

Abstract
Background
Prostaglandin D2 (PGD2) signaling via prostaglandin D2 receptor 2 (DP2) contributes to atopic and non-atopic asthma. Inhibiting DP2 has shown therapeutic benefit in certain subsets of asthma patients, improving eosinophilic airway inflammation. PGD2 metabolites prolong the inflammatory response in asthmatic patients via DP2 signaling. The role of PGD2 metabolites on eosinophil and ILC2 activity is not fully understood.
Methods
Eosinophils and ILC2s were isolated from peripheral blood of atopic asthmatic patients. Eosinophil shape change, ILC2 migration and IL-5/IL-13 cytokine secretion were measured after stimulation with seven PGD2 metabolites in presence or absence of the selective DP2 antagonist fevipiprant.
Results
Selected metabolites induced eosinophil shape change with similar nanomolar potencies except for 9α,11β-PGF2. Maximal values in forward scatter of eosinophils were comparable between metabolites. ILC2s migrated dose-dependently in the presence of selected metabolites except for 9α,11β-PGF2 with EC50 values ranging from 17.4 to 91.7 nM. Compared to PGD2, the absolute cell migration was enhanced in the presence of Δ12-PGD2, 15-deoxy-Δ12,14-PGD2, PGJ2, Δ12-PGJ2 and 15-deoxy-Δ12,14-PGJ2. ILC2 cytokine production was dose dependent as well but with an average sixfold reduced potency compared to cell migration (IL-5 range 108.1 to 526.9 nM, IL-13 range: 125.2 to 788.3 nM). Compared to PGD2, the absolute cytokine secretion was reduced in the presence of most metabolites. Fevipiprant dose-dependently inhibited eosinophil shape change, ILC2 migration and ILC2 cytokine secretion with (sub)-nanomolar potencies.
Conclusion
Prostaglandin D2 metabolites initiate ILC2 migration and IL-5 and IL-13 cytokine secretion in a DP2 dependent manner. Our data indicate that metabolites may be important for in vivo eosinophil activation and ILC2 migration and to a lesser extent for ILC2 cytokine secretion.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12931-021-01852-3.
Keywords: Eosinophil shape change; Type 2 innate lymphocyte cells; Asthma; PGD2; DP2; CRTH2; Fevipiprant; 13,14-Dihydro-15-keto-PGD2; PGD2 metabolites
Background
Allergic and non-allergic asthma are closely linked to increased production of prostaglandin D2 (PGD2) [1–3]. The main source of PGD2 are mast cells, which thereby orchestrate early and late immune responses of the innate and adaptive immune system. PGD2 primarily signals through prostaglandin D2 receptors 1 (DP1) and 2 (DP2, also known as chemoattractant receptor-homologous molecule expressed on Th2 cells [CRTH2]). Activation of DP1 is associated with increased smooth muscle relaxation, vasodilation, vascular permeability as well as reduced leukocyte chemotaxis and cytokine secretion [4, 5]. In contrast, DP2 signaling leads to activation, chemokinesis, migration, and cytokine production of various leukocytes such as eosinophils, basophils, T cells or type 2 innate lymphoid cells (ILC2s) [6–11].
In vivo and in vitro, PGD2 is rapidly degraded either enzymatically or spontaneously to various PGD2 metabolites of the D, F and J series (Fig. 1) [12, 13]. The persistence of degradation products in plasma and urine may indicate a biological relevance either by preventing or prolonging in vivo receptor signaling [6, 14–16]. Enzymatic degradation of PGD2 leads to 13,14-dihydro-15-keto-PGD2 (DK-PGD2) which is a highly selective DP2 agonist [14, 15]. In plasma, PGD2 is mainly metabolized to ∆12-PGD2 and ∆12-PGJ2 which preferably bind to DP2 [12, 15]. Dehydration of PGD2 and its spontaneous degradation product 9-deoxy-PGD2 (PGJ2) lead to formation of 15-deoxy-∆12,14-PGD212,17 and 15-deoxy-∆12,14-PGJ2 [7, 17, 18], respectively, which accumulate in low concentrations and as well preferably bind to DP2 [15]. A major enzymatically derived product of the PGD2 metabolism in vivo is 9α,11β-PGF2 which is e.g. found in urine and plasma of asthmatics following allergen challenge [14, 19, 20]. Since most metabolites have selective agonistic properties for DP2 over DP1 they are considered to contribute to inflammatory DP2 signaling [15, 21].
Fig. 1.
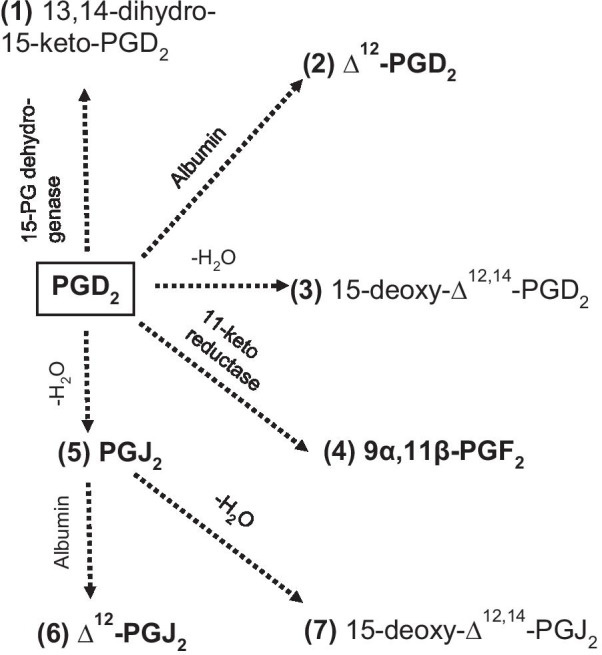
Selected metabolism of prostaglandin D2. Major metabolites are represented in bold. PGD2, metabolite 2 and 3 belong to the D-series while metabolites 5, 6 and 7 belong to the J-series of prostaglandins. Metabolite 4 belongs to the F-series of prostaglandins
Targeting the DP2 pathway is of significant interest for severe, uncontrolled asthmatic patients who do not or only partly respond to current available treatment options [1, 11, 22, 23]. Eosinophils and ILC2s are two major players in atopic asthma expressing DP2. PGD2 and metabolites activate eosinophils changing their cellular shapes [15, 18, 19], as well as inducing actin polymerization [21], CD11b expression [21] and migration [6, 12]. ILC2 numbers are increased in the airways of asthmatics and promote eosinophilia by secretion of type 2 cytokines [24, 25]. PGD2 induces interleukin 5 (IL-5) and IL-13 secretion, cell migration, cell aggregation as well as the expression of adhesion molecules in ILC2s in a DP2 dependent manner [26, 27]. Further, ILC2s produce endogenous PGD2 which is degraded to PGJ2, ∆12-PGJ2, 15-deoxy-∆12,14-PGJ2 and DK-PGD2 in culture supernatants [28].
