 2.1 mg/kg/day) significantly decreased total and spinal BMD (Fig. 1A), cancellous bone trabecular thickness (Tb.Th), and cortical bone area (BA/TA) in lumbar vertebra of WT mice (Fig. 1B, C), although no changes in cancellous BV/TV were detected. The high GC dose also decreased cortical thickness (Ct.Th). The decreased thickness of cortical bone was driven by thinning of the dorsal surface. Glucocorticoid excess also reduced the mechanical properties of WT bones, at both the structural level (ultimate force and energy to ultimate load) and at the material level (toughness), although modulus and ultimate stress were not altered (Fig. 1D). In contrast, Sost−/− mice were protected from glucocorticoid-induced decrease in bone mass, changes in bone architecture in cancellous and cortical sites, as well as the reduction in bone strength (Fig. 1). Ultimate stress is an estimate of the strength of the material—calculated as ultimate force normalized by the amount of bone. Therefore, the lack of changes in ultimate stress by glucocorticoids in either genotype indicates that the decrease in ultimate force is driven by structural changes as opposed to changes in tissue properties (such as mineralization or collagen alterations).
Fig. 1.
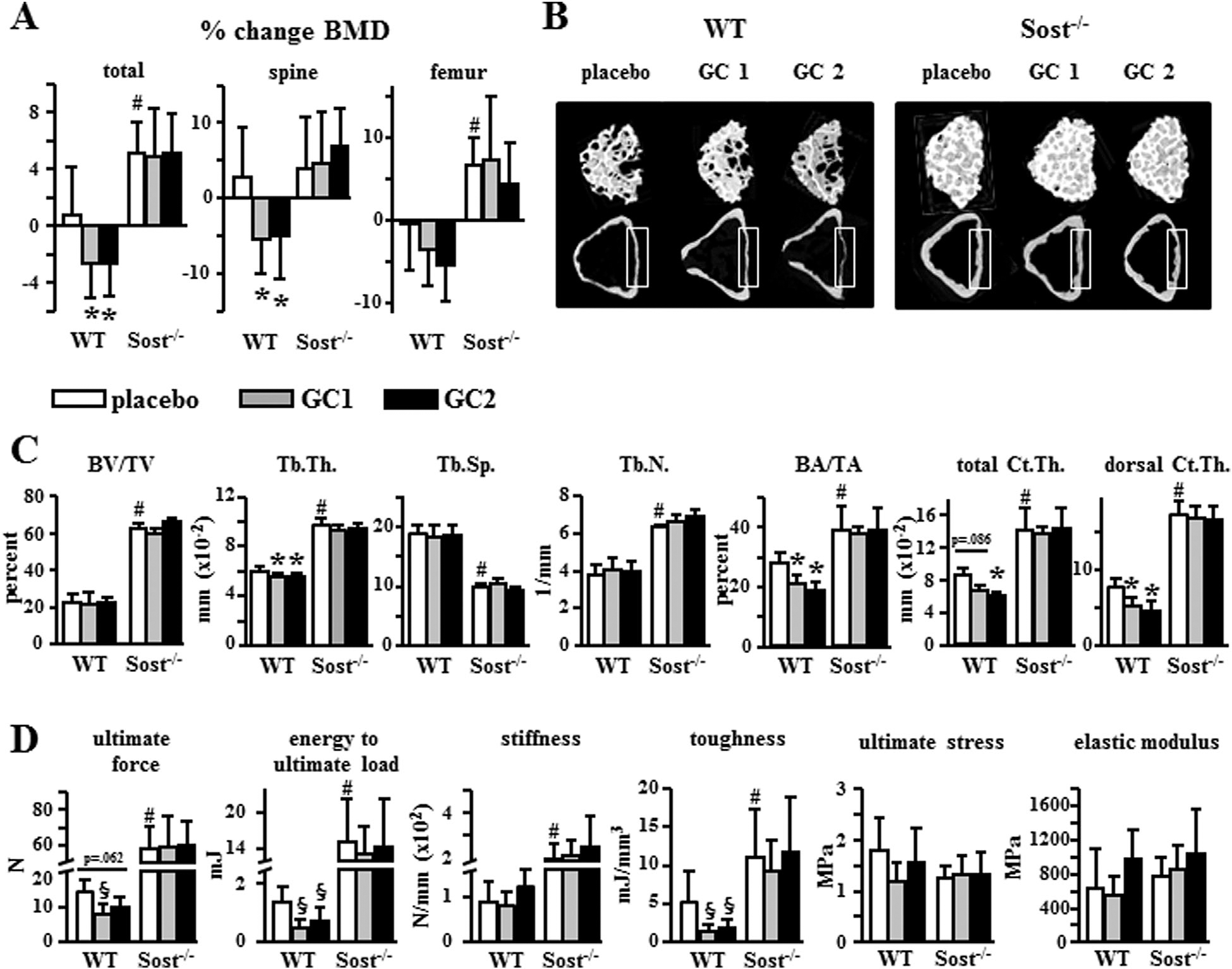
Sost−/− mice are protected from bone loss and decreased bone strength induced by glucocorticoids. (A) Percent change in BMD for WT or Sost−/− mice treated with placebo, 1.4 mg/kg/day prednisolone (GC1), or 2.1 mg/kg/day prednisolone (GC2) for 28 days, measured by DXA. n = 7–10. (B) Representative reconstructed μCT images of L6 vertebral cancellous bone (3D, top row) and cortical bone (2D, lower row). The dorsal surface of cortical bone for which thickness was also quantified is indicated by the gray boxes. (C) BV/TV, Tb.Th, Tb.Sp, and Tb.N, cortical BA/TA, total Ct.Th, and thickness of the dorsal surface of cortical bone (dorsal Ct.Th) are shown. n = 7–10. (D) Biomechanical properties were measured in vertebral bone (L6) by axial compression testing; n = 6–9. Bars represent means ± SD. *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus placebo-treated WT mice, by two-way ANOVA; §p < 0.05 versus corresponding placebo-treated mice by one-way ANOVA, Tukey post hoc test. BV/TV = bone volume/tissue volume; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; Tb.N = trabecular number; BA/TA = bone area/total area; Ct.Th = thickness of cortical bone.
The protection from loss of tissue mass by Sost/sclerostin deficiency was specific to bone, because Sost−/− mice exhibited glucocorticoid-induced sarcopenia similar to WT mice (Fig. 2). Thus, both WT and Sost−/− mice treated with prednisolone displayed decreased lean body mass, an index of skeletal muscle mass, with a loss of 6% to 9% in lean body mass over the 4-week treatment (Fig. 2A). In addition, the weight of the fast twitch type II–dominated muscles from glucocorticoid-treated mice of both genotypes was equally decreased about 22% to 23% and 13% to 15% for gastrocnemius and tibialis anterior, respectively (Fig. 2B). On the other hand, glucocorticoids did not decrease in either genotype the weight of the slow twitch type I–dominated soleus muscle, a muscle type shown to be less sensitive to the action of the hormones.(46) These changes in bone and muscle mass induced by glucocorticoids occurred in the absence of alterations in total mouse body weight (Fig. 2C).
Fig. 2.
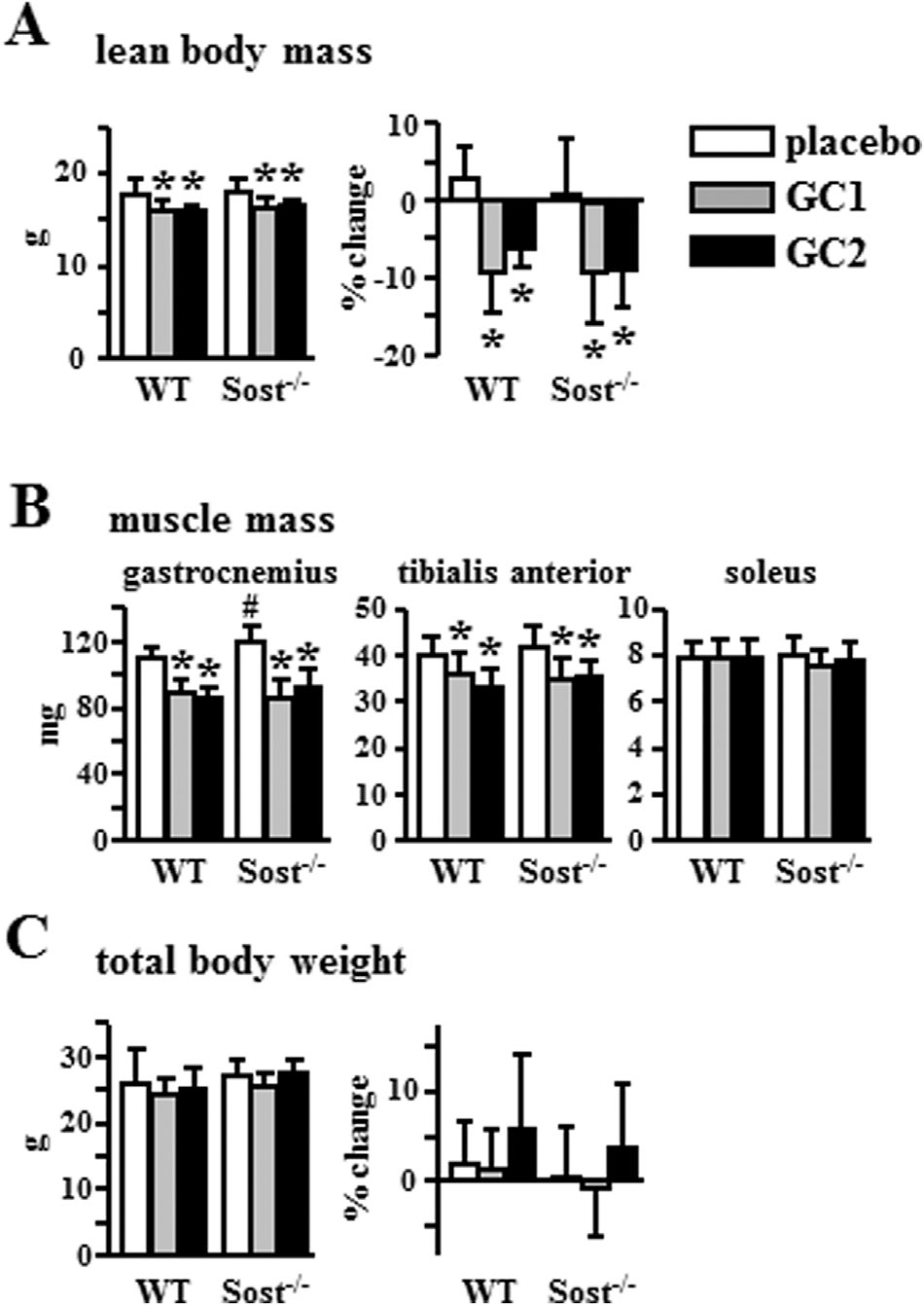
Sost/sclerostin deficiency does not prevent glucocorticoid-induced sarcopenia. Final and percent change in lean body mass (A), the mass of the indicated muscles (B), and final and percent change in total body weight (C) are shown; n = 8–10. Bars represent means ± SD. *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus placebo-treated WT mice by two-way ANOVA, Tukey post hoc test.
Sost/sclerostin deficiency does not prevent glucocorticoid-induced decrease in bone formation, increase in osteoblast/osteocyte apoptosis, or reduced mineral deposition
Consistent with the recognized bone anabolism induced by Wnt/β-catenin activation, Sost−/− mice exhibited increased bone formation indexes at the tissue level (mineralizing surface [MS/BS]; mineral apposition rate [MAR]; and bone formation rate [BFR/BS]) and in the circulation (P1NP and alkaline phosphatase [ALP]); as well as increased mRNA expression in bone of the osteoblast marker osteocalcin, OCN (Fig. 3A–D).
Fig. 3.

Sost/sclerostin deficiency does not prevent glucocorticoid-induced decrease in bone formation or increase in osteoblast/osteocyte apoptosis. (A) Representative images of fluorochrome incorporation and dynamic histomorphometric data measured in longitudinal sections of lumbar vertebra (L1–L3). MS/BS, MAR, and BFR/BS are shown. Scale bars = 50 μm, n = 8–10. P1NP, n = 6–10 (B), and alkaline phosphatase, n = 7–10 (C) were measured in blood collected 2 or 4 weeks after pellet implantation. (D) OCN gene expression was quantified by qPCR in L5 lumbar vertebra and femur. n = 7–10 for WT and n = 6–8 for Sost−/− mice. mRNA levels were normalized to the housekeeping gene Rplp2. (E) Apoptosis of Ob and Ot was quantified in cancellous and cortical bone in longitudinal sections of lumbar vertebrae (L1–L3) stained for TUNEL. n = 6. Representative images of cortical bone are shown. Scale bar = 20 μm. (F) Quantification of mineralization in cultures of calvaria-derived primary osteoblastic cells from WT or Sost−/− mice treated with veh or dex for 1 or 2 weeks stained using the OsteoImage Mineralization Assay Kit (RFU 492 nm/520 nm excitation/emission fluorescence). Scale bars = 400 μm. n = 7–8. Bars represent means ± SD. (A–E) *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus placebo-treated WT mice by two-way ANOVA, Tukey post-hoc test. (F) *p < 0.05 versus corresponding vehicle-treated primary osteoblasts by Student’s t test. MS/BS = mineralizing bone surface per bone surface; MAR = mineral apposition rate; BFR/BS = bone formation rate per bone surface; OCN = Osteocalcin; Rplp2 = ribosomal protein large P2; Ob = osteoblasts; Ot = osteocytes; veh = vehicle; dex = dexamethasone.