Fevipiprant is a potent and selective DP2 antagonist that has shown therapeutic benefit in certain subsets of asthma patients in phase 2 clinical trials [29–31]. In a single center mechanistic phase 2 clinical trial in patients with persistent eosinophilic asthma, fevipiprant not only reduced airway inflammation, but also improved epithelial integrity and reduced airway smooth muscle mass [31, 32]. In two recently published phase 3 studies in severe asthmatics, although neither trial showed a statistically significant reduction in asthma exacerbations, consistent and modest reductions in exacerbations rates were observed in both studies with a high dose of fevipiprant [33]. In two Phase 3 studies in moderate asthmatics, no significant improvements were observed in lung function or other asthma related outcomes, such as daytime symptom score or quality of life [34]. Previously, the inhibitory effects of fevipiprant in primary human cells have been characterized with PGD2 activation [26, 35, 36].
Here, we demonstrate the potencies of seven PGD2 metabolites to induce ILC2 migration and IL-5 and IL-13 cytokine secretion. We also reproduce previously reported effects of the PGD2 metabolites on eosinophil shape change. ILC2 as well as eosinophil activation was then blocked with fevipiprant demonstrating the DP2 dependency of cell activation.
Methods
Study design
Fifteen atopic asthmatic volunteers (eight male/seven female; aged 18–65 years, average 34.5 ± 10.6 years; BMI 19–32 kg/m2, no oral steroids for > four weeks) were enrolled into the study. Subjects differed between eosinophil (n = 8) and ILC2 (n = 7) experiments and also between activation and inhibition experiments, respectively. Whole blood (200–500 ml) was collected in 3.8% trisodium citrate and was processed within one hour after blood withdrawal. Eosinophil levels in peripheral blood had to be > 0.15 × 106/ml for the eosinophil shape change.
Reagents
PGD2, 13,14-dihydro-15-keto-PGD2, PGJ2, Δ12-PGJ2, Δ12-PGD2, 15-deoxy-Δ12,14-PGJ2, 15-deoxy-Δ12,14-PGD2, 9α,11β-PGF2 were purchased from Cayman Chemicals (Biomol GmBH, Hamburg, Germany). Fevipiprant (GST0000013789) was provided by Novartis Pharma AG (Basel, Switzerland). Reagents were dissolved in sterile-filtered Hybri-Max dimethylsulfoxide (DMSO, Sigma-Aldrich, Taufkirchen, Germany).
Granulocyte isolation
Blood was stored on ice until processing. Granulocytes were isolated as described [18, 37]. In brief, 200 ml blood was diluted 1:3 in Dulbecco’s Phosphate Buffered Saline (DPBS). The blood suspension was incubated with 4% (w/v) dextran-T500 (dilution 5:1, VWR, Hannover, Germany) for 30 min on ice. The upper phase was layered on Ficoll-Paque® (Sigma-Aldrich, Taufkirchen, Germany) and centrifuged (25 min, 300×g, 18 °C). Granulocyte pellets each were resuspended in 500 µl DPBS and erythrocytes were lysed for 40 s with 20 ml ice cold, sterile, endotoxin-free distilled water. The reaction was stopped with 20 ml DPBS (2 × concentrated). After centrifugation (10 min, 300×g, 18 °C), cell pellets were washed with 50 ml DPBS. Granulocytes were resuspended in assay buffer (0.1% bovine serum albumin (BSA) in DPBS, Sigma-Aldrich, Taufkirchen, Germany) to a cell density of 6.25 × 106/ml.
Eosinophil shape change
Granulocytes (80 µl) and assay buffer (10 µl) were incubated in a water bath (5 min, 37 °C). Metabolite solutions (10 µl, final concentration (conc.) 0.01 nM, 0.1 nM, 0.5 nM, 1 nM, 5 nM, 10 nM, 100 nM, 1 µM, 1% DMSO in assay buffer) were added (5 min, 37 °C). The incubation was stopped with 250 µl of 0.25% BD Cell-Fix™ Solution (1:10 with sterile water followed by 1:4 with assay buffer, BD Biosciences, Heidelberg, Germany) and immediate placement on ice (≥ 5 min). Flow cytometric analysis was performed with a Navios 3/10 (Beckman Coulter). Granulocytes were determined by FSC and SSC properties. Eosinophils were discriminated from neutrophils by autofluorescence properties at 560 nm (FL2). 1000 eosinophils were acquired. Eosinophil shape change was calculated as percentage increase of the mean FSC units.
For DP2 inhibition experiments, granulocytes (80 µl) and fevipiprant (10 µl, final conc. 0.01 nM, 0.05 nM, 0.1 nM, 1 nM, 5 nM, 10 nM, 100 nM, 500 nM, 1 µM, 10 µM) were incubated in a water bath (5 min, 37 °C). Metabolite solution (10 µl, EC70 concentration) was added for 5 min (37 °C). The reaction was stopped and cells were analyzed as described above.
Type 2 innate lymphoid cell isolation
Blood was stored at RT until processing. Whole blood (500 ml) was diluted 1:2 in DPBS. PBMCs were isolated using Ficoll-Paque® and SepMate-50 PBMC Isolation tubes (STEMCELL Technologies, Grenoble, France) following manufacturer’s protocol. PBMCs were pooled, washed with 50 ml DPBS and centrifuged (5 min, 300×g, 4 °C). T cells, B cells and monocytes depletion enriched ILC2s using CD3, CD14 and CD19 MACS separation beads (Miltenyi Biotech, Bergisch Gladbach, Germany) and LD columns following manufacturer’s protocol. Enriched cells were centrifuged (5 min, 300×g, 4 °C) and resuspended in staining buffer containing 5% fetal bovine serum (FBS) and 2 mM ethylenediaminetetraacetic acid (EDTA) in DPBS. Cells were stained for 15 min at RT with a PerCP-Cy5.5-labeled lineage cocktail (CD4, CD8, CD14, CD16, CD19, CD34, CD123, FcεRI), CD11b-FITC, CD56-FITC, CD3-BV510, CD127-BV421, CD45-Alexa Fluor 700 and CD294-PE. View supplement for more detailed information. CD45+, Lineage-, CD11b-, CD56-, CD3-, CD127+, CD294+ cells were sorted with an FACS ARIA Fusion (BD Bioscience) into 96 U bottom well plates (Corning, Amsterdam, Netherlands).