In spite of the elevated bone formation exhibited by the Sost-deficient mice under basal conditions, glucocorticoids decreased MAR and BFR/BS to a similar extent compared to placebo in WT and Sost−/− mice (Fig. 3A). Specifically, BFR/BS was reduced by about 30% to 40% in both WT and Sost−/− mice. Further, circulating P1NP and ALP were decreased by glucocorticoids in both genotypes at 2 weeks (Fig. 3B, C). In addition, glucocorticoids reduced P1NP in the Sost−/− mice. In addition, glucocorticoids markedly decreased OCN expression in bones from both WT and Sost−/− mice (Fig. 3D). Further, both WT and Sost−/− mice receiving prednisolone exhibited high prevalence of osteoblast and osteocyte apoptosis in cancellous and cortical bone (Fig. 3E), consistent with the relationship between decreased bone formation and promotion of osteoblast (and osteocyte) apoptosis by glucocorticoids.(18,20,32) Moreover, addition of the glucocorticoid dexamethasone to cultured primary calvarial osteoblasts decreased mineral deposition in cells isolated from WT or Sost−/− mice (Fig. 3F).
Sost/sclerostin deficiency prevents glucocorticoid-induced increase in bone resorption
Sost−/− mice exhibited overall unremarkable changes in osteoclasts and resorption compared to WT mice under basal conditions (Fig. 4A–C). Compared to WT mice, Sost-deficient mice showed no differences in osteoclast surface (Oc.S/BS), blood bone resorption marker CTX at 4 weeks or osteoclast marker TRAP5b at 2 and 4 weeks; and only a minor decrease in osteoclast number (N.Oc/BS) at 4 weeks and a transient increase in CTX at 2 weeks. Further, glucocorticoids increased osteoclast number (N.Oc/BS) and surface (Oc.S/BS) and raised CTX and TRAP5b circulating levels in WT mice. In contrast, glucocorticoids failed to do so at any dose or time point in Sost−/− mice.
Fig. 4.
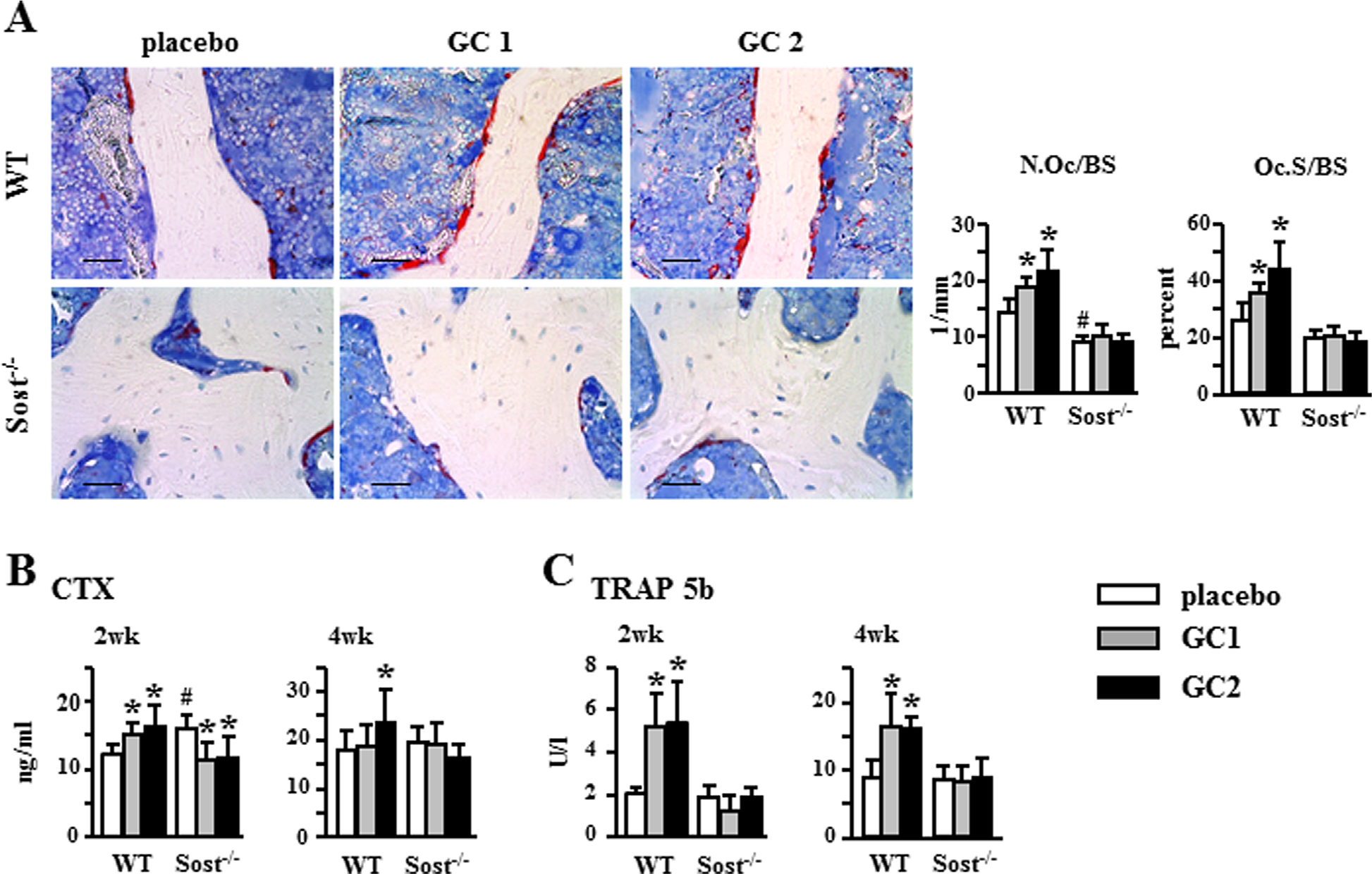
Sost−/− mice are protected from the increase in bone resorption induced by glucocorticoid. (A) Representative microscopy images of osteoclasts on cancellous bone surface in lumbar vertebra (L2) stained for TRAPase. N.Oc/BS and Oc.S/BS normalized to bone surface were measured. Scale bars = 30 μm, n = 5. CTX, n = 6–11 (B) and TRAP 5b, n = 6–7 (C) were measured in blood collected 2 and 4 weeks after pellet implantation. Bars represent means ± SD. *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus placebo-treated WT mice by two-way ANOVA, Tukey post hoc test. N.Oc/BS = osteoclast number; Oc.S/BS = osteoclast surface.