Type 2 innate lymphoid cell culture
Sorted ILC2s (100 cells/well) were expanded with human feeder PBMCs (100,000/well; 37 °C, 5% CO2) for three to five weeks. Culture medium contained RPMI 1640 Glutamax medium (Life Technologies, Darmstadt, Germany), 1% Pen/Strep (Life Technologies, Darmstadt, Germany), 10% h.i. human AB serum (Sigma-Aldrich, Taufkirchen, Germany), and 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Lonza, Wakersville, USA). The medium was supplemented with 100 U/ml rh-IL-2 (Life Technologies, Darmstadt, Germany), 25 ng/ml rh-IL-4 (Miltenyi Biotech, Bergisch Gladbach, Germany), 5 µg/ml phytohemagglutinin-M (PHA-M; Sigma-Aldrich, Taufkirchen, Germany).
Cell migration assay
Migration was assessed using 5.0 µm pore size, polycarbonate membrane, polystyrene 96-transwell plates (Corning, Amsterdam, Netherlands; Hölzel, Köln, Germany) following manufacturer’s protocols. Metabolites (lower well, final conc. 5 nM, 10 nM, 50 nM, 100 nM, 500 nM, 1 µM, 2 µM, 3 µM and 5 µM) and cells (upper well, ≤ 100,000/well) were incubated for 6 h (37 °C, 5% CO2) in culture medium without IL-2, IL-4 and PHA. Migrated cells (25 µl) were incubated with CellTiter-Glo® Luminescence Cell Viability Assay (Promega, Mannheim, Germany) following manufacturer’s protocol. Luminescence was measured using a Tecan infinite F200 pro.
For DP2 inhibition experiments, ILC2s were incubated with fevipiprant (final conc. 0.01 nM, 0.1 nM, 0.5 nM, 1 nM, 5 nM, 10 nM, 100 nM, 1 µM, 10 µM, for 1 h, 37 °C, 5% CO2) in culture medium without IL-2, IL-4 and PHA and cell migration was measured as described above using the EC70 concentration of respective metabolites.
Cytokine measurement
Cells (≤ 150,000/well) and metabolite solutions (final conc. 2.5 nM, 5 nM, 25 nM, 50 nM, 250 nM, 500 nM, 1 µM, 1.5 µM, 2.5 µM) were incubated for 24 h in culture medium without IL-2, IL-4 and PHA (U-bottom 96-well plates, 37 °C, 5% CO2). After centrifugation (5 min, 300×g, RT), the supernatant was collected and stored at − 80 °C until measurement.
For DP2 inhibition experiments, ILC2s were incubated with fevipiprant (final conc. 0.01 nM, 0.1 nM, 0.5 nM, 1 nM, 5 nM, 10 nM, 50 nM, 100 nM, 1 µM) for 1 h (37 °C, 5% CO2) and afterwards with the EC70 concentration of respective metabolites for 24 h. Cells were incubated as described above.
Supernatants were diluted 1:10 and IL-5 and IL-13 concentrations were measured by MSD immunoassays (V-PLEX Meso Dale Discovery, Rockville, MD, USA) following manufacturer’s protocol. Raw values are depicted in Additional file 1: Figs. S2 and S3.
Statistics
GraphPad Prism 9.0.1 was used for analysis. Four parameter non-linear agonist or inhibitor regression models were used fit curves and to calculate EC50/IC50 values, respectively. Fitting curves constraints are indicated in respective figure legends.
Maximal PGD2-induced responses were compared with respective maximal responses of metabolites using paired One-Way ANOVA with post-hoc Dunn’s multiple comparisons tests.
Results
PGD2 metabolites induce eosinophil shape change with similar potencies
PGD2 and seven PGD2 metabolites were evaluated for their ability to induce activation of eosinophils by measurement of cellular shape changes. All prostaglandins induced a concentration dependent shape change with nanomolar potency except for 9α,11β-PGF2 which was less potent (Additional file 1: Fig. S1). EC50 values of prostaglandins, described as mean ± standard error of the mean (SEM), fell into following rank order: PGD2: 0.7 ± 0.2 nM < Δ12-PGD2: 1.2 ± 1.8 nM < 15-deoxy-Δ12,14-PGD2: 1.5 ± 1.6 nM < PGJ2: 1.6 ± 3.8 nM < DK-PGD2: 2.7 ± 2.3 nM < Δ12-PGJ2: 5.6 ± 1.0 nM < 15-deoxy-Δ12,14-PGJ2: 12.0 ± 0.7 nM < 9α,11β-PGF2: > 1000. Similar EC50 values were reported by Sandig et al. but with different rank order (Table 1) [19].
Table 1.
EC50 values of PG-induced eosinophil shape change were calculated and are given as mean ± standard error of the mean (SEM), n = 2–3
| Eosinophil shape change EC50 [nM] | ||
|---|---|---|
| Metabolite | Measured values | Sandig et al. [19] |
| PGD2 | 0.7 ± 0.2 | 0.4 |
| DK-PGD2 | 2.7 ± 2.3 | 1.1 |
| ∆12-PGD2 | 1.2 ± 1.8 | 7.3 |
| 15-deoxy-∆12,14-PGD2 | 1.5 ± 1.6 | 2.4 |
| 9α,11β-PGF2 | > 1000 | 156.0 |
| PGJ2 | 1.6 ± 3.8 | 2.2 |
| ∆12-PGJ2 | 5.6 ± 1.0 | 3.7 |
| 15-deoxy-∆12,14-PGJ2 | 12.0 ± 0.7 | 8.4 |
Published reference values are given in the right column
Metabolites induced shape changes with similar maximal responses as compared to PGD2, which was observed by a comparison of FSCmax values normalized on 100% PGD2 shape change. Activation responses of PGD2 metabolites ranged between 98.0 and 102.9%, compared to the parent PGD2 (Fig. 2). Sandig et al. did not compare maximal responses but their results indicate similar responses for most metabolites as well while responses of Δ12-PGD2 and Δ12-PGJ2 seemed to have higher FCS values.
Fig. 2.