Glucocorticoids differentially alter the anti-catabolic versus anabolic/survival molecular signature of the Wnt/β-catenin pathway in a Sost/sclerostin-dependent manner
Treatment with glucocorticoids increased Sost mRNA and the prevalence of sclerostin positive osteocytes in WT mice; and, as expected, Sost or sclerostin expression was not detectable in bones from Sost−/− mice (Fig. 5A, B). Consistent with the recognized antagonistic function of Sost/sclerostin on Wnt/β-catenin activation, glucocorticoids decreased the expression of several target genes of the pathway, including OPG, Cx43, BMP4, cyclin D1, Axin2, and Smad6 in WT mice (Fig. 5C). However, glucocorticoids only decreased the expression of a set of these genes in Sost−/− mice, suggesting that sustained activation of Wnt/β-catenin signaling due to Sost/sclerostin deficiency only partially opposes glucocorticoid effects on gene expression. Overall, similar patterns of expression were found in bones from the appendicular (femur) and axial (lumbar vertebra) skeleton. Specifically, glucocorticoids decreased the expression of cyclin D1, Smad6, and Axin2 in both WT and Sost−/− mice. In contrast, they decrease the expression of OPG, Cx43, and BMP4 only in WT mice. Increased cyclin D1 is associated with bone anabolism and anti-apoptosis(36,47–49); and increased OPG is responsible for inhibition of osteoclast formation and resorption.(27,28,50) Thus, whereas glucocorticoids oppose all Wnt/β-catenin signaling in the presence of Sost/sclerostin, the hormones only oppose signaling associated with bone formation/survival in the absence of Sost/sclerostin.
Fig. 5.
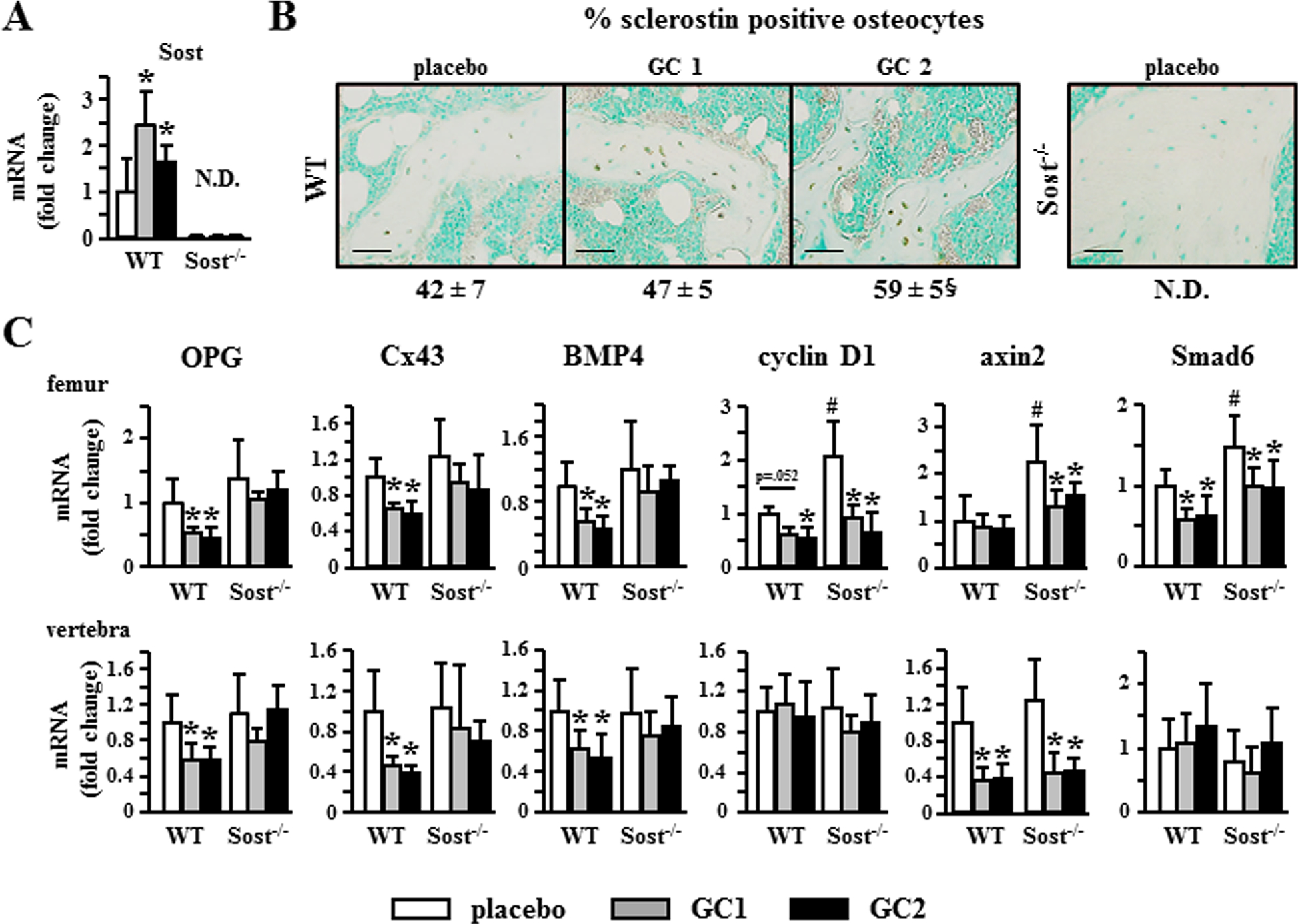
Glucocorticoids differentially alter Wnt/β-catenin signaling in a Sost/sclerostin–dependent manner. (A) Sost gene expression was quantified by qPCR in L5 lumbar vertebra. n = 7–10 for WT and n = 6–8 for Sost−/−. (B) The percentage of sclerostin-positive osteocytes was quantified in longitudinal sections of femoral bone stained with an anti-sclerostin antibody and is shown as percentage ± SD, n = 3–4. Scale bars = 50 μm. (C) Expression of the indicated genes normalized to the housekeeping gene Rplp2 in femur and lumbar vertebra (L5). n = 7–10 for WT and n = 6–8 for Sost−/−. Bars represent means ± SD. *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus placebo-treated WT mice by two-way ANOVA and Tukey post hoc test. §p < 0.05 versus placebo-treated WT mice, by one-way ANOVA and the Tukey post hoc test.
Genetic or pharmacologic activation of Wnt/β-catenin signaling blocks downregulation of OPG expression induced by glucocorticoids
The lack of downregulation of OPG in Sost−/− mice (Fig. 5C) resulted in lower RANKL/OPG ratio in prednisolone-treated Sost−/− mice compared to WT mice (Fig. 6A). The differential regulation of OPG in WT and Sost−/− mice was due to direct action of glucocorticoids on osteocytes as evidenced in ex vivo cultures of bones enriched in osteocytes. Genetic deletion of Sost or pharmacologic inhibition of sclerostin with the neutralizing antibody in ex vivo bone organ cultures mimicked the in vivo regulation of OPG in Sost−/− mice. Thus, glucocorticoids increased Sost and decreased OPG expression, resulting in increased RANKL/OPG ratio in cultured bones from WT mice (Fig. 6B, C). In contrast, bones from Sost−/− mice were protected from glucocorticoid-induced decrease in OPG and increase in RANKL/OPG ratio (Fig. 6B). Similarly, glucocorticoids failed to decrease OPG and increase the RANKL/OPG ratio in WT bones in which sclerostin function was blocked with an anti-sclerostin antibody (Scl-Ab), even when Sost mRNA expression was still increased by glucocorticoids (Fig. 6C). RANKL expression was not consistently regulated by glucocorticoids either in vivo or ex vivo (Fig. 6), demonstrating that OPG expression is responsible for the differential regulation of the RANKL/OPG ratio observed in the presence or absence of Sost/sclerostin deficiency.
Fig. 6.
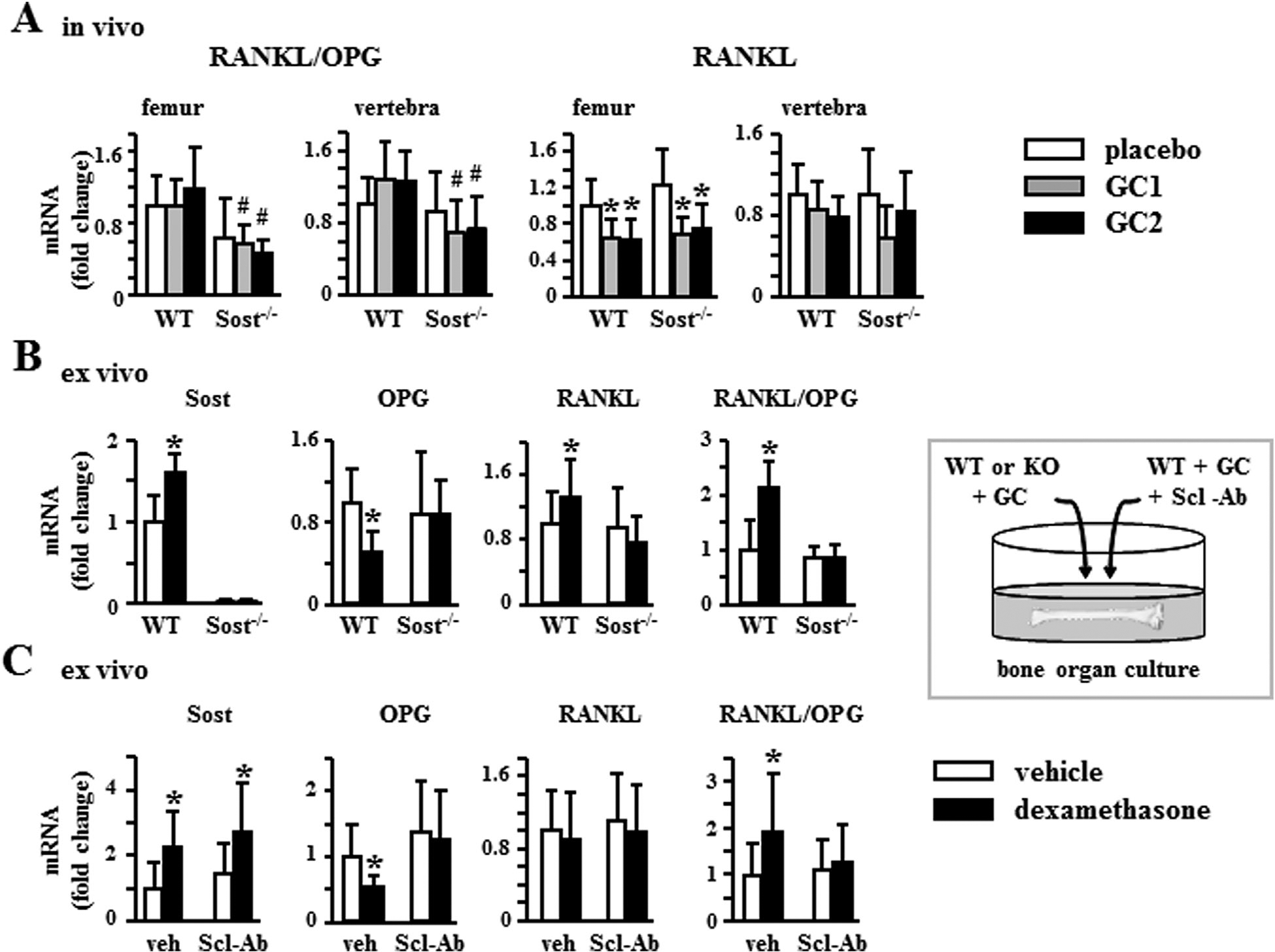
Genetic or pharmacologic inhibition of Sost/sclerostin blocks downregulation of OPG expression and the increase in RANKL/OPG ratio induced by glucocorticoid. (A) Expression of the indicated genes normalized to the housekeeping gene Rplp2 in femur and lumbar vertebra (L5). n = 7–10 for WT and n = 6–8 for Sost−/−. *p < 0.05 versus corresponding placebo-treated mice and #p < 0.05 versus WT mice treated with the same glucocorticoid dose by two-way ANOVA, Tukey post hoc test. (B, C) Gene expression in bones from WT or Sost−/− mice, n = 6 (B) or from WT mice cultured with or without anti-sclerostin antibody (Scl-Ab), n = 16 (C) were treated ex vivo with vehicle or dexamethasone for 6 hours. *p < 0.05 versus corresponding vehicle-treated bones, by paired t test. OPG = osteoprotegerin.
Discussion
Glucocorticoid-induced osteoporosis is a leading cause of bone fragility worldwide and better therapies are critically needed. This study provides insights into novel mechanisms of glucocorticoid action and the cellular and molecular basis by which activation of Wnt/β-catenin signaling interferes with the detrimental effect of glucocorticoid excess in bone, but not in muscle, using in vivo, ex vivo, and in vitro approaches.
We report that glucocorticoids hinder the expression of target genes of the Wnt/β-catenin pathway, regardless of whether they are associated with bone anabolism (ie, bone gain) or anti-catabolism (ie, inhibition of bone loss). Glucocorticoids exert this action, in part, by increasing the expression of Sost/sclerostin, the osteocyte-derived Wnt/β-catenin antagonist and potent inhibitor of bone formation. Future investigations are warranted to determine whether this phenomenon is due to direct regulation of transcription of the Sost gene or whether is exerted through indirect mechanisms. Nevertheless, Sost/sclerostin deficiency protected against the loss of bone mass, deterioration of microarchitecture, and reduction of extrinsic/structural and intrinsic/material mechanical properties induced by glucocorticoids. Remarkably, however, the bone protective effect of Sost/sclerostin deficiency against glucocorticoids was not due to an opposing action to increase bone formation and maintain anabolic signaling. Instead, it was due to preservation of the Wnt/β-catenin anti-catabolic cellular and molecular signature (Fig. 7). These results indicate that a pathway predominantly anabolic for bone is switched to anti-catabolic in the frame of glucocorticoid excess. Our findings suggest that therapeutic interventions activating Wnt/β-catenin signaling could effectively halt the high bone resorption responsible for the profound and rapid bone loss induced by glucocorticoids, which in humans can reach up to 12% during the first year of treatment.(10)
Fig. 7.