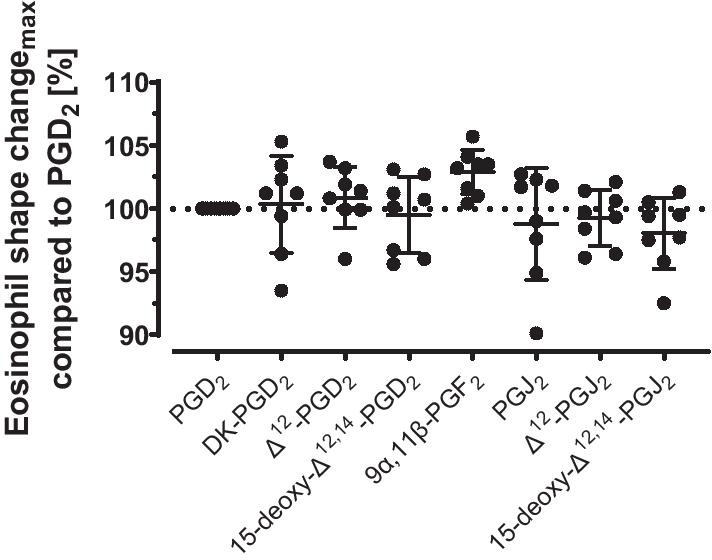
Comparison of maximal eosinophil shape change in the presence of PGD2 and selected metabolites. The mean forward scatter values in the presence of the highest PGD2 concentration was normalized to 100% shape change, n = 6–8. Values are given as mean ± SD
PGD2 metabolites induce ILC2 migration and IL-5 and IL-13 cytokine secretion
Next, PGD2 metabolites were evaluated for their potential to induce ILC2 migration and Type 2 cytokine secretion. The metabolites induced cell migration concentration dependently and with nanomolar potency except for 9α,11β-PGF2. 9α,11β-PGF2 increased migration in comparison to the medium control but dose independently (Fig. 3A). Migratory potencies were increased for the D-series of metabolites in comparison to the J-series with mean EC50 ± SEM values of PGD2 17.4 ± 3.9 nM, DK-PGD2 14.2 ± 3.4 nM, ∆12-PGD2 19.3 ± 3.2 nM, 15-deoxy-∆12,14-PGD2 21.8 ± 6.3 nM, PGJ2 66.3 ± 7.0 nM, ∆12-PGJ2 91.7 ± 9.2 nM and 15-deoxy-∆12,14-PGJ2 38.1 ± 5.4 nM (Table 2). Micromolar metabolite concentrations abolished cell migration.
Fig. 3.

A ILC2 migration in the presence of ascending concentrations of PGD2 and selected metabolites, n = 3. Values are given as mean ± SD. Nonlinear curve fit with constraints of bottom constant equal to zero and top constant equal to 100 was applied to derive EC50 and EC70. B Maximal cell migration of PGD2 and selected metabolites. Absolute numbers of migrated cells were compared and normalized to 100% PGD2, n = 7. Values are given as mean ± SD. DK-PGD2: ns, ∆12-PGD2: ns, 15-deoxy-∆12,14-PGD2: p = 0.05, 9α,11β-PGF2: ns, PGJ2: ns, ∆12-PGJ2: p = 0.03 and 15-deoxy-∆12,14-PGJ2: p = 0.01. *p < 0.05
Table 2.
EC50 values of PG-induced ILC2 migration and IL-5 and IL-13 cytokine secretion were calculated and are given as mean ± standard error of the mean (SEM), n = 3
| ILC2 migration and cytokine secretion EC50 [nM] | |||
|---|---|---|---|
| Metabolite | Migration | IL-5 secretion | IL-13 secretion |
| PGD2 | 17.4 ± 3.9 | 139.2 ± 15.5 | 131.9 ± 10.8 |
| DK-PGD2 | 14.2 ± 3.4 | 146.0 ± 18.0 | 159.2 ± 28.8 |
| ∆12-PGD2 | 19.3 ± 3.2 | 108.1 ± 11.8 | 125.2 ± 8.3 |
| 15-deoxy-∆12,14-PGD2 | 21.8 ± 6.3 | 175.4 ± 26.4 | 159.1 ± 13.9 |
| 9α,11β-PGF2 | –a | 526.9 ± 12,926.7 | 788.3 ± 173.7 |
| PGJ2 | 66.3 ± 7.0 | 244.4 ± 26.3 | 321.9 ± 43.8 |
| ∆12-PGJ2 | 91.7 ± 9.2 | 234.1 ± 30.6 | 343.2 ± 52.4 |
| 15-deoxy-∆12,14-PGJ2 | 38.1 ± 5.4 | 185.3 ± 21.5 | 226.2 ± 35.2 |
aCurve fit was ambiguous, EC50 value could not be determined
The maximal migration response measured by total number of migrated cells compared to PGD2 was increased for ∆12-PGD2, 15-deoxy-∆12,14-PGD2, PGJ2, ∆12-PGJ2 and 15-deoxy-∆12,14-PGJ2. In contrast DK-PGD2 and 9α,11β-PGF2 showed a non-significant trend to reduced maximal migration responses, (Fig. 3B).
IL-5 and IL-13 cytokine secretion of ILC2s was induced by PGD2 metabolites in a concentration dependent manner. ILC2 responses were comparable between IL-5 and IL-13 (Fig. 4A). Similar to cell migration, higher potencies for cytokine secretion were found for the D-series of metabolites compared to the J-series (Table 2). 9α,11β-PGF2 was able to induce cytokine secretion although less potent than the other metabolites and with high variability. The maximal cytokine secretion response was significantly lower in the presence of 9α,11β-PGF2 and PGJ2 compared to PGD2 while the maximal response was similar for DK-PGD2, ∆12-PGJ2 and 15-deoxy-∆12,14-PGJ2. Cytokine secretion was non-significantly enhanced for ∆12-PGD2 and 15-deoxy-∆12,14-PGD2 compared to PGD2 (Fig. 4B).
Fig. 4.