Sost/sclerostin deficiency prevents GC-induced bone loss. GC-induced osteoporosis is characterized by decreased bone formation, increased apoptosis of Ot and Ob, and increased Oc and bone resorption, which combined induced bone loss and fragility. Activation of Wnt/β-catenin signaling resulting from Sost/sclerostin deletion protects from glucocorticoid-osteoporosis by inhibiting bone resorption through sustained anti-catabolic signaling driven by OPG. GC = glucocorticoid; Ot = osteocytes; Ob = osteoblasts; Oc = osteoclasts; OPG = osteoprotegerin.
Consistent with our current findings in skeletally mature Sost/sclerostin–deficient mice, previous studies showed that pharmacologic inhibition of sclerostin with a neutralizing antibody opposed the lack of bone gain and the loss of strength induced by glucocorticoids in growing mice.(51,52) Although it was proposed that these effects were due to preservation of osteoblast activity,(52) mice treated with glucocorticoids and the anti-sclerostin antibody in these earlier studies exhibited lower circulating TRAP5b(51) or CTX-1,(52) but still markedly reduced bone formation markers osteocalcin and P1NP,(51) compared to the corresponding mice treated with glucocorticoids alone. Likewise, our in vivo studies show that sustained activation of the Wnt/β-catenin signaling in Sost-deficient mice abolishes the increase in resorption induced by glucocorticoids but not the decreased bone formation. Moreover, our ex vivo results demonstrate that glucocorticoids are unable to decrease OPG and increase the RANKL/OPG ratio in bones from Sost−/− mice or in WT bones treated with an anti-sclerostin antibody. Taken together, these findings demonstrate that Sost/sclerostin deficiency, either genetic or pharmacologically achieved with the neutralizing anti-sclerostin antibody, maintains bone mass and strength in conditions of glucocorticoid excess by inhibiting bone resorption, through sustained anti-catabolic signaling driven by OPG.
In our mouse model of glucocorticoid-induced osteoporosis, the bone loss in this early phase is accompanied by both decreased bone formation and increased bone resorption, with the latter being the only one opposed by Sost/sclerostin deficiency. In view of the striking reduction in bone formation that remains in both genotypes, we speculate that during the second phase of glucocorticoid-induced osteoporosis that occurs with prolonged treatment and that is driven by reduced bone formation, bone loss could occur even in the Sost KO mice. Future investigations are warranted to address this issue.
Although bone mass, shape, and microarchitecture are the major recognized contributors to bone strength, accumulating evidence supports also an important role of osteocyte viability.(18,37,53) Our findings demonstrate that Sost KO mice treated with glucocorticoids maintain structural and material mechanical properties despite increased osteocyte apoptosis, highlighting the relative contribution of these factors. Evidently, the high bone mass and strong architecture exhibited by the Sost KO mice compensate for the bone weakening effects of increased osteocyte apoptosis under glucocorticoid excess.
A clinical case reported that glucocorticoids stop the exaggerated bone gain and reduced the high circulating P1NP in a patient with Van Buchem disease, a condition that results from lack of sclerostin expression and in which continuous bone anabolism causes life-threatening increased intracranial pressure.(54) Prior to glucocorticoid intervention, the patient exhibited annual BMD gains of 4% to 9% in the lumbar spine and of 4% to 24% in the hip. Treatment with prednisone blunted the anabolic effect of Sost deficiency as evidenced by no gain in BMD after 2 years (–0.7% in lumbar spine and 0.4% in the hip). Our report showing that bone formation and Wnt/β-catenin anabolic signaling is still decreased in Sost/sclerostin–deficient mice treated with glucocorticoids provides a mechanistic explanation for these clinical findings. Taken together with the current study, these pieces of evidence demonstrate that glucocorticoids oppose the effects of Sost/sclerostin deficiency on bone formation in both humans and mice.
Inhibition of resorption with bisphosphonates is the current standard of care for glucocorticoid-induced osteoporosis.(8,10) These drugs protect from the loss of bone mass induced by glucocorticoids in animal models and patients. However, bone formation is decreased even further by bisphosphonates compared to glucocorticoids alone.(7,31,37,55) Treatment with anti-RANKL antibodies induces even more pronounced reductions in bone formation compared to treatment with bisphosphonates in patients receiving glucocorticoids.(56) Profound reduction in bone turnover is not desirable because it leads to accumulation of microdamage and advanced glycation end-products (AGEs), and the potential for developing osteonecrosis of the jaw with long-term treatments.(43,57–59) Severe suppression of turnover by bisphosphonates can also lead to reduced toughness,(57,59,60) the energy that bone tissue absorbs before failure, which in turn could be the cause of increased risk for low-energy atypical fractures.(61,62) In contrast to purely antiresorptive agents, Sost deficiency confers high bone formation per se; thus, Sost-deficient mice treated with glucocorticoids exhibit bone formation levels comparable to WT mice treated with placebo. Even when the reduction in P1NP induced by glucocorticoids in Sost KO mice is more severe than in WT mice, the reduced P1NP is similar to that of placebo-treated WT mice (Fig. 3B). Similarly, the reduced bone formation exhibited by Sost KO mice treated with glucocorticoids is equivalent to bone formation in placebo-treated WT mice (Fig. 3A). These findings suggest that Sost/sclerostin deficiency maintains tissue-level toughness by preserving modest amounts of bone formation while preventing glucocorticoid-induced increases in resorption. This mechanical finding points to a potential benefit compared to the current therapeutic approach to treat glucocorticoid-induced bone fragility.
Another unwanted consequence of excess glucocorticoids, either endogenous or iatrogenic, is muscle weakness, which reduces body balance and, when combined with lower bone mass, increases the risk of bone fractures. Because of their intimate association as a mechanical unit, changes in bone might impact skeletal muscle and vice versa. Our mouse model of glucocorticoid elevation faithfully reproduces the bone and skeletal muscle atrophy exhibited by humans. However, the bone preservation resulting from Sost/sclerostin deficiency in our studies did not protect from muscle atrophy; and conversely, the marked loss of muscle mass experienced by the Sost-deficient mice did not translate into apparent detrimental effects on bone volume or mechanical properties. These findings demonstrate that Sost/sclerostin deficiency protects exclusively bone, but not muscle, from the action of glucocorticoids, and show a lack of crosstalk between these two tissues in the frame of glucocorticoid-induced musculoskeletal atrophy. Future studies are warranted to investigate whether muscle-derived factors contribute to the low bone formation and high prevalence of osteoblast and osteocyte apoptosis still exhibited by Sost/sclerostin–deficient mice treated with glucocorticoids. Nevertheless, more recent findings of ours showed that glucocorticoids induce bone and muscle atrophy by distinct mechanisms upstream of the atrophy-related E3 ligases atrogin1 and MuRF1.(63) Taken together with the current study, these findings support the contention that combination of bone- and muscle-specific therapeutic interventions might be required to interfere with the damaging actions of glucocorticoids in the musculoskeletal system.
We show that glucocorticoids differentially regulate anabolic/survival versus anti-catabolic Wnt/β-catenin signaling depending on the expression of Sost/sclerostin. Thus, whereas cyclin D1 is downregulated and bone formation is inhibited regardless of whether Sost is expressed or not, the decrease in OPG and increase in bone resorption is strictly dependent on Sost expression. The basis for this differential regulation of Wnt/β-catenin target genes by glucocorticoids remains unknown. However, glucocorticoids might regulate some of these genes by interfering with the Wnt/β-catenin pathway itself, whereas they might control other genes either directly by repressing the activity of their promoters or through regulation of alternative pathways including kinase signaling or induction of ROS/endoplasmic reticulum (ER) stress.(31,49,64–67)
In closing, the current report demonstrates that the deleterious effects of glucocorticoids on the skeleton are linked to increased expression of the osteocyte-derived Wnt/β-catenin antagonist Sost/sclerostin and to downregulation of Wnt/β-catenin target genes; and that Sost/sclerostin deficiency prevents glucocorticoid-induced osteoporosis by anti-catabolic, not anabolic, actions.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (R01-AR059357 to TB; T32-AR065971 to AYS; and NHLBI T35 HL110854-01) and the Veterans Administration (1 I01 BX002104-01 to TB). We thank Dr. Xiaolin Tu, Jasmine Tzeggai, Kali Marie Kuhlenschmidt, and Kevin McAndrews for their assistance in tissue collection; Drew Brown for assistance with μCT analysis and mechanical testing; Anthony Acton, Jr. for measurement of serum alkaline phosphatase; Dr. Ziyue Liu for assistance with statistical analysis; Dr. Michael Ominsky from AMGEN for providing the Sost−/− mice; Dr. Alexander G. Robling for advice on μCT analysis; and Dr. David Burr for critical reading of the manuscript and insightful discussions.
Authors’ roles: LIP and TB designed research; AYS, MC, JDC, KWC, and MP performed research; AYS, MRA, LIP, and TB analyzed and interpreted data; AYS, LIP, and TB wrote the manuscript. TB takes responsibility for the integrity of the data analysis.
Footnotes
Disclosures
All authors state that they have no conflicts of interest.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Warriner AH, Saag KG. Glucocorticoid-related bone changes from endogenous or exogenous glucocorticoids. Curr Opin Endocrinol Diabetes Obes. 2013;20:510–6. [DOI] [PubMed] [Google Scholar]
- 2.Necela BM, Cidlowski JA. Mechanisms of glucocorticoid receptor action in noninflammatory and inflammatory cells. Proc Am Thorac Soc. 2004;1:239–46. [DOI] [PubMed] [Google Scholar]
- 3.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005;353:1711–23. [DOI] [PubMed] [Google Scholar]
- 4.Tauchmanovà L, Pivonello R, Di Somma C, et al. Bone demineralization and vertebral fractures in endogenous cortisol excess: role of disease etiology and gonadal status. J Clin Endocrinol Metab. 2006;91:1779–84. [DOI] [PubMed] [Google Scholar]
- 5.Vestergaard P, Lindholm J, Jorgensen JO, et al. Increased risk of osteoporotic fractures in patients with Cushing’s syndrome. Eur J Endocrinol. 2002;146:51–6. [DOI] [PubMed] [Google Scholar]
- 6.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31:266–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med. 1998;339:292–9. [DOI] [PubMed] [Google Scholar]
- 8.Rizzoli R, Adachi JD, Cooper C, et al. Management of glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2012;91:225–43. [DOI] [PubMed] [Google Scholar]
- 9.Lukert B. Glucocorticoid-induced osteoporosis. In: Marcus R, Felman D, Kelsey J, editors. Osteoporosis. San Diego, CA: Academic Press; 1996. p. 801–20. [Google Scholar]
- 10.Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70. [DOI] [PubMed] [Google Scholar]
- 11.Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367:1605–17. [DOI] [PubMed] [Google Scholar]
- 12.Weinstein RS. Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord. 2001;2:65–73. [DOI] [PubMed] [Google Scholar]
- 13.Van Staa TP. The pathogenesis, epidemiology and management of glucocorticoid-induced osteoporosis. Calcif Tissue Int. 2006;79: 129–37. [DOI] [PubMed] [Google Scholar]
- 14.Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A. Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol Metab. 2006;17:144–9. [DOI] [PubMed] [Google Scholar]
- 15.Overman RA, Yeh JY, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken). 2013;65:294–8. [DOI] [PubMed] [Google Scholar]
- 16.Weinstein RS, Chen JR, Powers CC, et al. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109:1041–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. J Clin Endocrinol Metab. 2000;85:2907–12. [DOI] [PubMed] [Google Scholar]
- 18.O’Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41. [DOI] [PubMed] [Google Scholar]
- 19.Reid IR. Glucocorticoid osteoporosis—mechanisms and management. Eur J Endocrinol. 1997;137:209–17. [DOI] [PubMed] [Google Scholar]
- 20.Plotkin LI, Weinstein RS, Parfitt AM, et al. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their lifespan and reduce bone density. Endocrinology. 2006;147:5592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hofbauer LC, Zeitz U, Schoppet M, et al. Prevention of glucocorticoid-induced bone loss in mice by inhibition of RANKL. Arthritis Rheum. 2009;60:1427–37. [DOI] [PubMed] [Google Scholar]
- 23.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–92. [DOI] [PubMed] [Google Scholar]
- 24.Balemans W, Ebeling M, Patel N, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10:537–43. [DOI] [PubMed] [Google Scholar]
- 25.Dixon JM, Cull RE, Gamble P. Two cases of Van Buchem’s disease. J Neurol Neurosurg Psychiatry. 1982;45:913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leupin O, Piters E, Halleux C, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glass DA, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. [DOI] [PubMed] [Google Scholar]
- 28.Holmen SL, Zylstra CR, Mukherjee A, et al. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280: 21162–8. [DOI] [PubMed] [Google Scholar]
- 29.Jilka RL, Bellido T, Almeida M, et al. Apoptosis in bone cells. In: Bilezikian JP, Raisz LG, Martin TJ, editors. Principles of bone biology. 3rd ed. San Diego, CA: Academic Press; 2008. p. 237–61. [Google Scholar]
- 30.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. [DOI] [PubMed] [Google Scholar]