A IL-5 (white circles) and IL-13 (black squares) cytokine secretion of ILC2s in the presence of ascending concentrations of PGD2 and selected metabolites, n = 3. Values are given as mean ± SD. Nonlinear curve fit with constraints of bottom constant equal to zero and top constant equal to 100 was applied to derive EC50 and EC70. B Maximal cytokine secretion of selected metabolites compared to PGD2. The absolute concentrations of secreted cytokines were compared and normalized to 100% PGD2, n = 6. Values are given as mean ± SD. 9α,11β-PGF2: IL-5 p = 0.006, IL-13 p = 0.02, PGJ2: IL-5 p = 0.03, IL-13 p = 0.007. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Selective DP2 inhibition of eosinophils and ILC2s
Isolated whole blood eosinophils were incubated with the selective DP2 antagonist fevipiprant prior to activation of cells with EC70 concentrations of PGD2 or respective metabolites (supplementary table 2). Fevipiprant inhibited eosinophil shape changes in dose dependent manners with sub-nanomolar potencies (Fig. 5A and B). IC50 values against metabolites were comparable and ranged between 0.1 and 0.9 nM for PGD2 and six metabolites (Table 3). Eosinophil shape changes induced by 15-deoxy-∆12,14-PGJ2 could be inhibited by fevipiprant, however, the dose–response was not sigmoidal.
Fig. 5.

Inhibition of PGD2 and metabolite-induced cell activation with DP2 inhibitor fevipiprant. Cells were incubated with ascending concentrations of fevipiprant and stimulated with EC70 concentrations of PGD2, DK-PGD2, ∆12-PGD2, 15-deoxy-∆12,14-PGD2, PGJ2, 9a,11b-PGF2, ∆12-PGJ2 and 15-deoxy-∆12,14-PGJ2, respectively. A, B Inhibition of eosinophil shape change, n = 3–5. Top constraints equal 100. C, D Inhibition of ILC2 migration, n = 4. Top constraints equal 100. E, F Inhibition of ILC2 IL-5 secretion, n = 4. Bottom constraints equal zero. (G,H) Inhibition of ILC2 IL-13 secretion, n = 4. Bottom constraints equal zero. Values are given as mean ± SEM
Table 3.
IC50 values of fevipiprant-inhibited eosinophil shape change, ILC2 migration and IL-5 and IL-13 cytokine secretion of ILC2s
| IC50 fevipiprant [nM] | ||||
|---|---|---|---|---|
| Metabolite | shape change (n = 3–5) | Migration (n = 4) | IL-5 (n = 4) | IL-13 (n = 4) |
| PGD2 | 0.9 ± 0.2 | 2.4 ± 0.6 | 4.2 ± 0.8 | 5.1 ± 1.1 |
| DK-PGD2 | 0.1 ± 0.0 | 1.9 ± 0.7 | 6.5 ± 1.6 | 8.5 ± 2.1 |
| ∆12-PGD2 | 0.1 ± 0.0 | 4.9 ± 1.3 | 4.8 ± 1.2 | 4.8 ± 1.3 |
| 15-deoxy-∆12,14-PGD2 | 0.5 ± 0.3 | 4.6 ± 2.0 | 4.5 ± 1.7 | 4.6 ± 1.4 |
| 9α,11β-PGF2 | 0.9 ± 0.1 | 10.9 ± 7.8 | –a | –a |
| PGJ2 | 0.5 ± 0.2 | 3.7 ± 0.6 | 4.0 ± 1.0 | 5.9 ± 3.6 |
| ∆12-PGJ2 | 0.6 ± 0.3 | 3.1 ± 0.5 | 3.7 ± 0.9 | 4.6 ± 1.6 |
| 15-deoxy-∆12,14-PGJ2 | –a | 3.8 ± 0.8 | 2.3 ± 0.9 | 2.6 ± 0.9 |
Values were calculated from 3 to 5 experiments and are given as mean ± standard error of the mean (SEM)
aCurve fit was ambiguous, IC50 values could not be determined
Similar to eosinophil activation, ILC2 migration (Fig. 5C and D) and cytokine secretion (Fig. 5E–H) could be inhibited by fevipiprant in a concentration dependent manner. In comparison to eosinophils, ILC2 related IC50 values were approximately one order of magnitude higher. Migration IC50 values ranged between 1.9 and 10.9 nM, IL-5 IC50 values ranged between 2.3 and 6.5 nM, and IL-13 IC50 values ranged between 2.6 and 8.5 nM (Table 3). 9a,11b-PGF2 induced cytokine secretion results were highly variable among donors and IC50 values could not be determined.
Discussion
In vivo and in vitro, PGD2 is rapidly degraded to various metabolites which presumably contribute to DP2 immune cell activation in large part due to their DP2 selectivity [15, 38]. Here, we show for the first time that ILC2s respond to PGD2 metabolites by migration and IL-5 and IL-13 secretion. We determined respective EC50 values, quantified cellular responses of eosinophils and ILC2s and compared the activation maximal responses with respective PGD2 responses. DP2 dependency was demonstrated using fevipiprant which abolished cell activities with nanomolar potencies.
The influence of prostaglandin metabolites on eosinophil cell activation and shape change is well known. Sandig et al. first reported EC50 values, which we could largely confirm [19]. EC50 concentrations resulted in different rank orders of potencies compared to ours, however, those differences were negligibly small. The potency of 9α,11β-PGF2 was clearly reduced in both studies compared to other metabolites. Since 9α,11β-PGF2 has an approximately 130 fold reduced binding affinity to DP2 compared to PGD2 [38], it can be assumed that the concentrations used were too low to sufficiently induce a shape changes in eosinophils. Merely concentrations of 1 µM demonstrated a shift in FCS properties (Additional file 1: Fig. S1). Comparing the maximal changes in the forward scatters indicated that all metabolites induced shape changes with similar maximal responses. This may imply their comparable agonistic function on eosinophil activation. This finding could be supported by further eosinophil activation data addressing e.g. cell surface CD11b expression or degranulation.
The selective relevance of DP2 for eosinophil activation has been reported for PGD2 and DK-PGD2 using various DP2 antagonists [19, 39, 40]. Nevertheless, blocking the activation of other metabolites was described only using the dual DP2 and thromboxane receptor (TP) antagonist ramatroban [19]. We complemented data affirming that metabolites induced eosinophil shape changes via DP2 selectively using fevipiprant [41].
Less is known about the role of PGD2 metabolites on ILC2 activation. Recently it was shown that ILC2s produce endogenous PGD2 upon alarmin activation which was metabolized to PGJ2, ∆12-PGJ2, DK-PGD2 and 15-deoxy-∆12,14-PGJ2 in culture supernatants [28]. However, so far only exogeneous and endogeneous PGD2 was reported to induce migration and IL-5 and IL-13 secretion in ILC2s [26–28]. Here, we show that ILC2s also migrate and release Type 2 cytokines in presence of DK-PGD2, PGJ2, Δ12-PGJ2, Δ12-PGD2, 15-deoxy-Δ12,14-PGJ2, and 15-deoxy-Δ12,14-PGD2. D-series metabolites were more potent than those of the J-series. 9α,11β-PGF2 was not able to induce ILC2 migration in the concentration range used. The comparably low binding affinity of 9α,11β-PGF2 to DP2 might have resulted in the low response. Varying activating potencies among the other metabolites might be related to differences in respective DP2 binding affinities as well [15, 38], however, EC50 rank orders of shape change, migration and cytokine release experiments were different. Moreover, quantifying the maximal assay responses among metabolites revealed that ILC2 cell migration was enhanced in presence of most metabolites compared to PGD2 while cytokine production was not enhanced. Interestingly, cell migration was strongest for the J-series metabolites Δ12-PGJ2, 15-deoxy-Δ12,14-PGJ2 and cytokine secretion was strongest for the D-series metabolites Δ12-PGD2, 15-deoxy-Δ12,14-PGD2. Our data may indicate that PGD2 metabolites contribute predominantly to ILC2 migration rather than cytokine secretion which needs to be confirmed in vivo. Although it is known that numbers of eosinophils and ILC2s are elevated in the airways of severe asthmatic patients [24] little is known about local distributions of PGD2 metabolite concentrations in human tissues. ILC2 numbers and PGD2 concentrations in the bronchoalveolar lavage (BAL) fluid were reported to correlate after allergen challenge of mild asthmatics [25]. Moreover, metabolic products of PGD2 were found in human serum [42], plasma, BAL [43] and urine [44, 45] samples, however, their contribution to the asthmatic disease is poorly understood [20]. Studies on eosinophils in mice showed that ∆12-PGJ2 mobilizes eosinophils from the bone marrow and facilitates cell extravasation [18] which fits with the observation that PGD2 is preferably converted to ∆12-PGD2 and ∆12-PGJ2 in blood plasma [12]. The contribution of metabolites to cell recruitment might be of biological relevance but remains speculative without further experiments. 9α,11β-PGF2 is another major degradation products found in human and showed low agonistic potency on eosinophils and ILC2s [42, 44, 46]. At micromolar concentrations, 9α,11β-PGF2 was able to activate cells, which probably do not represent physiological concentrations of the metabolite. Further, quantification of ILC2 responses indicated a reduced maximal response compared to PGD2. Therefore, it seems more likely that 9α,11β-PGF2 does not contribute to DP2 signaling in vivo but rather serves as an signal-inactivating degradation product.
Blocking with fevipiprant confirmed the DP2 dependency of ILC2 activities which were comparable to previous experiments performed with PGD2 [26]. Inhibition of 15-deoxy-Δ12,14-PGJ2 activation in eosinophils followed a non-sigmoidal dose–response principle which should be assessed more closely in future studies. Inhibiting PGD2 signaling has been studied in a wide range of clinical studies due to its key role in allergic inflammation. While fevipiprant itself is no longer being developed for asthma, our findings may be of relevance to other DP2 antagonists which remain under clinical investigation [22, 47].
Conclusion
PGD2 metabolites are effective DP2 agonists and promote eosinophil shape change, ILC2 cell migration and Type 2 cytokine secretion in vitro. They might contribute to ILC2 recruitment in vivo since predominantly ILC2 migration but not ILC2 cytokine secretion or eosinophil shape change was enhanced compared to PGD2.
Supplementary Information
Additional file 1: Table S1. Detailed information on flow cytometric antibodies used for cell sorting of ILC2s. Table S2. Calculated agonist EC70 values for PGD2 and seven selected PGD2 metabolites. Table S3. Characteristics of study subjects. All subjects had a history of allergic asthma since at least 12 months. BMI had to be between 19 to 32 kg/ m2. Figure S1. Eosinophil shape change induced by PGD2, DK-PGD2, ∆12-PGD2, 15-deoxy-∆12,14-PGD2, PGJ2, 9a,11b-PGF2, ∆12-PGJ2 and 15-deoxy-∆12,14-PGJ2. Granulocytes were isolated from whole blood of asthmatic patients and were incubated with increasing concentrations of metabolites, n=3, 9α,11β-PGF2: n=2. The mean fluorescence values of the forward scatter were determined by flow cytometry and the percentage of shape change above basal was calculated. Values are given as mean ± SD. Figure S2. Concentration of IL-5 cytokine secretion of ILC2s in the presence of ascending concentrations of PGD2 and selected metabolites, n=3. Values are given for three different subjects (circle, square and triangle). Figure S3. Concentration of IL-13 cytokine secretion of ILC2s in the presence of ascending concentrations of PGD2 and selected metabolites, n=3. Values are given for three different subjects (circle, square and triangle).
Acknowledgements
The authors would like to thank Professor Luzheng Xue (University of Oxford) for his support and help as well as the clinical staff of Fraunhofer ITEM for subject recruitment.
Abbreviations
- BAL
Bronchoalveolar lavage
- BMI
Body mass index
- BSA
Bovine serum albumin
- CD
Cluster of differentiation
- Conc.
Concentration
- CRTH2
Chemoattractant receptor-homologous molecule expressed on Th2 cells
- DK-PGD2
13,14-Dihydro-15-keto-prostaglandin D2
- DMSO
Dimethylsulfoxide
- DP
D-prostanoid receptor
- DPBS
Dulbecco’s Phosphate Buffered Saline
- EC50
Half maximal effective concentration
- EDTA
Ethylenediaminetetraacetic acid
- FSC
Forward scatter
- HEPES
4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid
- IC50
Half maximal inhibitory concentration
- IL
Interleukin
- ILC2
Type 2 innate lymphoid cells
- MFI
Mean fluorescence intensity
- PG
Prostaglandin
- PHA-M
Phytohemagglutinin-M
- SD
Standard deviation
- SEM
Standard error of mean
- SSC
Sideward scatter
Authors' contributions
All authors added and revised the manuscript. SC: Data curation, formal analysis, methodology, visualization, writing original draft, writing review and editing; CG: Data curation, formal analysis, methodology, visualization, writing review and editing; VJE: Conceptualization, project administration, writing review and editing; SDK: Conceptualization, project administration, writing review and editing; JMH: Conceptualization, writing review and editing; DAS: Conceptualization, project administration, writing review and editing; MM: Conceptualization, writing review and editing. All authors read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was supported by Novartis Pharma AG and was performed under a collaboration agreement between Novartis and Fraunhofer ITEM.
Availability of data and materials
The datasets supporting the conclusion of this article are available on reasonable request from MM and DAS.
Declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Hannover Medical School (7882_BO_S_2018). All subjects gave written informed consent.