- 31.Sato AY, Tu X, McAndrews KA, Plotkin LI, Bellido T. Prevention of glucocorticoid induced-apoptosis of osteoblasts and osteocytes by protecting against endoplasmic reticulum (ER) stress in vitro and in vivo in female mice. Bone. 2015;73:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102: 274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plotkin LI, Lezcano V, Thostenson J, et al. Connexin 43 is required for the anti-apoptotic effect of bisphosphonates on osteocytes and osteoblasts in vivo. J Bone Miner Res. 2008;23:1712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu X, Delgado-Calle J, Condon KW, et al. Osteocytes mediate the anabolic actions of canonical Wnt/b-catenin signaling in bone. Proc Natl Acad Sci U S A. 2015;112:E478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Brien CA, Plotkin LI, Galli C, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS One. 2008;3:e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhee Y, Allen MR, Condon K, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intra-cortical remodeling. J Bone Miner Res. 2011;26:1035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plotkin LI, Bivi N, Bellido T. A bisphosphonate that does not affect osteoclasts prevents osteoblast and osteocyte apoptosis and the loss of bone strength induced by glucocorticoids in mice. Bone. 2011;49:122–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tu X, Rhee Y, Condon KW, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aguirre JI, Plotkin LI, Stewart SA, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. [DOI] [PubMed] [Google Scholar]
- 40.Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of PTH in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83. [DOI] [PubMed] [Google Scholar]
- 41.Pacheco-Costa R, Hassan I, Reginato RD, et al. High bone mass in mice lacking Cx37 due to defective osteoclast differentiation. J Biol Chem. 2014;289:8508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill Gallant KM, Gallant MA, Brown DM, et al. Raloxifene prevents skeletal fragility in adult female Zucker Diabetic Sprague-Dawley rats. PLoS One. 2014;9:e108262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen MR, Iwata K, Phipps R, Burr DB. Alterations in canine vertebral bone turnover, microdamage accumulation, and biomechanical properties following 1-year treatment with clinical treatment doses of risedronate or alendronate. Bone. 2006;39:872–9. [DOI] [PubMed] [Google Scholar]
- 44.Ota K, Quint P, Ruan M, et al. Sclerostin is expressed in osteoclasts from aged mice and reduces osteoclast-mediated stimulation of mineralization. J Cell Biochem. 2013;114:1901–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bellido T, Ali AA, Plotkin LI, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278: 50259–72. [DOI] [PubMed] [Google Scholar]
- 46.Schakman O, Kalista S, Barbe C, Loumaye A, Thissen JP. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol. 2013;45:2163–72. [DOI] [PubMed] [Google Scholar]
- 47.Iyer S, Ambrogini E, Bartell SM, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123: 3409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iyer S, Han L, Bartell SM, et al. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem. 2014; 289:24069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou W, Yang S, Zhang T, et al. Hypoxia enhances glucocorticoid-induced apoptosis and cell cycle arrest via the PI3K/Akt signaling pathway in osteoblastic cells. J Bone Miner Metab. 2015;33: 615–24. [DOI] [PubMed] [Google Scholar]
- 50.Kramer I, Halleux C, Keller H, et al. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marenzana M, Greenslade K, Eddleston A, et al. Sclerostin antibody treatment enhances bone strength but does not prevent growth retardation in young mice treated with dexamethasone. Arthritis Rheum. 2011;63:2385–95. [DOI] [PubMed] [Google Scholar]
- 52.Yao W, Dai W, Jiang L, et al. Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporos Int. 2016;27:283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marcus R, Bouxsein ML. The nature of osteoporosis. In: Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Burlington, MA: Elsevier Academic Press; 2008. p. 27–36. [Google Scholar]
- 54.van Lierop AH, Hamdy NA, Papapoulos SE. Glucocorticoids are not always deleterious for bone. J Bone Miner Res. 2010;25:2796–800. [DOI] [PubMed] [Google Scholar]
- 55.Saag KG, Shane E, Boonen S, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357: 2028–39. [DOI] [PubMed] [Google Scholar]
- 56.Mok CC, Ho LY, Ma KM. Switching of oral bisphosphonates to denosumab in chronic glucocorticoid users: a 12-month randomized controlled trial. Bone. 2015;75:222–8. [DOI] [PubMed] [Google Scholar]
- 57.Mashiba T, Turner CH, Hirano T, et al. Effects of suppressed bone turnover by bisphosphonates on microdamage accumulation and biomechanical properties in clinically relevant skeletal sites in beagles. Bone. 2001;28:524–31. [DOI] [PubMed] [Google Scholar]
- 58.O’Ryan FS, Khoury S, Liao W, et al. Intravenous bisphosphonate-related osteonecrosis of the jaw: bone scintigraphy as an early indicator. J Oral Maxillofac Surg. 2009;67:1363–72. [DOI] [PubMed] [Google Scholar]
- 59.Allen MR, Burr DB. Mineralization, microdamage, and matrix: how bisphosphonates influence material properties of bone. Bonekey Osteovision. 2007;4:49–60. [Google Scholar]
- 60.Allen MR, Burr DB. Changes in vertebral strength-density and energy absorption-density relationships following bisphosphonate treatment in beagle dogs. Osteoporos Int. 2008;19:95–9. [DOI] [PubMed] [Google Scholar]
- 61.Aspenberg P, Schilcher J. Atypical femoral fractures, bisphosphonates, and mechanical stress. Curr Osteoporos Rep. 2014;12:189–93. [DOI] [PubMed] [Google Scholar]
- 62.Shane E, Burr D, Abrahamsen B, et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29:1–23. [DOI] [PubMed] [Google Scholar]
- 63.Sato AY, Au E, Richardson D, et al. Glucocorticoids induce bone and muscle atrophy by distinct mechanisms upstream of atrogin1 and MuRF1. J Bone Miner Res. 2015;30:S1.26678553 [Google Scholar]
- 64.Bellido T Antagonistic interplay between mechanical forces and glucocorticoids in bone: a tale of kinases. J Cell Biochem. 2010; 111:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Plotkin LI, Manolagas SC, Bellido T. Glucocorticoids induce osteocyte apoptosis by blocking focal adhesion kinase-mediated survival: evidence for inside-out signaling leading to anoikis. J Biol Chem. 2007;282:24120–30. [DOI] [PubMed] [Google Scholar]
- 66.Almeida M, Han L, Ambrogini E, Weinstein RS, Manolagas SC. Glucocorticoids and tumor necrosis factor (TNF) alpha increase oxidative stress and suppress WNT signaling in osteoblasts. J Biol Chem. 2011;286:44326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frenkel B, White W, Tuckermann J. Glucocorticoid-induced osteoporosis. Adv Exp Med Biol. 2015;872:179–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.