Consent for publication
Not applicable.
Competing interests
SC’s, CG’s, JMH’s and MM’s institution received funding from ALK, Allergopharma, Astellas, AstraZeneca, GlaxoSmithKline, Janssen Pharmaceutica NV, LETI, Novartis, and Sanofi-Aventis for clinical trial conduct outside the submitted work. JMH reports personal fees from Boehringer Ingelheim, CSL Behring, HAL, Merck, and Novartis for consultancy and lectures outside the submitted work. VJE and SK were employees of Novartis at the time of the study. DAS is an employee of Novartis.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Saskia Carstensen and Christina Gress have contributed equally
References
- 1.Kupczyk M, Kuna P. Targeting the PGD2/CRTH2/DP1 signaling pathway in asthma and allergic disease current status and future perspectives. Drugs. 2017;77:1281–1294. doi: 10.1007/s40265-017-0777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balzar S, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183:299–309. doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Domingo C, Palomares O, Sandham DA, Erpenbeck VJ, Altman P. The prostaglandin D2 receptor 2 pathway in asthma a key player in airway inflammation. Respir Res. 2018;19:189. doi: 10.1186/s12931-018-0893-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.García-Solaesa V, et al. The prostaglandin D2 receptor (PTGDR) gene in asthma and allergic diseases. Allergol Immunopathol. 2014;42:64–68. doi: 10.1016/j.aller.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Kostenis E, Ulven T. Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med. 2006;12:148–158. doi: 10.1016/j.molmed.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Hirai H, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. 2001;98:1942–1948. doi: 10.1182/blood.V98.6.1942. [DOI] [PubMed] [Google Scholar]
- 8.Mjösberg JM, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 9.Gervais FG, et al. Selective modulation of chemokinesis, degranulation, and apoptosis in eosinophils through the PGD2 receptors CRTH2 and DP. J Allergy Clin Immunol. 2001;108:982–988. doi: 10.1067/mai.2001.119919. [DOI] [PubMed] [Google Scholar]
- 10.Stubbs VEL, et al. Indomethacin causes prostaglandin D(2)-like and eotaxin-like selective responses in eosinophils and basophils. J Biol Chem. 2002;277:26012–26020. doi: 10.1074/jbc.M201803200. [DOI] [PubMed] [Google Scholar]
- 11.Peinhaupt M, Sturm EM, Heinemann A. Prostaglandins and their receptors in eosinophil function and as therapeutic targets. Front Med. 2017 doi: 10.3389/fmed.2017.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuligoi R, et al. PGD2 metabolism in plasma: kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem Pharmacol. 2007;74:107–117. doi: 10.1016/j.bcp.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki F, Hayashi H, Hayashi O. Transport of prostaglandin D2 into brain. Brain Res. 1986;385:321–328. doi: 10.1016/0006-8993(86)91079-6. [DOI] [PubMed] [Google Scholar]
- 14.Giles H, Leff P. The biology and pharmacology of PGD2. Prostaglandins. 1988;35:277–300. doi: 10.1016/0090-6980(88)90093-7. [DOI] [PubMed] [Google Scholar]
- 15.Gazi L, et al. Δ12-prostaglandin D2 is a potent and selective CRTH2 receptor agonist and causes activation of human eosinophils and Th2 lymphocytes. Prostaglandins Other Lipid Mediat. 2005;75:153–167. doi: 10.1016/j.prostaglandins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Pettipher R, Hansel TT, Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev. 2007;6:313–325. doi: 10.1038/nrd2266. [DOI] [PubMed] [Google Scholar]
- 17.Fitzpatrick FA, Wynalda MA. Albumin-catalyzed metabolism of prostaglandin D2. Identification of products formed in vitro. J Biol Chem. 1983;258:11713–11718. doi: 10.1016/S0021-9258(17)44287-6. [DOI] [PubMed] [Google Scholar]
- 18.Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Delta 12-prostaglandin J2, a plasma metabolite of prostaglandin D2, causes eosinophil mobilization from the bone marrow and primes eosinophils for chemotaxis. J Immunol. 2003;170:4752–4758. doi: 10.4049/jimmunol.170.9.4752. [DOI] [PubMed] [Google Scholar]
- 19.Sandig H, Andrew D, Barnes AA, Sabroe I, Pease J. 9α,11β-PGF2 and its stereoisomer PGF2α are novel agonists of the chemoattractant receptor, CRTH2. FEBS Lett. 2006;580:373–379. doi: 10.1016/j.febslet.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 20.Bochenek G, Nagraba K, Nizankowska E, Szczeklik A. A controlled study of 9α,11β-PGF2 (a prostaglandin D2 metabolite) in plasma and urine of patients with bronchial asthma and healthy controls after aspirin challenge. J Allergy Clin Immunol. 2003;111:743–749. doi: 10.1067/mai.2003.1387. [DOI] [PubMed] [Google Scholar]
- 21.Monneret G, Li H, Vasilescu J, Rokach J, Powell WS. 15-Deoxy-Δ12,14-prostaglandins D2 and J2 are potent activators of human eosinophils. J Immunol. 2002;168:3563–3569. doi: 10.4049/jimmunol.168.7.3563. [DOI] [PubMed] [Google Scholar]
- 22.Singh D, Ravi A, Southworth T. CRTH2 antagonists in asthma: current perspectives. Clin Pharmacol. 2017;9:165–173. doi: 10.2147/CPAA.S119295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kerstjens HAM, Gosens R. Prostaglandin D2: the end of a story or just the beginning? Lancet Respirat Med. 2020;20:2213–2600. doi: 10.1016/S2213-2600(20)30449-5. [DOI] [PubMed] [Google Scholar]
- 24.Smith SG, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137:75–86. doi: 10.1016/j.jaci.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 25.Winkler C, et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol. 2019;144:61–69. doi: 10.1016/j.jaci.2019.01.027. [DOI] [PubMed] [Google Scholar]
- 26.Hardman C, et al. Fevipiprant, a selective prostaglandin D2 receptor 2 antagonist, inhibits human group 2 innate lymphoid cell aggregation and function. J Allergy Clin Immunol. 2019;143:2326–2329. doi: 10.1016/j.jaci.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Xue L, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133:1184–1194. doi: 10.1016/j.jaci.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maric J, et al. Cytokine-induced endogenous production of prostaglandin D2 is essential for human group 2 innate lymphoid cell activation. J Allergy Clin Immunol. 2019;143:2202–2214. doi: 10.1016/j.jaci.2018.10.069. [DOI] [PubMed] [Google Scholar]
- 29.Erpenbeck VJ, et al. The oral CRTh2 antagonist QAW039 (fevipiprant): a phase II study in uncontrolled allergic asthma. Pulmonary Pharmacol Therap. 2016;39:54–63. doi: 10.1016/j.pupt.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Bateman ED, et al. Fevipiprant, an oral prostaglandin DP2 receptor (CRTh2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur Respir J. 2017 doi: 10.1183/13993003.00670-2017. [DOI] [PubMed] [Google Scholar]
- 31.Gonem S, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4:699–707. doi: 10.1016/S2213-2600(16)30179-5. [DOI] [PubMed] [Google Scholar]
- 32.Saunders R, et al. DP2 antagonism reduces airway smooth muscle mass in asthma by decreasing eosinophilia and myofibroblast recruitment. Sci Transl Med. 2019 doi: 10.1126/scitranslmed.aao6451. [DOI] [PubMed] [Google Scholar]
- 33.Brightling CE, et al. Effectiveness of fevipiprant in reducing exacerbations in patients with severe asthma (LUSTER-1 and LUSTER-2): two phase 3 randomised controlled trials. Lancet Respir Med. 2020;20:2213–2600. doi: 10.1016/S2213-2600(20)30412-4. [DOI] [PubMed] [Google Scholar]
- 34.Castro M, et al. Efficacy and safety of fevipiprant in patients with uncontrolled asthma: two replicate, phase 3, randomised, double-blind, placebo-controlled trials (ZEAL-1 and ZEAL-2) EClinicalMedicine. 2021;35:100847. doi: 10.1016/j.eclinm.2021.100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sykes DA, et al. Fevipiprant (QAW039), a slowly dissociating CRTh2 antagonist with the potential for improved clinical efficacy. Mol Pharmacol. 2016;89:593–605. doi: 10.1124/mol.115.101832. [DOI] [PubMed] [Google Scholar]
- 36.Xue L, et al. Fevipiprant inhibits DP2 mediated upregulation of tissue remodeling genes and autocrine prostaglandin D2 production in Tc2 cells. Am J Respir Crit Care Med. 2020;201:1. doi: 10.1164/rccm.201909-1741ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabroe I, et al. Differential regulation of eosinophil chemokine signaling via CCR3 and non-CCR3 pathways. J Immunol. 1999;162:2946–2955. [PubMed] [Google Scholar]
- 38.Sawyer N, et al. Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br J Pharmacol. 2002;137:1163–1172. doi: 10.1038/sj.bjp.0704973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy S, et al. GB001 potently inhibits PGD2 metabolite-induced DP2-mediated cell signaling and eosinophil activation. Eur Respir J. 2020;56:2902. [Google Scholar]
- 40.Pettipher R, et al. Pharmacologic profile of OC000459, a potent, selective, and orally active D prostanoid receptor 2 antagonist that inhibits mast cell-dependent activation of T helper 2 lymphocytes and eosinophils. J Pharmacol Exp Therap. 2012;340:473–482. doi: 10.1124/jpet.111.187203. [DOI] [PubMed] [Google Scholar]
- 41.Sandham DA, et al. Discovery of fevipiprant (NVP-QAW039), a potent and selective DP2 receptor antagonist for treatment of asthma. ACS Med Chem Lett. 2017;8:582–586. doi: 10.1021/acsmedchemlett.7b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nassiri M, Eckermann O, Babina M, Edenharter G, Worm M. Serum levels of 9α,11β-PGF2 and cyseinyl leukotrienes are useful biomarkers for anaphylaxis. J Allergy Clin Immunol. 2016;137:312–314. doi: 10.1016/j.jaci.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 43.Mastalerz L, et al. Induced sputum eicosanoids during aspirin bronchial challenge of asthmatic patients with aspirin hypersensitivity. Allergy. 2014;69:1550–1559. doi: 10.1111/all.12512. [DOI] [PubMed] [Google Scholar]
- 44.Liston TE, Roberts LJ. Metabolic fate of radiolabeled prostaglandin D2 in a normal human male volunteer. J Biol Chem. 1985;260:13172–13180. doi: 10.1016/S0021-9258(17)38853-1. [DOI] [PubMed] [Google Scholar]
- 45.Kolmert J, Dahlén S-E, Wheelock CE. Reply to Thomson: Exposure to active and passive tobacco smoke on urinary eicosanoid metabolites in type 2 asthma. Am J Respir Critic Care Med. 2021;203:1204–1205. doi: 10.1164/rccm.202101-0208LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liston TE, Jackson Roberts II L. Transformation of prostaglandin D2 to 9α,11β-(15S)-trihydroxyprosta-(5Z, 13E)-dien-1-oic acid (9α,11β-prostaglandin F2): a unique biologically active prostaglandin produced enzymatically in vivo in humans. Proc Natl Acad Sci USA. 1985;82:6030–6034. doi: 10.1073/pnas.82.18.6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asano K, et al. A Phase 2a Study of DP2 Antagonist GB001 for Asthma. J Allergy Clin Immunol. 2020;8:1275–1283. doi: 10.1016/j.jaip.2019.11.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Detailed information on flow cytometric antibodies used for cell sorting of ILC2s. Table S2. Calculated agonist EC70 values for PGD2 and seven selected PGD2 metabolites. Table S3. Characteristics of study subjects. All subjects had a history of allergic asthma since at least 12 months. BMI had to be between 19 to 32 kg/ m2. Figure S1. Eosinophil shape change induced by PGD2, DK-PGD2, ∆12-PGD2, 15-deoxy-∆12,14-PGD2, PGJ2, 9a,11b-PGF2, ∆12-PGJ2 and 15-deoxy-∆12,14-PGJ2. Granulocytes were isolated from whole blood of asthmatic patients and were incubated with increasing concentrations of metabolites, n=3, 9α,11β-PGF2: n=2. The mean fluorescence values of the forward scatter were determined by flow cytometry and the percentage of shape change above basal was calculated. Values are given as mean ± SD. Figure S2. Concentration of IL-5 cytokine secretion of ILC2s in the presence of ascending concentrations of PGD2 and selected metabolites, n=3. Values are given for three different subjects (circle, square and triangle). Figure S3. Concentration of IL-13 cytokine secretion of ILC2s in the presence of ascending concentrations of PGD2 and selected metabolites, n=3. Values are given for three different subjects (circle, square and triangle).
Data Availability Statement
The datasets supporting the conclusion of this article are available on reasonable request from MM and DAS.